
Abstract
Infectious diseases are a major driver of morbidity and mortality globally. Treatment of malaria, tuberculosis and human immunodeficiency virus infection are particularly challenging, as indicated by the ongoing transmission and high mortality associated with these diseases. The formulation of new and existing drugs in nano-sized carriers promises to overcome several challenges associated with the treatment of these diseases, including low on-target bioavailability, sub-therapeutic drug accumulation in microbial sanctuaries and reservoirs, and low patient adherence due to drug-related toxicities and extended therapeutic regimens. Further, nanocarriers can be used for formulating vaccines, which represent a major weapon in our fight against infectious diseases. Here we review the current burden of infectious diseases with a focus on major drivers of morbidity and mortality. We then highlight how nanotechnology could aid in improving existing treatment modalities. We summarize our progress so far and outline potential future directions to maximize the impact of nanotechnology on the global population.
Similar content being viewed by others

Main
Infections are a dominant contributor to the global disease burden. High mortality rates are associated with lower respiratory infections, diarrhoea, tuberculosis (TB), human immunodeficiency virus (HIV) infection and malaria (Fig. 1a)1. Mortality rates are highest in developing countries, where resources such as vaccines and anti-infectives may be limited (Fig. 1b)1. Temporal trends in mortality show that although the overall number of deaths is declining, the gap between deaths in high socio-demographic index (SDI) countries and low SDI countries remains substantial (Fig. 1c)1. Unfortunately, clinical trials for these infectious diseases (IDs) are also lagging compared with conditions such as cancer and cardiovascular diseases (Fig. 1d)1 and there is thus an urgent need to identify practical and impactful strategies that enable better treatment of IDs.

IDs represent a chronic healthcare burden around the world and as such are high priority targets for investigational drugs. a, The communicable diseases with the five highest mortality rates are lower respiratory infections, diarrhoeal diseases, TB, HIV infection and malaria. b, Low SDI countries are disproportionately affected by these top five communicable diseases. c, Temporal trends in mortality rate for the five diseases. Although mortality rates from these communicable diseases is declining, there is considerable disparity between low and high SDI countries. The solid lines represent the mean values and the dotted lines show the uncertainty interval. d, The number of clinical studies listed in clinicaltrials.gov for various diseases. Data in a–c are from ref. 1. Credit: Ohaiyoo/Adobe.
A host of challenges must be overcome to effectively manage IDs. The lack of safe and effective drugs is central to our inability to treat IDs2. In some cases, a lack of drug efficacy can be attributed to pathogen resistance, which then requires a more expensive drug regimen for cure3. Challenges to treating IDs are often compounded in low SDI countries. Poor patient adherence to therapies and the need for sustained patient monitoring are major obstacles to effective treatment4. Poor procurement practices, the inability to pay for drugs and poor stability of drug products at high temperature and humidity prevent access to effective treatments5. A concerted effort on scientific and policy levels is necessary to overcome these challenges.
Nanotechnology has the potential to transform both detection and treatment of a wide range of diseases. These technologies, involving systems with a diameter of about one-thousandth of the thickness of a hair, stand to substantially impact the globe’s main sources of morbidity and mortality. Nanosystems have been the subject of intensive research over the past few decades, resulting in Food and Drug Administration (FDA)-approved chemotherapeutics, anaesthetics, imaging agents, nutritional supplements and others6. Not surprisingly, nanotechnology has been extensively evaluated to improve the treatment of IDs. In this Review, we examine the opportunities presented by nanotechnology for the treatment of IDs and discuss preclinical and clinical progress in this area with a focus on HIV, TB and malaria. Additionally, we recognize the ongoing impact nanotechnology is having in the form of successful mRNA and protein vaccines for SARS-CoV-2. Finally, we discuss challenges to translating these technologies from the laboratory to the clinic. We do not include polymicrobial IDs, other monomicrobial IDs, metal nanoparticles7 and vaccination8,9—all of which are areas of intensive research.
Nanotechnology in treating IDs
The availability and correct use of safe and efficacious medications are imperative for treating IDs. Nanotechnology-based approaches have been the topic of intensive preclinical evaluation to improve the therapeutic index of ID drugs and simplify their use. We highlight promising preclinical studies and discuss challenges to their clinical translation.
Sustained systemic delivery of anti-infectives
The management of several IDs involves chronic treatment, which places a major burden on the patient and the health care system. HIV-infected individuals require lifelong treatment to ensure control of the viral burden10. To treat TB, a combination of pills must be consumed over a period of several months to years—often multiple times a day11. Prolonged and complicated dosing regimens with high pill burden result in lower patient adherence and, ultimately, failure of treatment. At times, providers administer medications only once a day for programmatic ease even though this may not achieve optimal drug levels. Hence there is interest in developing systems that reduce dosing frequency and ease the dosing regimen. Injectable nanocarriers that deliver drugs for sustained periods have been actively pursued. Broadly, these systems can be classified into two categories—systems that control drug release with an excipient (such as a polymer or lipid) and those that rely on the slow dissolution of poorly soluble drug crystals in interstitial fluid (Fig. 2).

The encapsulation of drugs in nanocarriers such as liposomes, polymer nanoparticles, or prepared drug nanocrystals can enable sustained drug delivery in a localized manner (left). These systems have been evaluated for treating local infections of the female reproductive tract, lungs and skin. Injectable nanocarriers are also being explored for systemic drug delivery (right).
Considerable research has focused on polyester-based drug delivery systems that degrade in the presence of physiological esterases (for example poly(lactide-co-glycolide) (PLGA) and poly(caprolactone)). Drug release occurs via bulk degradation of the polymer and drug diffusion12 and can be modified by altering the hydrophobicity of the monomer, polymer chain length and particle size13. Other polymers used for sustained release include poly(anhydrides), poly(orthoesters), poly(cyanoacrylates) and poly(amides)12.
Sustained release can also be obtained by encapsulating drugs in liposomes, which are lipid vesicles with the capacity to load both hydrophobic and hydrophilic drugs. NeXstar Pharmaceuticals developed MiKasomes—a liposomal formulation of amikacin14, a drug requiring frequent dosing and continuous therapeutic monitoring. In a rat model, intravenously administered Mikasomes distributed to several tissues, where they released drug gradually resulting in an eightfold increase in drug half-life. Despite early positive results in clinical trials for urinary tract infections and Mycobacterium infections15, development of the formulation was discontinued in 2000.
A drawback of some polymeric and liposomal formulations is that they require large quantities of excipients to control drug release. This increases the size of the injection and limits the amount of drug that can be administered. To circumvent this, aqueous dispersions of nano-milled drug crystals are being developed16,17. Following subcutaneous or intramuscular injection, drug absorption into systemic circulation is dictated by the rate of dissolution of the drug crystals in the interstitial fluid16. Hence, physicochemical properties of the drug and size of the drug crystals play an important role in determining drug release kinetics. A common technique to prolong drug absorption is to convert the drug into salt forms of lower solubility or into prodrugs that have greater hydrophobicity18.
An important consideration with sustained release systems is the potential for prolonged exposure to sub-therapeutic concentrations, which may lead to drug resistance19. Dwindling amounts of drug released during the terminal phase of the release period may lead to a ‘long pharmacologic tail’20, a period of slowly declining drug levels. Pulsatile release formulations, instead of sustained release formulations, may help address this issue. Alternatively, minimally invasive means of removing the depot at the end of the therapeutic period may be attractive.
Local delivery
As epithelial surfaces are common sites of entry and residence for pathogens, regional drug delivery to these surfaces holds great promise. Compared with systemic administration, local delivery to the site of infection is likely to enhance on-target (and minimize off-target) drug exposure. Of course, its utility is limited for systemic infections. Preclinical studies, predominantly in rodents, have identified several advantages of using nanoformulation for local therapies. Encapsulation of drugs in nanocarriers allows for sustained drug delivery, thereby reducing dosing frequency. Nanocarriers can be engineered to release drugs in the presence of certain triggers and provide temporal control on drug exposure. Nanocarriers may enhance drug uptake in cells and improve efficacy against intracellular pathogens. Finally, encapsulation in nanocarriers may protect labile drugs (such as nucleic acids) in harsh physiological conditions such as low pH or the presence of enzymes.
Vaginal drug delivery
HIV and herpes simplex virus (HSV) can enter the body via the vaginal tract. Prophylactic local administration of microbicides can reduce the chance of infection. Ensuring maximal coverage throughout the vaginal tract at the time of infection is likely to minimize the chances of infection. Several barriers impede complete coverage of the vaginal tract and prolonged residence of the drug delivery system21. First, formulations can leak out of the vaginal tract following application of a drug delivery system. Second, the vaginal mucosa is covered by a layer of mucous (a viscous biopolymer made of glycoproteins called mucins), which acts as a diffusional barrier trapping the drug particulate matter. Third, the mucous layer is shed frequently, and particles trapped within it can be lost22. Fourth, the vaginal surface has several folds or rugae reducing drug accessibility. Finally, the low pH and enzymes present in the vaginal fluid may degrade susceptible drugs21.
To enhance vaginal residence, a common theme has been to develop systems that are rapidly transported across the mucous into the mucosa. In a study from the Saltzman group23, the authors compared the vaginal uptake of dye-loaded PLGA nanoparticles that were surface coated with either avidin or poly(ethylene glycol) (PEG) or left uncoated. Leakage out of the vaginal tract was highest for uncoated nanoparticles (~13% of the dose within 0.5 h). Vaginal lavage, which presumably enabled collection of the mucous layer, was rich in the mucoadhesive avidin-coated nanoparticles. The highest tissue uptake was achieved with PEG-coated nanoparticles; however, tissue uptake was limited to the lower reproductive tract. Improved tissue uptake of PEGylated nanoparticles may be attributed to their higher diffusivity through the mucous layer—an observation shown using multiple particle tracking experiments by the Hanes lab24. An alternative strategy to enhance vaginal retention is to administer nanoparticles embedded in films. Although nanoparticle dispersions can rapidly leak out of the vagina, films may hold the nanoparticles in place for some duration. Cunha-Reis et al. demonstrated that nanoparticles loaded with an anti-HIV drug (efavirenz) and embedded in a water-soluble film showed lower leakage and enabled higher tissue concentrations than nanoparticle dispersions25. However, the improved tissue concentrations were observed only within the first hour of administration, and not at later times. These studies highlight two challenges with nanoparticle-based vaginal drug delivery systems. First, there is minimal nanoparticle uptake in the upper reproductive tract, bringing into question the coverage offered by these systems. Second, in rodent models, drug concentrations become undetectable at about 24 h post-dose, suggesting that sustained delivery may be possible for only a relatively short time. The issue of residence may be overcome by loading nanocarriers in macro-drug delivery systems such as vaginal rings, which have been clinically shown to have extended residence times.
