
Abstract
Germline mutations in the SAMD9 and SAMD9L genes, located in tandem on chromosome 7, are associated with a clinical spectrum of disorders including the MIRAGE syndrome, ataxia–pancytopenia syndrome and myelodysplasia and leukemia syndrome with monosomy 7 syndrome. Germline gain-of-function mutations increase SAMD9 or SAMD9L’s normal antiproliferative effect. This causes pancytopenia and generally restricted growth and/or specific organ hypoplasia in non-hematopoietic tissues. In blood cells, additional somatic aberrations that reverse the germline mutation’s effect, and give rise to the clonal expansion of cells with reduced or no antiproliferative effect of SAMD9 or SAMD9L include complete or partial chromosome 7 loss or loss-of-function mutations in SAMD9 or SAMD9L. Furthermore, the complete or partial loss of chromosome 7q may cause myelodysplastic syndrome in these patients. SAMD9 mutations appear to associate with a more severe disease phenotype, including intrauterine growth restriction, developmental delay and hypoplasia of adrenal glands, testes, ovaries or thymus, and most reported patients died in infancy or early childhood due to infections, anemia and/or hemorrhages. SAMD9L mutations have been reported in a few families with balance problems and nystagmus due to cerebellar atrophy, and may lead to similar hematological disease as seen in SAMD9 mutation carriers, from early childhood to adult years. We review the clinical features of these syndromes, discuss the underlying biology, and interpret the genetic findings in some of the affected family members. We provide expert-based recommendations regarding diagnosis, follow-up, and treatment of mutation carriers.
Similar content being viewed by others


Introduction
Aberrations of chromosome 7 in myeloid malignancies was first described in the 1960’s, but the exact gene(s) in this region driving myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) development remain elusive [1]. Besides total chromosomal loss (monosomy 7) or loss of the long arm of chromosome 7 (7q-), different commonly deleted regions (CDR) have been identified, including 7q22 (CDR1), 7q34 (CDR2), and 7q35-36 (CDR3) [2,3,4]. Interestingly, aberrations of chromosome 7 are found in 30–40% of pediatric MDS patients [5, 6], but only in 10% of adult MDS, for unknown reasons [7]. Despite decades of research striving to identify the gene(s) on 7q contributing to myeloid malignancies, very few mutations in the remaining haploinsufficient alleles have been found: in essence, only recurrent mutations in CUX1 [4, 8] and EZH2 [9] have been identified. This indicates that haploinsufficiency for genes located in the CDR1-3 is the principal mechanism driving MDS development in these cases.
For the myelodysplasia and leukemia syndrome with monosomy 7 (MLSM7; OMIM #252270), the presence of a sibling pair both diagnosed with a myeloid neoplasm displaying monosomy 7 is a prerequisite for diagnosis [10]. No candidate gene(s) has been ascertained for MLSM7 and the disease does not follow a clear monogenic pattern [11]. It has even been suggested that the predisposing locus is not located on chromosome 7 due to retainment of different parental chromosomes 7 between siblings with leukemic bone marrows [12].
The ataxia–pancytopenia (ATXPC) syndrome (OMIM #159550) was first described in 1978 by Dr. Frederik Li in a family with neurologic symptoms and pancytopenia of unknown origin, with some family members developing a myeloid neoplasm with monosomy 7 [13], Exclusion and linkage analyses combined with whole exome sequencing (WES) of an ATXPC kindred revealed mutations in the gene SAMD9L as the cause of this syndrome, and mutations in this gene were also found in the original family described by Dr. Li [15].
We were part of a collaborative team describing two families—from Sweden and Finland, respectively—with SAMD9L mutations and neurological symptoms and fluctuating cytopenias, infections, and MDS [16, 17]. In light of these recent findings, it is unlikely that ATXPC and MLSM7 really represent two distinct entities, but merely reflect a variation in the penetrance and severity of symptoms in one and the same disease spectrum that includes germ line mutations in e.g., GATA2, SAMD9L, and SAMD9, leading to monosomy 7. In this review, we summarize the disease phenotype in SAMD9L and SAMD9 mutation carriers, and we attempt to provide recommendations for the identification, initial screening, follow-up and treatment of patients with, based on the limited data currently available about these disorders.
SAMD9L
Function of SAMD9L
SAMD9L, a protein widely expressed across human tissues, contains a SAM domain that binds RNA as well as oligomerizes both with SAM-containing and non-SAM-containing proteins. The exact role of SAMD9L is currently unknown, but the gene has a general antiproliferative function and has been demonstrated to function as tumor suppressor in breast, hepatocellular and in squamous cell carcinoma, being repressed by the p53 pathway [18]. In hematopoetic tissue SAMD9L regulates cell proliferation by being a crucial component of a protein complex that facilitates the degradation of cytokine receptors through the homotypic fusion of endosomes[19].
