
Abstract
The approval of tisagenlecleucel and axicabtagene ciloleucel represents a breakthrough in the field of immune and cellular therapy for hematologic malignancies. These anti-CD19 chimeric antigen receptor-T cells (CAR) proved to be highly effective in the treatment of relapsed/refractory B-cell acute lymphoblastic leukemia (B-ALL) and specific histologic subtypes of B-cell non-Hodgkin lymphomas. This expert review aims to summarize the current available research evidence in this field, with a special focus on the different challenges faced by treating physicians, and we also provide future perspectives.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout

Similar content being viewed by others

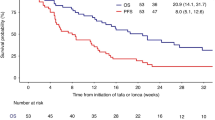
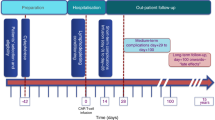
References
Buechner J, Kersten MJ, Fuchs M, Salmon F, Jäger U. Chimeric antigen receptor-T cell therapy: practical considerations for implementation in. Eur HemaSphere. 2018;2:e18.
June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–5.
Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 2019;16:372–85.
Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer. 2013;13:525–41.
Park JH, Brentjens RJ. Adoptive immunotherapy for B-cell malignancies with autologous chimeric antigen receptor modified tumor targeted T cells. Discov Med. 2010;9:277–88.
Scheuermann RH, Racila E. CD19 antigen in leukemia and lymphoma diagnosis and immunotherapy. Leuk Lymphoma. 1995;18:385–97.
Sommermeyer D, Hill T, Shamah SM, Salter AI, Chen Y, Mohler KM, et al. Fully human CD19-specific chimeric antigen receptors for T-cell therapy. Leukemia. 2017;31:2191–9.
Sadelain M. CAR therapy: the CD19 paradigm. J Clin Investig. 2015;125:3392–400.
Castella M, Boronat A, Martin-Ibanez R, Rodriguez V, Sune G, Caballero M, et al. Development of a novel anti-CD19 chimeric antigen receptor: a paradigm for an affordable CAR T cell production at academic institutions. Mol Ther Methods Clin Dev. 2019;12:134–44.
Fielding AK, Richards SM, Chopra R, Lazarus HM, Litzow MR, Buck G, et al. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood. 2007;109:944–50.
Tavernier E, Boiron JM, Huguet F, Bradstock K, Vey N, Kovacsovics T, et al. Outcome of treatment after first relapse in adults with acute lymphoblastic leukemia initially treated by the LALA-94 trial. Leukemia. 2007;21:1907–14.
Bhojwani D, Pui CH. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013;14:e205–217.
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–28.
Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. New Engl J Med. 2018;378:449–59.
Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129:3322–31.
Shimabukuro-Vornhagen A, Godel P, Subklewe M, Stemmler HJ, Schlosser HA, Schlaak M, et al. Cytokine release syndrome. J Immunother Cancer. 2018;6:56.
Frey NV, Porter DL. CAR T-cells merge into the fast lane of cancer care. Am J Hematol. 2016;91:146–50.
Jackson HJ, Rafiq S, Brentjens RJ. Driving CAR T-cells forward. Nat Rev Clin Oncol. 2016;13:370–83.
Hay KA, Gauthier J, Hirayama AV, Voutsinas JM, Wu Q, Li D, et al. Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood. 2019;133:1652–63.
Novartis Pharmaceuticals Corporation. KYMRIAH [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2017.
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. New Engl J Med. 2018;378:439–48.
Wierda WG, Bishop MR, Oluwole OO, Logan AC, Baer MR, Donnellan WB, et al. Updated phase 1 results of Zuma-3: Kte-C19, an Anti-CD19 chimeric antigen receptor T cell therapy, in adult patients with relapsed/refractory acute lymphoblastic leukemia. Blood. 2018;132:897.
Lee DW, Wayne AS, Huynh V, Handgretinger R, Pieters R, Michele G, et al. 1008PDZUMA-4 preliminary results: phase 1 study of KTE-C19 chimeric antigen receptor T cell therapy in pediatric and adolescent patients (pts) with relapsed/refractory acute lymphoblastic leukemia (R/R ALL). Ann Oncol. 2017;28.
Maffini E, Saraceni F, Lanzavolume F. Treatment of adult patients with relapsed/refractory B-Cell philadelphia-negative acute lymphoblastic leukemia. Clin Hematol Int. 2019;1:85–93.
Jacoby E. Relapse and Resistance to CAR-T Cells and Blinatumomab in Hematologic Malignancies. Clin Hematol Int. 2019;1:79–84.
