
Abstract
Immune-mediated inflammatory diseases are various groups of conditions that result in immune system disorders and increased cancer risk. Despite the identification of causative cytokines and pathways, current clinical treatment for immune-mediated inflammatory diseases is limited. In addition, immune-mediated inflammatory disease treatment can increase the risk of cancer. Several previous studies have demonstrated that Toxoplasma gondii manipulates the immune response by inhibiting or stimulating cytokines, suggesting the potential for controlling and maintaining a balanced immune system. Additionally, T. gondii also has the unique characteristic of being a so-called “Trojan horse” bacterium that can be used as a drug delivery system to treat regions that have been resistant to previous drug delivery therapies. In this study, we reviewed the potential of T. gondii in drug development and as a delivery system through current research on inflammation-regulating mechanisms in immune-mediated inflammatory diseases.
Similar content being viewed by others


Introduction
Immune-mediated inflammatory diseases (IMIDs) are clinically heterogeneous groups of diseases that include rheumatoid arthritis (RA), spondyloarthritis (SpA) disease spectrum, connective tissue disorders, cutaneous inflammatory conditions (including psoriasis and atopic dermatitis), inflammatory bowel disease (IBD), asthma and autoimmune neurological diseases1, and their incidence rate has increased over the past several decades2. Furthermore, several IMIDs, including IBD, colitis-associated colorectal cancer (CAC), celiac disease, small bowel adenocarcinoma (SBA), primary sclerosing cholangitis, and hepatobiliary cancer, have been associated with an increased risk of developing cancer3.
IMIDs are characterized by an imbalance in inflammatory cytokines and dysregulation of the normal immune response. IMIDs share inflammatory pathways initiated by dendritic cells (DCs) and subsets of CD4+T cells, such as T helper type 1 (Th1) cells, T helper type 2 (Th2) cells, T helper type 17 (Th17) cells, T follicular helper (Tfh) cells, B regulatory (Breg) cells, and T regulatory (Treg) cells4,5,6,7. In addition, DCs release interleukin-12 (IL-12) and IL-23 to stimulate Th1 cells, Th17 cells, and Th22 cells to increase the levels of proinflammatory cytokines, including IL-17, interferon-γ (IFN-γ), tumor necrosis factor (TNF) and IL-228,9. Furthermore, a recent study reported that psoriatic patients have elevated levels of inflammatory serum markers, such as beta-defensin 2, lipocalin 2, IL-8, IL-22, and calprotectin10. In particular, the expression levels of proinflammatory genes, including CCL2, CCL5, CCL22, VEGF, IL-15, IL-15, TNF, and S100A12, are elevated in the skin and blood of psoriatic patients9. Furthermore, recent research has indicated that the Janus kinase/signal transduction and activator of transcription (JAK/STAT) signaling pathway is involved in IMIDs. Diverse proinflammatory cytokines within IMIDs and tumor environments activate JAK/STAT signaling through transduction. The production of type l interferons (IFNs), IL-6, IL-12, and IL-23 is specifically associated with the class I and II cytokine families11. For example, Th17 cells, which secrete cytokines such as IL-17 and IL-22, are differentiated by IL-6-activated JAK/STAT signaling12. Furthermore, the activation of the JAK/STAT pathway by IL-6 activates different intracellular signaling pathways, including the mitogen-activated protein kinase pathway and the phosphoinositide 3-kinase (PI3K)/protein kinase B (or Akt)/mechanistic target of the rapamycin (mTOR) pathway13. In contrast, anti-inflammatory cytokines such as IL-4 and IL-10 can be effective modulators of IMIDs14,15.
In this review, we described the immune dysregulation that various IMIDs cause, examined the current treatment and drawbacks of IMIDs, and focused on identifying new drugs available for use in immune-mediated diseases.
Treatment and drawbacks of IMIDs
Traditional clinical treatments for IMIDs include nonsteroidal anti-inflammatory drugs (NSAIDs), glucocorticoids, and disease-modifying antirheumatic drugs (DMARDs)16. NSAIDs are commonly used to treat arthritis and headache and have analgesic, antipyretic, and anti-inflammatory properties17. NSAIDs work by inhibiting cyclooxygenase (COX), an enzyme that transforms arachidonic acid into prostaglandins, which are mediators of inflammation and pain18. The effects of NSAIDs are analgesic and anti-inflammatory. However, NSAIDs cannot alter the course of IMIDs or prevent damage on their own19. Glucocorticoids have potent anti-inflammatory effects and are used to relieve pain and suppress inflammation. However, glucocorticoids are only temporarily used due to their severe side effects20. To control the disease process, researchers recommend combining NSAID therapy with DMARDs such as methotrexate (MTX) and leflunomide21. MTX is a folic acid-like substance that inhibits intracellular metabolism by preventing the binding of dihydrofolic acid to dihydrofolate reductase22. Leflunomide inhibits pyrimidine synthesis and T-cell proliferation23, demonstrating an effect comparable to that of MTX in clinical trials. Leflunomide is administered to patients who have contraindications to MTX or who have not demonstrated any MTX-related effects. In addition, combination therapy with leflunomide is highly effective24. Several studies indicate that TNF is a therapeutic target in IMIDs25,26. Furthermore, anti-TNF medications such as infliximab, adalimumab, etanercept, golimumab, and certolizumab pegol significantly alleviate signs and symptoms in patients with IMIDs27. Recently, studies have shown that small molecules that inhibit the intracellular JAK/STAT signaling pathway are bioavailable. Currently, five JAK inhibitors (JAKis), namely, tofacitinib, baricitinib, peficitinib, upadacitinib, and filgotinib, are authorized for use in the treatment of one or more IMIDs28. However, all of these treatments have disadvantages and side effects. NSAIDs and selective COX-2 inhibitors increase the risk of stomach ulcers and cardiovascular disease29. Glucocorticoids also induce severe adverse effects, including ulcers and bleeding in the gastrointestinal tract, infection, immunosuppression, and bone damage30. Furthermore, MTX and leflunomide are associated with hepatotoxicity, pulmonary toxicity, hematologic abnormalities, skin rash, hair loss, weight loss, thrombocytopenia, and diarrhea31. Recent research by Li et al. indicated that anti-TNFα biopharmaceuticals increase the risk of serious infections, cardiovascular events, and32 cancer. Furthermore, Boyadzhieva et al. confirmed that JAKis have adverse effects, including infectious events, embolism, and thrombosis33. Most IMIDs are incurable and are accompanied by complications, including cardiovascular disease, metabolic and bone disorders, and cognitive deficits, which increase mortality1. Moreover, IMIDs result from the immune response by aberrant cytokine production, while tumors develop when the immune system does not respond to malignant cells. Therefore, several cancers and cancer treatments have serious adverse effects that increase the risk of IMIDs.
In contemporary medicine, the development of pharmaceuticals faces obstacles such as adverse drug effects, drug toxicity, and drug resistance. Thus, recently, biological drugs derived from the microbiome or parasites have been used to treat various diseases, reduce adverse drug effects and improve treatment outcomes.
Immune response to Toxoplasma gondii infection
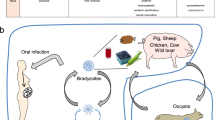
T. gondii is a protozoan parasite that infects approximately one-third of the human population worldwide34. T. gondii causes long-term chronic infection in the tissues of warm-blooded animals and humans and exists as a latent infection state primarily in the tissues of the central nervous system (CNS)35. Furthermore, T. gondii has developed self-preservation mechanisms to manipulate the immune systems of its hosts, such as by inducing an immune response or immune system evasion. Toll-like receptors (TLRs) are the first to respond to T. gondii infection. Gopal et al. reported that the mRNA expression levels of TLR2, TLR4, TLR9, and TLR11 were significantly increased in the MODE-K cell line in response to T. gondii ME49 strain infection36. Moreover, Hamie et al. reported that TLR7, TLR11, and TLR12 expression was significantly upregulated in the brains of imiquimod-induced interconversion mice37. Another study reported that UNC93 homolog B1 (UNC93B1), which plays an important role in producing IL-12 and IFN-γ, is involved in the function of TLRs (TLR3. TLR7, TLR9, TLR11, TLR12, and TLR13)38,39. TLR stimulation activates a signaling cascade involving the TLR-associated adaptor protein MyD88 and the production of IL-12 by dendritic cells (DCs)40. The production of IL-12 induces the differentiation of CD4+ T cells into Th1 cells expressing the transcription factor T-bet, which is needed for IFN-γ production41,42,43. Interestingly, Benson et al. demonstrated that TLR2, TLR4, and TLR9 play crucial roles in regulating IFN-γ responses in C57BL/6 mice infected with T. gondii ME49, and TLR11 deficiency has a slight effect on IFN-γ production in T. gondii during oral infection. In contrast, the IL-12 response was not elevated by TLR2, TLR4, or TLR9, and only TLR11 activated the production of IL-12 in DCs from T. gondii ME49 strain-infected mice44. Moreover, Yarovinsky et al. reported that the activation of the MyD88-dependent downstream immune signaling pathway through TLR11 is associated with TLR1245. Raetz et al. demonstrated that TLR11 and TLR12 directly bind to T. gondii profilin (TgPRF) and assemble to form a heterodimeric complex and that IFN regulatory factor 8 (IRF8) plays a crucial role in the production of IL-12 through the regulation of TLR11 and TLR1246. Koblansky et al. showed that the production of IL-12p40 was decreased in macrophages obtained from TLR12-deficient mice stimulated with TgPRF. Furthermore, the likelihood of T. gondii infection was significantly greater in TLR12-deficient mice than in WT mice, and the survival rate was decreased47. However, despite TLRs playing a crucial role in the detection of T. gondii, TLR11 is a pseudogene, and TLR12 is not expressed in humans45,48. TLR7 is a receptor for detecting the RNA of pathogens, such as RNA viruses and bacterial RNA. TLR9 recognizes bacterial and viral DNA containing the cytosine–phosphate–guanine (CpG) dideoxynucleotide motif and induces proinflammatory cytokine production by DCs45,49. Numerous studies have reported that the innate and adaptive immune systems respond to T. gondii infection by recruiting various cell types, such as inflammatory monocytes, antigen-presenting cells (APCs), T cells, neutrophils (NEs), and natural killer (NK) cells50,51. Several in vivo studies have demonstrated the requirement for T cells and the cytokine IFN-γ for preventing parasite reactivation. Disruption of CD8+ T cells alone was associated with 50% mortality; in contrast, no mortality was observed when CD4+ T cells were exhausted alone52,53,54. Moreover, the maintenance of memory CD8+ T cells is dependent on CD4+ T-cell help, which is mediated predominantly by IL-2 and IL-21 production55. Another study reported that brain pathology or mortality is significantly enhanced in C57BL/6 mice chronically infected with an ME49 strain of T. gondii treated with an anti-CD4 or anti-CD8 mAb alone52. In contrast, mice treated with a combination of anti-CD4 and anti-CD8 antibodies showed augmented pathology and mortality similar to those of anti-IFN-γ-treated mice52. These results indicate that although CD8+ T cells play a dominant role in preventing chronic infection, their maintenance is dependent on the critical help provided by CD4+ T cells. CD4+ T cells are crucial for the induction of primary CD8+ T-cell differentiation by converting APC into an effective stimulator56 (Fig. 1). Furthermore, Khan et al. demonstrated that the exhaustion of CD4+ T cells by increased expression of the transcription factor BLIMP-1 caused CD8+ T dysfunction, leading to the reactivation of latent infection in chronically infected hosts55 (Fig. 1). Furthermore, innate immune cells, such as neutrophils, NK cells, and innate lymphoid cells, sequentially and mutually lead to the production of IFN-γ57. IFN-γ-mediated immune responses play a crucial role in host protection, and Th2-type immune responses are also involved in resistance to T. gondii infection58,59. All IL-4-deficient mice died late in toxoplasmosis infection, whereas all control mice survived59. A histological study showed that significantly more cysts and areas of acute focal inflammation involved tachyzoites in the brains of IL-4-deficient mice than in those of the control mice in the late stage of T. gondii infection59. Furthermore, the production of IFN-γ stimulated by T. gondii antigens was significantly elevated in the spleen cells of control mice compared with those of IL-4-deficient mice59. These results indicate that IL-4 plays a role in inducing IFN-γ production at the late stage of T. gondii infection. Additionally, T. gondii infection activates nucleotide-binding domain leucine-rich repeat containing receptors (NLRs), which in turn trigger inflammasome assembly and the subsequent activation of caspase-1 and IL-1β secretion, resulting in pyroptosis in various cell types60.