Heterozygous germline mutations in SAMD9L in ATXPC syndrome
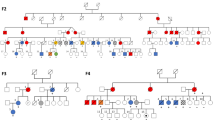
To date, four families with the ATXPC syndrome and SAMD9L mutations have been described (Fig. 1a) [15,16,17,18]. In total, 25 mutation carriers have been identified. Eighteen had documented transient or permanent cytopenia, and eight of these had recurrent or severe infections or hemorrhages. Five individuals developed hematological malignancies at ages 1.5, 4, 7, 10, and 56 years. Except for one 4-year-old and one 16-year-old mutation carrier, all displayed either gait disturbance or nystagmus, or both (Table 1; Fig. 2). All mutations identified so far have been missense and none have been recurrent between families. The first observation led to the hypothesis by Chen et al. that germline mutations in SAMD9L result in a gain-of-function (GOF) [15], not a loss-of-function (LOF) and haploinsufficiency. In the latter theory, LOF/haploinsufficiency for SAMD9L in ATXPC would be the first hit towards leukemic transformation, followed by monosomy 7 or 7q- that includes other genes involved in the pathogenesis of MDS. This idea was however proven wrong by functional assays ascertaining that the germline mutations actually are GOF aberrations increasing the antiproliferative effect of SAMD9L[16].

Disease-associated and somatic reversion mutations in SAMD9L and SAMD9. a Overview of the SAMD9L gene and protein (blue), including the SAM domain (green). Positions of all identified disease-associated germ line gain-of-function mutations (red) and somatic reversion loss-of-function mutations (lilac) reported to date are indicated. b Overview of the SAMD9 gene and protein (blue), including the SAM domain (green). Positions of all identified disease-associated de novo (black) and germ line (red) gain-of-function mutations as well as somatic reversion loss-of-function mutations (lilac) reported to date are indicated

Known physical manifestations of the MIRAGE and ATXPC syndrome. The SAMD9L or SAMD9 mutations’ antiproliferative effect may underly the general intrauterine growth reduction or organ-specific hypoplasias or atrophies observed in these syndromes. In SAMD9L-associated disease the non-hematological disease manifestations may be mild and patients may not report symptoms, but some degree of nervous system involvement was noted almost all ATXPC patients
SAMD9L mutations are lost due to aberrations of chromosome 7 during progression towards MDS
The SAMD9L gene is located on chromosome 7q21.2 in the MDS CDR1 [19]. In the three patients identified to date with inherited GOF SAMD9L mutations who developed MDS, the mutant allele was lost due to aberrations of chromosome 7 [15, 16]. Importantly, a clear correlation between metaphases carrying the aberration of chromosome 7 and the change in the ratio between wild type (wt) and mutant SAMD9L ratio could be observed with disease progression [16]. One patient developed a der(1;7) which resulted in loss of genetic material on chromosome 7, including the mutant SAMD9L allele [16]. The other two patients that developed MDS lost the mutant SAMD9L copy by monosomy 7 and deletion of 7q, respectively [15, 16]. Again, we could clearly show a correlation between the metaphases carrying the aberration of chromosome 7 and the change in the variant allele frequency (VAF) towards the wt allele [16].
Reversion of the germline SAMD9L mutation
Sequencing revealed a mutant SAMD9L VAF lower than the expected 50% in the blood in some of the unaffected family members. Furthermore, SNP array B-allele frequencies demonstrated clear skewing towards the wt allele, without net genomic loss in the region on chromosome 7q21.2. This implies that the low mutant VAF was due to a copy neutral loss of heterozygosity (CN-LOH) of chromosome 7q [16]. Chen et al. also observed decreased frequency of the missense mutant SAMD9L in two of their study subjects and suspected hematopoietic mosaicism. Long-term in vitro culturing of transformed lymphocytic lines from heterozygous carriers demonstrated that CN-LOH of 7q, resulting in net loss of mutant allele, occurred after 3 weeks to 6 months in culture. This is most likely due to the selective growth advantage attributed to hematopoietic precursor cells that have lost their mutated copy and duplicated the wt counterpart of SAMD9L by uniparental disomy (UPD) [15].
Moreover, in some individuals the GOF mutation was retained but instead counteracted by frameshift/stop-gain mutations in SAMD9L in cis (Fig. 1 and Fig. 3) [16]. Normally frameshift/stop-gain mutations would result in destruction of the mRNA from the mutant allele by nonsense-mediated decay but this requires that the mutation occurs in a defined distance to intron/exon boundaries [20]. In the case of SAMD9L, functionally being a one exon gene, nonsense-mediated decay is thus likely not responsible for any LOF. As an additional mechanism of reversion, we identified in cis mutations in the same codon that carried the GOF mutation (Fig. 1a). These resulted in amino acid changes probably inducing less GOF, a hypothesis supported by proliferative effects seen in cells positive for in cis mutations when overexpressing the different SAMD9L variants in 293T cells [16].