Despite their limitations, vaginal delivery with nanosystems has shown therapeutic potential in murine models. Ensign and colleagues showed that PEGylated PLGA nanoparticles dosed in a hypotonic solution were rapidly taken up into vaginal folds via advection26. Acyclovir monophosphate nanoparticles protected ~54% of the mice that were infected with HSV-2 30 min post-treatment. In contrast, the soluble drug protected only 16% of the mice from viral challenge. In a mouse model of lethal HSV-2 infection, intravaginal administration of oligofectamine-complexed small interfering RNA (siRNA) targeting genes encoding viral envelope and DNA binding proteins resulted in improved survival compared with untreated controls27. Interestingly, PLGA nanoparticles containing cationic spermidine have been used for siRNA-based knockdown of the same target, and have shown better protection than lipofectamine lipoplexes28. The authors found that the PLGA nanoparticles had a similar knockdown efficiency to lipoplexes, but elicited lower inflammation (as observed by measuring neutrophil invasion using immunohistochemistry). They attributed the improved protective activity of PLGA nanoparticles to their ability to cause minimal inflammation. It must be noted that in all studies described here, exposure to the pathogen was within a few minutes to hours of treatment. Such treatment schedules are difficult to reduce to practice in resource-limited settings10, underscoring the need for long-acting local systems. Even so, the recent regulatory success of Starpharma’s VivaGel, a dendrimer antimicrobial with demonstrated efficacy against HIV, HSV and bacterial vaginosis and formulated both as gels and as a surface coating for condoms, highlight the vast potential for growth and the versatility of nanotechnological systems in this area.
Pulmonary delivery
Pulmonary delivery is an attractive route for the treatment of respiratory infections as it may enable greater on-target drug exposure. However, formulations that cannot penetrate mucous layers and biofilms have low pulmonary bioavailability due to rapid enzymatic deactivation, sequestration and elimination by coughing22,29. In the lungs, mucous is secreted by goblet cells within the epithelium of mucous membranes. Biofilms are produced when microbes aggregate on a surface and produce an extracellular matrix comprising high-molecular-weight polysaccharides (such as alginate and N-acetyl glucosamine), DNA and proteins30.
Greater local bioavailability of therapeutics has been observed following pulmonary administration compared with oral or intravenous administration. In mouse studies, pulmonary delivery of amorphous itraconazole nanoparticles resulted in a tenfold higher drug concentration in the lungs compared with oral administration of its commercial formulation (Sporanox) and the unformulated drug31. In a murine Aspergillus fumigatus infection model, the same group observed an increased median survival in the pulmonary nanoparticle treated group (7.5 days) compared with oral Sporanox treated group (5 days)32. These benefits cannot be attributed completely to nanoformulation as changing the route of administration from oral to pulmonary is likely to contribute heavily to this improvement. However, the poor aqueous solubility of itraconzole may have precluded the pulmonary administration of the unformulated drug. In a study with ciprofloxacin (a drug with higher water solubility), Wong et al. compared the activity of liposome-encapsulated and free drug upon pulmonary administration33. The half-life of the liposome-encapsulated drug in mouse lungs was about double that of the free drug (~3 h versus ~1.5 h). Importantly, in a Francisella tularensis infection model, survival of mice in the untreated and free-drug-treated groups was similar, in that all mice succumbed to the infection within 14 days. In contrast, there were no deaths observed in mice treated with pulmonary liposomes. Interestingly, the authors found the liposomes to be effective even when administered via the intravenous route. Direct comparison of the pharmacokinetics of intravenous and pulmonary liposomes would have yielded important information regarding on-target and off-target exposure.
Bacteria regulate their cooperative growth and form biofilms by secreting and sensing molecules that signal for virulence. Inhibitors of this quorum sensing phenomenon have the potential to disrupt biofilms. However, the mucous covering, especially in patients suffering from cystic fibrosis, retards drug delivery to the bacterial biofilms. To overcome mucous resistance in the respiratory tract, Nafee and colleagues encapsulated the quorum sensing inhibitor in lipid nanoparticles surface coated with polysorbate 80—a co-polymer surfactant containing PEG34. In comparison with the free drug, the nanoparticle-encapsulated quorum sensing inhibitor showed better in vitro control of virulence as measured by quantifying levels of a bacterial product (pyocyanin). Confocal microscopy also demonstrated that polysorbate 80-coated nanoparticles diffused across porcine mucous. However, transport of the quorum sensing inhibitor in its free-drug form and nanoparticulate form was not assessed. Although these studies introduce an interesting concept, in vivo evaluation in a model of cystic fibrosis will be critical.
Another biofilm-disrupting approach involves the use of nitrous oxide-releasing polymeric nanoparticles. To enable sustained release of nitric oxide, which has a short half-life, Duong et al.35 conjugated nitric oxide with star-shaped polymers. Using a bacterial reporter strain, the authors confirmed sustained release of nitric oxide from the star-shaped polymer nanoparticles in vitro. Furthermore, they found that nanoparticles were able to decrease Pseudomonas aeruginosa biofilm formation by >70% compared with untreated controls35. However, in vivo validation was not performed.
Mucolytic agents, such as N-acetyl cysteine, can be used to improve the penetration of nanocarriers by reducing disulphide bonds in mucous. N-acetyl cysteine enhanced the diffusivity of DNA-loaded poly(lysine) polyplexes in the sputum of cystic fibrosis patients by more than fivefold36. To validate the benefit of N-acetyl cysteine in vivo, the authors treated mice with inflammatory lipopolysaccharide from P. aeruginosa, which led to enhanced mucous secretion in the lungs. Compared with healthy mice, the transfection efficiency of the polyplex in mice treated with lipopolysaccharide was reduced by about tenfold. Impressively, pre-treatment with N-acetyl cysteine restored transfection efficiency in the inflammation model. This study demonstrates the inhibitory effects of mucous on drug delivery to lungs and the benefits of combining mucolytics with nanotherapeutics.
Topical delivery
Chronic and non-healing wounds are a source of increasing healthcare cost and mortality37. These wounds occur due to breakdowns in the wound repair pathway and chronic infection caused by opportunistic pathogens. Unlike acute infections, which occur due to the presence of a large burden of individual (planktonic) pathogens and have a more serious symptomatology, chronic infections are localized and recur periodically. Furthermore, while acute infections may be managed effectively by medications, chronic infections do not respond to conventional antibiotic therapy. Interestingly, a study found that 60% of clinical samples from chronic infection sites contained biofilms compared with 6% found in acute infections38. These results suggest that the presence of biofilms plays a role in chronic infections and non-healing wounds, and strategies that overcome biofilm resistance may help treat these infections.
Topical drug delivery shares both challenges and therapeutic targets with pulmonary delivery—biofilm-forming opportunistic pathogens such as Staphylococcus aureus and P. aeruginosa have been implicated in both lung and skin infections. As such, many principles of engineering biofilm-penetrating nanosystems for pulmonary delivery can be leveraged for topical delivery. Sustained release of nitric oxide was achieved by fabricating nanoparticles containing reducing sugars and sodium nitrite. These nanoparticles were capable of releasing nitric oxide for over a month. In murine skin infection models of S. aureus and methicillin-resistant S. aureus biofilms, nitric oxide-releasing nanoparticles decreased overall wound bacterial burden compared with both unloaded nanoparticles and the control group39.
One strategy for treating wound infections is the application of silver solutions using a cotton gauge. However, due to the short retention and sudden exposure to high concentrations, the treatment needs to be applied very frequently and it can be toxic to host cells40. One proposed solution is to encapsulate porous silver microparticles in a poly(lactide) nanofibre scaffold. It was hypothesized that the slower release of silver from the scaffold may minimize its toxicity to human cells while maintaining its anti-infective properties. The scaffold did enable the slow release of silver; however, toxicity to host cells was not reduced40. Further work in this field could be valuable.
Targeted delivery to sites of infection
There is considerable interest in designing systems that enable targeted drug delivery to sites of infection. This is motivated by cases of low drug penetration into infected tissues (for example, low penetration of antiretrovirals into the brain and lymph nodes10 and anti-TB drugs into cavitary lesions41), drug distribution into sites of toxicity (such as aminoglycosides in the ear42) and killing of commensal microbiota43. Encapsulation in nanocarriers offers the possibility of achieving drug targeting (Fig. 3), but there is considerable room for improvement. Improved on-target accumulation occurs because of enhanced nanocarrier permeability at sites of infection compared with uninfected tissue. Nanocarrier surfaces can also be functionalized with ligands that bind to infected tissues or microbes. This latter strategy has been termed active targeting or ligand-mediated targeting. Strategies that do not rely on specific ligands have thus been dubbed passive targeting.

Targeting delivery of drugs to sites of infection can improve efficacy and reduce toxicity. a, On systemic injection, serum proteins are adsorbed on the surface of nanocarriers (opsonization; i) leading to phagocytosis by macrophages (ii). Hence, nanocarriers have been used for targeting drugs to macrophages, which are common sites of infection. b, Vascular permeability at sites of infection is higher than that observed in non-infected tissue. Increased vascular permeability is due to pathogens (shown as red and green shapes), which activate the kallikrein-kinin pathway, and due to inflammation, which results in enhanced reactive oxygen species. While entry of free drug (yellow circles) is possible at both uninfected and infected sites, entry of nanocarriers (blue circles) at uninfected sites is limited. This phenomenon has been exploited for targeting anti-infectives to sites of infection. c, Coating nanocarriers with ligands that bind receptors on the surface of infected host cells (right) or pathogens (left) have also been used for increasing drug targeting.
Passive targeting
Nanoencapsulation of anti-infectives has been pursued for two divergent goals—targeting drugs to macrophages and to infected tissues. The mechanisms by which nanocarriers deposit drugs at these two sites, and hence formulation strategies, are distinct.
Macrophages are common targets for bacteria (such as Mycobacterium tuberculosis), fungi (for example Aspergillus species) and viruses (such as HIV). As nanocarriers are predominantly cleared by these cells (Fig. 3a), they have been widely used for targeting drugs to macrophages. Administering the antiretroviral azidothymidine in poly(hexacyanoacrylate) nanoparticles improved its accumulation in reticuloendothelial system (RES) organs such as the liver, lungs and spleen44. In rats, 60% of the drug dose was found in the RES organs 8 h after treatment with nanoparticles. In contrast, after treatment with the soluble drug alone, only 12% of the drug was recovered in the RES tissues.
Nanocarriers have been used to enhance drug accumulation at sites of infection other than the RES organs. This is based on the observation that blood vessels at infection sites are leaky, promoting greater nanocarrier entry (Fig. 3b). To study vascular permeability at infection sites, Evan’s blue was injected intravenously into guinea pigs following intradermal injection of microbial peptidases. As the dye binds to albumin in the blood, accumulation of this dye in tissues has been used as a marker of vascular permeability. Although there was no accumulation of the dye at sites of saline injection, dye accumulation was elevated at sites of peptidase injection. These effects were observed with enzymes isolated from Candida albicans45, Pseudomonas aeruginosa, Aspergillus meleus and so on46. There have also been several studies that show higher accumulation of nanocarriers at sites of infection compared with non-infected sites (reviewed in ref. 47). In these studies, following a unilateral bacterial infection in the calf muscle of rats, the animals were treated intravenously with radiolabelled liposomes. In one study48, there was an ~40-fold greater liposome concentration (per cent injected dose per gram of tissue) in the abscess compared with the muscle. The change in vascular permeability and drug targeting depended, in part, on the infectious agent.
