
Abstract
Our understanding of host-microbe interactions has broadened through numerous studies over the past decades. However, most investigations primarily focus on the dominant members within ecosystems while neglecting low-abundance microorganisms. Moreover, laboratory animals usually do not have microorganisms beyond bacteria. The phenotypes observed in laboratory animals, including the immune system, have displayed notable discrepancies when compared to real-world observations due to the diverse microbial community in natural environments. Interestingly, recent studies have unveiled the beneficial roles played by low-abundance microorganisms. Despite their rarity, these keystone taxa play a pivotal role in shaping the microbial composition and fulfilling specific functions in the host. Consequently, understanding low-abundance microorganisms has become imperative to unravel true commensalism. In this review, we provide a comprehensive overview of important findings on how low-abundance commensal microorganisms, including low-abundance bacteria, fungi, archaea, and protozoa, interact with the host and contribute to host phenotypes, with emphasis on the immune system. Indeed, low-abundance microorganisms play vital roles in the development of the host’s immune system, influence disease status, and play a key role in shaping microbial communities in specific niches. Understanding the roles of low-abundance microbes is important and will lead to a better understanding of the true host-microbe relationships.
Similar content being viewed by others

Introduction
Our understanding of host-microbe interactions has grown based on the work of many researchers in past decades. These studies have revealed the various roles of commensal bacteria in host phenotypes, including obesity and diabetes1,2,3,4,5, brain development and behavior6,7,8, protection against pathogenic infection, diseases9,10,11, aging, and response to cancer therapy12. However, the majority of investigations primarily focus on the dominant members within ecosystems, typically emphasizing highly abundant bacteria and viruses (including bacteriophages) while neglecting low-abundance microorganisms. This tendency persists, particularly in studies employing high-throughput sequencing approaches13. Furthermore, it is important to note that laboratory animals maintained under clean conditions usually lack microorganisms beyond bacteria, such as fungi and helminths. Indeed, the microbiome-associated phenotypes observed in laboratory animals, including the immune system, are markedly different from those observed in the real world because the commensal microbiome present in the natural environment exhibits a considerably more intricate and diverse community structure. As an example, laboratory mice and humans show distinct immune profiles; however, by cohousing laboratory mice with pet-store mice or rewilding laboratory mice, it is possible to recapitulate human immune traits14,15,16. Intriguingly, recent studies have unveiled the beneficial roles of certain helminths, also known as parasitic worms, in host physiology through helminth-bacterial interactions, although we do not cover helminths in this review because they are non-commensal and they are not microorganisms17,18,19,20. In brief, helminth infection (e.g., Trichuris muris) induces immune responses that play a crucial role in maintaining the integrity of the epithelial barrier. These responses involve the activation of type 2 cytokines and IL-22, which synergistically increase goblet cell mucus production and antimicrobial peptide expression. Moreover, helminth infection brings about alterations in bacterial composition that favor a beneficial microbial milieu. Specifically, helminth inhibits the proliferation of inflammatory Bacteroides species while promoting the growth of protective bacteria such as Clostridiales; this reshaping of the bacterial community contributes significantly to the amelioration of inflammatory bowel disease (IBD) severity.
Recently, numerous studies have directed their attention toward low-abundance microbes within the community. These studies emphasize the fact that low abundance does not mean less important using the “keystone species” concept. Despite their low abundances, these keystone taxa play a pivotal role in shaping the microbial composition and performing specific functions within the host, as supported by recent research21,22. Therefore, a better understanding of low-abundance microorganisms is essential to elucidate true host-microbe interactions. In this review, we describe how low-abundance commensal microorganisms interact with the host and contribute to the host’s health. Specifically, we highlight recent important findings on the roles of low-abundance bacteria, fungi, archaea, and protozoa in the context of host phenotypes, including the development of the immune system, the association with diseases, and interactions with commensal bacteria.