Overview of the genetic reversion mechanisms associated with gain-of-function SAMD9/SAMD9L mutations and their effect on hematopoiesis. Three different mechanisms of genetic reversion have been associated with gain-of-function mutations in the SAMD9 and SAMD9L genes, often even in the same patient. Firstly, uniparental isodisomy by homologous recombination of the long arm of chromosome 7 results in replacement of the mutant allele with a wt copy and restores hematopoiesis. Secondly, somatic loss-of-function mutations in cis inactivates the GOF mutation and also restores hematopoiesis. Lastly, monosomy 7, del(7q) or der(1;7) all are “adaption by aneuploidy”, where the mutant allele is eliminated by total or partial loss of chromosome 7, on which SAMD9/SAMD9L are located, however, not restoring hematopoiesis but leading to MDS
Strikingly, some patients carried multiple clones with different genetic mechanisms of reversion and in one case a patient even carried a reversion by CN-LOH and additionally an MDS clone with aberrations of chromosome 7 [16]. These findings show the tremendous selection pressure of the cells to neutralize the GOF SAMD9L mutation and the advantage inferred to the cells that have undergone genetic reversion (Fig. 3). Family members that did not develop MDS all showed genetic reversions in some form or another, which is relevant in regard to the diagnostic criteria that at least two siblings have to be affected by MDS with aberration of chromosome 7 to make the diagnosis MLSM7. It appears likely that the common occurrence of genetic reversion has led to underdiagnosis of MLSM7. It can also be speculated, as in the study by Shannon et al. that investigated three families with MLSM7 and found retainment of mixed parental alleles in leukemic bone marrow between siblings, actually reflected mosaicism of clones harboring mutational reversal events and 7q-, respectively [12].
The origin of SAMD9L mutations in hematopoietic hierarchy and development
SAMD9L is most highly expressed in NK cells and monocytes and subsequently symptomatic carriers usually present with low numbers of NK cells and monocytes [16]. To investigate the effect of the genetic reversion in mature cells; B-, T-, NK cells, monocytes, and granulocytes were isolated from two individuals affected by either CN-LOH or reversion mutations. Detection of allelic ratios of the wt and mutant SAMD9L showed that NK cells showed a higher percentage of gene reversion, indirectly arguing for the essential gene function of SAMD9L in NK-cell differentiation [16].
To investigate when the reversion of the inherited GOF SAMD9L mutations occurred, mutant VAF in peripheral blood (PB) at birth (using Guthrie cards) was investigated. Equal fractions of wt and mutant SAMD9L in PB directly after birth indicated that the low mutant SAMD9L VAF detected later in life was not caused by mosaicism at birth [16], but instead by a clonal genetic event that occurred most likely between 1 and 4 years of age. This also coincided with a time when many individuals were experiencing a transient pancytopenia, probably undergoing genetic reversion leading to a fitness advantage of the reverted cells, resulting in their expansion and recovery of the PB counts.
Cell sorting of hematopoietic stem cells (HSC), multi-potent progenitors (MPP), common myeloid progenitors, common lymphoid progenitors and granulocyte–monocyte progenitors followed by analysis of the mutant VAF using digital droplet PCR was performed to investigate at which cellular level the reversion has occurred. Surprisingly, this analysis suggested that the molecular reversion occurred at the MPP rather than at the HSC level [16], although more patients would have to be studied to draw more definitive and generalized conclusions. We hypothesize that the reversion of the inherited SAMD9L mutation might result in increased proliferation of MPPs (at least when compared to mutant cells), leading to clonal expansion. Alternatively, a rare HSC clone would represent the initial revertant, with the correction leading to a selective recruitment of these HSCs into differentiation but with no evident advantage in terms of self-renewal. In both cases, MPPs must associate with a high maintenance capacity. While most hematopoietic homeostasis has previously been attributed to HSCs, mainly based on transplantation experiments, such scenarios appear in line with more recent lineage-tracing studies and selective HSC-depletion strategies in mice, demonstrating substantial long-term contribution of MPPs not only in steady-state but also the context of stress hematopoiesis [21,22,23,24].
SAMD9L knock-out mice develop MDS
It has been suggested that monosomy 7, 7q-, or 7q21 microdeletions increase the signaling through the cytokine cascade by the loss of SAMD9 and SAMD9L, causing delays in homotypic endosome fusion in which these proteins partake, resulting in persistence of ligand-bound receptors [1]. To test this, a Samd9l knockout mouse was generated. Interestingly, both hetero- and homozygous Samd9l knockout mice developed an MDS-like phenotype with long latency. Of note, the latency was not significantly different between mice carrying one or two functional copies of the gene, arguing that haploinsufficiency alone might be sufficient for MDS development [19]. However, whether the other Samd9l allele was lost in heterozygous mice during transformation to MDS was unfortunately not investigated. Hence, it remains an open question as to what extent the Samd9l mouse model mimics the MDS phenotype observed in ATXPC patients.