TLR11 and TLR12 are the first molecules to recognize TgPRF. TLR7 and TLR9 are receptors for detecting T. gondii RNA and genomic DNA, respectively. UNC93B1 binds to endolysosomal TLRs in the endoplasmic reticulum, and stimulation of TLRs activates MyD88-dependent signaling. The MyD88-dependent signaling pathway, which depends on the activation of IRF8, induces IL-12 production in DCs. TLR2, TLR4, and TLR9 play central roles in the production of IFN-γ. However, the production of IL-12 is induced by TLR11 and TLR12. Activated IL-2 induces the differentiation of CD4+ T cells into Th1 cells expressing T-bet, which is involved in IFN-γ production. In addition, the production of IL-2 and IL-21 by CD4+ T cells helps maintain memory CD8+ T cells.
Interestingly, to resist the immune response, T. gondii has successfully evolved strategies to evade the host immune system and migrate to immune-privileged sites, such as the CNS. T. gondii prevents the host immune system from entering through multiple mechanisms, including transcriptional regulation and interference with cell signaling, resulting in the secretion of inflammatory cytokines, modulation of matrix metalloproteases (MMPs), production of microbicidal molecules and apoptosis61,62,63,64. In particular, T. gondii secretes effector proteins, including rhoptry proteins (ROPs) and dense granules (GRAs), from secretory organelles; these proteins alter host cell signaling pathways and protect T. gondii throughout infection65.
T. gondii-associated diseases
Primary and chronic T. gondii infection is generally harmless and asymptomatic in most immunocompetent individuals66. However, T. gondii infection is most dangerous during pregnancy and in people with compromised immune systems, such as those with human immunodeficiency virus (HIV)67. If a pregnant woman contracts a primary infection, it is transmitted to the fetus through the amniotic fluid and placenta, resulting in congenital toxoplasmosis68. Congenital toxoplasmosis is responsible for fetal death and infant hydrocephalus, cerebral calcifications, and ocular toxoplasmosis (OT)69. Typically, prenatally acquired infections frequently cause OT. However, several studies have reported that postnatally acquired infection is also associated with OT70,71. The clinical presentation of OT depends on the anatomical location of the lesion72. Furthermore, de-la-Torre et al. reported that specific cytokine patterns, such as inflammation, recurrence, and infection with a T. gondii strain, are correlated with the clinical presentation of OT73. Knight et al. reported that IL-4, IL-6, CCL2, CXCL-2, and CXCL-8 were increased in Müller cells infected with T. gondii RH or the Prugniaud strain. In contrast, IFN-γ and IL-12 expression is not altered by T. gondii infection74. Furthermore, various studies have reported that the levels of Th1 cells and inflammatory cytokines, such as IL-2, IL-6, IL-17, and IFN-γ, are elevated in the aqueous humor of OT patients. In contrast, the Th2 cytokine IL-13 was poorly expressed73.
Acute infection and reactivation of T. gondii are associated with multiple sclerosis (MS) and neurological disorders by inducing inflammatory lesions in the CNS in patients with severely compromised immune systems. MS is an inflammatory disease of the CNS that is primarily driven by a chronic autoimmune response. In recent years, several studies have reported an association between T. gondii infection and MS incidence. Except for one study, four out of five studies revealed a negative association between T. gondii infection and MS; however, one study that reported a positive association had an insufficient sample size, particularly because of a lack of significance. More recently, Nicoletti et al.75 demonstrated a significant negative association between T. gondii seropositivity and MS in a population-based case‒control study that included 164 patients and 481 controls. Furthermore, Cicero et al. reported a significant negative association between T. gondii and MS via meta-analysis. The incidence of clinical symptoms and the expression levels of cytokines such as TNF-α and INF-γ were lower in T. gondii RH strain-infected MS mice than in MS mice76.
Parkinson’s disease (PD) and Alzheimer’s disease (AD) are the most common age-related neurodegenerative disorders. Numerous epidemiological and experimental studies have demonstrated the association between T. gondii infection and the development of PD and AD. Associations between T. gondii and PD were based on data collected from eight studies. Two of the eight studies showed a significant association based on T. gondii infection determined via IgG-ELISA, and one revealed a significant association based on T. gondii infection defined through PCR. However, an increase in the incidence of T. gondii infection in PD patients compared with that in control individuals might not indicate a risk factor for PD. In fact, PD patients are at increased risk of T. gondii infection due to decreased personal and food hygiene habits. Furthermore, Çelik et al. and Oskouei et al. did not observe significant differences between T. gondii infection and PD77,78. Several studies have suggested that T. gondii infection is a risk factor for the development of AD. Galeh et al. investigated the effects of chronic T. gondii infection on cognitive impairment in an amyloid beta 1–42 (Aβ1-42)-induced AD rat model established with the Types I (RH), II (PRU), and III (VEG) strains and in combination. The RH strain plays a detrimental role in AD pathogenesis. In contrast, chronic infection with the PRU strain or a combination of the PRU and VEG strains significantly improved cognitive deficits in Alzheimer’s disease rat models79.
In summary, the possible association between T. gondii infection and disease risk has still not been fully characterized, and conversely, a negative correlation between T. gondii infection and disease status has been reported. Therefore, these results should be interpreted cautiously, and further studies are needed to confirm the association between T. gondii and disease.
Anti-inflammatory effects of T. gondii
Multiple studies have indicated that T. gondii modulates the cytokine response by either inhibiting or augmenting it, resulting in an antagonized immune response. Recently, Charles et al. reported that CD4+ T-cell activation is suppressed by the cell surface protein programmed death ligand 1 (PD-L1) in mouse retinal pigment epithelial cells and leukocytes80. Additionally, Washino et al. reported that IL-17 mRNA expression is inhibited in CD4+ T cells isolated from the mesenteric lymph nodes and spleen of IL-1Ra-deficient induced-RA model mice infected with T. gondii. Moreover, the mRNA expression of RORγt, a key transcription factor for Th17 cells on CD4+ cells, was decreased in infected mice compared to control mice, indicating that T. gondii regulates transcription factors to reduce Th17 cell polarization81. Furthermore, Kim et al. reported that the ratio of CD4+ cells, macrophages, and NK cells classified as splenocytes is significantly reduced in T. gondii-infected mice82. Similarly, Butcher et al. demonstrated that T. gondii-infected macrophages do not produce TNF-α or IL-12 compared to lipopolysaccharide (LPS)-treated peritoneal macrophages and several macrophage lines, including J774, RAW264.7, and THP-1 cells. Furthermore, LPS-induced TNF-α production was reduced in macrophages infected with T. gondii83. In addition to its effect on macrophages, LPS stimulation does not fully increase TNF-α levels in T. gondii-infected NEs compared to those in T. gondii-infected NEs treated with LPS alone84.
