
Abstract
In a registry-based analysis of 135 patients with “myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions” (MLN-TK; FIP1L1::PDGFRA, n = 78; PDGFRB, diverse fusions, n = 26; FGFR1, diverse, n = 9; JAK2, diverse, n = 11; ETV6::ABL1, n = 11), we sought to evaluate the disease-defining characteristics. In 81/135 (60%) evaluable patients, hypereosinophilia (>1.5 × 109/l) was observed in 40/44 (91%) FIP1L1::PDGFRA and 7/7 (100%) ETV6::ABL1 positive patients but only in 13/30 (43%) patients with PDGFRB, FGFR1, and JAK2 fusion genes while 9/30 (30%) patients had no eosinophilia. Monocytosis >1 × 109/l was identified in 27/81 (33%) patients, most frequently in association with hypereosinophilia (23/27, 85%). Overall, a blast phase (BP) was diagnosed in 38/135 (28%) patients (myeloid, 61%; lymphoid, 39%), which was at extramedullary sites in 18 (47%) patients. The comparison between patients with PDGFRA/PDGFRB vs. FGFR1, JAK2, and ETV6::ABL1 fusion genes revealed a similar occurrence of primary BP (17/104, 16% vs. 8/31 26%, p = 0.32), a lower frequency (5/87, 6% vs. 8/23, 35%, p = 0.003) of and a later progression (median 87 vs. 19 months, p = 0.053) into secondary BP, and a better overall survival from diagnosis of BP (17.1 vs. 1.7 years, p < 0.0008). We conclude that hypereosinophilia with or without monocytosis and various phenotypes of BP occur at variable frequencies in MLN-TK.
Similar content being viewed by others
Introduction
The recently published World Health Organization (WHO) 2022 classification and the International Consensus Classification of Myeloid and Lymphoid Neoplasms (ICC-MLN) define a distinct subcategory of myeloid neoplasms as “myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions” (MLN-TK) [1, 2]. This category name has changed from the previous “myeloid/lymphoid neoplasms with eosinophilia and rearrangement of PDGFRA, PDGFRB or FGFR1, or with PCM1::JAK2” (MLN-eo) [3], to specify the underlying molecular genetic changes and to include cases with ETV6::ABL1, FLT3 fusions or other TK fusion genes. MLN-TK are driven by rearrangements/fusion genes with involvement of PDGFRA, PDGFRB, FGFR1, JAK2, ABL1, or FLT3. This definition implicates eosinophilia as a recurrent finding, which therefore serves as the main trigger for initiation of distinct cytogenetic and molecular analyses conferring to the identification of disease-defining underlying TK fusion genes. Beside eosinophilia, blast phase (BP) in bone marrow (BM) or at extramedullary sites (extramedullary disease, EMD) of myeloid or lymphoid origin, initially often diagnosed as “myelosarcoma” or “high-grade lymphoma”, is present at diagnosis (primary BP) or develops during follow-up (secondary BP).
To date, more than 70 different TK fusion genes with recurrent involvement of at least six TK (PDGFRA, PDGFRB, FGFR1, JAK2, ABL1, FLT3) have been identified in clinically and morphologically distinct MLN with or without eosinophilia [4]. Targeted treatment with TK inhibitors (TKI) such as imatinib is highly effective in patients with PDGFRA and PDGFRB fusion genes, e.g., FIP1L1::PDGFRA or ETV6::PDGFRB [5,6,7,8], resulting in excellent long-term survival. In contrast, TK fusion genes with involvement of FGFR1 or JAK2 are associated with a more aggressive phenotype and clinical course with variable sensitivity to currently available TKI [4, 9,10,11,12,13,14].
Single case reports and small series described absence of eosinophilia and/or presence of monocytosis in association with distinct TK fusion genes. Due to the absence of eosinophilia, the diagnosis of a TK-fusion driven MLN may therefore be delayed or even completely missed. Within the “German Registry for Disorders of Eosinophils and Mast Cells (GREM)”, we sought to evaluate incidence, the incidence of eosinophilia and monocytosis and the phenotype and prognosis of BP within this distinct subcategory of myeloid neoplasms.
Patients and methods
Patients
Within the GREM, we identified 135 patients with diagnosis of a MLN-TK. The involved TK included PDGFRA (all FIP1L1::PDGFRA positive, n = 78), PDGFRB (diverse fusion partners, n = 26), FGFR1 (diverse fusion partners, n = 9), JAK2 (PCM1 or BCR as fusion partners, n = 11) and ABL1 (ETV6::ABL1, n = 11, Table 1). Patients with primary ETV6::ABL1 positive ALL were not included. The 135 patients were recruited from approximately 60 participating hematology centers and hematologists in private practice. Fifteen patients had been diagnosed with a suspected eosinophilia-associated myeloid neoplasm prior to 2002 and were subsequently tested FIP1L1::PDGFRA positive in 2003. Sixty-seven patients were recruited between 2003 and 2012, 53 patients between 2013 and 2022. We repeatedly reported on treatment of various MLN-TK with specific TKI, e.g., PDGFRA/PDGFRB fusion genes with imatinib [7, 15], JAK2 fusion genes with ruxolitinib [10, 16] and ETV6::ABL1 fusion gene with imatinib, nilotinib and dasatinib [16].