Children’ Oncology Group. Study of efficacy and safety of tisagenlecleucel in HR B-ALL EOC MRD positive patients (CASSIOPEIA). ClinicalTrials.gov Identifier: NCT03876769.
Crump M, Neelapu SS, Farooq U, Van Den Neste E, Kuruvilla J, Westin J, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130:1800–8.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Reagan PM, Miklos DB, et al. A comparison of one year outcomes in ZUMA-1 (axicabtagene ciloleucel) and SCHOLAR-1 in patients with refractory, aggressive non-hodgkinlymphoma (NHL). Blood. 2017;130:579.
Lulla PD, Hill LC, Ramos CA, Heslop HE. The use of chimeric antigen receptor T cells in patients with non-Hodgkin lymphoma. Clin Adv Hematol Oncol. 2018;16:375–86.
YESCARTA [package insert]. Santa Monica, CA: Kite Pharma, Inc; 2017.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. New Engl J Med. 2017;377:2531–44.
Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019;20:31–42.
Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. New Engl J Med. 2019;380:45–56.
Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak O, et al. Chimeric antigen receptor T cells in refractory B-Cell Lymphomas. New Engl J Med. 2017;377:2545–54.
Abramson JS, Gordon LI, Palomba ML, Lunning MA, Arnason JE, Forero-Torres A, et al. Updated safety and long term clinical outcomes in TRANSCEND NHL 001, pivotal trial of lisocabtagene maraleucel (JCAR017) in R/R aggressive NHL. J Clin Oncol. 2018;36:7505–7505.
Hirayama AV, Gauthier J, Hay KA, Voutsinas JM, Wu Q, Gooley T, et al. The response to lymphodepletion impacts PFS in patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR T cells. Blood. 2019;133:1876–87.
Neelapu SS. CAR-T efficacy: is conditioning the key? Blood. 2019;133:1799–800.
Sauter C, Riviere I, Senechal B, Ni A, Bernal Y, Wang X, et al. A phase I trial of 19-28Z car-T cells post-high dose therapy and autologous transplantation (HDT-ASCT) for relapsed and refractory (R/R) B-cell non-hodgkin lymphoma (B-NHL). Hematological Oncol. 2017;35:138–9.
Oluwole OO, Bishop MR, Gisselbrecht C, Gordon LI, Kersten MJ, Maloney DG, et al. ZUMA-7: A phase 3 randomized trial of axicabtagene ciloleucel (Axi-Cel) versus standard-of-care (SOC) therapy in patients with relapsed/refractory diffuse large B cell lymphoma (R/R DLBCL). J Clin Oncol. 2018;36:TPS7585–TPS7585.
Abramson JS, McGree B, Noyes S, Plummer S, Wong C, Chen Y-B, et al. Anti-CD19 CAR T cells in CNS diffuse large-B-cell lymphoma. New Engl J Med. 2017;377:783–4.
Chong EA, Svoboda J, Dwivedy Nasta S, Landsburg DJ, Winchell N, Napier E, et al. Sequential Anti-CD19 directed chimeric antigen receptor modified T-cell therapy (CART19) and PD-1 blockade with pembrolizumab in patients with relapsed or refractory B-cell non-hodgkin lymphomas. Blood. 2018;132:4198.
Hirayama AV, Gauthier J, Hay KA, Sheih A, Cherian S, Chen X, et al. Efficacy and toxicity of JCAR014 in combination with durvalumab for the treatment of patients with relapsed/refractory aggressive B-cell non-hodgkin lymphoma. Blood. 2018;132:1680.
Svoboda J, Rheingold SR, Gill SI, Grupp SA, Lacey SF, Kulikovskaya I, et al. Nonviral RNA chimeric antigen receptor-modified T cells in patients with Hodgkin lymphoma. Blood. 2018;132:1022–6.
Ramos CA, Bilgi M, Gerken CP, Dakhova O, Mei Z, Grilley BJ, et al. CD30-chimeric antigen receptor (CAR) T cells for therapy of hodgkin lymphoma (HL). Blood. 2018;132:680.
Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. New Engl J Med. 2013;369:32–42.
Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. New Engl J Med. 2014;370:997–1007.
Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–8.
Flinn IW, Hillmen P, Montillo M, Nagy Z, Illes A, Etienne G, et al. The phase 3 DUO trial: duvelisib vs ofatumumab in relapsed and refractory CLL/SLL. Blood. 2018;132:2446–55.
Goede V, Fischer K, Busch R, Engelke A, Eichhorst B, Wendtner CM, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. New Engl J Med. 2014;370:1101–10.