Enhanced circulation time can lead to more passes across the target and non-target tissues. Modelling studies suggest that if permeability into the target tissue is greater, longer circulation times can improve targeting efficiency49. Longer circulation times can be achieved by reducing the uptake of nanocarriers by macrophages. Hence, strategies orthogonal to those used to target drugs to macrophages are required. One approach involves modifying the surface of the nanocarriers with hydrophilic polymers such as PEG, which is thought to provide steric hindrance to opsonization50. The effect of circulation half-life on infection site accumulation was studied in a rat model of unilateral lung Klebsiella pneumoniae infection51,52. Gallium-67-labelled liposomes with different half-lives were administered to infected rats. Liposomes, regardless of their circulation half-life showed higher accumulation in the infected lung compared with the uninfected lung, validating the enhanced permeability discussed above. Furthermore, the accumulation of liposomes was proportional to the intensity of infection. Importantly, in rats that had the most severe lung infection, PEG-coated liposomes (half-life = 27 h) resulted in approximately fourfold higher concentrations compared with uncoated liposomes (half-life = 19 h)51. The benefit of PEG coating was reduced at low infection severity.
Ligand-mediated (active) and trigger-mediated targeted drug delivery
The expression of certain targets/receptors in diseased areas may be elevated. Conjugating ligands that bind these targets to the surface of nanocarriers can increase their accumulation in diseased sites (Fig. 3c). Ligand-conjugated nanocarriers can also increase their cell uptake, aiding in the treatment of intracellular pathogens.
The mannose receptor is expressed on tissue-resident macrophages and dendritic cells, and serves to clear pathogens expressing mannosylated glycoproteins53. Nanocarriers functionalized with sugar moieties can highjack this uptake mechanism and enhance uptake into tissue-resident macrophages. Ciprofloxacin-loaded mannose-functionalized liposomes were administered as a pulmonary spray to rats, and drug exposure in alveolar macrophages was studied54. Mannose-functionalized liposomes showed a 1.5-fold higher exposure in alveolar macrophages compared with unmodified liposomes. In comparison with the free drug, the targeted liposomes showed a 23-fold higher bioavailability in the alveolar macrophages. However, these studies were carried out in an uninfected model, and therapeutic efficacy was not studied.
Nanocarriers targeting epitopes displayed on the surface of the pathogen can improve drug delivery into infection sites. Arias et al. encapsulated pentamidine in PLGA nanoparticles for treating Trypanosoma brucei, the causative agent of African sleeping disease55. The nanoparticles were functionalized with a fragment of an antibody called a nanobody. These fragments were 7.5% of the mass of the original antibody, which enabled them to penetrate the glycoprotein coat on the microbe and access its epitope. Nanoparticle-based treatment enabled a tenfold dose reduction in vivo.
In vivo phage display screening has identified cyclic peptides that bind the surface of S. aureus56. The distribution of vancomycin-loaded nanoparticles functionalized with this fluorescein-tagged peptide was measured by confocal microscopy and indicated preferential accumulation in S. aureus-infected lungs. In a 20 day efficacy study, 100% of the mice administered the peptide-functionalized nanoparticles survived, compared with roughly 40% survival in the non-functionalized-nanoparticle and free-vancomycin cohorts. These data suggest that targeted nanoparticles enable greater therapeutic efficacy and may allow reduction of antibiotic doses.
The use of antibody-based targeting has also been employed in hyperthermia-based treatment of S. aureus infection. Kim et al.57 decorated streptavidin-coated magnetic nanoparticles with biotinylated anti-protein A monoclonal antibodies. Binding of the antibody-functionalized nanoparticles to cultures of S. aureus was quantified by flow cytometry and demonstrated a 2.5-fold increase in binding compared with the IgG-coated nanoparticles. Local injection of anti-protein A-functionalized nanoparticles was observed to be nearly twice as effective at killing S. aureus in murine cutaneous infection models compared with the control57. Similar antibody-targeted nanoparticles have been described in the literature58,59 but did not include determination of in vivo efficacy. Considerable inquiry is being conducted on similar ligand-based targeting in the context of drug delivery to cancers and specific tissues, but further development of this technology in the space of ID is required.
Drug molecules released while nanocarriers are in circulation are subject to non-selective distribution. Hence, there is value in developing systems that are binary, whereby no drug is released in systemic circulation and then all of the content is released upon encountering the pathogen. Nanocarriers that disintegrate rapidly in the presence of bacterial phosphatase60 (made from diblock copolymers of poly(phosphoesters) and PEG) and lipase61 (made from triblock copolymers of poly(phosphoester), poly(caprolactone) and PEG) have been used for pathogen-triggered release of vancomycin. In a zebrafish model of methicillin-resistant S. aureus infection, the phosphatase-sensitive nanoparticles enhanced survival by 20 percentage points over free-drug controls. Stimuli-sensitive nanoparticles have also been used for treatment of sepsis, where the goal was to reduce bacterial load and inflammation in the lungs62. Nanoparticles were made with a cationic polyester (poly(β-amino ester)) to enable their degradation in the presence of bacterial enzymes and reduced pH, commonly found at sites of infection. The nanoparticles were surface functionalized with an antibody that binds intercellular adhesion molecule-1, a protein upregulated on the surface of endothelial cells of infected tissues. The nanoparticles were dually loaded with an antibiotic and an anti-inflammatory agent. In a mouse model of pulmonary P. aeruginosa infection, treatment with targeted nanoparticles improved survival over the free-drug control by nearly 50 percentage points. A common limitation of these studies is that comparison of the stimuli-sensitive nanoparticles with non-degradable nanoparticles was not performed. Polymers such as poly(β-amino esters) also degrade rapidly even in the absence of enzymes, further underscoring the need for including the non-degradable nanoparticle controls.
Although nanoencapsulation may enhance drug accumulation at sites of infection, there are certain limitations when translating from rodent models to humans. It is not clear if enhanced permeability is limited to rodent models or if it is observed in humans as well. Furthermore, some studies show that the degree of targeting is directly correlated with the severity of infection51,52. Hence, whether targeting can be achieved to treat low titres of persistent infection is unclear. It may also be interesting to compare the numbers of bacteria/viruses used in disease models in rats to those found in humans. If the numbers used in rodents are inordinately high, the data obtained thus far may need reinterpretation. Although drug accumulation at sites of infection is increased in rodent models, a substantial fraction of the drug is at off-target locations. Drugs with toxicity at these non-target sites are not good candidates for nanoencapsulation. It must also be determined whether the increase in on-target accumulation is high enough to justify reformulation. Metrics such as the drug targeting index (which compares the ratio of nanocarrier bioavailability at sites of efficacy and toxicity to those of the drug)49 may prove valuable in this regard. For triggered-release systems, some drug leakage is generally observed even in the absence of triggers. The specificity of release is unclear as even host enzymes may act as triggers. Moreover, due to the non-specific biodistribution of nanocarriers that typically precedes triggering, drug release at non-target sites cannot be avoided. Most importantly, it must be noted that the intravenous route of administration may not be practical in resource-limited settings or in mass drug administration campaigns. Hence, it will be critical to understand whether these targeting strategies are applicable if the nanocarriers are administered via clinically relevant routes of administration.
Overcoming resistance to anti-infectious agents
Pathogen resistance is a critical and ever-growing obstacle to treatment of IDs. Resistance results from the secretion of drug-inactivating enzymes and/or the pathogens switching to a state of reduced metabolic activity, forming biofilms, adopting an intracellular life cycle or being obligate intracellular pathogens3.
Poor drug diffusion across the host cell membrane and its active efflux via drug transporters can reduce activity against intracellular pathogens63. Another complexity is that both drug molecules and pathogens are unevenly distributed within host cells, and in the absence of colocalization, efficacy is compromised (Fig. 4a). For example, aminoglycosides largely accumulate in the lysosomes and are likely to be ineffective against cytosolic pathogens64. Hence, strategies that enhance intracellular accumulation of drugs and targeting to subcellular locations may improve efficacy.

Nanocarrier systems can be designed to evade physical and chemical mechanisms for microbial drug resistance. a, Pathogens can escape the action of anti-infectives by residing in intracellular foci that are poorly accessible to drugs147. b, Highly polar drugs can have low uptake into cells, and drug internalized in the cell can subsequently be removed by efflux transporters. Both factors can protect intracellular pathogens. Nanocarriers have been used to improve cellular uptake of drugs and improve their activity. c, Nanocarriers can be loaded or surface functionalized with moieties that disrupt quorum sensing or the biofilm matrix. d, Nanocarriers can protect drug-degrading enzymes present in the biofilm. e, Co-encapsulation of drug combinations in nanocarriers has been shown to produce improved efficacy over co-administration of the free-drug combination.
Encapsulation in nanocarriers offers opportunities to increase cell uptake of anti-infectives and alter their intracellular disposition (Fig. 4b). Lutwyche et al. attempted to improve the delivery of gentamicin to cytoplasmic strains such as Salmonella typhirium and Listeria monocytogenes65. Gentamycin was encapsulated in fusogenic liposomes that are stable under neutral conditions but disintegrate under the acidic conditions of the endolysosomes. Upon disintegration, the lipids fuse with the endosomal membrane, destabilizing it and releasing the drug in the cytoplasm. To validate the acid-mediated breakdown of the liposomes, the authors mixed two liposomal formulations—one formulation containing a fluorescence resonance energy transfer pair, and one formulation without. Owing to the proximity of the fluorescent molecules, this mixture had low baseline fluorescence. However, upon acidification of the mixture, the liposomes disintegrated and the lipids mixed, resulting in dequenching of the fluorescence. In an in vitro model of recombinant S. typhirium infection, gentamicin-loaded pH-sensitive liposomes showed threefold higher activity than the free drug. Despite higher cell uptake of pH-insensitive liposomes, they showed inferior activity to the pH-sensitive formulation. To understand the mechanism, authors loaded pH-sensitive and insensitive liposomes with a marker that fluoresced upon release and metabolism by cellular enzymes. Confocal microscopy showed that only pH-sensitive liposomes released the marker intracellularly in murine macrophages. Marker release was abrogated by bafilomycin, an inhibitor of endosome acidification. This study showed excellent mechanistic details of the role of each formulation component, and the need for intracellular targeting.
Clemens et al. developed mesoporous silica nanoparticles loaded with isoniazid for treating intracellular M. tuberculosis66. The pores of the nanoparticles were plugged with β-cyclodextrin, which prevents the drug from leaking out at a neutral pH. However, at an acidic pH, the plug is lost, resulting in drug release. Sémiramoth and colleagues conjugated penicillin G to hydrophobic squalene using a pH-sensitive ester bond67. Nanoparticles formed from this conjugate were rapidly taken up by cells and released drug in the endolysosomes. This resulted in improved activity over the free drug.