In the current analysis, OS was analyzed in the cohort of 135 patients (male 126/135; median age 49 years, range 19–80), in either chronic phase (CP) from time of diagnosis (n = 110, including 13 patients with progression to secondary BP) or in BP from time of diagnosis of BP (n = 38, primary BP, n = 25; secondary BP, n = 13). Of note, the 13 patients with secondary BP are included in both cohorts (Table 2). Data on absolute and relative counts of eosinophils and monocytes at time of diagnosis were available for 81 patients: FIP1L1::PDGFRA (n = 44), PDGFRB (n = 16), FGFR1 (n = 6), JAK2 (PCM1::JAK2, n = 7, BCR::JAK2, n = 1) or ETV6::ABL1 (n = 7, Table 1). All patients gave written informed consent. Data collection was compliant with the Declaration of Helsinki and approved by the ethics committee of the Medical Faculty Mannheim at the University Heidelberg, Germany.
Cytogenetics and molecular analyses
Cytogenetics and fluorescence in situ hybridization (FISH) analyses were performed on BM according to standard procedures. Specific nested reverse transcription polymerase chain reaction (RT-PCR) was performed for confirmation of suspected fusion genes in all patients [9, 17, 18].
Statistical analyses
All clinical and laboratory parameters including peripheral blood cell counts are expressed as median and range. Overall survival (OS) was determined from date of diagnosis to date of death or last contact and calculated by using the Kaplan–Meier method. Pearson correlation analysis was performed for the correlation between two parameters. Differences in the distribution of continuous variables between categories were analysed by Mann–Whitney test (for comparison of two groups). For categorical variables, Fisher’s exact test was used. P < 0.05 (two-sided) were considered significant. All statistical analyses were performed using GraphPad Prism Software, Inc. version 7 and SPSS (version 28.0; IBM-Corporation, Armonk, NY, USA).
Results
Incidence and phenotype of blast phase
A BP of myeloid (23/38, 61%) or lymphoid (15/38, 39%) origin was diagnosed in 38/135 (28%) patients (Tables 1 and 2). BP in BM (≥20% blast cells, n = 20, 53%; myeloid, n = 14; lymphoid, n = 6) was primary in 10/20 (50%) or secondary in 10/20 (50%) patients while EMD (n = 18) was primary in 15/18 (83%; myeloid, n = 7; lymphoid, n = 8) or secondary in only 3/18 patients (17%; myeloid, n = 2; lymphoid, n = 1, Table 3). Independent of phenotype or time point of occurrence of EMD, the phenotype in BM or peripheral blood (PB) was myeloid in all cases. A lineage discordance between BM/PB and EMD was therefore observed in 9/18 (50%) patients (Table 2). Compared to patients with FGFR1, JAK2 and ETV6::ABL1 fusion genes, PDGFRA/PDGFRB fusion positive patients had a lower frequency of primary (17/104, 16% vs. 8/31 26%, p = 0.32) and secondary BP (5/87, 6% vs. 8/23, 35%, p = 0.003), which occurred at later time points (median 87 months, range 9–189 vs. 19 months, range 10–36; p = 0.053).
Survival
In MLN with PDGFRA/PDGFRB fusion genes, 16/104 (15%) patients had died after a median follow-up of 9.2 years (range 0–28.6, Fig. 1A, B). While in CP (n = 87), 8 patients died because of comorbidity (n = 5; PDGFRA, n = 2, PDGFRB, n = 3), resistance/progression (n = 1; PDGFRB, n = 1), resistance/allogeneic SCT/GvHD (n = 1) and cardiac involvement (n = 1). Five patients progressed into secondary BP (PDGFRA, n = 4, PDGFRB, n = 1). Causes of death in BP (n = 8; primary, n = 6; secondary, n = 2) included resistance/relapse (n = 4; PDGFRA, n = 2, PDGFRB, n = 2), comorbidity (n = 3; PDGFRA, n = 2, PDGFRB, n = 1) and intracerebral bleeding (n = 1; PDGFRA, n = 1). Mutations conferring resistance to imatinib were identified in 2 patients (PDGFRA T674I, n = 2).

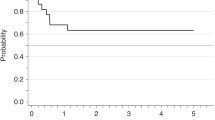
A Overall survival (OS) from time of diagnosis of 135 patients with various tyrosine kinase fusion genes (PDGFRA, n = 78; PDGFRB, n = 26; FGFR1, n = 9; JAK2, n = 11; ETV6::ABL1, n = 11) independent of disease phase. B OS of patients with PDGFRA/PDGFRB (FIP1L1::PDGFRA, n = 65; PDGFRB, n = 22) and FGFR1, JAK2 and ETV6::ABL1 (FGFR1, n = 3; JAK2, n = 11; ETV6::ABL1, n = 9) fusion genes in chronic phase (including 13 patients with progression into secondary blast phase), median follow-up 9.7 years (0–34.0) and 2.2 years (0.1–6.3), respectively (p < 0.0001). C OS of patients with PDGFRA/PDGFRB (FIP1L1::PDGFRA, n = 17; PDGFRB, n = 5) and FGFR1, JAK2 and ETV6::ABL1 (FGFR1, n = 7; JAK2, n = 3; ETV6::ABL1, n = 6) fusion genes from diagnosis of blast phase (primary BP, n = 25, secondary BP, n = 13), median OS 17.1 years (range 0.2–22) vs. 1.7 years (range 0.1–5.5; p = 0.0008). D OS of patients after allogeneic stem cell transplantation in prior chronic (n = 12) or blast phase (n = 13) independent of underlying TK fusion gene.