Several studies have indicated that T. gondii stimulates the secretion of anti-inflammatory cytokines. Yan et al. reported an increase in IL-4 mRNA expression and secretion in T. gondii-infected RAW264.7 macrophages compared to those in the control group85. Hunter et al. demonstrated that the mRNA expression of IL-4 was detected concurrently with the onset of inflammation 10 days after infection with the RRA strain of T. gondii in the brains of C57BL/10 Sc Sn mice. Furthermore, of all the mice, 2 were still IL-4 positive on Day 2086. Suzuki et al. reported that T. gondii cysts, foci of acute inflammation, and tachyzoite-specific mRNAs were significantly more common in the brains of IL-4−/− mice than in those of controls in the early stage of T. gondii infection59. In the early stages of T. gondii infection, the complex reaction of rapidly dividing tachyzoites and the immune response can often lead to host death, and IL-4 plays a protective role by reducing the products of Th1 cells87. Another study reported that IL-4, IL-10, and transforming growth factor-β (TGF-β) levels gradually and significantly increased in the T. gondii acute infection stage of C57BL/6 mice. Furthermore, the transcript levels of anti-inflammatory cytokines (IL-4, IL-10, and TGF-β) were increased in T. gondii-infected mouse brains compared with those in control mouse brains88. Additionally, compared with that in noninfected control mice, IL-10 expression in the intestines of infected mice was elevated89. Furthermore, Bliss et al.90 demonstrated that the expression of IL-10 was detected by confocal fluorescence microscopy in T. gondii-infected macrophages and confirmed that IL-10+ cells were F4/80+ by FACS. Elevated levels of IL-10 were detected in the lymph nodes and CNS of CBA/Ca mice that developed progressive toxoplasmic encephalitis (TE)91. Beaman et al. reported that STAT3 is activated by IL-6 and IL-10, which antagonize the defense mechanism and result in enhanced susceptibility to toxoplasmosis. Furthermore, activated STAT3 reduces IL-12 production to evade immunosurveillance; however, T. gondii also directly activates STAT3 to escape host immunity. T. gondii also upregulates SOCS3, which blocks the phosphorylation of STAT3 by inhibiting JAK-2 directly. The depletion of SOCS3 resulted in elevated T. gondii-induced STAT3 signaling, and macrophages and neutrophils with SOCS3 deletion had reduced levels of IL-12 and IFN-γ and succumbed to toxoplasmosis. These results indicate that SOCS3 might have additional mechanisms to restrict the phosphorylation of STAT3. In addition, T. gondii antagonizes IFN-γ production by interfering with NF-κB activation and STAT1 activation, which are inhibited by SOCS192 (Fig. 2). Zhang et al. demonstrated that IL-5 expression is increased in the brain and spleen of C57BL/6 mice infected with the ME49 strain of T. gondii compared with those in the control group. Moreover, IL-5 KO mice exhibited decreased survival rates and weight loss during T. gondii infection93. In contrast, Nickdel et al. reported that, compared with WT mice infected by the same route, IL-5-deficient mice infected with the RRA strain T. gondii through the oral route had significantly greater survival rates, irrespective of sex. Moreover, splenocytes obtained from WT mice produced significantly more IL-12 and IFN-γ than those obtained from IL-5−/− mice when stimulated with TLA. Conversely, compared with those in WT mice, the plasma IL-12 and IFN-γ levels in IL-5-deficient mice infected with T. gondii orally were significantly greater on Days 6 and 8, respectively94. These results indicate that IL-5 plays different roles in influencing the immune response and disease progression between acute and chronic T. gondii infection. TGF-β is an anti-inflammatory cytokine that plays an important role in antagonizing TNFα, TNFβ, IFNγ, and IL-1295,96. Several studies have reported that TGF-β-deficient mice develop inflammatory autoimmune disease97,98,99. Nagineni et al. reported that TGF-β1 and TGF-β2 mRNA levels are increased in T. gondii-infected human retinal pigment epithelial cultures (HRPE). Furthermore, soluble extracts of T. gondii significantly increase the secretion of both TGF-β1 and TGF-β2100.

After invasion of the host cell, T gondii alters host signaling pathways and gene expression, antagonizing immune responses. STAT3 activated by IL-6 and IL-10 is associated with antagonizing host defense. T. gondii also directly activates STAT3 to antagonize the host immune response. T. gondii also upregulates SOCS3, which binds JAK-2 and inhibits the phosphorylation of STAT3. The depletion of SOCS3 enhanced T. gondii-induced STAT3 signaling, resulting in reduced levels of IL-12 and IFN-γ. Taken together, these results indicate that SOCS3 might have additional mechanisms to limit the phosphorylation of STAT3. In addition, T. gondii antagonizes IFN-γ production by interfering with NF-κB activation and STAT1 activation, which are inhibited by SOCS1.
In summary, T. gondii infection leads to anti-inflammatory cytokine production through an altered host immune system by manipulating host gene transcription and regulating signaling pathways.
Reversing the immune response by T. gondii
In recent decades, multiple studies have indicated that inflammation is involved in tumorigenesis. Chronic inflammation initiates tumor progression through genetic modifications, such as the accumulation of DNA mutations, epigenetic alterations, or both101. Furthermore, inflammatory cytokines, such as TNF-α, IL-1, and IL-6; growth factors; chemokines; and proteases secreted by tumor-associated lymphocytes and macrophages increase tumor cell proliferation, survival, migration, and invasion, thus promoting tumor growth and metastasis102. Furthermore, tumor-associated macrophages (TAMs) produce inflammatory mediators involved in tumor angiogenesis and lymphangiogenesis103 and secrete immune-suppressive soluble cytokines, including TGF-β and IL-10104. Interestingly, inflammation has a questionable benefit in relation to cancer. In contrast, TNF functions as an endogenous tumor promoter. TNF inhibits tumor growth by inducing apoptosis and inhibiting nuclear factor κB (NF-κB) signaling105. Furthermore, cell death through CD8+ cytotoxic T cells and natural killer (NK) cells, as well as tumor cell senescence induced by CD4+ Th1 cells that produce IFN-γ and TNF-α, inhibits tumor growth106. Another study revealed that IFN-γ signaling and antigen presentation are essential for the lysis mediated by CD8+ cytotoxic T cells in mouse colon carcinoma cells and that TNF is a key factor in antitumor activity107. Consequently, the immune response reawakened by T. gondii can increase susceptibility to cancer treatment or lead to the development of an effective treatment.
According to previous research, immune cells, including NEs, DCs, macrophages, and T cells, produce TNF-α in response to T. gondii infection. Bliss et al. reported that NEs treated with the T. gondii antigen produced IL-12p70 and TNF-α. Additionally, elevated TNF-α induces macrophage-inflammatory MIP-1α and MIP-1β gene expression via autocrine stimulation in T. gondii-infected human peripheral blood neutrophils108. Additionally, T. gondii recruits NEs through a multitude of immature DC chemotactic chemokines. Furthermore, T. gondii-cascade neutrophils release chemokines that induce DC CD40 and CD86 upregulation in a manner dependent on T. gondii-induced neutrophil TNF-α production109. The ovarian tumor protease (OTU) domain, ubiquitin aldehyde binding 1 (OTUB1), and deubiquitinase increase in DCs after infection with T. gondii and LPS stimulation. Mulas et al. demonstrated that TgPRF and LPS-induced activation of NF-κB increased the production of IL-6, IL-12, and TNF-α in DCs110. Similarly, Corrêa et al. reported that T. gondii infection increases the expression of proinflammatory cytokines, including IL-12, IL-1β, IFN-γ, and TNF-α, in macrophages from P2X7+/+ mice111. Mennechet et al. demonstrated that IFN-γ and TNF-α levels were elevated in primed lamina propria CD4+ T cells isolated from mice infected with T. gondii. Furthermore, the expression of C-C chemokine mRNAs, such as RANTES, monocyte chemoattractant protein-1 (MCP-1), and MCP-3, and CXC chemokine mRNAs, such as IP-10, was significantly upregulated in response to infection with T. gondii112.
Although the suppressive effect of the increased immune response induced by T. gondii infection in different cancer types has not yet been fully elucidated, several studies have shown that T. gondii contributes to host antitumor immunity. Wu et al. demonstrated that T. gondii infection induces apoptosis and inhibits cell proliferation in various cancer cell lines, including the breast cancer cell line MCF-7, prostate cancer cell line DU145, esophageal cancer cell line EC-109, and lung cancer cell line A549113,114. In addition, a previous study showed that T. gondii infection modulates the expression of tumor-related factors, thus improving the antitumor capacity of hosts. TP53 is a tumor suppressor gene that is frequently mutated in various cancers. T. gondii infection upregulates the expression of Ladd45, which induces cell cycle arrest and apoptosis and is involved in the p53 signaling pathway. This study showed that DCC, Smad3, Smad4, hMLH1, hMSH2, and hMSH3, which are involved in the colorectal cancer pathway, were regulated by T. gondii infection. Furthermore, T. gondii infection increases the RASSF1 tumor suppressor gene in non-small cell lung cancer (NSCLC) patients. After T. gondii infection, the expression of the breast cancer-related genes CCND1 and PRCA2 decreased and increased, respectively115.