Reverse engineering of bacterial cell uptake mechanisms on the surface of nanocarriers has been used to improve intracellular delivery. Invasin, a Yersinia pseudotuberculosis protein, binds integrins on host cells, causes reorganization of its cytoskeleton and enables bacterial uptake into membrane enclosed structures. Labouta and colleagues68 reverse engineered this mechanism on the surface of liposomes by conjugating them to the C-terminal fragment of the invasin—InvA497. In vitro confocal microscopy studies in Hep-2 cells showed that InvA497-functionalized liposomes had increased cell binding (>30-fold) compared with albumin-functionalized liposomes, and this was abrogated in competition experiments. InvA497-funtionalized gentamycin-loaded liposomes caused a 30% reduction in bacterial load in an in vitro model of Y. pseudotuberculosis infection, whereas albumin-functionalized liposomes had no effect68. This group has shown that surface functionalization with InvA497 is beneficial for enhancing both in vitro cell uptake and antimicrobial efficacy of polymeric nanoparticles69. This is a highly innovative concept that may benefit from in vivo evaluation. Furthermore, in the event that these systems are applied orally, it may be important to understand the stability of the peptides in the enzyme-rich gastrointestinal environment. If the nanocarriers are administered systemically, it will be important to understand immune response to these systems as they are functionalized with peptides found in bacteria, and its effect on their pharmacokinetics and tolerability.
Intracellular delivery is of particular importance with molecules such as messenger RNA, which have gained much attention due their application in vaccination. Most polymer-based nucleic acid and protein delivery systems are inspired by initial work done by Langer and Folkman70, which showed that macromolecules such as proteins could be encapsulated in small polymer-based carriers—a system deemed unlikely to work due to the use of organic solvents during synthesis and the perceived imperviousness of polymers to macromolecules71. Following this initial proof-of-concept with polymers, encapsulation of RNA in lipid carriers72, and their utility in vaccination73, was demonstrated. A key feature of nanocarriers is that they aid in the transport of mRNA across the cell membrane, enable endolysosomal escape and release the nucleic acid in the cytoplasm74. In one example75, self-replicating mRNA encoding an antigen protein was complexed with a cationic dendrimer-based polymer. Animals treated with mRNA nanoparticles produced antibody-based and CD8+ T-cell immune responses, and were completely protected from lethal challenges of Ebola virus, H1N1 influenza and Toxoplasma gondii. mRNA vaccines based on lipid nanocarriers have also been tested in the clinic by companies such as Moderna, Acuitas Therapeutics, Pfizer, BioNTech, GlaxoSmithKline, Sanofi, Translate Bio and others. Initial results with lipid-nanoparticle-formulated mRNA vaccines against H10N8 and H7N9 influenza viruses and SARS-CoV-2 indicate that these vaccines are well tolerated and that they produce a robust humoral response76,77. Specifically, mRNA-based vaccines have shown >90% efficacy against SARS-CoV-2. The stability of mRNA-based vaccines during long-term storage and transport could also be major obstacles to their application in resource-limited settings. Peptide-based antigen assembled into nanoparticles have also been developed for vaccination against SARS-CoV-278.
As bacteria and nanocarriers can accumulate in intracellular vesicles, nanoencapsulation has been used for intracellular targeting of antibiotics. Using confocal and transmission electron microscopy, Couvreur and colleagues showed that poly(cyanoacrylate) nanoparticles colocalized with S. typhirium in phagosomes of macrophages, resulting in enhanced intracellular targeting of ampicillin79. In another study, Toti et al. found that PLGA nanoparticles loaded with a fluorescent dye accumulated with chlamydial inclusion bodies in infected human lung epithelial cells80. In vitro studies revealed that antibiotics (azithromycin and rifampicin) were effective if applied immediately after infection, but not if applied after 24 or 48 h. However, encapsulation in nanoparticles partly restored their activities even when applied at 24 and 48 h after infection.
Nanoparticles can prevent, disrupt and inhibit or disperse bacteria from biofilm infections30 (Fig. 4c). By forming a diffusion barrier, the extracellular polymeric matrix in the biofilm protects bacteria from high antibiotic concentrations, often leading to chronic infections. Teirlinck et al. showed that laser irradiation of gold nanoparticles resulted in the formation of nanobubbles that disrupted this diffusional barrier81. Local disruption of the biofilms increased the permeability and therefore susceptibility to the antibiotic tobramycin. Other examples of nanocarrier-mediated biofilm disruption are discussed earlier in this Review in the context of pulmonary drug delivery.
The evolution of antibiotic-degrading enzymes is a major mechanism causing bacterial resistance. These enzymes, such as β-lactamases and aminoglycoside-modifying enzymes, are readily transferred through horizontal gene transfer and have been implicated in the pattern of increasing drug resistance3. Nanoparticles have been used to protect antibiotic agents from enzymatic degradation and resensitize resistant bacteria to their bactericidal effects (Fig. 4d). Shaaban et al. developed poly(caprolactone) nanoparticles loaded with imipenem82 and found that drug-loaded nanoparticles conferred 250- and 62.5-fold decreases in minimal inhibitory concentrations compared with free imipenem in in vitro cultures of imipenem-resistant K. pneumoniae and P. aeruginosa, respectively. Furthermore, to verify the ability of the nanocarriers to protect the drug from carbapenem-degrading enzymes, the imipenem formulations were tested with and without the addition of exogenous carbapenemase in Escherischia coli. The results demonstrated that while free imipenem had negligible effect when co-administered with the enzyme, the nanoparticles had comparable bactericidal effects regardless of whether carbapenemase had been co-administered. Liposomes have also been used for protecting drugs against bacterial enzymes83. When co-administered with exogenous β-lactamase, piperacillin-loaded liposomes provided twofold growth inhibition compared with the free drug co-administered with β-lactamase. The authors posited that the lactamases were either unable to penetrate the liposome or were sterically hindered by the liposomal surface, but were unable to confirm these hypotheses experimentally. Further testing in animal biofilm models and mechanistic confirmation must be done to reliably demonstrate the ability of nanoparticles to protect therapeutic payloads from enzymatic degradation.
Multiple antimicrobial agents can be packaged within the same nanoparticle to increase the likelihood of overcoming existing drug resistance mechanisms instead of using one drug alone (Fig. 4e). Schiffelers and colleagues evaluated the efficacy of gentamicin and ceftazidime as single agents and in combination when formulated as free-drug solutions and as liposomes84. Testing was performed in a rat unilateral lung infection model with drug-sensitive and drug-resistant K. pneumoniae. In the drug-sensitive model, ten doses of the soluble drug combination treatment resulted in complete survival. Impressively, only a single dose of the liposomal combination produced the same effect. In the drug-resistant model, the dose of the drug combination was increased compared with that used in the drug-sensitive model—the dose of ceftazidime was 4-fold higher, and that of gentamicin was 66-fold higher. Ten treatments at these elevated doses produced complete survival in the drug-resistant model. However, only two treatments with substantially lower doses of the liposomal combination also produced 100% survival. This system is attractive for two reasons—it enabled dose reduction and the treatment of drug-resistant infection; and it dramatically reduced the number of administration events required to produce efficacy. This latter feature is of great value in resource-limited settings.
Advanced preclinical and clinical nanotechnologies
In this section, we highlight the most advanced nanotechnologies in major IDs—HIV infection, TB and malaria. The reader will glean that nanotechnology has been most actively studied in the clinic and in large animals for the treatment and prevention of HIV infection (Table 1). In contrast, for malaria and TB, nanotechnology has been pursued with less gusto. For these IDs, work has mainly been limited to preclinical trials in rodents. We also hope to convey that the most advanced nanotechnologies are non-complex, yet highly impactful. Hence, perhaps for nanotechnology to benefit the patient, it needs to satisfy certain tenets such as simplicity in design, clinical need and financial enthusiasm.
HIV infection
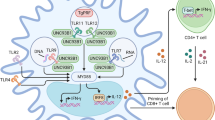
HIV infection is one of the major IDs contributing to high rates of morbidity and mortality globally85. HIV primarily targets CD4+ T cells (Fig. 5) and weakens the patient’s immune system making the individual susceptible to opportunistic infections. Although access to antiretroviral therapy has continued to improve for people living with the disease around the world—reducing morbidity, mortality and ongoing transmission—more than 900,000 people continue to die of acquired immunodeficiency syndrome each year. In 2017, there were 1.8 million people who became newly infected with HIV. Moreover, new HIV infections are increasing at an alarming rate in some populations, including people of colour, men who have sex with men and younger women, as well as in certain geographic regions including Russia and other eastern European countries86,87.

Fusion of the viral membrane with the host cell membrane enables entry of viral contents into the host cells. Viral RNA is reverse transcribed in complementary DNA (cDNA), which can be integrated into the host genome. This is followed by the formation of new copies of the viral RNA and proteins, which assemble into new viral particles that can infect new cells. Antiretrovirals work by inhibiting one or more steps of this pathway10.
Long-acting injectable antiretrovirals are the most clinically advanced nanotechnology in HIV treatment. Long-acting injectable nanoparticles reduce dosing frequency, which stands to improve patient adherence. They also allow drugs to be administered to dysphagic patients, for example individuals infected with HIV and suffering from opportunistic oesophageal infections88. Moreover, there has been some interest in developing long-acting nanoparticles for prevention—including both pre-exposure prophylaxis (which requires long-term administration of two medications to achieve protection) and post-exposure prophylaxis (where administration of a single injection or long-acting oral medication would be especially valuable)89,90. Specifically, nanoparticles of two antiretrovirals, cabotegravir and rilpivirine, are being actively pursued in the clinic. Cabotegravir16 and rilpivirine17 were chosen owing to their efficacy at low oral doses and low water solubility.
The pharmacokinetics, safety and efficacy of rilpivirine and cabotegravir nanosuspensions (surfactant-stabilized drug nanocrystals with a mean diameter of 200 nm) have been evaluated in clinical studies. Both nanosuspensions were safe and devoid of major adverse effects88. Estimated half-lives of rilpivirine and cabotegravir in nanosuspensions were 44–61 days (ref. 91) and 25–54 days (ref. 91), respectively, enabling administration every 1–3 months. In contrast, half-lives of rilpivirine and cabotegravir in tablets have been estimated to be ~1–2 days (refs. 91,92). In Phase IIb clinical trials, the combination of rilpivirine and cabotegravir nanosuspensions enabled disease suppression equivalent to oral tablets of cabotegravir and abcavir-lamivudine93.
Two other injectable nanoparticles have shown promising results in non-human primates. One of these formulations was developed for the prolonged systemic delivery of dolutegravir, which has relatively high water solubility18,94. A water-insoluble prodrug of dolutegravir was produced by esterification with myristic acid and formulated into nanoparticles. In rhesus macaques, prodrug nanoparticles maintained therapeutic concentrations of dolutegravir for ~40 days and extended the half-life of dolutegravir to ~19.5 days. It is believed that the nanoparticles are engulfed by macrophages, leading to intracellular activation of the prodrug, followed by release of the active moiety from the macrophages18,94. Lipid-based nanoparticles loaded with three to four antiretrovirals including lopinavir, ritonavir and tenofovir have also been studied in non-human primates95,96. Nanoencapsulation enhanced circulation times of the antiretrovirals and increased their uptake into the mononuclear cells in peripheral blood and lymph nodes of macaques. This is especially notable for antiretrovirals, as HIV persists in macrophages for prolonged durations, even in patients receiving antiretroviral therapy.