In MLN with FGFR1, JAK2 and ETV6::ABL1 fusion genes (n = 31; BP, n = 16), 11 patients died of which 10 patients were previously diagnosed with BP. The median OS from diagnosis of BP was 1.7 years (range 0.1–5.5, Fig. 1C). Overall, the incidence of BP was significantly lower (22/104, 21% vs. 16/31, 52%; P = 0.002) and overall survival from diagnosis of BP was significantly better (17.1 vs. 1.7 years, P < 0.0008) in patients with PDGFRA/PDGFRB fusions than with other TK fusion genes (Table 1, Fig. 1C).
Allogeneic stem cell transplantation
Allogeneic stem cell transplantation (SCT) was performed in 25 patients (Table 4) at a significant lower frequency in patients with PDGFRA/PDGFRB fusion genes (9/104, 9%, CP, n = 3; BP, n = 6) than in patients with FGFR1, JAK2 and ETV6::ABL1 fusion genes (16/31, 52%, CP, n = 9; BP, n = 7). After allogeneic SCT, 10/12 CP patients are alive at median 3.0 (range 0.3–10.5) years and 2/12 patients (PDGFRA, n = 1; JAK2, n = 1) died because of relapse at 0.5 and 1.5 years. In BP, 7/13 patients are alive at median 4.7 (range 0.1–6.5) years and 6/13 patients died at median 0.9 (range 0.6–1.4) years (Fig. 1D).
Age, gender, partner genes, and eosinophilia in association with various TK fusion genes
FIP1L1::PDGFRA
At diagnosis, the median age was 45.5 years (range 19–70), 43/44 (98%) patients were male. Leukocytosis >10 × 109/l was present in 29/44 (66%) patients (median 14 × 109/l, range 5–156), eosinophilia (median 6.4 × 109/l, range 0.9–30.1) was >0.5 × 109/l or >1.5 × 109/l in 44/44 (100%) and 40/44 (90%) patients, respectively (Table 5).
PDGFRB fusion genes
In 16 patients (male 14/16; median age 53 years, range 20–80), eleven different partner genes of PDGFRB were identified. Only ETV6 was a recurrent fusion partner (ETV6, n = 5; CDCC88C, n = 1; CCDC6, n = 1; CEP120, n = 1; CPSF6, n = 1; GIT2, n = 1; GPIAP1, n = 1; MYO18A, n = 1; PRKG2, n = 1; SPECC1, n = 1; TP53BP1, n = 1; uncharacterized partner, n = 1). Leukocytosis >10 × 109/l was present in 13/16 (81%) patients (median 28 × 109/l, range 4–127). Eosinophils (median 1.6 × 109/l, range 0.2–12.0) were ≤0.5 × 109/l, >0.5 × 109/l and >1.5 × 109/l in 4/16 (25%), 12/16 (75%) and 8/16 (50%) patients, respectively.
FGFR1 fusion genes
In six patients with FGFR1 fusion genes (male 5/6; median age 58 years, range 49–77), ZMYM2 (n = 3) and BCR (n = 2) were recurrent partner genes (FGFR1OP::FGFR1, n = 1). Leukocytosis >10 × 109/l was observed in 5/6 (83%) patients (median 64.5 × 109/l, range 4.8–173.0). Eosinophils (median 0.6 × 109/l, range 0–2.5) were ≤0.5 × 109/l, >0.5 × 109/l and >1.5 × 109/l in 3/6 (50%), 3/6 (50%) and 2/6 (33%) patients, respectively.
JAK2 fusion genes
All 8 patients with JAK2 fusion genes (PCM1, n = 7; BCR, n = 1; male 7/8; median age 69 years, range 29–73) presented with leukocytosis (median 25.9 × 109/l, range 10.5–55.0). Eosinophils (median 1.5 × 109/l, range 0–4.6) were ≤0.5 × 109/l, >0.5 × 109/l and >1.5 × 109/l in 2/8 (25%), 6/8 (75%) and 4/8 (50%) patients, respectively.
ETV6::ABL1 fusion gene
All ETV6::ABL1 positive patients (n = 7; male 6/7; median age 30 years, range 20–74) presented with leukocytosis >10 × 109/l (median 62 × 109/l, range 20.9–143). Hypereosinophilia >1.5 × 109/l (median 5.6 × 109/l, range 2.0–7.1) was observed in 7/7 (100%) cases.
Hypereosinophilia >1.5 × 109/l
Overall, hypereosinophilia >1.5 × 109/l was observed in 60/81 (74%) evaluable patients, most frequently in patients with FIP1L1::PDGFRA (40/44, 90%) and ETV6::ABL1 (7/7, 100%) fusion genes. In contrast, it was only observed in 13/30 (43%) patients with PDGFRB, FGFR1 or JAK2 fusion genes Absence of eosinophilia was restricted to 9/30 (30%) patients with PDGFRB, FGFR1 and JAK2 fusion genes (Table 5). In those 9 patients, primary diagnoses included atypical chronic myeloid leukemia (PDGFRB, n = 1; FGFR1, n = 1), myelodysplastic/myeloproliferative neoplasm (PDGFRB, n = 1; FGFR1, n = 1), myeloproliferative neoplasm unclassified (PDGFRB, n = 2; JAK2 n = 1), myelofibrosis (JAK2, n = 1) and mixed phenotype acute leukemia (FGFR1, n = 1). In all 9 patients, the underlying fusion gene was indicated by a characteristic reciprocal translocation.