Nagamachi et al. provided substantial evidence that SAMD9L is involved in endocytosis and receptor recycling affecting signaling through the KIT receptor [19]. This observation probably explains the advantage of Samd9l−/− and Samd9l+/− cells in the setting of a competitive transplantation, even though the advantage is rather small and is probably enhanced by the competition in the post-transplantation setting and/or by infections post transplantation that trigger interferon associated Samd9l expression. The observation that cells with reduced or absent levels of SAMD9L have a growth advantage compared to wt cells fits well with the observation that GOF SAMD9L mutations are leading to growth inhibition [16].
Neurological phenotype of the ATXPC syndrome
Some degree of nervous system involvement has been documented in almost all carriers of pathogenic SAMD9L mutations reported to date (Fig. 2). Balance problems and nystagmus are most common, followed by mild pyramidal signs [17]. A few patients developed mild cerebellar-type dysarthria. Other cerebellar motor signs such as impairment of rapid alternating movements, dysmetria, cerebellar tremor were typically less pronounced than the balance problems, or entirely absent, why the usage of the term “ataxia” is actually debatable [17]. Moreover, we have identified paracentral retinal dysfunction in two mutation carriers and additional reported patients had clinical signs and symptoms highly suggestive of this. We therefore suggest that part of the dysmetria reported in the other families could be motor problems caused by limited visual control. Behavioral symptoms have been described in carriers of specific SADM9L mutations, especially during childhood and adolescence.
Magnetic resonance imaging of individuals with ATXPC revealed cerebellar atrophy in all patients and white matter hyperintensities in some. Cerebellar atrophy started during early childhood and was slowly progressive. The cerebellar atrophy is surprisingly pronounced compared to the generally mild balance impairment. Neuropathological examination of four individuals with ATXPC has been reported and brains showed pronounced loss of cerebellar Purkinje cells [13,14,15]. Granule cell loss was found in three patients but was less pronounced. There was depletion of white matter in the cerebellum in two individuals [13, 14] and in the hippocampus in one patient [15]. While the cerebellar atrophy can be readily explained by loss of Purkinje cells, cerebellar white matter and granule cells, these patients have been autopsied at ages when there commonly are no or no marked white matter hyperintensities in MRI. Thus, the exact neuropathological correlate of the white matter hyperintensities remains unknown, but we hypothesize that SAMD9L mutations may slow down the physiological myelination that physiologically occurs in childhood and adolescence.
SAMD9
Normal function of SAMD9
SAMD9, a highly conserved gene contigous to SAMD9L, shares a common gene structure to its paralogue and both genes encode proteins with 58% amino acid identity. Most mammals have both Samd9 and Samd9l. However, mice have only Samd9l, and cows encode only Samd9 suggesting that the genes partially complement each other’s functions [25]. SAMD9, like SAMD9L, is involved in control of cell proliferation and functions as a tumor suppressor in some cancers. Deleterious mutations in the SAMD9 gene are known to cause normophosphatemic familial tumoral carcinosis (NFTC), a rare autosomal recessive disease. NFTC is characterized by abnormal inflammation of the skin and gingiva that is induced by excessive signaling via interferon pathways—in line with this it has been experimentally demonstrated that the function SAMD9 is tightly regulated by interferon-γ [26].
SAMD9 mutations cause the MIRAGE syndrome
Recent work has identified mutations in SAMD9 in children with myelodysplasia, infection, restriction of growth, adrenal hypoplasia, genital phenotypes, and enteropathy, named MIRAGE syndrome (OMIM #617053) (Figs. 1b and 2; Table 1) [27]. In the initial report the majority of the affected children died in early childhood, but two individuals with an expanded lifespan developed MDS with mononsomy 7, resulting in a loss of the mutant SAMD9 allele. A recent study also reported that in seven MIRAGE children in whom parental DNA was available, the mutations were de novo and located in highly conserved residues, often affecting arginines (6 of 8) with half of all mutations clustering in a hotspot of codons 982 and 983 of SAMD9 (Fig. 1b) [28]. The conclusion is therefore that SAMD9 mutations underlying the MIRAGE syndrome are GOF and the negative effect on cellular growth puts a selection pressure on the cells to lose their mutated copy. In addition, this argues that the functions of these gene homologs are very similar. However, besides the similar hematologic phenotype, it remains elusive why the mutations in SAMD9L result in cerebellar atrophy while mutations in SAMD9 are associated with complex multi-organ defects. We hypothesize that possible neurological manifestations in SAMD9-associated disease may have remained unnoticed because most patients died at a very young age when the nervous system is still immature and mild balance problems or visual problems would not have been evident.