Long-acting injectable antiretrovirals are widely considered breakthrough interventions; however, certain challenges remain. Despite most advanced nanosuspensions containing >90% drug, clinical use of this technology has been limited to drugs that have a daily oral dose of <25–50 mg. Patients treated with injectable nanosuspensions are exposed to the drug for at least a month, and there is no mechanism to withdraw exposure in cases of adverse reactions. This problem has been addressed in clinical trials by providing patients with a lead-in period of oral drug therapy before the injection. An alternative approach was recently tested in mice and involves injecting a solution of the drug and polymer in an organic solvent subcutaneously97. A surgically removable drug depot (~0.75 cm diameter in mice) forms as the organic solvent diffuses out of the injection site. Finally, in most clinical trials, the nanosuspensions have been administered by medical practitioners, and it is likely that patients will need to visit their caregiver for each injection. It remains to be seen whether the need for chronic visits to a clinic deters patient adherence. The ability to self-medicate at home with infrequently administered oral dosage forms may help address this issue. For this, we have developed an orally administered gastric retentive dosage form that can reduce dosing frequency from daily to weekly98. However, personal preferences may be highly varied and providing multiple options to the patient may aid in maximizing patient adherence.
Oral antiretroviral nanoparticles have been developed for paediatric use with the intent to replace tablets (which are difficult to swallow) and alcohol-containing solutions (which contain harmful excipients). These nanoparticles are produced by a novel emulsion template freeze-drying approach and have been loaded with lopinavir and efavirenz99. In rats, the optimized nanoparticle formulation was bioequivalent to lopinavir dissolved in alcohol and propylene glycol99. The authors believe that the nanoparticles are probably absorbed via the lymphatic vessels in the gastrointestinal tract. However, further work may be required to validate this observation. Efavirenz and lopinavir nanoparticles made using this strategy are currently awaiting clinical testing (NCT02631473).
Malaria
Malaria, caused by the single-celled complex protozoan of the Plasmodium genus (Fig. 6), is the most prevalent parasitic disease in the world. In 2017, malaria cases rose for the second year in a row, reaching 219 million cases100. Around 90% of all malaria cases and deaths are in Africa, where the disease costs the continent’s economy US$12 billion yr−1 in direct losses and reduces GDP growth by 1.3% (ref. 101). Despite over 3 billion people being at risk of infection, funding for the global malaria response has plateaued since 2010, reaching only US$3.1 billion in 2017, which is less than half of the US$6.6 billion yr−1 funding target for 2020100. As part of the investment, both short-term and long-term solutions to malaria must be considered, such as using nanotechnology for treatment and vaccination. A vital consideration for development of new approaches will be their cost-effectiveness, given the target product profiles of US$1 day−1 for treatment of malaria.

The infection is spread to a human through the bite of a female Anopheles mosquito, which injects the parasite Plasmodium in the form of sporozoites that migrate through the skin into the circulation. Within minutes, the sporozoites reach the liver, where they invade hepatocytes and proliferate to form merozoites. Each infected hepatocyte releases thousands of merozoites into the bloodstream, beginning a cycle of invasion of RBCs. Merozoites replicate asexually through ring, trophozoite and schizont stages within RBC vacuoles to form new daughter cell merozoites that egress and infect other RBCs. Meanwhile, a small fraction of merozoites sexually differentiate into male and female gametocytes, which are ingested into the midgut of a mosquito from peripheral blood when it bites an infected human. The gametocytes further develop into gametes, zygotes, ookinetes, oocysts and finally sporozoites, which can resume the cycle of human infection when they migrate to the mosquito’s salivary glands and are injected into another human during a blood meal148.
Malaria is mainly treated using chemotherapy, with artemisinin-based combination therapy being integral to curing the disease102. Unfortunately, due to the low uptake of artemisinin in parasite-infected red blood cells (RBCs), these drugs provide only symptomatic relief at low doses103. Killing parasites in the RBCs requires prolonged treatment with high doses, which results in toxicities, potential for missed doses and development of resistance104. Given that blood stage parasites are the main therapeutic target, researchers are developing various approaches to target antimalarials to infected RBCs105,106,107, thereby maximizing their therapeutic window. Covalent attachment of heparin and monoclonal antibodies to liposomes has been proven to selectively target infected RBCs in vitro105,108. These functionalized liposomes preferentially adhere to surface proteins decorating infected RBCs and reduce the overall parasitaemia. Further studies of parasite resistance and in vivo testing of these systems will be needed, while potentially prioritizing heparin–liposome formulations due to their cheaper cost. Although liposomal drug delivery is an established platform with proven biocompatibility, the oral route of administration is typically not used due to instability in the gastrointestinal tract109. Therefore, researchers must continue to develop other targeting strategies to maximize the benefit to those living in malaria-endemic regions, where oral administration is preferred. These can include designing nanocarriers to selectively target other Plasmodium stages, such as at the parasite stage in the liver or during the transmissible sexual stages.
Nanotechnology can also improve the oral bioavailability of poorly water-soluble antimalarials, such artemether and tafenoquine. NanoAbsorb is a solid microemulsion pre-concentrate formulation that can safely deliver the widely used drug artemether with higher antimalarial efficacy in vivo than the marketed formulation Larither110. Another example is the use of self-microemulsifying drug delivery systems, which can be filled in hard gelatine capsules and rapidly transform into microemulsions in gastrointestinal fluids111. Long-chain-triglyceride-based self-microemulsifying systems containing β-artemether were demonstrated to be safe and have a notable improvement in the antimalarial activity in vivo compared with Larither. An oral lipid-based nano-emulsion of tafenoquine with sizes <20 nm enhanced drug solubility, bioavailability and efficacy in vivo while also reducing the toxicity112. These approaches, using oily vehicles to enhance solubilization in the gastrointestinal tract, improve drug bioavailability and patient adherence.
Nanoformulation of multiple combined drugs is an efficient method for designing innovative therapeutics and can prevent resistance of the malaria parasite. Polymer–drug conjugates were synthesized into nanoparticles with substituted poly(phosphazenes) and the combination of primaquine and dihydroartemisinin. The combination therapy exhibited promising antimalarial efficacy in vivo against a resistant parasite strain113. Artemether and lumefantrine are well accepted as a combination therapy for treating uncomplicated malaria, yet the current marketed formulation degrades in the gastrointestinal tract, leading to unpredictable pharmacokinetics. Using co-loaded nanostructured lipid carriers with these drugs, researchers showed that the systems resulted in greater clearance of parasites from infected mice and had a clear benefit over individual drug-loaded carriers in terms of survival period and parasitaemia progression114.
There is only one malaria vaccine candidate that has received a positive regulatory assessment: RTS,S/AS01 (RTS,S) (Mosquirix), which is an injectable vaccine that provides partial protection against malaria in young children115. The antigen is formulated as virus-like particle and combined with a liposomal adjuvant system. In July 2015, European regulators approved the use of the vaccine, although it is at most 50% effective115. Vaccine development is an active area of research, and a recent study used spontaneous nanoliposome antigen particleization to show efficacy of a multistage, multi-antigen vaccine in mice and rabbits116. Further research and development with affordable large-scale production is necessary for a successful malaria vaccine, and nanotechnology could play a critical role in providing new approaches to protect all ages.
TB
TB, which claims the lives of over 3,500 people every day, is the world’s leading killer among IDs117. According to the World Health Organization (WHO), 10 million people developed TB in 2017 with a global economic burden amounting to US$12 billion annually117,118. The burden of TB is concentrated in Asia and Africa—only 6% of global cases were in the WHO European Region and WHO Region of the Americas117. Furthermore, M. tuberculosis (Fig. 7) is the most critical pathogen in the global antimicrobial resistance crisis119. Unless radical action is taken, drug-resistant strains of M. tuberculosis will account for 25% of antimicrobial resistance-related deaths and cost the global economy US$16.7 trillion by the year 2050119.

TB is caused by the inhalation of droplet nuclei containing M. tuberculosis that enter the respiratory tract and infect the alveoli of the lungs. During the first stage, alveolar macrophages recognize, engulf and attempt to destroy the bacilli. The second stage of symbiosis occurs when the bacilli grow logarithmically within infected alveolar macrophages that are unable to stop the bacilli from growing. Eventually, the infected areas transform into a granuloma, a hallmark of TB characterized by a wall of lymphocytes and severe chest pain. This third stage of the disease results in a solid caseous centre, where the bacteria can survive for years. Most humans infected with TB do not exhibit progression of the disease, as it remains in a latent state. It is estimated that 1.7 billion people, or 23% of the world’s population, have latent TB. However, some infected individuals progress to the fourth and final stage of the disease, where the caseous centre liquefies and cavitates to fill the lungs with free-floating bacteria and spread in the lungs, causing pulmonary TB. These bacteria can disseminate to more distant tissues and organs via the lymphatic system or bloodstream to result in extrapulmonary TB. The cycle is complete when an infected TB individual coughs, sneezes, speaks or sings to release highly transmissible infectious droplet nuclei into the air for a susceptible individual to inhale149.
WHO guidelines recommend treating drug-susceptible TB for at least 6 months with an oral drug regimen of four antibiotics taken daily11. Because of the prolonged and frequent dosing, and side effects, patients find it difficult to adhere to these regimens and are at risk of developing drug-resistant strains4. Several of the current antibiotics are poorly soluble, unstable in gastric acid and unable to penetrate the alveolar macrophages where the bacilli reside120. Given that bedaquiline was the first new drug approved in more than 40 years (ref. 121) and the dearth of others in the pipeline, reformulating existing drugs in nanocarriers is an attractive way of having an immediate impact on the treatment of TB.
Several oral nanotechnology-based systems have been designed with existing TB drugs and tested in animal models. Solid lipid nanoparticles have been tested in rodents with the goal of improving the bioavailability of rifampicin and the combination of rifampicin, isoniazid and pyrazinamide122,123. In the lungs and spleen of infected mice, no tubercle bacilli could be detected after five oral doses of drug-loaded solid lipid nanoparticles administered every tenth day, whereas 46 daily doses of oral free drugs were required to achieve the same therapeutic benefit123. Polymer-based nanoparticles (PLGA and sodium alginate) have also been developed to improve the oral bioavailability of rifampicin, isoniazid, ethambutol, pyrazinamide and streptomycin124,125,126,127.
Inhalable nanocarriers of TB drugs have been a major focus of research given that pulmonary TB is the most common TB form128. Researchers have developed nebulized solid lipid particles129 and sodium alginate nanoparticles130 incorporating rifampicin, isoniazid and pyrazinamide to improve targeted bioavailability in the lungs and reduce hepatotoxicity. Interestingly, pulmonary administration of rifampicin-loaded solid lipid nanoparticles in rats enabled delivery to alveolar macrophages131. These examples highlight the promise of pulmonary administration of nanocarriers, although they must be designed for delivery with portable and easy-to-operate cost-effective devices such as inhalers.