Monocytosis >1.0 × 109/l
Irrespective of the underlying TK fusion gene, monocytosis >1.0 × 109/l was observed in 27/81 (33%) patients (FIP1L1::PDGFRA, 12/44, 27%; PDGFRB, 5/16, 31%; FGFR1, 2/6, 33%; JAK2, 2/8, 25%; ETV6::ABL1, n = 6/7, 85%) with relative monocytosis ≥10% being present in 6/27 (22%) patients (Table 5). In FIP1L1::PDGFRA positive patients, a significant association was noted between the absolute number of eosinophils and monocytes (r = 0.52, p = 0.0002). Monocytosis was present in 6/7 (85%) ETV6::ABL1 positive patients, all 6 patients also had hypereosinophilia >1.5 × 109/l. Overall, monocytosis >1.0 × 109/l was significantly associated with hypereosinophilia >1.5 × 109/l (23/27, 85%) but was without significant impact on progression or OS after a median follow-up of 7.2 years (range 0.1–33.1).
Serum tryptase
Due to the known association between increased basic serum tryptase levels (normal <11.4 µg/l) and PDGFRA/PDGFRB fusion genes, serum tryptase levels were available from 43/104 PDGFRA/PDGFRB positive patients. The serum tryptase level was ≥11.4 µg/l in 31/43 (72%) and ≥20 µg/l in 23/43 (53%) patients, the median level was 22.9 µg/l (range 3–183). The formal need to adjust normal ranges in patients with hereditary alpha-tryptasemia (HaT) could not be performed because none of the patients was retrospectively tested [19].
Discussion
Common features of the vast majority of TK-fusion driven myeloid neoplasms include an underlying chronic myeloid neoplasm with a high incidence of concurrent primary BP or progression to secondary BP. BP can be myeloid or lymphoid and is identified in the BM or at extramedullary sites (EMD). In the EMD, initial diagnosis frequently states “myelosarcoma” or “T-cell lymphoma”, while in the BM, the differentiation between a de novo myeloid/lymphoid/biphenotypic acute leukemia and a myeloid or lymphoid BP also remains challenging. We have reported on several patients with suspected primary lymphoma or de novo acute leukemia in which the underlying TK fusion gene was only identified because of poor response to intensive chemotherapy or even allogeneic SCT and persisting eosinophilia [7].
In the currently reported cohort of 135 MLN-TK patients, incidence, phenotype and prognosis of BP was highly variable within the various cohorts of MLN-TK. BP occurred equally distributed either in the BM or as EMD in approximately 30% of patients. It was primary in approximately 70% of patients with a lower relative frequency of 16% in patients with PDGFRA/PDGFRB fusion genes as compared to 26% in patients with FGFR1, JAK2 and ETV6::ABL1 fusion genes. In patients with PDGFRA/PDGFRB fusion genes, secondary BP only occurred in 6% of patients after a median of 87 months because >90% of patients achieved durable complete hematologic, complete cytogenetic (PDGFRB) and complete molecular (FIP1L1::PDGFRA) remissions on imatinib.
In contrast, neither ponatinib on FGFR1 [20], ruxolitinib on JAK2 [10, 16] nor imatinib/nilotinib/dasatinib on ETV6:ABL1 fusions [16] have shown a similar efficacy than imatinib on PDGFRA/PDGFRB fusions (Fig. 1A–C). Of interest, the FIGHT-203 study presented promising results on pemigatinib in patients with FGFR1 fusions in CP and to a lesser extent in BP [21]. In a recent literature review of a heterogenous cohort of 66 PCM1::JAK2 positive patients, Kaplan et al. reported on 11 ruxolitinib-treated patients [12]. However, the authors did not draw conclusions on its effect on survival because of the small cohort and because analysis on survival was complicated by the fact that 5 of these patients received a subsequent allogeneic SCT with a 5-year survival of 75% [12]. In consequence, patients with FGFR1, JAK2, and ETV6::ABL1 fusion genes progressed more often (35%) and faster (median 19 months) into secondary BP than patients with PDGFRA/PDGFRB fusion genes. The inferior prognosis of patients with FGFR1, JAK2, and ETV6::ABL1 fusion genes with a median 5-year survival of approximately 50–60% is therefore related to the more aggressive phenotype and the lack of effective and durable conventional treatment [16, 22,23,24,25,26]. In line with recently published data on patients with FGFR1 [25] and JAK2 fusion genes [12], data confirm that the poor prognosis of primary and secondary BP can only be overcome by allogeneic SCT (Table 4 and Fig. 1A–D).