Several studies have reported the application of nonreplicating T. gondii uracil auxotrophs (NRTUAs), which are considered “immunotherapy attenuated vaccine strains”, in cancer treatment. Although NRTUAs are nontoxic and are rapidly cleared by the immune system in approximately 5 days, they are still able to significantly inhibit tumor progression even after they are killed. Baird et al. reported that NRTUAs with carbamoyl phosphate synthetase II (CPS-II) knockout invade CD8+ T cells and NK cells to induce antitumor immune activity. NRTUA treatment altered IL-12, IFN-γ, and the CXCR3-stimulating cytokine-involved CD8+ T-cell population in the tumor microenvironment (TME), subsequently inhibiting melanoma tumor growth in mice116. Moreover, RH-Δcps predominantly invades immunosuppressive CD11c+ APCs and converts to a CD80- and CD86-expressing immunostimulatory phenotype in the ovarian carcinoma microenvironment. In response to increased levels of T-cell receptor costimulatory molecules, CD11c+ cells reawaken the ability to cross-present and prime CD8+ T cells. Moreover, RH-Δcps treatment significantly reduced the tumor size and enhanced the survival rate of ovarian tumor-bearing mice. These results demonstrated that T. gondii effectively inhibits ovarian tumor progression by restoring the T-cell population in tumor-bearing mice117. Sanders et al. reported that RH-Δcps treatment significantly enhances infiltrating CD4+ and CD8+ T-cell activation and elevates the number of circulating tumor-specific T cells in the TME in pancreatic cancer patients. Furthermore, RH-Δcps treatment had significant therapeutic effects on pancreatic tumor-bearing mice, as indicated by changes in IL-12 and IFN-γ production, MyD88 signaling, and CD8+ T-cell populations118. Another NRTUA, orotidine-5’-monophosphate decarboxylase (OMPDC) knockout, revealed vaccine features that might induce significant immune responses by enhancing the expression of inflammatory cytokines such as IL-12p40 and IFN-γ in mouse serum and the TME. Moreover, RH-Δompdc restricted tumor growth, reduced lung metastasis, and increased the survival rate of 4T1 breast tumor-bearing mice119. Bahwal et al. reported that the combination of RH-ΔompdcΔup and an anti-PD-1 antibody induced a significant antitumor immune response and synergistically restricted tumor growth through the enhancement of CD8+ T-cell infiltration induced by DC-secreted IL-12 and IFN-γ production in pancreatic ductal adenocarcinoma (PDAC) tumors120. Li et al. reported that ME49-Δompdc inhibits the growth of B16F1 melanoma by eliciting a Th1 immune response, with high levels of IL-12, TNF-α, and IFN-γ and CD8+ T-cell activation in C57BL/6 mice121 (Fig. 3).

gondii infection might reverse the immunosuppressive state of the TME to stimulate effective therapeutic immunity. T. gondii recruits macrophages that produce proinflammatory cytokines, and the produced IFN-γ and TNF-α enhance CD4+ T-cell priming. In addition, T. gondii invades CD11c+ APCs and converts DCs to a CD80- and CD86-expressing immunostimulatory phenotype via MyD88 signaling. IL-12 and IFN-γ production restricts tumor growth through enhancement of CD8+ T-cell infiltration. In addition, T. gondii infection significantly enhances infiltrating CD4+ T-cell activation and helps maintain CD8+ T-cell activation. Finally, macrophages, CD4+ T cells, and CD8+ T cells are recruited to the TME to restrict tumor growth synergistically via secreted IL-12 and IFN-γ.
In conclusion, T. gondii has multiple functions associated with host and antitumor immune responses by reversing tumor-associated immunosuppression. However, because the pathological mechanisms differ among various carcinomas, further research is needed to fully understand the antitumor effect of T. gondii.
Immune response to T. gondii antigens
T. gondii microneme proteins (MICs), ROPs, GRAs, surface antigens (SAGs), and SAG-1-related sequences (SRSs) are antigens that play crucial roles in parasite infection and maintaining intracellular host-parasite relationships. First, the MICs located at the tip of T. gondii mediate the motility and adhesion of the host cell. When the parasite is transmitted to a host cell, the apical membrane antigen 1 (AMA1)-rhoptry neck protein (RON) pairs form a moving junction, and the subsequent ROPs involved in the formation of the parasitophorous vacuole membrane (PVM) are secreted simultaneously with invasion. After the invasion and establishment of the PVM, GRA secretion continues and is involved in the maturation of the PVM and in the maintenance of host-parasite interactions for intracellular survival and replication122,123,124.
Recent studies have reported that T. gondii antigens influence the immune response. Benevides et al. showed that IFN-γ and TNF-α levels were significantly elevated in the ileum of T. gondii-infected mice [76]. However, the levels of the IFN-γ and TNF mRNAs were lower for the soluble antigen of T. gondii tachyzoites (STAg) than for those of T. gondii-infected mice. Furthermore, pretreatment with soluble STAg decreased the number of CD4+ T cells expressing proinflammatory cytokines in the lamina propria (LP) and increased the number of CD8+ T cells in the intraepithelium of the small intestine125. Moreover, STAg increases the expression of suppressor of cytokine signaling 2 (SOCS2) in DCs through the action of endogenous lipoxin A4 (LXA4), which is dependent on 5-lipoxygenase (5-LOX), resulting in a reduction in the secretion of IL-2 and in the secretion of C-C chemokine receptor 5 (CCR5)126. According to another study, TNF-α release and IL-1β secretion are reduced by pretreatment of LPS-stimulated Ana-1 macrophages with T. gondii excretory/secretory antigens (TgESAs)127. These findings suggest that ThAgs, a component of T. gondii, modulate immune modulation. MIC1 and MIC4 interact with TLR2 and TLR4 and activate NF-κB to induce the expression of proinflammatory cytokines, such as IL-12, TNF-α, and IL-6, in mouse bone marrow-derived macrophages (BMDMs). Interestingly, MIC1 and MIC4 also stimulate the secretion of the anti-inflammatory cytokine IL-10 by transporting TLR4 into endosomes, although the underlying mechanisms are unknown128.
Recently, Chen et al. demonstrated that ROP16 interacts with STAT3 and STAT6 in host cells to promote their phosphorylation, resulting in a cascade of downstream signals for host cell manipulation129. The phosphorylation of STAT6 by ROP16 induces arginase-1 (Arg-1), which is involved in anti-inflammatory effects, tumor immunity, and immunosuppression-related diseases130. Arg-1 is also a key effector and marker of M2a macrophages through STAT3 and STAT6131. M2 macrophages secrete anti-inflammatory cytokines, such as IL-10 and IL-1ra, which inhibit the host immune response to T. gondii infection81,89,132. Furthermore, ROP16 promotes the differentiation of M2 macrophages through STAT6 and STAT3 activation by inducing IL-4 and IL-10, respectively133. Specifically, ROP16 types I and III (ROP16 I/III) activate M2 macrophages to ameliorate IBD134. Similarly, the deletion of ROP5, ROP18, and GRA7 was associated with resistance to IFN-γ-activated immunity-related GTPases (IRGs), such as Irgm1/m3, which degrade PV to promote parasite clearance, resulting in increased PV clearance and presentation of soluble PV antigen by MHC I molecules in macrophages or DCs from C57BL/6 mice135. Kim et al. reported that GRA9 has the potential to ameliorate sepsis by increasing anti-inflammatory and antibacterial effects through the conversion of M1 macrophages into M2 macrophages. Furthermore, GRA9 inhibits NLRP3 inflammasome assembly and suppresses the activation of pro-IL-1β and pro-IL-18 in BMDMs136. The T. gondii antigens SAG-1 and SAG-2 inhibit the mRNA expression of Th1 cytokines and proinflammatory cytokines, including IL-1, IL-6, GM-CSF, and TNF-α. Furthermore, the levels of SAG-1 and SAG-2. increased. Treatment with T. gondii infection and an anti-IFN-gamma mAb inhibited the expression of TNF-alpha and iNOS137.
Several studies have indicated that T. gondii antigens stimulate an antitumor immune response. PVM-associated T. gondii antigens, such as ROP5, ROP17, and ROP18, are essential for inducing an immune response. Furthermore, although the antitumor mechanisms of ROP35 and ROP38 are independent of the ROP5, ROP17, and ROP18 complexes, PVM-associated ROP35 and ROP38 are essential for the antitumor response. Furthermore, GRA2, GRA12, and GRA24 significantly inhibited the antitumor response to ovarian tumors138. Li et al. reported that GRA15 induced M1 macrophage differentiation; suppressed multiple properties of hepatocellular carcinoma, including proliferation, migration, and invasion; and decreased the expression of MMP-2 and MMP-9. In addition to tumor tissues and growth suppression, TNF-α and IL-12 mRNA levels are increased in splenocytes, while the expression of the IL-6 and IL-10 mRNAs is downregulated139. Another study reported that GRA15 decreases cell viability and induces apoptosis in JEG-3 choriocarcinoma cells by regulating genes associated with apoptosis and endoplasmic reticulum stress genes140. Seo et al. reported that GRA16 inhibits upstream Akt signaling and suppresses NF-B signaling in a PP2A-dependent manner. In NSCLC, cotreatment with irinotecan, an anticancer drug, and GRA16 induced cell cycle arrest and inhibited cell proliferation141 (Fig. 4).

T. gondii antigens, including MICs, ROPs, GRAs, and SAGs, manipulate host cell signaling pathways. a In DCs, STAg induces the expression of SOCS2 via LXA4 and consequently suppresses IL-2 expression and CCR5 secretion. ROP5 and ROP18 suppress MHC I antigen presentation by resisting IFN-γ and IRG. b In macrophages, MIC1 and MIC4 bind to TLR4; induce the secretion of IL-12, TNF-α, and IL-6 through NF-κB; and translocate TLR4 to endosomes to produce the anti-inflammatory cytokine IL-10. SAG-1 and SAG-2 induce IL-1/IL-6/GM-CSF/TNF-α transcription in macrophages. GRA9 disrupts NLRP3 inflammasome assembly by blocking the binding of ASC to NLRP3 and consequently suppressing IL-1β and IL-18 secretion. ROP5, ROP18, and GRA7 inhibit MHC I antigen presentation by resisting IFN-γ and IRG. ROP16 activates STAT3- and STAT6-dependent M2 macrophages and induces the expression of the M2 macrophage marker Arg-1 and the secretion of IL-1 and IL-1ra.
In summary, antigens are expected to play an important role in regulating the immune response of T. gondii. However, despite the presence of numerous antigens of T. gondii, only immune responses to a few antigens have been identified. Therefore, further research on the function of other T. gondii antigens and their underlying mechanisms is needed.