Buonocore et al. describes eight MIRAGE patients with intrauterine growth restriction and severe testicular dysfunction leading to female external genitalia in six of the eight children, while the remaining two children had atypical external genitalia (Figs. 1b and 2) [28]. The majority of 19 de novo MIRAGE patients identified to date have had signs of bone marrow failure (various cytopenias) and infections in their medical history. Four children in total have had a longer life span and developed MDS with aberrations of chromosome 7. Strikingly, in the three non-MDS patients in which cytogenetics were performed, hematopoietic cells showed some form of aberration of chromosome 7, either monosomy 7 or 7q-. These three patients had an initial VAF around 50% but showed a lower VAF in a later sample indicating expansion of clones that that had lost their GOF SAMD9 mutation due to monosomy 7 or 7q-. The reason that these patients did not develop MDS could be time related, since all died at 5–21 months of age. Similar to the observations made for GOF SAMD9L mutations in ATXPC, GOF SAMD9 mutations were also reverted by other genetic mechanisms such as frame-shift or stop gain mutations in the blood of four individuals with MIRAGE syndrome (Fig. 3), including patients with non-manifest hematopoietic phenotype [28].
Inherited SAMD9 mutation causing a milder phenotype
Schwartz et al. have recently identified a family with an inherited SAMD9 mutation, in which three children developed MDS with monosomy 7 at an early age [29]. While the mother was completely unaffected, both by MDS and by genetic reversion, all three children displayed clonal somatic reversion in the blood by different mechanisms. One affected child had a CN-LOH of chromosome 7 and simultaneous monosomy 7, while the other two children had monosomy 7 as well as in cis mutations in SAMD9, arguing for neutralization of the germline GOF variant by both mechanisms.
It can only be speculated that the differences between de novo and familial SAMD9 mutations is the severity of the GOF. Weaker GOF variants are probably tolerated while more severe ones can only occur de novo due to embryonic lethality or is not fixated in the population due to infertility by the accompanying genital malformation. However, it is unclear in the reported family, why the mother did not develop any phenotype and did not show any genetic reversion. Unfortunately, RNA was not available for testing of uni- or biallelic expression of SAMD9 (J. Klco, Personal communication, May 2017). Importantly, this latest publication also provides additional evidence that different clones which have undergone genetic reversion by different mechanism, exist in the same patient and compete with each other resulting in the end in an outgrowth of the MDS clone with monosomy 7 [29].
Diagnosis and follow-up
Screening for SAMD9 and SAMD9L mutations
All patients with pediatric MDS with monosomy 7, del 7q and der(1;7) and probably all adult MDS patients with these chromosomal aberrations should be tested with gene panels not only covering GATA2 associated disorders, but also including SAMD9 and SAMD9L mutations on genetic material not obtained from blood cells. In our experience skin fibroblasts are the best source of germline DNA, whereas buccal swabs on some occasions have showed non representative skewing towards wt copies, likely due contamination of blood cells that have undergone reversion by CN-LOH [30]. Accompanying monocytopenia and low or absent NK cells shall even raise more suspicion about GATA2 deficiency syndrome, ATXPC and MIRAGE syndromes, but this should not be a prerequisite for genetic testing because many patients with ATXPC and MIRAGE have “reverted” the GOF mutation resulting in a recovery of cell numbers in the PB.
With diagnosis of MIRAGE or ATXPC syndrome in one family member, a thorough family history should be taken and members of the family should, after consent, be offered genetic counseling and testing regarding the mutation identified in the family. Counseling should include the information that they may be mutation carriers, possibly at risk for MDS or cytopenias with potentially severe consequences, even if they have not experienced any symptoms previously. In ATXPC families, relatively subtle neurological signs may indicate carrier status. We consider testing family members because they may either be affected, at risk for developing MDS to our present knowledge, or since they might represent potential HSC donors for a family member with ATXPC syndrome. It should also be noted that some of the patients in the described families were suffering from clinical symptoms associated with low cell numbers of macrophages/monocytes as well as NK cells. So the recommendation for chest X-ray to exclude macrophage deficiency related alveolar proteinosis and screening vaccination for NK-cell deficiency related HPV infection is at the moment based on our clinical experience with a very limited patient cohort. However, we still think that the benefits of these screenings outweighs their risks and would recommend them until better data is available. In addition, for every carrier of pathogenic SAMD9 or SAMD9L mutations without MDS we recommend frequent (every 6–12 months) follow-up physician visits and laboratory tests, as suggested in Table 2 [31].