Consistent hypereosinophilia >1.5 × 109/l in more than 90% of patients was only observed in association with FIP1L1::PDGFRA and ETV6::ABL1 fusion genes, although we acknowledge an obvious ascertainment bias in that only cases with eosinophilia are routinely screened for distinct TK fusion genes, particularly FIP1L1::PDGFRA. In patients with PDGFRB fusion genes, hypereosinophilia >1.5 × 109/l was present in only 50% of patients and even absent (≤0.5 × 109/l) in 25% of patients. Lack of eosinophilia was also evident in patients with FGFR1 fusions (Table 5). These findings are in line with literature reports on the impact of the FGFR1 partner gene on phenotype [27]. While ZMYM2::FGFR1 positive patients frequently present with the combination of a T-cell lymphoma/T-ALL/mixed phenotype acute leukemia and eosinophilia, BCR::FGFR1 positive patients usually present with a MPN/CML-like phenotype but a much lower incidence of hypereosinophilia >1.5 × 109/l, in the literature overall only reported in 3/21 evaluable patients [11, 28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50].
Diagnosis of a TK fusion gene driven MLN may be missed due to the lack of eosinophilia and subsequent diverse morphological diagnoses [5]. A significant proportion of patients were not initially diagnosed as MLN-TK or chronic eosinophilic leukemia but rather as subtype of MDS/MPN or MPN unclassified and decisive diagnostic assays such as cytogenetic analysis, FISH analysis or specific RT-PCR were not performed or only with delay. Moreover, newly available NGS technologies such as targeted RNA-sequencing, whole transcriptome or whole genome sequencing have revealed an increasing number of cytogenetically cryptic [51] or cytogenetically difficult to identify fusion genes, e.g., ETV6::ABL1 and several fusion genes with involvement of PDGFRB [52].
A rarely recognized feature of MLN-TK is monocytosis >1.0 × 109/l which was identified in about one third of patients. It was clearly clustered in patients with hypereosinophilia >1.5 × 109/l and consequently in patients with FIP1L1::PDGFRA or ETV6::ABL1 fusion genes. However, only approximately 20% of these patients also had relative monocytosis ≥10%. Even with taking into account the new cut-off values for diagnosis of chronic myelomonocytic leukemia with absolute monocytosis of ≥0.5 × 109/l and relative monocytosis of ≥10%, these numbers did not substantially change. The data, therefore, clearly indicate that a MLN-TK may cause monocytosis but accompanying features include hypereosinophilia and relative monocytosis <10% in the vast majority of patients (Table 5).
Besides MLN-TK, the concurrent presence of significant eosinophilia and monocytosis is also a typical feature in patients with advanced systemic mastocytosis [53, 54]. While being the disease-defining characteristic for chronic myelomonocytic leukemia, monocytosis is also identified in other myeloid neoplasms, potentially as marker of poor prognosis, e.g., in polycythemia vera, myelofibrosis and systemic mastocytosis [55, 56]. These data therefore also underscore the current guidelines for diagnosis of chronic myelomonocytic leukemia, other myelodysplastic/myeloproliferative neoplasms and myeloproliferative neoplasms unclassified that the primary genetic work-up should not only exclude BCR::ABL1 positive chronic myeloid leukemia but also cases of MLN-TK. Due to the excellent prognosis of imatinib-treated patients with PDGFRA/PDGFRB [5, 57, 58] fusion genes, monocytosis had no obvious impact on progression and survival.
FIP1L1::PDGFRA and ETV6::ABL1 fusion genes share striking clinical and morphological similarities including male predominance, the relative frequency of eosinophilia and monocytosis, the median absolute number of eosinophils, and presentation or progression to BP including EMD. Of interest, progression to lymphoid BP in the BM seems to be a rare event for both fusion genes. Compared to FIP1L1::PDGFRA, the responses of ETV6::ABL1 positive patients to imatinib, nilotinib or dasatinib are less frequent and less durable [16]. In the current update of our own cohort, 6/11 patients were initially treated with imatinib but more durable remissions were only observed on primary or secondary treatment with nilotinib (n = 2), dasatinib (n = 3) or after allogeneic SCT (n = 1). Not included in our series, but important to note is that the MLN-TK subcategory also includes very rare fusion genes with involvement of other TK such as FLT3 [52, 59].
In summary, the relative frequency of the defining characteristics of MLN-TK such as myeloid or lymphoid BP and/or eosinophilia occur at markedly variable frequencies according to the underlying fusion gene. Monocytosis is a potentially important marker which frequently occurs in association with significant eosinophilia. Careful attention must be paid to these subtle characteristics to avoid missing a diagnosis of a TKI-sensitive MLN-TK.
Data availability
Freely available to any researcher
References
Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36:1703–19.
Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International consensus classification of myeloid neoplasms and acute leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140:1200–28.
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. WHO press; 2016.
Reiter A, Gotlib J. Myeloid neoplasms with eosinophilia. Blood. 2017;129:704–14.
Jawhar M, Naumann N, Schwaab J, Baurmann H, Casper J, Dang TA, et al. Imatinib in myeloid/lymphoid neoplasms with eosinophilia and rearrangement of PDGFRB in chronic or blast phase. Ann Hematol. 2017;96:1463–70.
Gotlib J, Cools J. Five years since the discovery of FIP1L1-PDGFRA: what we have learned about the fusion and other molecularly defined eosinophilias. Leukemia. 2008;22:1999–2010.