There is no effective vaccine that comprehensively protects against TB infection. Bacillus Calmette–Guerin (BCG), which was introduced in 1921, is the sole approved TB vaccine, but it offers only limited protection132. Therefore, researchers are attempting to improve the existing BCG vaccine and derive new vaccines to prevent TB133. When guinea pigs were administered an aerosolized formulation of BCG nanomicroparticles, they generated better immune protection and enhanced resistance to TB infection than when they were immunized with the standard parenteral BCG formulation134. The nanomicroparticles are a single-particle form with two nanometre-scale axes and a third of micrometre dimension. These dimensions facilitate better aerosolization compared with particles with similar dimensions in all axes. Pulmonary administration of the common TB antigen 85B conjugated to poly(propylene sulphide) nanoparticles demonstrated improved protection in mice after they were challenged with aerosolized M. tuberculosis (ref. 135). Researchers are also exploring chitosan, a biodegradable and biocompatible polycationic polymer, for its ability to bind and protect DNA from nuclease degradation and for its mucoadhesive properties. A nano-chitosan-based recombinant DNA vaccine elevated the immunologic and protective effects against TB in a mouse model136, and chitosan has a role as a vaccine adjuvant in the prevention of TB137.
It is also recommended that people who are infected with TB but do not have active disease (latent TB) receive treatment to eradicate the mycobacteria and prevent progress to active disease138. These preventative regimens can last for 9 months and require multidrug therapy. Given that most people with TB infection do not have symptoms, it can be challenging for them to complete their regimens139. Nanotechnology can potentially simplify dosing and minimize adverse events in this population, thereby greatly contributing to the elimination of TB.
Challenges and outlook
The examples above highlight many promising therapeutic strategies for developing nanomedicines to treat and prevent IDs. However, to have the maximum impact, nanotechnology solutions will need to overcome several financial, manufacturing and regulatory challenges. First, reducing the cost will be the major hurdle in developing new drugs and nanotechnology-based systems. For global health solutions, development of new drugs is expensive. Reformulating existing drugs in nanocarriers may help to achieve similar targets of enhanced efficacy and safety at a considerably lower cost. However, one currently approved nano-system (an amphotericin B liposomal formulation) is not cost-effective in developing countries140. Excipients used in nanosystems, such as lyoprotectants, can increase the cost of many treatments. Second, the synthesis and storage conditions of some nanoparticles may not be conducive to conditions in low-resource countries141. Researchers will need to enable the reproducibility and bulk production while considering the environmental effects of these nanosystems. Nanomedicines are likely to be three-dimensional constructs of multiple components with preferred spatial arrangements. Subtle changes in process or composition can adversely affect the complex composition of nanomedicines142,143. Third, regional and national differences within and between regulatory authorities, especially when running multi-centric international trials, will be a challenge during the clinical trial stage144. Finally, patient acceptability of these nanosystems will need to be addressed. In 2006, the International Centre for Technology Assessment and other consumer groups filed a legal petition against the FDA for perceived lack of initiative in regulating nanomaterial-containing products under their jurisdiction. The FDA created a Nano Task Force in response145. To achieve successful translation of nanomedicines, a network of stakeholders will need to converge. This includes academics, investors from industry and the government, and contract research and manufacturing organizations. Nanotechnology is a multi-disciplinary field that also requires intellectual property and commercialization strategies to grow146. Nonetheless, the rapid translation of nanotechnologies from the bench to the people, and the impact that these systems have had on the SARS-CoV-2 pandemic provide great promise. This example suggests that similar feats may be possible in treating other infectious diseases as well.
Conclusion
IDs are a major driver of morbidity and mortality globally, and their impact on low SDI countries is particularly grave. Simplifying the use of medicines and making drugs safer and more efficacious can improve patients’ quality of life and reduce disease burden. In this Review, we highlighted how nanotechnology-based approaches can enable oral drug administration, infrequent drug administration and drug targeting to sites of infection, ultimately improving treatment efficacy. Finally, we discussed examples of nanotechnologies that are in clinical trials for the treatment of HIV and in preclinical development for the treatment of malaria and TB. Innovative and cost-effective nanotechnologies that take into consideration the challenges that are encountered low SDI countries are most likely to benefit patients.
References
Roth, G. A. et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the global burden of disease Study 2017. Lancet 392, 1736–1788 (2018).
Kinch, M. S., Patridge, E., Plummer, M. & Hoyer, D. An analysis of FDA-approved drugs for infectious disease: antibacterial agents. Drug Discov. Today 19, 1283–1287 (2014).
Munita, J. M. & Arias, C. A. Mechanisms of antibiotic resistance. Microbiol. Spectrum 4, VMBF-0016-2015. (2016).
Sabaté, E. Adherence to Long-Term Therapy: Evidence for Action (WHO, 2003).
Pheage, T. Dying from Lack of Medicines (United Nations, 2016); https://www.un.org/africarenewal/magazine/december-2016-march-2017/dying-lack-medicines
Anselmo, A. C. & Mitragotri, S. Nanoparticles in the clinic. Bioeng. Transl. Med. 1, 10–29 (2016).
Huh, A. J. & Kwon, Y. J. ‘Nanoantibiotics’: a new paradigm for treating infectious diseases using nanomaterials in the antibiotics resistant era. J. Control. Release 156, 128–145 (2011).
Irvine, D. J., Hanson, M. C., Rakhra, K. & Tokatlian, T. Synthetic nanoparticles for vaccines and immunotherapy. Chem. Rev. 115, 11109–11146 (2015).
Pardi, N., Hogan, M. J., Porter, F. W. & Weissman, D. mRNA vaccines—a new era in vaccinology. Nat. Rev. Drug Discov. 17, 261–279 (2018).
Kirtane, A. R., Langer, R. & Traverso, G. Past, present, and future drug delivery systems for antiretrovirals. J. Pharm. Sci. 105, 3471–3482 (2016).
Treatment of Tuberculosis: Guidelines 4th edn (WHO, 2010); https://www.who.int/tb/publications/2010/9789241547833/en/
Langer, R. Drug delivery and targeting. Nature 392, 5–10 (1998).
Makadia, H. K. & Siegel, S. J. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 3, 1377–1397 (2011).
Fielding, R. M., Lewis, R. O. & Moon-McDermott, L. Altered tissue distribution and elimination of amikacin encapsulated in unilamellar, low-clearance liposomes (MiKasome). Pharm. Res. 15, 1775–1781 (1998).
Schiffelers, R., Storm, G. & Bakker-Woudenberg, I. Liposome-encapsulated aminoglycosides in pre-clinical and clinical studies. J. Antimicrob. Chemother. 48, 333–344 (2001).
Trezza, C., Ford, S. L., Spreen, W., Pan, R. & Piscitelli, S. Formulation and pharmacology of long-acting cabotegravir. Curr. Opin. HIV AIDS 10, 239–245 (2015).
Williams, P. E., Crauwels, H. M. & Basstanie, E. D. Formulation and pharmacology of long-acting rilpivirine. Curr. Opin. HIV AIDS 10, 233–238 (2015).
Mcmillan, J. et al. Pharmacokinetics of a long-acting nanoformulated dolutegravir prodrug in rhesus macaques. Antimicrob. Agents Chemother. 62, e01316-17 (2017).
Wistrand-Yuen, E. et al. Evolution of high-level resistance during low-level antibiotic exposure. Nat. Commun. 9, 1599 (2018).
Landovitz, R. J., Kofron, R. & McCauley, M. The promise and pitfalls of long-acting injectable agents for HIV prevention. Curr. Opin. HIV AIDS 11, 122–128 (2016).
das Neves, J., Nunes, R., Machado, A. & Sarmento, B. Polymer-based nanocarriers for vaginal drug delivery. Adv. Drug Deliv. Rev. 92, 53–70 (2015).
Leal, J., Smyth, H. D. C. & Ghosh, D. Physicochemical properties of mucus and their impact on transmucosal drug delivery. Int. J. Pharm. 532, 555–572 (2017).
Cu, Y., Booth, C. J. & Saltzman, W. M. In vivo distribution of surface-modified PLGA nanoparticles following intravaginal delivery. J. Control. Release 156, 258–264 (2011).
Lai, S. K. et al. Rapid transport of large polymeric nanoparticles in fresh undiluted human mucus. Proc. Natl Acad. Sci. USA 104, 1482–1487 (2007).
Cunha-Reis, C. et al. Nanoparticles-in-film for the combined vaginal delivery of anti-HIV microbicide drugs. J. Control. Release 243, 43–53 (2016).
Ensign, L. M. et al. Mucus-penetrating nanoparticles for vaginal drug delivery protect against herpes simplex virus. Sci. Transl. Med. 4, 138ra79 (2012).
Palliser, D. et al. An siRNA-based microbicide protects mice from lethal herpes simplex virus 2 infection. Nature 439, 89–94 (2006).
Steinbach, J. M., Weller, C. E., Booth, C. J. & Saltzman, W. M. Polymer nanoparticles encapsulating siRNA for treatment of HSV-2 genital infection. J. Control. Release 162, 102–110 (2012).
Roy, I. & Vij, N. Nanodelivery in airway diseases: challenges and therapeutic applications. Nanomedicine 6, 237–244 (2010).
Han, C. et al. Recent developments in the use of nanoparticles for treatment of biofilms. Nanotechnol. Rev. 6, 383–404 (2017).
Vaughn, J. M. et al. Single dose and multiple dose studies of itraconazole nanoparticles. Eur. J. Pharm. Biopharm. 63, 95–102 (2006).
Alvarez, C. A. et al. Aerosolized nanostructured itraconazole as prophylaxis against invasive pulmonary aspergillosis. J. Infect. 55, 68–74 (2007).
Wong, J. P. et al. Liposome delivery of ciprofloxacin against intracellular Francisella tularensis infection. J. Control. Release 92, 265–273 (2003).
Nafee, N. et al. Antibiotic-free nanotherapeutics: ultra-small, mucus-penetrating solid lipid nanoparticles enhance the pulmonary delivery and anti-virulence efficacy of novel quorum sensing inhibitors. J. Control. Release 192, 131–140 (2014).
Duong, H. T. T. et al. Nanoparticle (star polymer) delivery of nitric oxide effectively negates Pseudomonas aeruginosa biofilm formation. Biomacromolecules 15, 2583–2589 (2014).
Suk, J. S. et al. N-acetylcysteine enhances cystic fibrosis sputum penetration and airway gene transfer by highly compacted DNA nanoparticles. Mol. Ther. 19, 1981–1989 (2011).
Sen, C. K. et al. Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen. 17, 763–771 (2009).
James, G. A. et al. Biofilms in chronic wounds. Wound Repair Regen. 16, 37–44 (2008).
Martinez, L. R. et al. Antimicrobial and healing efficacy of sustained release nitric oxide nanoparticles against Staphylococcus aureus skin infection. J. Invest. Dermatol. 129, 2463–2469 (2009).
Mohiti-Asli, M., Pourdeyhimi, B. & Loboa, E. G. Skin tissue engineering for the infected wound site: biodegradable PLA nanofibers and a novel approach for silver ion release evaluated in a 3D coculture system of keratinocytes and Staphylococcus aureus. Tissue Eng. Pt C 20, 790–797 (2014).