A unique characteristic of T. gondii for drug delivery
Drug development is a costly and time-consuming process. In an effort to improve the safety and efficacy of previously developed drugs, numerous studies, including studies on drug delivery systems (DDSs), have been conducted. DDSs aim to efficiently deliver drugs to target locations, minimize adverse effects, and improve safety and efficacy to achieve the maximum therapeutic effect. Drug delivery consists of two concepts: first, a prodrug that has no medicinal effect outside the body but is metabolized by metabolic enzymes when it enters the body; and second, the direct delivery of drugs to target molecules based on a DDS142. Similarly, the second concept consists of a DDS using a polymer such as a synthetic polymer, micelle, liposome, or antibody and a DDS using convergence technology. Although the development of convergence technology has affected DDSs, novel technologies are still needed.
However, further studies are needed to develop new DDSs for the treatment of dementia, Parkinson’s disease (PD), and brain tumors, which are all diseases of the CNS caused by abnormalities or degeneration. These diseases are often difficult to treat via drug delivery. The blood‒brain barrier (BBB), a crucial immunological feature of the human CNS, plays a crucial role in blocking external toxic substances and restricting drug permeation143. In addition to brain diseases, the TME also has difficulties in drug delivery. The reason is that tumor blood vessels that supply nutrients to tumors are more prevalent at the tumor and host border than in central regions144. Therefore, the injection of drugs through blood vessels has a major limitation; drugs are delivered only to the periphery of blood vessels and rarely reach the center of the tumor.
A unique characteristic of T. gondii is its ability to metastasize to various organs, including the eyes, heart, liver, lungs, lymph nodes, and muscles. Furthermore, T. gondii can reach the brain, invade the CNS, and cause TE, establishing a continuous chronic infection in nerves and other brain cells145,146. There are three possible ways that T. gondii invades the CNS from the blood. First, in the BBB paracellular pathway, T. gondii tachyzoites accumulate in the peripheral circulation and cell-free T. gondii tachyzoites cross the BBB by disrupting tight junction proteins145,146. Second, during BBB endothelial cell lysis, free T. gondii and T. gondii released from positive monocytes can invade cerebrovascular endothelial cells (ECs), replicate in ECs, and subsequently impair the entry of ECs into the CNS147,148. The third way is to hijack immune cells; during viral or bacterial infections or neuroinflammatory diseases, DCs enter the perivascular, cerebrospinal fluid (CSF), and parenchymal regions of the brain through the BBB149. T. gondii successfully uses DCs and monocytes as vehicles for protection across the BBB and invasion of the brain150,151. Courret et al. demonstrated that spread from the intestine to the brain is predominantly mediated by CD11c− CD11b+ monocytes, which are preferentially infected via the blood and, to a lesser extent, CD11c+ CD11b+/− DCs152. This migration within infected immune cells is called the “Trojan horse” strategy (Fig. 5).

Three modes by which T. gondii invades the CNS from the blood. a T. gondii accesses the brain by disrupting tight junction proteins. b Direct infection and lysis of endothelial cells in the BBB by T. gondii. c The “Trojan horse” strategy, trafficking within infected immune cells.
Recent research has focused on immune cells as a drug delivery mechanism. Immune cells migrate within the TME and interact with other immune cells153. Circulating cell-mediated drug delivery, often specifically termed the Trojan horse strategy, has led to new strategies for delivering drugs to tumor centers or delivering drugs to difficult regions using host immune cells. For example, peripheral blood leukocytes are preferentially recruited to the TME, where they enhance tumor progression by inducing tissue remodeling and affecting angiogenesis154. NEs, the most abundant type of leukocyte, are reported to infiltrate inflamed brain tumors. Xue et al. demonstrated that liposomes that contain paclitaxel (PTX) and use NEs as “Trojan horses” can cross the BBB and suppress the relapse of glioma in mice whose tumor has been resected surgically155. Furthermore, He et al. reported that inflammatory monocyte-based biomimetic drug delivery systems loaded with legumain-activated nanoparticles inhibit the proliferation, migration, and invasion of breast cancer cells and penetrate metastatic tumors, resulting in significantly inhibited delivery to lung metastases156. Choi et al. demonstrated that nanoparticle-containing monocytes differentiated into macrophages and that the nanoparticle-laden macrophages penetrated the center of the T47D tumor spheroid. This result indicated that the tumor environment recruits monocytes and that nanoparticle-based drug delivery through monocytes might be exploited for therapeutic purposes157. Moreover, Lee et al. reported the use of inflammatory CD11b+ cells as active carriers to deliver doxorubicin-loaded nanoparticles into poorly vascularized regions of tumors158.
Although the application of Trojan horse drugs is promising, current therapy depends on adoptive cell transfer. In adoptive cell therapy, target cells are extracted from internal organs, cultivated ex vivo, and subsequently infused into patients159. However, ex vivo isolation and expansion consume time, and according to Marrache et al.160, the cytokine response of monocytes or macrophages is not always reproducible, preventing practical clinical translation. Furthermore, innate immune cells, such as NEs and monocytes, generally exhibit short lifespans that are less than expected in vivo161. To resolve these issues, drugs need flexible, functional groups that can be injected directly into the blood circulation and, depending on the target, conjugated to specific or diverse cell carriers.
Thus, the ability of T. gondii to infect various immune cell types, such as monocytes, DCs, and macrophages, and to infect Trojan horses has demonstrated its potential as a drug delivery tool.
Conclusion
IMIDs are a broad group of diseases caused by immune imbalances, such as abnormal cytokine production. However, currently, no definitive clinical treatment for IMIDs exists. Additionally, IMID treatment, which suppresses abnormal cytokine elevation, may increase the risk of developing cancer. The evidence provided in this manuscript leads to the conclusion that, due to their immunomodulatory capacity, T. gondii or T. gondii antigens could be new materials for treating IMIDs. First, T. gondii acts like a double-edged sword. T. gondii infection enhances the production of anti-inflammatory cytokines. However, conversely, NRTUA vaccination effectively controls tumor growth by reversing tumor-associated immunosuppression through an increase in proinflammatory cytokines. Furthermore, T. gondii antigens, such as MIC, ROP, and GRA, play roles in both stimulating the production of proinflammatory cytokines and inhibiting proinflammatory cytokines. Taken together, these findings suggest that T. gondii may maintain a balanced immune system by inducing a more stable and controlled immune response. Second, T. gondii has unique characteristics for drug delivery. T. gondii can infiltrate the BBB using immune cells, as in Trojan horses. Drug delivery via T. gondii employs immune cells that are involved in various diseases, and this approach might be applied to various diseases for which existing drug delivery has been difficult, including cancer. However, despite the presence of numerous T. gondii antigens, few T. gondii antigens have been identified. Thus, further studies are needed on the immune response to additional T. gondii antigens or synergistic effects of antigens and their molecular mechanisms. Ultimately, understanding and applying the immune response regulatory mechanisms of T. gondii may provide new perspectives on the potential for developing drugs for inflammatory diseases, cancer, and other intractable diseases.
References
McInnes, I. B. & Gravallese, E. M. Immune-mediated inflammatory disease therapeutics: past, present and future. Nat. Rev. Immunol. 21, 680–686 (2021).
David, T., Ling, S. F. & Barton, A. Genetics of immune-mediated inflammatory diseases. Clin. Exp. Immunol. 193, 3–12 (2018).
He, M. M. et al. Immune-mediated diseases associated with cancer risks. JAMA Oncol. 8, 209–219 (2022).
Schulze-Koops, H. & Kalden, J. R. The balance of Th1/Th2 cytokines in rheumatoid arthritis. Best. Pract. Res Clin. Rheumatol. 15, 677–691 (2001).
Wang, J., He, L., Li, W. & Lv, S. A role of IL-17 in rheumatoid arthritis patients complicated with atherosclerosis. Front. Pharm. 13, 828933 (2022).
Wehr, P., Purvis, H., Law, S. C. & Thomas, R. Dendritic cells, T cells and their interaction in rheumatoid arthritis. Clin. Exp. Immunol. 196, 12–27 (2019).
Yu, M., Cavero, V., Lu, Q. & Li, H. Follicular helper T cells in rheumatoid arthritis. Clin. Rheumatol. 34, 1489–1493 (2015).
Lowes, M. A., Suarez-Farinas, M. & Krueger, J. G. Immunology of psoriasis. Annu. Rev. Immunol. 32, 227–255 (2014).
Orlando, G. et al. Psoriasis and cardiovascular diseases: an immune-mediated cross talk? Front Immunol. 13, 868277 (2022).
Sokolova, M. V. et al. A set of serum markers detecting systemic inflammation in psoriatic skin, entheseal, and joint disease in the absence of C-reactive protein and its link to clinical disease manifestations. Arthritis Res. Ther. 22, 26 (2020).
Mok, C. C. The Jakinibs in systemic lupus erythematosus: progress and prospects. Expert Opin. Investig. Drugs 28, 85–92 (2019).
Banerjee, S., Biehl, A., Gadina, M., Hasni, S. & Schwartz, D. M. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs 77, 521–546 (2017).
Tzeng, H. T., Chyuan, I. T. & Lai, J. H. Targeting the JAK-STAT pathway in autoimmune diseases and cancers: a focus on molecular mechanisms and therapeutic potential. Biochem. Pharm. 193, 114760 (2021).
Muller-Ladner, U. et al. Activation of the IL-4 STAT pathway in rheumatoid synovium. J. Immunol. 164, 3894–3901 (2000).
van Roon, J. et al. Interleukin 10 treatment of patients with rheumatoid arthritis enhances Fc gamma receptor expression on monocytes and responsiveness to immune complex stimulation. J. Rheumatol. 30, 648–651 (2003).
Li, P., Zheng, Y. & Chen, X. Drugs for autoimmune inflammatory diseases: from small molecule compounds to anti-tnf biologics. Front. Pharm. 8, 460 (2017).
Rainsford, K. D. Anti-inflammatory drugs in the 21st century. Subcell. Biochem. 42, 3–27 (2007).
Hawkey, C. J. COX-1 and COX-2 inhibitors. Best. Pract. Res Clin. Gastroenterol. 15, 801–820 (2001).