Metzgeroth G, Schwaab J, Gosenca D, Fabarius A, Haferlach C, Hochhaus A, et al. Long-term follow-up of treatment with imatinib in eosinophilia-associated myeloid/lymphoid neoplasms with PDGFR rearrangements in blast phase. Leukemia. 2013;27:2254–6.
Cheah CY, Burbury K, Apperley JF, Huguet F, Pitini V, Gardembas M, et al. Patients with myeloid malignancies bearing PDGFRB fusion genes achieve durable long-term remissions with imatinib. Blood. 2014;123:3574–7.
Reiter A, Walz C, Watmore A, Schoch C, Blau I, Schlegelberger B, et al. The t(8;9)(p22;p24) is a recurrent abnormality in chronic and acute leukemia that fuses PCM1 to JAK2. Cancer Res. 2005;65:2662–7.
Schwaab J, Knut M, Haferlach C, Metzgeroth G, Horny HP, Chase A, et al. Limited duration of complete remission on ruxolitinib in myeloid neoplasms with PCM1-JAK2 and BCR-JAK2 fusion genes. Ann Hematol. 2015;94:233–8.
Verstovsek S, Subbiah V, Masarova L, Yin CC, Tang G, Manshouri T, et al. Treatment of the myeloid/lymphoid neoplasm with FGFR1 rearrangement with FGFR1 inhibitor. Ann Oncol. 2018;29:1880–2.
Kaplan HG, Jin R, Bifulco CB, Scanlan JM, Corwin DR. PCM1-JAK2 fusion tyrosine kinase gene-related neoplasia: a systematic review of the clinical literature. Oncologist. 2022;27:e661–e70.
Rumi E, Milosevic JD, Selleslag D, Casetti I, Lierman E, Pietra D, et al. Efficacy of ruxolitinib in myeloid neoplasms with PCM1-JAK2 fusion gene. Ann Hematol. 2015;94:1927–8.
Lierman E, Selleslag D, Smits S, Billiet J, Vandenberghe P. Ruxolitinib inhibits transforming JAK2 fusion proteins in vitro and induces complete cytogenetic remission in t(8;9)(p22;p24)/PCM1-JAK2-positive chronic eosinophilic leukemia. Blood. 2012;120:1529–31.
Metzgeroth G, Walz C, Score J, Siebert R, Schnittger S, Haferlach C, et al. Recurrent finding of the FIP1L1-PDGFRA fusion gene in eosinophilia-associated acute myeloid leukemia and lymphoblastic T-cell lymphoma. Leukemia. 2007;21:1183–8.
Schwaab J, Naumann N, Luebke J, Jawhar M, Somervaille TCP, Williams MS, et al. Response to tyrosine kinase inhibitors in myeloid neoplasms associated with PCM1-JAK2, BCR-JAK2 and ETV6-ABL1 fusion genes. Am J Hematol. 2020;95:824–33.
Tirado CA, Sebastian S, Moore JO, Gong JZ, Goodman BK. Molecular and cytogenetic characterization of a novel rearrangement involving chromosomes 9, 12, and 17 resulting in ETV6 (TEL) and ABL fusion. Cancer Genet Cytogenet. 2005;157:74–7.
Zhou MH, Gao L, Jing Y, Xu YY, Ding Y, Wang N, et al. Detection of ETV6 gene rearrangements in adult acute lymphoblastic leukemia. Ann Hematol. 2012;91:1235–43.
Valent P, Akin C, Hartmann K, Alvarez-Twose I, Brockow K, Hermine O, et al. Updated diagnostic criteria and classification of mast cell disorders: a consensus proposal. Hemasphere. 2021;5:e646.
Kreil S, Adès L, Bommer M, Stegelmann F, Ethell ME, Lubking A, et al. Limited efficacy of ponatinib in myeloproliferative neoplasms associated with FGFR1 fusion genes. Blood. 2015;126:2812.
Gotlib G, Kiladjian J, Vannucchi A, Rambaldi A, Reiter A, Shomali W, et al. A phase 2 study of pemigatinib (FIGHT-203; INCB054828) in patients with myeloid/lymphoid neoplasms (MLNs) with fibroblast growth factor receptor 1 (FGFR1) rearrangement (MLN FGFR1). Blood. 2021;138:385.
Chen M, Wang K, Cai X, Zhang X, Chao H, Chen S, et al. Myeloid/lymphoid neoplasm with CEP110-FGFR1 fusion: An analysis of 16 cases show common features and poor prognosis. Hematology. 2021;26:153–9.
Patterer V, Schnittger S, Kern W, Haferlach T, Haferlach C. Hematologic malignancies with PCM1-JAK2 gene fusion share characteristics with myeloid and lymphoid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, and FGFR1. Ann Hematol. 2013;92:759–69.
Strati P, Tang G, Duose DY, Mallampati S, Luthra R, Patel KP, et al. Myeloid/lymphoid neoplasms with FGFR1 rearrangement. Leuk Lymphoma. 2018;59:1672–6.
Hernandez-Boluda JC, Pereira A, Zinger N, Gras L, Martino R, Nikolousis E, et al. Allogeneic hematopoietic cell transplantation in patients with myeloid/lymphoid neoplasm with FGFR1-rearrangement: a study of the Chronic Malignancies Working Party of EBMT. Bone Marrow Transplant. 2022;57:416–22.