Treatment of MDS in SAMD9L and SAMD9 mutation carriers
Most difficult, regarding both syndromes, is to give recommendations on whom, when and how to perform hematopoietic stem cell transplantation (HSCT), because of a lack of evidence and extensive clinical experience. The 4-year ATXPC patient in the Swedish family developed severe neurological problems after allogeneic HSCT from a matched unrelated donor, although we still remain uncertain if there was a causal relationship [16]. Conditioning was performed according to the EWOG-MDS guidelines on HSCT in Childhood MDS and JMML, essentially using anti-thymocyte globulin, fludarabine and thiotepa for conditioning. Due to the fact that we were not aware of the definite diagnosis prior to the transplantation, a thorough neurological examination and a brain MRI were not performed during the pre-transplantation evaluation. However, MRI of the brain after the allogeneic HSCT showed pronounced cerebellar atrophy that correlated with onset of ataxia and nystagmus. This hypoplasia could be induced by a CMV infection the patient suffered during infancy, with prolonged problems due to his primary deficiency [32]. Moreover, no worsening of neurological symptoms in the transplanted boy in the Finnish family was observed [16]. However, it could be speculated that the conditioning triggered early onset of ATXPC related cerebellar atrophy, possibly by chemotherapy inducing an interferon response and that this in combination with the cellular damage imposed by these treatments will prevent cellular repair. Whether the same would apply to cellular damage induced by irradiation is of course unknown, but we would currently advise physicians to avoid higher doses of irradiation, especially to the brain, in patients with ATXPC syndrome.
With the currently available information it is impossible to draw any evidence-based conclusions, but we are worried that the only curative treatment for patients with ATXPC that develop MDS might aggravate the neurological symptoms and will severely affect their quality of life. Unfortunately, one can argue for both an early and a late time point for allogeneic HSCT. When transplanting before transformation to MDS, non-myeloablative regimens similar to the ones used for Fanconi’s anemia or GATA2 deficiency syndrome can probably be used [33, 34], even though evidence for this assumption are missing at the moment. This idea is based on the observation that like in GATA2 deficiency syndrome, the competitiveness of HSC is greatly reduced in ATXPC and MIRAGE syndrome and monocytes and NK cells are also reduced in number and possibly function. These effects reduce the risk for rejection and confer an advantage of the incoming HSCs in competition for niches in the bone marrow. Such reduced intensity conditioning will hopefully lessen any risk of developing cerebellar atrophy associated with allogeneic HSCT. However, once transformation to MDS with monosomy 7 has occurred, most transplant physicians would recommend a myeloablative conditioning, which at least in our patient possibly could have triggered cerebellar atrophy. We are reluctant to advice against this, but a careful risk-benefit analysis should of course be made, and of note should be that non-myeloablative conditioning regimens for HSCT after transformation are now in use for Fanconi anemia [35].
Treatment of neurological symptoms in ATXPC
The neurological symptoms of ATXPC are generally mild and very slowly progressive, causing far less disability than other types of genetic cerebellar syndromes [36,37,38]. For the neurologist, considering the diagnosis and enquiring about any of the non-neurological disease features in the patient or family members is paramount. Once the diagnosis is verified, the patient can be reassured that the neurological disease usually will be slowly progressive and often remain mild to moderate until high age. Rehabilitation programs may be beneficial for those who experience balance problems. Awareness and specific measures for the less well-known form of vision impairment due to retinal dysfunction may reduce overall impairment. Access to dedicated neurology, ophthalmology and rehabilitation services should be provided. In the presently known families, there was complete or near-complete penetrance of neurological signs in SAMD9L mutation carriers. Thus, including neurological examination for, at the very least, balance and eye-movement abnormalities, in a hematology workup for cytopenia or MDS may faster lead to a suspicion of SAMD9L-related disease.
Conclusions
GOF mutations in SAMD9L, located in the chromosome 7q, are responsible for the ATXPC syndrome. In all of the patients affected by myelodysplasia, aberrations of chromosome 7 resulted in a loss of the mutated SAMD9L allele during MDS transformation. Analysis of mutation carriers who went through a hematopoietic crisis at a young age, and who showed no symptoms at time of analysis, revealed CN-LOH of chromosome 7q not present at birth. Additionally, a LOF mutation in the same codon as the germline SAMD9L mutation occurred in one of these patients. De novo as well as familial germ-line mutations in the SAMD9 gene have been discovered to cause the MIRAGE syndrome. The majority of MIRAGE patients died in infancy, but some developed a progressive loss of the mutated SAMD9 allele through monosomy 7 or 7q-, and secondary LOF mutations. The latter eliminated the growth-restricting effects on the mutant SAMD9 protein in hematopoietic cells. However, a number of patients developed MDS with monosomy 7 or 7q- similar to what was observed in ATXPC. This illustrates that the elevated risk for MDS and AML in both syndromes are likely driven by monosomy 7 or 7q-, as a reversion mechanism gone awry, resulting in loss of the growth restricting imposed by the GOF mutation, but by secondary loss of genes in CDR1-3 on chromosome 7q contribute to leukemogenesis.