D’Souza A, Fretham C. Current uses and outcomes of hematopoietic cell transplantation (HCT): CIBMTR Summary Slides. 2018. https://www.cibmtr.org.
Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7:303ra139.
Geyer MB, Riviere I, Senechal B, Wang X, Wang Y, Purdon TJ, et al. Autologous CD19-targeted CAR T cells in patients with residual CLL following initial purine analog-based therapy. Mol Ther. 2018;26:1896–905.
Turtle CJ, Hay KA, Hanafi LA, Li D, Cherian S, Chen X, et al. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-specific chimeric antigen receptor-modified T cells after failure of ibrutinib. J Clin Oncol. 2017;35:3010–20.
Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood. 2016;127:1117–27.
Gill SI, Vides V, Frey NV, Metzger S, Brien M, Hexner E, et al. Prospective clinical trial of anti-CD19 CAR T cells in combination with ibrutinib for the treatment of chronic lymphocytic leukemia shows a high response rate. Blood. 2018;132:298.
Gauthier J, Hirayama AV, Hay KA, Li D, Lymp J, Sheih A, et al. Comparison of efficacy and toxicity of CD19-specific chimeric antigen receptor T-cells alone or in combination with ibrutinib for relapsed and/or refractory CLL. Blood. 2018;132:299.
Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127:3321–30.
Azoulay E, Shimabukuro-Vornhagen A, Darmon M, von Bergwelt-Baildon M. Critical care management of chimeric antigen receptor T cell-related toxicity. Be aware and prepared. Am J respiratory Crit care Med. 2019;200:20–3.
Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–95.
Teachey DT, Bishop MR, Maloney DG, Grupp SA. Toxicity management after chimeric antigen receptor T cell therapy: one size does not fit ‘ALL’. Nat Rev Clin Oncol. 2018;15:218.
Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. 2017;7:1404–19.
Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov. 2018;8:958–71.
Sanchez-Escamilla M, Yanez San Segundo L, Urbano-Ispizua A, Perales MA. CAR T cells: the future is already present. Med Clin. 2019;152:281–6.
Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25:625–38.
Kansagra AJ, Frey NV, Bar M, Laetsch TW, Carpenter PA, Savani BN, et al. Clinical utilization of chimeric antigen receptor T-cells (CAR-T) in B-cell acute lymphoblastic leukemia (ALL)-an expert opinion from the European Society for blood and marrow transplantation (EBMT) and the American Society for Blood and Marrow Transplantation (ASBMT). Bone marrow Transpl. 2019. [Epub ahead of print].
Garfall AL, Stadtmauer EA, Hwang WT, Lacey SF, Melenhorst JJ, Krevvata M, et al. Anti-CD19 CAR T cells with high-dose melphalan and autologous stem cell transplantation for refractory multiple myeloma. JCI insight. 2019;4:127684.
Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-Cell Therapy bb2121 in relapsed or refractory multiple myeloma. New Engl J Med. 2019;380:1726–37.
Author information
Authors and Affiliations
Contributions
All authors contributed substantially to the conception, writing, critical review and final approval of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
MAP reports honoraria from Abbvie, Bellicum, Bristol-Myers Squibb, Incyte, Merck, Novartis, Nektar Therapeutics, and Takeda. He serves on DSMBs for Servier and Medigene, and the scientific advisory boards of MolMed and NexImmune. He has received research support for clinical trials from Incyte, Kite (Gilead) and Miltenyi. Christian Chabanon reports honoraria from Kite/Gilead and Novartis. The remaining authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mohty, M., Gautier, J., Malard, F. et al. CD19 chimeric antigen receptor-T cells in B-cell leukemia and lymphoma: current status and perspectives. Leukemia 33, 2767–2778 (2019). https://doi.org/10.1038/s41375-019-0615-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-019-0615-5
This article is cited by
-
CD19 CAR T cells for B cell malignancies: a systematic review and meta-analysis focused on clinical impacts of CAR structural domains, manufacturing conditions, cellular product, doses, patient’s age, and tumor types
BMC Cancer (2024)
-
Acute lymphoblastic leukaemia
Nature Reviews Disease Primers (2024)
-
Valrubicin-loaded immunoliposomes for specific vesicle-mediated cell death in the treatment of hematological cancers
Cell Death & Disease (2024)
-
Patient-reported outcome (PRO) instruments used in patients undergoing adoptive cell therapy (ACT) for the treatment of cancer: a systematic review
Systematic Reviews (2023)
-
Indications for haematopoietic cell transplantation for haematological diseases, solid tumours and immune disorders: current practice in Europe, 2022
Bone Marrow Transplantation (2022)