Bindu, S., Mazumder, S. & Bandyopadhyay, U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: a current perspective. Biochem. Pharm. 180, 114147 (2020).
Oray, M., Abu Samra, K., Ebrahimiadib, N., Meese, H. & Foster, C. S. Long-term side effects of glucocorticoids. Expert Opin. Drug Saf. 15, 457–465 (2016).
Hodkinson, B., Magomero, K. R. & Tikly, M. Combination leflunomide and methotrexate in refractory rheumatoid arthritis: a biologic sparing approach. Ther. Adv. Musculoskelet. Dis. 8, 172–179 (2016).
Friedman, B. & Cronstein, B. Methotrexate mechanism in treatment of rheumatoid arthritis. Jt. Bone Spine 86, 301–307 (2019).
Breedveld, F. C. & Dayer, J. M. Leflunomide: mode of action in the treatment of rheumatoid arthritis. Ann. Rheum. Dis. 59, 841–849 (2000).
Kalden, J. R. et al. Use of combination of leflunomide with biological agents in treatment of rheumatoid arthritis. J. Rheumatol. 32, 1620–1631 (2005).
Silva, L. C., Ortigosa, L. C. & Benard, G. Anti-TNF-α agents in the treatment of immune-mediated inflammatory diseases: mechanisms of action and pitfalls. Immunotherapy 2, 817–833 (2010).
Zhu, Y. X., Kortuem, K. M. & Stewart, A. K. Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma. Leuk. Lymphoma 54, 683–687 (2013).
Stevenson, M. et al. Adalimumab, etanercept, infliximab, certolizumab pegol, golimumab, tocilizumab and abatacept for the treatment of rheumatoid arthritis not previously treated with disease-modifying antirheumatic drugs and after the failure of conventional disease-modifying antirheumatic drugs only: systematic review and economic evaluation. Health Technol. Assess. 20, 1–610 (2016).
Nash, P. et al. Points to consider for the treatment of immune-mediated inflammatory diseases with Janus kinase inhibitors: a consensus statement. Ann. Rheum. Dis. 80, 71–87 (2021).
Varga, Z., Sabzwari, S. R. A. & Vargova, V. Cardiovascular risk of nonsteroidal anti-inflammatory drugs: an under-recognized public health issue. Cureus 9, e1144 (2017).
Ethgen, O., de Lemos Esteves, F., Bruyere, O. & Reginster, J. Y. What do we know about the safety of corticosteroids in rheumatoid arthritis? Curr. Med. Res. Opin. 29, 1147–1160 (2013).
Karadag, A. S. et al. Pulmonary fibrosis developed secondary to methotrexate use in a patient with psoriasis vulgaris. North Clin. Istanb. 2, 159–161 (2015).
Li, J. et al. Risk of adverse events after Anti-TNF treatment for inflammatory rheumatological disease. a meta-analysis. Front. Pharm. 12, 746396 (2021).
Boyadzhieva, Z., Ruffer, N., Burmester, G., Pankow, A. & Krusche, M. Effectiveness and safety of JAK inhibitors in autoinflammatory diseases: a systematic review. Front. Med. (Lausanne) 9, 930071 (2022).
Chemoh, W. et al. Toxoplasma gondii infection: what is the real situation? Exp. Parasitol. 135, 685–689 (2013).
Tenter, A. M., Heckeroth, A. R. & Weiss, L. M. Toxoplasma gondii: from animals to humans. Int. J. Parasitol. 30, 1217–1258 (2000).
Gopal, R., Birdsell, D. & Monroy, F. P. Regulation of toll-like receptors in intestinal epithelial cells by stress and Toxoplasma gondii infection. Parasite Immunol. 30, 563–576 (2008).
Hamie, M. et al. Imiquimod targets toxoplasmosis through modulating host toll-like receptor-MyD88 signaling. Front. Immunol. 12, 629917 (2021).
Lee, B. L. et al. UNC93B1 mediates differential trafficking of endosomal TLRs. Elife 2, e00291 (2013).
Melo, M. B. et al. UNC93B1 mediates host resistance to infection with Toxoplasma gondii. PLoS Pathog. 6, e1001071 (2010).
Scanga, C. A. et al. Cutting edge: MyD88 is required for resistance to Toxoplasma gondii infection and regulates parasite-induced IL-12 production by dendritic cells. J. Immunol. 168, 5997–6001 (2002).
Däubener, W., Mackenzie, C. & Hadding, U. Establishment of T-helper type 1- and T-helper type 2-like human Toxoplasma antigen-specific T-cell clones. Immunology 86, 79–84 (1995).
Sanecka, A. & Frickel, E. M. Use and abuse of dendritic cells by Toxoplasma gondii. Virulence 3, 678–689 (2012).
Suzuki, Y., Orellana, M. A., Schreiber, R. D. & Remington, J. S. Interferon-gamma: the major mediator of resistance against Toxoplasma gondii. Science 240, 516–518 (1988).
Benson, A., Pifer, R., Behrendt, C. L., Hooper, L. V. & Yarovinsky, F. Gut commensal bacteria direct a protective immune response against Toxoplasma gondii. Cell Host Microbe 6, 187–196 (2009).
Yarovinsky, F. Innate immunity to Toxoplasma gondii infection. Nat. Rev. Immunol. 14, 109–121 (2014).
Raetz, M. et al. Cooperation of TLR12 and TLR11 in the IRF8-dependent IL-12 response to Toxoplasma gondii profilin. J. Immunol. 191, 4818–4827 (2013).
Koblansky, A. A. et al. Recognition of profilin by Toll-like receptor 12 is critical for host resistance to Toxoplasma gondii. Immunity 38, 119–130 (2013).
Andrade, W. A. et al. Combined action of nucleic acid-sensing Toll-like receptors and TLR11/TLR12 heterodimers imparts resistance to Toxoplasma gondii in mice. Cell Host Microbe 13, 42–53 (2013).
Seeber, F. & Steinfelder, S. Recent advances in understanding apicomplexan parasites. F1000Res 5, F1000 (2016)
Dupont, C. D., Christian, D. A. & Hunter, C. A. Immune response and immunopathology during toxoplasmosis. Semin. Immunopathol. 34, 793–813 (2012).
Sasai, M., Pradipta, A. & Yamamoto, M. Host immune responses to Toxoplasma gondii. Int. Immunol. 30, 113–119 (2018).
Gazzinelli, R., Xu, Y., Hieny, S., Cheever, A. & Sher, A. Simultaneous depletion of CD4+ and CD8+ T lymphocytes is required to reactivate chronic infection with Toxoplasma gondii. J. Immunol. 149, 175–180 (1992).
Landrith, T. A., Harris, T. H. & Wilson, E. H. Characteristics and critical function of CD8+ T cells in the Toxoplasma-infected brain. Semin. Immunopathol. 37, 261–270 (2015).
Suzuki, Y., Conley, F. K. & Remington, J. S. Importance of endogenous IFN-gamma for prevention of toxoplasmic encephalitis in mice. J. Immunol. 143, 2045–2050 (1989).
Khan, I. A., Hwang, S. & Moretto, M. Toxoplasma gondii: CD8 T cells cry for CD4 Help. Front. Cell Infect. Microbiol. 9, 136, https://doi.org/10.3389/fcimb.2019.00136 (2019).
Bennett, S. R., Carbone, F. R., Karamalis, F., Miller, J. F. & Heath, W. R. Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J. Exp. Med. 186, 65–70 (1997).
Sasai, M. & Yamamoto, M. Innate, adaptive, and cell-autonomous immunity against Toxoplasma gondii infection. Exp. Mol. Med. 51, 1–10 (2019).
Suzuki, Y., Sa, Q., Gehman, M. & Ochiai, E. Interferon-gamma- and perforin-mediated immune responses for resistance against Toxoplasma gondii in the brain. Expert Rev. Mol. Med. 13, e31 (2011).
Suzuki, Y. et al. IL-4 is protective against development of toxoplasmic encephalitis. J. Immunol. 157, 2564–2569 (1996).
Yoon, C., Ham, Y. S., Gil, W. J. & Yang, C.-S. The strategies of NLRP3 inflammasome to combat Toxoplasma gondii. Front. Immunol. 13, 1002387 (2022).
Brasil, T. R., Freire-de-Lima, C. G., Morrot, A. & Vetö Arnholdt, A. C. Host-toxoplasma gondii coadaptation leads to fine tuning of the immune response. Front. Immunol. 8, 1080 (2017).
Lima, T. S. & Lodoen, M. B. Mechanisms of human innate immune evasion by Toxoplasma gondii. Front. Cell Infect. Microbiol. 9, 103 (2019).
Mammari, N. et al. Toxoplasma gondii modulates the host cell responses: an overview of apoptosis pathways. Biomed. Res Int 2019, 6152489 (2019).
Ólafsson, E. B., Varas-Godoy, M. & Barragan, A. Toxoplasma gondii infection shifts dendritic cells into an amoeboid rapid migration mode encompassing podosome dissolution, secretion of TIMP-1, and reduced proteolysis of extracellular matrix. Cell Microbiol. 20, e12808 (2018).
Hunter, C. A. & Sibley, L. D. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat. Rev. Microbiol. 10, 766–778 (2012).
Hill, D. & Dubey, J. P. Toxoplasma gondii: transmission, diagnosis and prevention. Clin. Microbiol. Infect. 8, 634–640 (2002).
Lampejo, T. Toxoplasma gondii infection in HIV-infected pregnant women: epidemiology and risks of mother-to-child transmission. Pan. Afr. Med. J. 42, 275 (2022).
Sterkers, Y. et al. Novel interpretation of molecular diagnosis of congenital toxoplasmosis according to gestational age at the time of maternal infection. J. Clin. Microbiol. 50, 3944–3951, https://doi.org/10.1128/jcm.00918-12 (2012).
Saso, A. et al. Fifteen-minute consultation: management of the infant born to a mother with toxoplasmosis in pregnancy. Arch. Dis. Child Educ. Pract. Ed. 105, 262–269 (2020).