References
Honda H, Nagamachi A, Inaba T. -7/7q- syndrome in myeloid-lineage hematopoietic malignancies: attempts to understand this complex disease entity. Oncogene. 2015;34:2413–25.
Asou H, Matsui H, Ozaki Y, Nagamachi A, Nakamura M, Aki D, Inaba T. Identification of a common microdeletion cluster in 7q21.3 subband among patients with myeloid leukemia and myelodysplastic syndrome. Biochem Biophys Res Commun. 2009;383:245–51.
Jerez A, Sugimoto Y, Makishima H, Verma A, Jankowska AM, Przychodzen B, et al. Loss of heterozygosity in 7q myeloid disorders: clinical associations and genomic pathogenesis. Blood. 2012;119:6109–17.
Hosono N, Makishima H, Jerez A, Yoshida K, Przychodzen B, McMahon S, et al. Recurrent genetic defects on chromosome 7q in myeloid neoplasms. Leukemia. 2014;28:1348–51.
Hasle H, Kerndrup G, Jacobsen BB. Childhood myelodysplastic syndrome in Denmark: incidence and predisposing conditions. Leukemia. 1995;9:1569–72.
Hasle H, Arico M, Basso G, Biondi A, Cantu Rajnoldi A, Creutzig U. et al. Myelodysplastic syndrome, juvenile myelomonocytic leukemia, and acute myeloid leukemia associated with complete or partial monosomy 7. European Working Group on MDS in Childhood (EWOG-MDS). Leukemia. 1999;13:376–85.
Haase D. Cytogenetic features in myelodysplastic syndromes. Ann Hematol. 2008;87:515–26.
Hindersin S, Niemeyer CM, Germing U, Gobel U, Kratz CP. Mutation analysis of CUTL1 in childhood myeloid neoplasias with monosomy 7. Leuk Res. 2007;31:1323–4.
Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42:665–7.
Gilchrist DM, Friedman JM, Rogers PC, Creighton SP. Myelodysplasia and leukemia syndrome with monosomy 7: a genetic perspective. Am J Med Genet. 1990;35:437–41.
Kamiyama R, Shibata T, Mori W. Two autopsy cases of atypical myeloproliferative disorder with group C monosomy occurring in siblings. Acta Pathol Jpn. 1973;23:815–35.
Shannon KM, Turhan AG, Chang SS, Bowcock AM, Rogers PC, Carroll WL, et al. Familial bone marrow monosomy 7. Evidence that the predisposing locus is not on the long arm of chromosome 7. J Clin Invest. 1989;84:984–9.
Li FP, Potter NU, Buchanan GR, Vawter G, Whang-Peng J, Rosen RB. A family with acute leukemia, hypoplastic anemia and cerebellar ataxia: association with bone marrow C-monosomy. Am J Med. 1978;65:933–40.
Li FP, Hecht F, Kaiser-McCaw B, Baranko PV, Potter NU. Ataxia-pancytopenia: syndrome of cerebellar ataxia, hypoplastic anemia, monosomy 7, and acute myelogenous leukemia. Cancer Genet Cytogenet. 1981;4:189–96.
Chen DH, Below JE, Shimamura A, Keel SB, Matsushita M, Wolff J, et al. Ataxia-pancytopenia syndrome is caused by missense mutations in SAMD9L. Am J Hum Genet. 2016;98:1146–58.
Tesi B, Davidsson J, Voss M, Rahikkala E, Holmes TD, Chiang SCC, et al. Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood. 2017;129:2266–79.
Gorcenco S, Komulainen-Ebrahim J, Nordborg K, Suo-Palosaari M, Andreasson S, Krüger J, et al. Ataxia-pancytopenia syndrome with SAMD9L mutations. Neurol Genet. 2017;3:e183.
Phowthongkum P, Chen DH, Raskind WH, Bird T. SAMD9L-related ataxia-pancytopenia syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews(R). The publisher is University of Washington, Seattle, WA; 1993 ISSN: 2372-0697.
Nagamachi A, Matsui H, Asou H, Ozaki Y, Aki D, Kanai A, et al. Haploinsufficiency of SAMD9L, an endosome fusion facilitator, causes myeloid malignancies in mice mimicking human diseases with monosomy 7. Cancer Cell. 2013;24:305–17.
Kervestin S, Jacobson A. NMD: a multifaceted response to premature translational termination. Nat Rev Mol Cell Biol. 2012;13:700–12.
Busch K, Klapproth K, Barile M, Flossdorf M, Holland-Letz T, Schlenner SM, et al. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature. 2015;518:542–6.
Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho YJ, et al. Clonal dynamics of native haematopoiesis. Nature. 2014;514:322–7.
Schoedel KB, Morcos MN, Zerjatke T, Roeder I, Grinenko T, Voehringer D, et al. The bulk of the hematopoietic stem cell population is dispensable for murine steady-state and stress hematopoiesis. Blood. 2016;128:2285–96.