Gotlib J. Available and emerging therapies for bona fide advanced systemic mastocytosis and primary eosinophilic neoplasms. Hematology Am Soc Hematol Educ Program. 2022;2022:34–46.
Umino K, Fujiwara SI, Ikeda T, Toda Y, Ito S, Mashima K, et al. Clinical outcomes of myeloid/lymphoid neoplasms with fibroblast growth factor receptor-1 (FGFR1) rearrangement. Hematology. 2018;23:470–7.
Barnes EJ, Leonard J, Medeiros BC, Druker BJ, Tognon CE. Functional characterization of two rare BCR-FGFR1(+) leukemias. Cold Spring Harb Mol Case Stud. 2020;6:a004838.
Vega F, Medeiros LJ, Bueso-Ramos CE, Arboleda P, Miranda RN. Hematolymphoid neoplasms associated with rearrangements of PDGFRA, PDGFRB, and FGFR1. Am J Clin Pathol. 2015;144:377–92.
Kasbekar M, Nardi V, Dal Cin P, Brunner AM, Burke M, Chen YB, et al. Targeted FGFR inhibition results in a durable remission in an FGFR1-driven myeloid neoplasm with eosinophilia. Blood Adv. 2020;4:3136–40.
Demiroglu A, Steer EJ, Heath C, Taylor K, Bentley M, Allen SL, et al. The t(8;22) in chronic myeloid leukemia fuses BCR to FGFR1: transforming activity and specific inhibition of FGFR1 fusion proteins. Blood. 2001;98:3778–83.
Pini M, Gottardi E, Scaravaglio P, Giugliano E, Libener R, Baraldi A, et al. A fourth case of BCR-FGFR1 positive CML-like disease with t(8;22) translocation showing an extensive deletion on the derivative chromosome 8p. Hematol J. 2002;3:315–6.
Lee SG, Park TS, Lee ST, Lee KA, Song J, Kim J, et al. Rare translocations involving chromosome band 8p11 in myeloid neoplasms. Cancer Genet Cytogenet. 2008;186:127–9.
Richebourg S, Theisen O, Plantier I, Parry A, Soenen-Cornu V, Lepelley P, et al. Chronic myeloproliferative disorder with t(8;22)(p11;q11) can mime clonal cytogenetic evolution of authentic chronic myelogeneous leukemia. Genes Chromosomes Cancer. 2008;47:915–8.
Patnaik MM, Gangat N, Knudson RA, Keefe JG, Hanson CA, Pardanani A, et al. Chromosome 8p11.2 translocations: prevalence, FISH analysis for FGFR1 and MYST3, and clinicopathologic correlates in a consecutive cohort of 13 cases from a single institution. Am J Hematol. 2010;85:238–42.
Baldazzi C, Iacobucci I, Luatti S, Ottaviani E, Marzocchi G, Paolini S, et al. B-cell acute lymphoblastic leukemia as evolution of a 8p11 myeloproliferative syndrome with t(8;22)(p11;q11) and BCR-FGFR1 fusion gene. Leuk Res. 2010;34:e282–5.
Kim SY, Oh B, She CJ, Kim HK, Jeon YK, Shin MG, et al. 8p11 Myeloproliferative syndrome with BCR-FGFR1 rearrangement presenting with T-lymphoblastic lymphoma and bone marrow stromal cell proliferation: a case report and review of the literature. Leuk Res. 2011;35:e30–4.
Wakim JJ, Tirado CA, Chen W, Collins R. t(8;22)/BCR-FGFR1 myeloproliferative disorder presenting as B-acute lymphoblastic leukemia: report of a case treated with sorafenib and review of the literature. Leuk Res. 2011;35:e151–3.
Dolan M, Cioc A, Cross NC, Neglia JP, Tolar J. Favorable outcome of allogeneic hematopoietic cell transplantation for 8p11 myeloproliferative syndrome associated with BCR-FGFR1 gene fusion. Pediatr Blood Cancer. 2012;59:194–6.
Haslam K, Langabeer SE, Kelly J, Coen N, O'Connell NM, Conneally E. Allogeneic hematopoietic stem cell transplantation for a BCR-FGFR1 myeloproliferative neoplasm presenting as acute lymphoblastic leukemia. Case Rep Hematol. 2012;2012:620967.
Morishige S, Oku E, Takata Y, Kimura Y, Arakawa F, Seki R, et al. A case of 8p11 myeloproliferative syndrome with BCR-FGFR1 gene fusion presenting with trilineage acute leukemia/lymphoma, successfully treated by cord blood transplantation. Acta Haematol. 2013;129:83–9.
Shimanuki M, Sonoki T, Hosoi H, Watanuki J, Murata S, Mushino T, et al. Acute leukemia showing t(8;22)(p11;q11), myelodysplasia, CD13/CD33/CD19 expression and immunoglobulin heavy chain gene rearrangement. Acta Haematol. 2013;129:238–42.
Matikas A, Tzannou I, Oikonomopoulou D, Bakiri M. A case of acute myelogenous leukaemia characterised by the BCR-FGFR1 translocation. BMJ Case Rep. 2013;2013:bcr2013008834.