Atmaca, L. S., Simsek, T. & Batioglu, F. Clinical features and prognosis in ocular toxoplasmosis. Jpn. J. Ophthalmol. 48, 386–391, https://doi.org/10.1007/s10384-003-0069-0 (2004).
Gilbert, R., Tan, H. K., Cliffe, S., Guy, E. & Stanford, M. Symptomatic toxoplasma infection due to congenital and postnatally acquired infection. Arch. Dis. Child 91, 495–498 (2006).
Daher, D. et al. Comprehensive overview of toxoplasma gondii-induced and associated diseases. Pathogens 10, 1351 (2021).
Greigert, V., Bittich-Fahmi, F. & Pfaff, A. W. Pathophysiology of ocular toxoplasmosis: facts and open questions. PLoS Negl. Trop. Dis. 14, e0008905 (2020).
Knight, B. C. et al. Expression analysis of immune response genes of Müller cells infected with Toxoplasma gondii. J. Neuroimmunol. 179, 126–131 (2006).
Nicoletti, A. et al. Toxoplasma gondii and multiple sclerosis: a population-based case-control study. Sci. Rep. 10, 18855 (2020).
Motavallihaghi, S., Ghaemipanaeian, M., Soleimani Asl, S., Foroughi-Parvar, F. & Maghsood, A. H. Toxoplasma gondii attenuates the ethidium bromide induced demyelination lesions in multiple sclerosis model rats. Int. Immunopharmacol. 120, 110379 (2023).
Bayani, M., Riahi, S. M., Bazrafshan, N., Ray Gamble, H. & Rostami, A. Toxoplasma gondii infection and risk of Parkinson and Alzheimer diseases: a systematic review and meta-analysis on observational studies. Acta Trop. 196, 165–171 (2019).
Fallahi, S. et al. Parkinson’s disease and Toxoplasma gondii infection: sero-molecular assess the possible link among patients. Acta Trop. 173, 97–101 (2017).
Mikaeili Galeh, T. et al. Controversial effects of diverse types of toxoplasma gondii on the anxiety-like behavior and cognitive impairments in the animal model of Alzheimer’s disease. Iran. J. Psychiatry Behav. Sci. 16, e122961 (2022).
Charles, E. et al. CD4 T-cell suppression by cells from Toxoplasma gondii-infected retinas is mediated by surface protein PD-L1. Infect. Immun. 78, 3484–3492 (2010).
Washino, T., Moroda, M., Iwakura, Y. & Aosai, F. Toxoplasma gondii infection inhibits Th17-mediated spontaneous development of arthritis in interleukin-1 receptor antagonist-deficient mice. Infect. Immun. 80, 1437–1444 (2012).
Kim, W. H. et al. Suppression of CD4 T-Cells in the spleen of mice infected with Toxoplasma gondii KI-1 tachyzoites. Korean J. Parasitol. 48, 325–329 (2010).
Butcher, B. A., Kim, L., Johnson, P. F. & Denkers, E. Y. Toxoplasma gondii tachyzoites inhibit proinflammatory cytokine induction in infected macrophages by preventing nuclear translocation of the transcription factor NF-kappa B. J. Immunol. 167, 2193–2201 (2001).
Bennouna, S., Sukhumavasi, W. & Denkers, E. Y. Toxoplasma gondii inhibits toll-like receptor 4 ligand-induced mobilization of intracellular tumor necrosis factor alpha to the surface of mouse peritoneal neutrophils. Infect. Immun. 74, 4274–4281 (2006).
Yan, K. et al. Inhibitory effects of inonotus obliquus polysaccharide on inflammatory response in Toxoplasma gondii-Infected RAW264.7 macrophages. Evid. Based Complementary Altern. Med. 2021, 2245496 (2021).
Hunter, C. A., Roberts, C. W. & Alexander, J. Kinetics of cytokine mRNA production in the brains of mice with progressive toxoplasmic encephalitis. Eur. J. Immunol. 22, 2317–2322 (1992).
Roberts, C. W. et al. Different roles for interleukin-4 during the course of Toxoplasma gondii infection. Infect. Immun. 64, 897–904 (1996).
Hwang, Y. S. et al. Characteristics of infection immunity regulated by toxoplasma gondii to maintain chronic infection in the brain. Front. Immunol. 9, 158 (2018).
Nickdel, M. B. et al. Intestinal pathology during acute toxoplasmosis is IL-4 dependent and unrelated to parasite burden. Parasite Immunol. 26, 75–82 (2004).
Bliss, S. K., Butcher, B. A. & Denkers, E. Y. Rapid recruitment of neutrophils containing prestored IL-12 during microbial infection. J. Immunol. 165, 4515–4521 (2000).
Hunter, C. A., Litton, M. J., Remington, J. S. & Abrams, J. S. Immunocytochemical detection of cytokines in the lymph nodes and brains of mice resistant or susceptible to toxoplasmic encephalitis. J. Infect. Dis. 170, 939–945 (1994).
Whitmarsh, R. J. et al. A critical role for SOCS3 in innate resistance to Toxoplasma gondii. Cell Host Microbe 10, 224–236 (2011).
Zhang, Y. & Denkers, E. Y. Protective role for interleukin-5 during chronic Toxoplasma gondii infection. Infect. Immun. 67, 4383–4392 (1999).
Nickdel, M. B., Roberts, F., Brombacher, F., Alexander, J. & Roberts, C. W. Counter-protective role for interleukin-5 during acute Toxoplasma gondii infection. Infect. Immun. 69, 1044–1052 (2001).
Hunter, C. A., Bermudez, L., Beernink, H., Waegell, W. & Remington, J. S. Transforming growth factor-beta inhibits interleukin-12-induced production of interferon-gamma by natural killer cells: a role for transforming growth factor-beta in the regulation of T cell-independent resistance to Toxoplasma gondii. Eur. J. Immunol. 25, 994–1000 (1995).
Langermans, J. A., Nibbering, P. H., Van Vuren-Van Der Hulst, M. E. & Van Furth, R. Transforming growth factor-beta suppresses interferon-gamma-induced toxoplasmastatic activity in murine macrophages by inhibition of tumour necrosis factor-alpha production. Parasite Immunol. 23, 169–175 (2001).
Aoki, C. A. et al. Transforming growth factor beta (TGF-beta) and autoimmunity. Autoimmun. Rev. 4, 450–459 (2005).
Letterio, J. J. et al. Autoimmunity associated with TGF-beta1-deficiency in mice is dependent on MHC class II antigen expression. J. Clin. Invest. 98, 2109–2119 (1996).
Marie, J. C., Liggitt, D. & Rudensky, A. Y. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity 25, 441–454 (2006).
Nagineni, C. N., Detrick, B. & Hooks, J. J. Transforming growth factor-beta expression in human retinal pigment epithelial cells is enhanced by Toxoplasma gondii: a possible role in the immunopathogenesis of retinochoroiditis. Clin. Exp. Immunol. 128, 372–378 (2002).
Kay, J., Thadhani, E., Samson, L. & Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair (Amst.) 83, 102673 (2019).
Beyaert, R. et al. Cancer risk in immune-mediated inflammatory diseases (IMID). Mol. Cancer 12, 98 (2013).
Chen, Y., Tan, W. & Wang, C. Tumor-associated macrophage-derived cytokines enhance cancer stem-like characteristics through epithelial-mesenchymal transition. Onco Targets Ther. 11, 3817–3826 (2018).
Lin, Y., Xu, J. & Lan, H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J. Hematol. Oncol. 12, 76 (2019).
Wang, X. et al. 17-allylamino-17-demethoxygeldanamycin synergistically potentiates tumor necrosis factor-induced lung cancer cell death by blocking the nuclear factor-kappaB pathway. Cancer Res. 66, 1089–1095 (2006).
Müller-Hermelink, N. et al. TNFR1 signaling and IFN-gamma signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell 13, 507–518 (2008).
Kearney, C. J. et al. Tumor immune evasion arises through loss of TNF sensitivity. Sci. Immunol. 3, eaar3451 (2018).
Bliss, S. K., Marshall, A. J., Zhang, Y. & Denkers, E. Y. Human polymorphonuclear leukocytes produce IL-12, TNF-alpha, and the chemokines macrophage-inflammatory protein-1 alpha and -1 beta in response to Toxoplasma gondii antigens. J. Immunol. 162, 7369–7375 (1999).
Bennouna, S., Bliss, S. K., Curiel, T. J. & Denkers, E. Y. Cross-talk in the innate immune system: neutrophils instruct recruitment and activation of dendritic cells during microbial infection. J. Immunol. 171, 6052–6058 (2003).
Mulas, F. et al. The deubiquitinase OTUB1 augments NF-κB-dependent immune responses in dendritic cells in infection and inflammation by stabilizing UBC13. Cell Mol. Immunol. 18, 1512–1527 (2021).
Corrêa, G. et al. Inflammatory early events associated to the role of P2X7 receptor in acute murine toxoplasmosis. Immunobiology 222, 676–683 (2017).
Mennechet, F. J. et al. Lamina propria CD4+ T lymphocytes synergize with murine intestinal epithelial cells to enhance proinflammatory response against an intracellular pathogen. J. Immunol. 168, 2988–2996 (2002).
Chen, J., Liao, W. & Peng, H. Toxoplasma gondii infection possibly reverses host immunosuppression to restrain tumor growth. Front. Cell Infect. Microbiol. 12, 959300 (2022).
Wu, X. et al. [Impact of Toxoplasma gondii on the proliferation and apoptosis of tumor cell lines]. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing. Za Zhi 30, 157–159 (2012).
Lu, G. et al. Transcriptome sequencing investigated the tumor-related factors changes after T. gondii infection. Front. Microbiol. 10, 181 (2019).