Säwén P, Lang S, Mandal P, Rossi DJ, Soneji S, Bryder D. Mitotic history reveals distinct stem cell populations and their contributions to hematopoiesis. Cell Rep. 2016;14:2809–18.
Li CF, MacDonald JR, Wei RY, Ray J, Lau K, Kandel C, et al. Human sterile alpha motif domain 9, a novel gene identified as down-regulated in aggressive fibromatosis, is absent in the mouse. BMC Genom. 2007;8:92.
Hershkovitz D, Gross Y, Nahum S, Yehezkel S, Sarig O, Uitto J, et al. Functional characterization of SAMD9, a protein deficient in normophosphatemic familial tumoral calcinosis. J Invest Dermatol. 2011;131:662–9.
Narumi S, Amano N, Ishii T, Katsumata N, Muroya K, Adachi M, et al. SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet. 2016;48:792–7.
Buonocore F, Kuhnen P, Suntharalingham JP, Del Valle I, Digweed M, Stachelscheid H, et al. Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans. J Clin Invest. 2017;127:1700–13.
Schwartz JR, Wang S, Ma J, Lamprecht T, Walsh M, Song G, et al. Germline SAMD9 mutation in siblings with monosomy 7 and myelodysplastic syndrome. Leukemia. 2017;31:1827–30.
Thiede C, Prange-Krex G, Freiberg-Richter J, Bornhauser M, Ehninger G. Buccal swabs but not mouthwash samples can be used to obtain pretransplant DNA fingerprints from recipients of allogeneic bone marrow transplants. Bone Marrow Transplant. 2000;25:575–7.
Churpek JE, Godley LA. How I diagnose and manage individuals at risk for inherited myeloid malignancies. Blood. 2016;128:1800–13.
Averill LW, Kandula VV, Akyol Y, Epelman M. Fetal brain magnetic resonance imaging findings in congenital cytomegalovirus infection with postnatal imaging correlation. Semin Ultrasound CT MR. 2015;36:476–86.
Cuellar-Rodriguez J, Gea-Banacloche J, Freeman AF, Hsu AP, Zerbe CS, Calvo KR, et al. Successful allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Blood. 2011;118:3715–20.
Dufour C, Svahn J. Fanconi anaemia: new strategies. Bone Marrow Transplant. 2008;41:S90–95.
Talbot A, Peffault de Latour R, Raffoux E, Buchbinder N, Vigouroux S, Milpied N, et al. Sequential treatment for allogeneic hematopoietic stem cell transplantation in Fanconi anemia with acute myeloid leukemia. Haematologica. 2014;99:e199–200.
Schmitz-Hubsch T, Coudert M, Giunti P, Globas C, Baliko L, Fancellu R, et al. Self-rated health status in spinocerebellar ataxia--results from a European multicenter study. Mov Disord. 2010;25:587–95.
Ygland E, Taroni F, Gellera C, Caldarazzo S, Duno M, Soller M, et al. Atypical Friedreich ataxia in patients with FXN p.R165P point mutation or comorbid hemochromatosis. Park Relat Disord. 2014;20:919–23.
Wictorin K, Bradvik B, Nilsson K, Soller M, van Westen D, Bynke G, et al. Autosomal dominant cerebellar ataxia with slow ocular saccades, neuropathy and orthostatism: a novel entity? Park Relat Disord. 2014;20:748–54.
Acknowledgements
The authors are very grateful to all of the family members for their time, patience and willingness to provide samples and want to assure them that without their contribution and help, the unraveling of this rare syndrome would never have been possible.
Author contributions
JD, AP, UT, DB, LN, and JC co-wrote the manuscript and approved the final version. JC initiated this review and JD coordinated rounds of revisions and edited the manuscript for content.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Davidsson, J., Puschmann, A., Tedgård, U. et al. SAMD9 and SAMD9L in inherited predisposition to ataxia, pancytopenia, and myeloid malignancies. Leukemia 32, 1106–1115 (2018). https://doi.org/10.1038/s41375-018-0074-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-018-0074-4
This article is cited by
-
Pediatric Pancytopenia and Monosomy 7: A Case Report of SAMD9L-Associated Disease
Journal of Clinical Immunology (2024)
-
CRISPR screening in human hematopoietic stem and progenitor cells reveals an enrichment for tumor suppressor genes within chromosome 7 commonly deleted regions
Leukemia (2022)
-
Pediatric Germline Predisposition to Myeloid Neoplasms
Current Hematologic Malignancy Reports (2022)
-
Approach Toward Germline Predisposition Syndromes in Patients with Hematologic Malignancies
Current Hematologic Malignancy Reports (2022)
-
Genomic profiling identifies distinct genetic subtypes in extra-nodal natural killer/T-cell lymphoma
Leukemia (2022)