Sarathy, J. P. et al. Prediction of drug penetration in tuberculosis lesions. ACS Infect. Dis. 2, 552–563 (2016).
Huth, M. E., Ricci, A. J. & Cheng, A. G. Mechanisms of aminoglycoside ototoxicity and targets of hair cell protection. Int. J. Otolaryngol. 2011, 937861 (2011).
Tedijanto, C., Olesen, S. W., Grad, Y. H. & Lipsitch, M. Estimating the proportion of bystander selection for antibiotic resistance among potentially pathogenic bacterial flora. Proc. Natl Acad. Sci. USA 115, E11988–E11995 (2018).
Löbenberg, R., Araujo, L., von Briesen, H., Rodgers, E. & Kreuter, J. Body distribution of azidothymidine bound to hexyl-cyanoacrylate nanoparticles after i.v. injection to rats. J. Control. Release 50, 21–30 (1998).
Kaminishi, H., Tanaka, M., Cho, T., Maeda, H. & Hagihara, Y. Activation of the plasma kallikrein-kinin system by Candida albicans proteinase. Infect. Immun. 58, 2139–2143 (1990).
Molla, A., Yamamoto, T., Akaike, T., Miyoshi, S. & Maeda, H. Activation of hageman factor and prekallikrein and generation of kinin by various microbial proteinases. J. Biol. Chem. 264, 10589–10594 (1989).
Laverman et al. Liposomes for scintigraphic detection of infection and inflammation. Adv. Drug Deliv. Rev. 37, 225–235 (1999).
Laverman, P. et al. Microscopic localization of PEG-liposomes in a rat model of focal infection. J. Control. Release 75, 347–355 (2001).
Siegel, R. A., Kirtane, A. R. & Panyam, J. Assessing the benefits of drug delivery by nanocarriers: a partico/pharmacokinetic framework. IEEE Trans. Biomed. Eng. 64, 2176–2185 (2017).
Gref, R. et al. Biodegradable long-circulating polymeric nanospheres. Science 263, 1600–1603 (1994).
Bakker-Woudenberg, I. A. et al. Liposomes with prolonged blood circulation and selective localization in Klebsiella pneumoniae-infected lung tissue. J. Infect. Dis. 168, 164–171 (1993).
Bakker-Woudenberg, I. A., Lokerse, A. F., ten Kate, M. T. & Storm, G. Enhanced localization of liposomes with prolonged blood circulation time in infected lung tissue. Biochim. Biophys. Acta 1138, 318–326 (1992).
Azad, A. K., Rajaram, M. V. S. & Schlesinger, L. S. Exploitation of the macrophage mannose receptor (CD206) in infectious disease diagnostics and therapeutics. J. Cytol. Mol. Biol. 1, 1000003 (2014).
Chono, S., Tanino, T., Seki, T. & Morimoto, K. Efficient drug targeting to rat alveolar macrophages by pulmonary administration of ciprofloxacin incorporated into mannosylated liposomes for treatment of respiratory intracellular parasitic infections. J. Control. Release 127, 50–58 (2008).
Arias, J. L. et al. Nanobody conjugated PLGA nanoparticles for active targeting of African trypanosomiasis. J. Control. Release 197, 190–198 (2015).
Hussain, S. et al. Antibiotic-loaded nanoparticles targeted to the site of infection enhance antibacterial efficacy. Nat. Biomed. Eng. 2, 95–103 (2018).
Kim, M.-H. et al. Magnetic nanoparticle targeted hyperthermia of cutaneous Staphylococcus aureus infection. Ann. Biomed. Eng. 41, 598–609 (2013).
Luo, Y. et al. Targeted nanoparticles for enhanced X-ray radiation killing of multidrug-resistant bacteria. Nanoscale 5, 687–694 (2013).
Millenbaugh, N., Baskin, J., DeSilva, M., Elliott, W. R. & Glickman, R. Photothermal killing of Staphylococcus aureus using antibody-targeted gold nanoparticles. Int. J. Nanomed. https://doi.org/10.2147/IJN.S76150 (2015).
Xiong, M.-H. et al. Bacteria-responsive multifunctional nanogel for targeted antibiotic delivery. Adv. Mater. 24, 6175–6180 (2012).
Xiong, M.-H. et al. Lipase-sensitive polymeric triple-layered nanogel for ‘on-demand’ drug delivery. J. Am. Chem. Soc. 134, 4355–4362 (2012).
Zhang, C. Y., Gao, J. & Wang, Z. Bioresponsive nanoparticles targeted to infectious microenvironments for sepsis management. Adv. Mater. 30, 1803618 (2018).
Carryn, S. et al. Intracellular pharmacodynamics of antibiotics. Infect. Dis. Clin. North Am. 17, 615–634 (2003).
Tulkens, P. & Trouet, A. The uptake and intracellular accumulation of aminoglycoside antibiotics in lysosomes of cultured rat fibroblasts. Biochem. Pharmacol. 27, 415–424 (1978).
Lutwyche, P. et al. Intracellular delivery and antibacterial activity of gentamicin encapsulated in pH-sensitive liposomes. Antimicrob. Agents Chemother. 42, 2511–2520 (1998).
Clemens, D. L. et al. Targeted Intracellular delivery of antituberculosis drugs to Mycobacterium tuberculosis-infected macrophages via functionalized mesoporous silica nanoparticles. Antimicrob. Agents Chemother. 56, 2535–2545 (2012).
Sémiramoth, N. et al. Self-assembled squalenoylated penicillin bioconjugates: an original approach for the treatment of intracellular infections. ACS Nano 6, 3820–3831 (2012).
Labouta, H. I. et al. Bacteriomimetic invasin-functionalized nanocarriers for intracellular delivery. J. Control. Release 220, 414–424 (2015).
Castoldi, A. et al. Aspherical and spherical InvA497-functionalized nanocarriers for intracellular delivery of anti-infective agents. Pharm. Res. 36, 22 (2019).
Langer, R. & Folkman, J. Polymers for the sustained release of proteins and other macromolecules. Nature 263, 797–800 (1976).
Langer, R. Controlling the movement of molecules. Q. Rev. Biophys. 52, e5 (2019).
Ostro, M. J., Giacomoni, D. & Dray, S. Incorporation of high molecular weight RNA into large artificial lipid vesicles. Biochem. Biophys. Res. Commun. 76, 836–842 (1977).
Martinon, F. et al. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur. J. Immunol. 23, 1719–1722 (1993).
Kowalski, P. S., Rudra, A., Miao, L. & Anderson, D. G. Delivering the messenger: advances in technologies for therapeutic mRNA Delivery. Mol. Ther. 27, 710–728 (2019).
Chahal, J. S. et al. Dendrimer-RNA nanoparticles generate protective immunity against lethal Ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc. Natl Acad. Sci. USA 113, E4133–E4142 (2016).
Jackson, L. A. et al. An mRNA vccine against SARS-CoV-2—preliminary report. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2022483 (2020).
Feldman, R. A. et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 37, 3326–3334 (2019).
Tian, J.-H. et al. SARS-CoV-2 spike glycoprotein vaccine candidate NVX-CoV2373 immunogenicity in baboons and protection in mice. Nat. Commun. 12, 372 (2021).
Pinto-Alphandary, H. et al. Intracellular visualization of ampicillin-loaded nanoparticles in peritoneal macrophages infected in vitro with Salmonella typhimurium. Pharm. Res. 11, 38–46 (1994).
Toti, U. S. et al. Targeted delivery of antibiotics to intracellular chlamydial infections using PLGA nanoparticles. Biomaterials 32, 6606–6613 (2011).
Teirlinck, E. et al. Laser-induced vapour nanobubbles improve drug diffusion and efficiency in bacterial biofilms. Nat. Commun. 9, 4518 (2018).
Shaaban, M. I., Shaker, M. A. & Mady, F. M. Imipenem/cilastatin encapsulated polymeric nanoparticles for destroying carbapenem-resistant bacterial isolates. J. Nanobiotechnol. 15, 29 (2017).
Nacucchio, M. C., Bellora, M. J., Sordelli, D. O. & D’Aquino, M. Enhanced liposome-mediated activity of piperacillin against staphylococci. Antimicrob. Agents Chemother. 27, 137–139 (1985).
Schiffelers, R. M. et al. In vivo synergistic interaction of liposome-coencapsulated gentamicin and ceftazidime. J. Pharmacol. Exp. Ther. 298, 369–375 (2001).
Miles to Go: Closing Gaps, Breaking Barriers, Righting Injustices (United Nations AIDS, 2018); http://www.unaids.org/en/resources/documents/2018/global-aids-update
Beyrer, C., Wirtz, A. L., O’Hara, G., Léon, N. & Kazatchkine, M. The expanding epidemic of HIV-1 in the Russian Federation. PLoS Med. 14, e1002462 (2017).
HIV in the United States and Dependent Areas (Centers for Disease Control and Prevention, 2019); https://www.cdc.gov/hiv/statistics/overview/ataglance.html
Swindells, S., Flexner, C., Fletcher, C. V. & Jacobson, J. M. The critical need for alternative antiretroviral formulations, and obstacles to their development. J. Infect. Dis. 204, 669–674 (2011).
Olsthoorn, A. V. et al. Barriers to the uptake of postexposure prophylaxis among Nairobi-based female sex workers. AIDS 1, 99–103 (2015).
Wheelock, A. et al. Are Thai MSM willing to take PrEP for HIV prevention? An analysis of attitudes, preferences and acceptance. PLoS ONE 8, e54288 (2013).
Verloes, R. et al. Safety, tolerability and pharmacokinetics of rilpivirine following administration of a long-acting formulation in healthy volunteers. HIV Med. 16, 477–484 (2015).
Ford, S. L. et al. Lack of pharmacokinetic interaction between rilpivirine and integrase inhibitors dolutegravir and GSK1265744. Antimicrob. Agents Chemother. 57, 5472–5477 (2013).
Margolis, D. A. et al. Cabotegravir plus rilpivirine, once a day, after induction with cabotegravir plus nucleoside reverse transcriptase inhibitors in antiretroviral-naive adults with HIV-1 infection (LATTE): a randomised, phase 2b, dose-ranging trial. Lancet Infect. Dis. 15, 1145–1155 (2015).
Sillman, B. et al. Creation of a long-acting nanoformulated dolutegravir. Nat. Commun. 9, 443 (2018).
Freeling, J. P., Koehn, J., Shu, C., Sun, J. & Ho, R. J. Y. Anti-HIV drug-combination nanoparticles enhance plasma drug exposure duration as well as triple-drug combination levels in cells within lymph nodes and blood in primates. AIDS Res. Hum. Retrov. 31, 107–114 (2015).
Freeling, J. P., Koehn, J., Shu, C., Sun, J. & Ho, R. J. Y. Long-acting three-drug combination anti-HIV nanoparticles enhance drug exposure in primate plasma and cells within lymph nodes and blood. AIDS 28, 2625–2627 (2014).
Kovarova, M. et al. Nanoformulations of rilpivirine for topical pericoital and systemic coitus-independent administration efficiently prevent HIV transmission. PLoS Pathog. 11, e1005075 (2015).