Baird, J. R. et al. Immune-mediated regression of established B16F10 melanoma by intratumoral injection of attenuated Toxoplasma gondii protects against rechallenge. J. Immunol. 190, 469–478 (2013).
Baird, J. R. et al. Avirulent Toxoplasma gondii generates therapeutic antitumor immunity by reversing immunosuppression in the ovarian cancer microenvironment. Cancer Res. 73, 3842–3851 (2013).
Sanders, K. L., Fox, B. A. & Bzik, D. J. Attenuated toxoplasma gondii stimulates immunity to pancreatic cancer by manipulation of myeloid cell populations. Cancer Immunol. Res. 3, 891–901 (2015).
Xu, L. Q. et al. A uracil auxotroph Toxoplasma gondii exerting immunomodulation to inhibit breast cancer growth and metastasis. Parasit. Vectors 14, 601 (2021).
Bahwal, S. A. et al. Attenuated Toxoplasma gondii enhances the antitumor efficacy of anti-PD1 antibody by altering the tumor microenvironment in a pancreatic cancer mouse model. J. Cancer Res. Clin. Oncol. 148, 2743–2757 (2022).
Li, Y., Zhang, Y., Xia, N., Zhou, T. & Shen, B. Antitumor effects of a Toxoplasma mutant lacking lactate dehydrogenases. Parasitol. Res. 120, 3335–3339 (2021).
Rastogi, S., Cygan, A. M. & Boothroyd, J. C. Translocation of effector proteins into host cells by Toxoplasma gondii. Curr. Opin. Microbiol. 52, 130–138 (2019).
Zhang, Y., Lai, B. S., Juhas, M. & Zhang, Y. Toxoplasma gondii secretory proteins and their role in invasion and pathogenesis. Microbiol. Res. 227, 126293 (2019).
Zhu, J., Wang, Y., Cao, Y., Shen, J. & Yu, L. Diverse roles of TgMIC1/4/6 in the Toxoplasma infection. Front. Microbiol. 12, 666506 (2021).
Benevides, L. et al. Toxoplasma gondii soluble tachyzoite antigen triggers protective mechanisms against fatal intestinal pathology in oral infection of C57BL/6 mice. PLoS One 8, e75138 (2013).
Machado, F. S. et al. Anti-inflammatory actions of lipoxin A4 and aspirin-triggered lipoxin are SOCS-2 dependent. Nat. Med. 12, 330–334 (2006).
Wang, S. et al. Toxoplasma gondii excretory/secretory antigens (TgESAs) suppress pro-inflammatory cytokine secretion by inhibiting TLR-induced NF-κB activation in LPS-stimulated murine macrophages. Oncotarget 8, 88351–88359 (2017).
Ricci-Azevedo, R., Mendonça-Natividade, F. C., Santana, A. C., Alcoforado Diniz, J. & Roque-Barreira, M. C. Microneme proteins 1 and 4 from Toxoplasma gondii Induce IL-10 production by macrophages through TLR4 endocytosis. Front. Immunol. 12, 655371 (2021).
Butcher, B. A. et al. Toxoplasma gondii rhoptry kinase ROP16 activates STAT3 and STAT6 resulting in cytokine inhibition and arginase-1-dependent growth control. PLoS Pathog. 7, e1002236 (2011).
You, J. et al. The oncogenic role of ARG1 in progression and metastasis of hepatocellular carcinoma. Biomed. Res. Int. 2018, 2109865 (2018).
Choi, J. W. et al. Pyropia yezoensis glycoprotein promotes the M1 to M2 macrophage phenotypic switch via the STAT3 and STAT6 transcription factors. Int. J. Mol. Med. 38, 666–674 (2016).
Rőszer, T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediators Inflamm. 2015, 816460 (2015).
Chen, L. et al. The Toxoplasma gondii virulence factor ROP16 acts in cis and trans, and suppresses T cell responses. J. Exp. Med. 217, e20181757 (2020).
Xu, Y. W. et al. Toxoplasma ROP16(I/III) ameliorated inflammatory bowel diseases via inducing M2 phenotype of macrophages. World J. Gastroenterol. 25, 6634–6652 (2019).
Rommereim, L. M. et al. Rhoptry and dense granule secreted effectors regulate CD8(+) T cell recognition of toxoplasma gondii infected host cells. Front. Immunol. 10, 2104 (2019).
Kim, J. S. et al. Toxoplasma gondii GRA9 regulates the activation of NLRP3 inflammasome to exert anti-septic effects in mice. Int. J. Mol. Sci. 21, 8437 (2020).
Gazzinelli, R. T., Eltoum, I., Wynn, T. A. & Sher, A. Acute cerebral toxoplasmosis is induced by in vivo neutralization of TNF-alpha and correlates with the down-regulated expression of inducible nitric oxide synthase and other markers of macrophage activation. J. Immunol. 151, 3672–3681 (1993).
Fox, B. A., Sanders, K. L., Rommereim, L. M., Guevara, R. B. & Bzik, D. J. Secretion of rhoptry and dense granule effector proteins by nonreplicating toxoplasma gondii uracil auxotrophs controls the development of antitumor immunity. PLoS Genet. 12, e1006189 (2016).
Li, Y. et al. Macrophages polarized by expression of ToxoGRA15(II) inhibit growth of hepatic carcinoma. Front. Immunol. 8, 137 (2017).
Wei, W. et al. Toxoplasma gondii dense granule protein 15 induces apoptosis in choriocarcinoma JEG-3 cells through endoplasmic reticulum stress. Parasit. Vectors 11, 251 (2018).
Seo, S. H., Kim, S. G., Shin, J. H., Ham, D. W. & Shin, E. H. Toxoplasma GRA16 inhibits NF-κB activation through PP2A-B55 upregulation in non-small-cell lung carcinoma cells. Int. J. Mol. Sci. 21, 6642 (2020).
Moses, M. A., Brem, H. & Langer, R. Advancing the field of drug delivery: taking aim at cancer. Cancer Cell 4, 337–341 (2003).
Dong, X. Current strategies for brain drug delivery. Theranostics 8, 1481–1493 (2018).
Nagy, J. A., Chang, S. H., Dvorak, A. M. & Dvorak, H. F. Why are tumour blood vessels abnormal and why is it important to know? Br. J. Cancer 100, 865–869 (2009).
Elsheikha, H. M., Marra, C. M. & Zhu, X. Q. Epidemiology, pathophysiology, diagnosis, and management of cerebral toxoplasmosis. Clin. Microbiol. Rev. 34, e00115-19 (2021).
Wang, Q., Zhong, Y., Chen, N. & Chen, J. From the immune system to mood disorders especially induced by Toxoplasma gondii: CD4(+) T cell as a bridge. Front. Cell Infect. Microbiol 13, 1078984, (2023).
Harun, M. S. R., Taylor, M., Zhu, X. Q. & Elsheikha, H. M. Transcriptome profiling of Toxoplasma gondii-infected human cerebromicrovascular endothelial cell response to treatment with monensin. Microorganisms 8, 842 (2020).
Konradt, C. et al. Endothelial cells are a replicative niche for entry of Toxoplasma gondii to the central nervous system. Nat. Microbiol. 1, 16001 (2016).
Sagar, D. et al. Mechanisms of dendritic cell trafficking across the blood-brain barrier. J. Neuroimmune Pharmacol. 7, 74–94 (2012).
Mendez, O. A. & Koshy, A. A. Toxoplasma gondii: Entry, association, and physiological influence on the central nervous system. PLoS Pathog. 13, e1006351 (2017).
Yan, Z. et al. Four chemotherapeutic compounds that limit blood-brain-barrier invasion by toxoplasma gondii. Molecules 27, 5572 (2022).
Courret, N. et al. CD11c- and CD11b-expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood 107, 309–316 (2006).
Combes, F., Meyer, E. & Sanders, N. N. Immune cells as tumor drug delivery vehicles. J. Control. Release 327, 70–87 (2020).
Koodie, L. et al. Morphine inhibits migration of tumor-infiltrating leukocytes and suppresses angiogenesis associated with tumor growth in mice. Am. J. Pathol. 184, 1073–1084 (2014).
Xue, J. et al. Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nat. Nanotechnol. 12, 692–700 (2017).
He, X. et al. Inflammatory monocytes loading protease-sensitive nanoparticles enable lung metastasis targeting and intelligent drug release for anti-metastasis therapy. Nano Lett. 17, 5546–5554 (2017).
Choi, M. R. et al. A cellular Trojan Horse for delivery of therapeutic nanoparticles into tumors. Nano Lett. 7, 3759–3765 (2007).
Lee, S. H. et al. Deep tumor penetration of drug-loaded nanoparticles by click reaction-assisted immune cell targeting strategy. J. Am. Chem. Soc. 141, 13829–13840 (2019).
Cho, Y., Han, J. & Kim, W. Recent advances and future directions in immunotherapeutics for hepatocellular carcinoma. J. Liver Cancer 19, 1–11 (2019).
Marrache, S., Tundup, S., Harn, D. A. & Dhar, S. Ex vivo programming of dendritic cells by mitochondria-targeted nanoparticles to produce interferon-gamma for cancer immunotherapy. ACS Nano 7, 7392–7402 (2013).
Pillay, J. et al. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood 116, 625–627 (2010).
Acknowledgements
This work was supported by a National Research Foundation of Korea grant funded by the Korean government (MSIP) (grant no. 2019R1I1A2A01064237 and 2021R1A4A5032463) and by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI22C0884). C.Y. was supported by the research fund of Hanyang University (HY-2023-0536).
Author information
Authors and Affiliations
Contributions
C.Y., Y.S.H., W.J.G., and C.-S.Y. designed, conceptualized, and wrote the manuscript. All the authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yoon, C., Ham, Y.S., Gil, W.J. et al. Exploring the potential of Toxoplasma gondii in drug development and as a delivery system. Exp Mol Med 56, 289–300 (2024). https://doi.org/10.1038/s12276-024-01165-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-024-01165-7