Wang W, Tang G, Kadia T, Lu X, Li Y, Huang L, et al. Cytogenetic evolution associated with disease progression in hematopoietic neoplasms With t(8;22)(p11;q11)/BCR-FGFR1 rearrangement. J Natl Compr Canc Netw. 2016;14:708–11.
Khodadoust MS, Luo B, Medeiros BC, Johnson RC, Ewalt MD, Schalkwyk AS, et al. Clinical activity of ponatinib in a patient with FGFR1-rearranged mixed-phenotype acute leukemia. Leukemia. 2016;30:947–50.
Landberg N, Dreimane A, Rissler M, Billstrom R, Agerstam H. Primary cells in BCR/FGFR1-positive 8p11 myeloproliferative syndrome are sensitive to dovitinib, ponatinib, and dasatinib. Eur J Haematol. 2017;99:442–8.
Montenegro-Garreaud X, Miranda RN, Reynolds A, Tang G, Wang SA, Yabe M, et al. Myeloproliferative neoplasms with t(8;22)(p11.2;q11.2)/BCR-FGFR1: a meta-analysis of 20 cases shows cytogenetic progression with B-lymphoid blast phase. Hum Pathol. 2017;65:147–56.
Liu JJ, Meng L. 8p11 Myeloproliferative syndrome with t(8;22)(p11;q11): a case report. Exp Ther Med. 2018;16:1449–53.
Villafuerte-Gutierrez P, Lopez Rubio M, Herrera P, Arranz E. A case of myeloproliferative neoplasm with BCR-FGFR1 rearrangement: favorable outcome after haploidentical allogeneic transplantation. Case Rep Hematol. 2018;2018:5724960.
Konishi Y, Kondo T, Nakao K, Asagoe K, Otsuka Y, Nishikori M, et al. Allogeneic hematopoietic stem cell transplantation for 8p11 myeloproliferative syndrome with BCR-FGFR1 gene rearrangement: a case report and literature review. Bone Marrow Transplant. 2019;54:326–9.
Cross NCP, Hoade Y, Tapper WJ, Carreno-Tarragona G, Fanelli T, Jawhar M, et al. Recurrent activating STAT5B N642H mutation in myeloid neoplasms with eosinophilia. Leukemia. 2018;33:415–25.
Jawhar M, Naumann N, Knut M, Score J, Ghazzawi M, Schneider B, et al. Cytogenetically cryptic ZMYM2-FLT3 and DIAPH1-PDGFRB gene fusions in myeloid neoplasms with eosinophilia. Leukemia. 2017;31:2271–3.
Kluin-Nelemans HC, Reiter A, Illerhaus A, van Anrooij B, Hartmann K, Span LFR, et al. Prognostic impact of eosinophils in mastocytosis: analysis of 2350 patients collected in the ECNM Registry. Leukemia. 2020;34:1090–101.
Schwaab J, Umbach R, Metzgeroth G, Naumann N, Jawhar M, Sotlar K, et al. KIT D816V and JAK2 V617F mutations are seen recurrently in hypereosinophilia of unknown significance. Am J Hematol. 2015;90:774–7.
Tefferi A, Shah S, Mudireddy M, Lasho TL, Barraco D, Hanson CA, et al. Monocytosis is a powerful and independent predictor of inferior survival in primary myelofibrosis. Br J Haematol. 2018;183:835–8.
Barraco D, Cerquozzi S, Gangat N, Patnaik MM, Lasho T, Finke C, et al. Monocytosis in polycythemia vera: clinical and molecular correlates. Am J Hematol. 2017;92:640–5.
Legrand F, Renneville A, MacIntyre E, Mastrilli S, Ackermann F, Cayuela JM, et al. The spectrum of FIP1L1-PDGFRA-associated chronic eosinophilic leukemia: new insights based on a survey of 44 cases. Medicine (Baltimore). 2013;92:e1–e9.
Rohmer J, Couteau-Chardon A, Trichereau J, Panel K, Gesquiere C, Ben Abdelali R, et al. Epidemiology, clinical picture and long-term outcomes of FIP1L1-PDGFRA-positive myeloid neoplasm with eosinophilia: Data from 151 patients. Am J Hematol. 2020;95:1314–23.
Walz C, Erben P, Ritter M, Bloor A, Metzgeroth G, Telford N, et al. Response of ETV6-FLT3-positive myeloid/lymphoid neoplasm with eosinophilia to inhibitors of FMS-like tyrosine kinase 3. Blood. 2011;118:2239–42.
Acknowledgements
The authors thank all physicians who contributed clinical data of their patients into the “German Registry on Disorders of Eosinophils and Mast Cells (GREM)”.
Funding
This work was supported by the “Deutsche José Carreras Leukämie-Stiftung e.V.” (Grant No. 08 R/2020), Germany. Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
GM and AR wrote the manuscript. All authors contributed to the manuscript, provided major intellectual contributions, reviewed and revised its content, and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Metzgeroth, G., Steiner, L., Naumann, N. et al. Myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions: reevaluation of the defining characteristics in a registry-based cohort. Leukemia 37, 1860–1867 (2023). https://doi.org/10.1038/s41375-023-01958-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-023-01958-1
This article is cited by
-
Progression of myeloproliferative neoplasm with BCR::JAK2 fusion to acute leukemia of ambiguous lineage
Annals of Hematology (2024)