Kirtane, A. R. et al. Development of an oral once-weekly drug delivery system for HIV antiretroviral therapy. Nat. Commun. 9, 2 (2018).
Giardiello, M. et al. Accelerated oral nanomedicine discovery from miniaturized screening to clinical production exemplified by paediatric HIV nanotherapies. Nat. Commun. 7, 13184 (2016).
World Malaria Report 2018 (WHO, 2018).
Financing Malaria Strategic Plans in Africa in 2018-2020 (RBM Parternship to End Malaria, 2018).
World Malaria Report 2017 (WHO, 2017); http://www.who.int/malaria/publications/world-malaria-report-2017/report/en/
Fidock, D. A. Priming the antimalarial pipeline. Nature 465, 297–298 (2010).
Baird, J. K. Effectiveness of antimalarial drugs. N. Engl. J. Med. 352, 1565–1577 (2005).
Marques, J. et al. Adaptation of targeted nanocarriers to changing requirements in antimalarial drug delivery. Nanomedicine13, 515–525 (2017).
Moles, E. et al. ImmunoPEGliposomes for the targeted delivery of novel lipophilic drugs to red blood cells in a falciparum malaria murine model. Biomaterials 145, 178–191 (2017).
Urban, P. & Fernandez-Busquets, X. Nanomedicine against malaria. Curr. Med. Chem. 21, 605–629 (2014).
Urbán, P., Estelrich, J., Cortés, A. & Fernàndez-Busquets, X. A nanovector with complete discrimination for targeted delivery to Plasmodium falciparum-infected versus non-infected red blood cells in vitro. J. Control. Release 151, 202–211 (2011).
Allen, T. M. & Cullis, P. R. Liposomal drug delivery systems: from concept to clinical applications. Adv. Drug Deliv. Rev. 65, 36–48 (2013).
Joshi, M., Pathak, S., Sharma, S. & Patravale, V. Solid microemulsion preconcentrate (NanOsorb) of artemether for effective treatment of malaria. Int. J. Pharm. 362, 172–178 (2008).
Mandawgade, S. D., Sharma, S., Pathak, S. & Patravale, V. B. Development of SMEDDS using natural lipophile: application to β-artemether delivery. Int. J. Pharm. 362, 179–183 (2008).
Melariri, P. et al. Oral lipid-based nanoformulation of tafenoquine enhanced bioavailability and blood stage antimalarial efficacy and led to a reduction in human red blood cell loss in mice. Int. J. Nanomed. 10, 1493–1503 (2015).
Kumar, S., Singh, R. K., Sharma, R., Murthy, R. S. R. & Bhardwaj, T. R. Design, synthesis and evaluation of antimalarial potential of polyphosphazene linked combination therapy of primaquine and dihydroartemisinin. Eur. J. Pharm. Sci. 66, 123–137 (2015).
Parashar, D., Aditya, N. P. & Murthy, R. S. R. Development of artemether and lumefantrine co-loaded nanostructured lipid carriers: physicochemical characterization and in vivo antimalarial activity. Drug Deliv. 23, 123–129 (2016).
RTS,S Clinical Trials Partnership. A phase 3 trial of RTS,S/AS01 malaria vaccine in African infants. N. Engl. J. Med. 367, 2284–2295 (2012).
Huang, W.-C. et al. A malaria vaccine adjuvant based on recombinant antigen binding to liposomes. Nat. Nanotechnol. https://doi.org/10.1038/s41565-018-0271-3 (2018).
Global Tuberculosis Report 2018 (WHO, 2018).
Kim, J. Y., Shakow, A., Castro, A. Vande, C. & Farmer, P. Tuberculosis Control: The burden of tuberculosis: Economic burden (2) (World Health Organization, 2003).
O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations (2016).
Jain, R. et al. in Handbook of Nanomaterials for Industrial Applications Ch. 33 (Elsevier, 2018).
Cohen, J. Approval of novel TB drug celebrated—with restraint. Science 339, 130 (2013).
Singh, H., Jindal, S., Singh, M., Sharma, G. & Kaur, I. P. Nano-formulation of rifampicin with enhanced bioavailability: development, characterization and in-vivo safety. Int. J. Pharm. 485, 138–151 (2015).
Pandey, R., Sharma, S. & Khuller, G. K. Oral solid lipid nanoparticle-based antitubercular chemotherapy. Tuberculosis 85, 415–420 (2005).
Ahmad, Z. & Khuller, G. Alginate-based sustained release drug delivery systems for tuberculosis. Expert Opin. Drug Deliv. 5, 1323–1334 (2008).
Pandey, R. & Khuller, G. K. Nanoparticle-based oral drug delivery system for an injectable antibiotic—streptomycin. Evaluation in a murine tuberculosis model. Chemotherapy 53, 437–441 (2007).
Pandey, R., Zahoor, A., Sharma, S. & Khuller, G. K. Nanoparticle encapsulated antitubercular drugs as a potential oral drug delivery system against murine tuberculosis. Tuberculosis 83, 373–378 (2003).
Sharma, A., Pandey, R., Sharma, S. & Khuller, G. K. Chemotherapeutic efficacy of poly (DL-lactide-co-glycolide) nanoparticle encapsulated antitubercular drugs at sub-therapeutic dose against experimental tuberculosis. Int. J. Antimicrob. Agents 24, 599–604 (2004).
Costa, A. et al. The formulation of nanomedicines for treating tuberculosis. Adv. Drug Deliv. Rev. 102, 102–115 (2016).
Pandey, R. & Khuller, G. K. Solid lipid particle-based inhalable sustained drug delivery system against experimental tuberculosis. Tuberculosis 85, 227–234 (2005).
Zahoor, A., Sharma, S. & Khuller, G. K. Inhalable alginate nanoparticles as antitubercular drug carriers against experimental tuberculosis. Int. J. Antimicrob. Agents 26, 298–303 (2005).
Chuan, J. et al. Enhanced rifampicin delivery to alveolar macrophages by solid lipid nanoparticles. J. Nanopart. Res. 15, 1634 (2013).
Hawn, T. R. et al. Tuberculosis vaccines and prevention of infection. Microbiol. Mol. Biol. Rev. 78, 650–671 (2014).
Kaufmann, S. H. E., Weiner, J. & von Reyn, C. F. Novel approaches to tuberculosis vaccine development. Int. J. Infect. Dis. 56, 263–267 (2017).
Garcia-Contreras, L. et al. Immunization by a bacterial aerosol. Proc. Natl Acad. Sci. USA 105, 4656–4660 (2008).
Ballester, M. et al. Nanoparticle conjugation and pulmonary delivery enhance the protective efficacy of Ag85B and CpG against tuberculosis. Vaccine 29, 6959–6966 (2011).
Feng, G. et al. Enhanced immune response and protective effects of nano-chitosan-based DNA vaccine encoding T cell epitopes of Esat-6 and FL against Mycobacterium tuberculosis infection. PLoS ONE 8, e61135 (2013).
Liu, Q. et al. Preparation and evaluation of antigen/N-trimethylaminoethylmethacrylate chitosan conjugates for nasal immunization. Vaccine 32, 2582–2590 (2014).
Latent Tuberculosis Infection: Updated and Consolidated Guidelines for Programmtic Management (WHO, 2018).
Van Ginderdeuren, E., Bassett, J., Hanrahan, C., Mutunga, L. & Van Rie, A. Health system barriers to implementation of TB preventive strategies in South African primary care facilities. PLoS ONE 14, e0212035 (2019).
Sundar, S. & Jaya, J. Liposomal amphotericin B and leishmaniasis: dose and response. J. Glob. Infect. Dis. 2, 159–166 (2010).
Mitchell, S. L. & Carlson, E. E. Tiny things with enormous impact: nanotechnology in the fight against infectious disease. ACS Infect. Dis. https://doi.org/10.1021/acsinfecdis.8b00138 (2018).
Desai, N. Challenges in development of nanoparticle-based therapeutics. AAPS J. 14, 282–295 (2012).
Ioannidis, J. P. A., Kim, B. Y. S. & Trounson, A. How to design preclinical studies in nanomedicine and cell therapy to maximize the prospects of clinical translation. Nat. Biomed. Eng. 2, 797–809 (2018).
Satalkar, P., Elger, B. S., Hunziker, P. & Shaw, D. Challenges of clinical translation in nanomedicine: a qualitative study. Nanomedicine 12, 893–900 (2016).
Nanotechnology: A Report of the US Food and Drug Administration Nanotechnology Task Force (USFDA, 2007).
Bhatia, P., Vasaikar, S. & Wali, A. A landscape of nanomedicine innovations in India. Nanotechnol. Rev. 7, 131–148 (2018).
Armstead, A. L. & Li, B. Nanomedicine as an emerging approach against intracellular pathogens. Int. J. Nanomed. 6, 3281–3293 (2011).
Aly, A. S. I., Vaughan, A. M. & Kappe, S. H. I. Malaria parasite development in the mosquito and infection of the mammalian host. Annu. Rev. Microbiol. 63, 195–221 (2009).
Pai, M. et al. Tuberculosis. Nat. Rev. Dis. Prim. 2, 16076 (2016).
Acknowledgements
A.R.K. received financial support from a PhRMA Foundation postdoctoral fellowship. M.V. was supported in part by an MIT Tata Centre Grant and a National Science Foundation Graduate Research Fellowship. R.L. and G.T. were supported in part by Bill and Melinda Gates Foundation grant number OPP1179091, NIH grant number EB000244 and the MIT Tata Centre. G.T. was supported in part by the Karl van Tassel (1925) Career Development Professorship at MIT, the Department of Mechanical Engineering, MIT and the Division of Gastroenterology, Brigham and Women’s Hospital.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
A.R.K., M.V., R.L. and G.T. are co-inventors on multiple patent applications describing gastric resident systems for extended drug release with a focus on global health applications. R.L. has a financial interest in Moderna, Inc, and Alnylam, Inc., which are biotechnology companies focused on RNA therapeutics including vaccines against SARS-CoV-2. R.L. and G.T. have a financial interest in Lyndra Therapeutics, Inc., a biotechnology company focused on the development of encapsulated gastric resident systems for extended drug delivery.
Additional information
Peer review information Nature Nanotechnology thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kirtane, A.R., Verma, M., Karandikar, P. et al. Nanotechnology approaches for global infectious diseases. Nat. Nanotechnol. 16, 369–384 (2021). https://doi.org/10.1038/s41565-021-00866-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41565-021-00866-8
This article is cited by
-
Nanotechnology’s frontier in combatting infectious and inflammatory diseases: prevention and treatment
Signal Transduction and Targeted Therapy (2024)
-
Efficacy of co-loading Ag nanoparticles and metronidazole in PEG–gelatin-based sponges for the treatment of chronic wounds
Polymer Bulletin (2024)
-
Bioinspired nanomaterials for the treatment of bacterial infections
Nano Research (2024)
-
Revisiting the smart metallic nanomaterials: advances in nanotechnology-based antimicrobials
World Journal of Microbiology and Biotechnology (2024)
-
Nanozymes as Catalytic Marvels for Biomedical and Environmental Concerns: A Chemical Engineering Approach
Journal of Cluster Science (2024)