Low-abundance bacteria
In many microbiome studies, low-abundance bacteria (usually referring to a taxon that accounts for less than 1% of the relative abundance) have often been neglected in comparison to high-abundance “dominant” bacteria, primarily due to their rarity in the environment. Furthermore, a significant number of researchers opt to filter out the low-abundance taxa from their microbiome studies (e.g., 16S rRNA gene sequencing analysis) during the quality control and filtering steps prior to downstream analysis13. Despite their low abundances, the number of taxa and diversity of low-abundance bacteria is much higher than that of high-abundance bacteria in the host23. Multiple studies have consistently emphasized the substantial contributions of low-abundance bacteria to host phenotypes, underscoring the necessity to allocate comparable attention to these less prevalent members alongside dominant members when investigating host-microbiome interactions. In this review, we present up-to-date information on the role of low-abundance microbes in regulating host physiology and disease pathology while highlighting the knowledge gap in this area (Table 1).
Roles of low-abundance bacteria in the host immune system
The pivotal role of commensal gut bacteria in immune development is widely recognized. For example, Bacteroides fragilis has been recognized to induce systemic Th1 cells through the utilization of bacterial polysaccharides. Segmented filamentous bacteria (SFB) or Candidatus Savagella, previously named Candidatus Arthromitus, induce the differentiation of intestinal Th17 cells via the presence of flagellins24,25,26. Notably, even low-abundance bacteria make significant contributions to host immune development.
Eggerthella lenta, previously known as Eubacterium lentum, is a human commensal in the intestine and is present at a very low level in healthy subjects27. Interestingly, this prevalent gut commensal plays opposite roles in the context of immune responses. Alexander et al. reported that E. lenta promotes Th17 activities by reducing the inhibition of Rorγt in mice27. They also showed that the cardiac glycoside reductase 2 (Cgr2) enzyme encoded by this bacterium is sufficient for IL-17A induction and that dietary arginine blocks E. lenta-induced intestinal inflammation. In contrast, another study demonstrated that this particular bacterium possesses the ability to convert lithocholic acid (LCA) into 3-oxolithocholic acid (3-oxoLCA) and isolithocholic acid (isoLCA), which, in turn, suppress Th17 cell differentiation28. Moreover, the anti-inflammatory metabolites 3-oxoLCA and isoLCA were inversely associated with Crohn’s disease (CD).
The genus Helicobacter is one of the major histocompatibility complex (MHC) class II-associated bacteria in the intestine. It was reported that mice naturally colonized with Helicobacter species such as H. mastomyrinus and H. typhlonius show higher epithelial MHC class II expression than Helicobacter-free mice, and the relative abundance of Helicobacter species is positively correlated with epithelial MHC class II expression in the mouse intestine. In addition, cohousing of Helicobacter-free mice with Helicobacter-harboring mice increased MHC class II expression29. In our previous study, we demonstrated the potential of low-abundance bacteria in the context of MHC class II expression using a dilution strategy. In this study, the cecal microbiome of mice was diluted to eliminate low-abundance bacteria, and the undiluted and diluted microbiome was engrafted into the intestines of germ-free mice. Loss of low-abundance bacteria resulted in low expression of multiple genes involved in the MHC class II antigen presentation pathway and lower MHC class II-expressing cell counts in the small intestine. Notably, the relative abundance of the family Erysipelotrichaceae was positively correlated with MHC class II expression23. The findings from multiple independent studies provide consistent evidence of the immunogenic properties exhibited by Erysipelotrichaceae30,31,32. Palm et al. classified and selected intestinal bacteria based on immunoglobulin A (IgA) coating. They revealed that high IgA coating is related to colitogenic bacteria and that unclassified Erysipelotrichaceae is one of the high IgA-coating bacteria31. Dinh et al. showed a positive correlation between the relative abundance of Erysipelotrichaceae and tumor necrosis factor alpha (TNF-α)32. Taken together, these studies highlight the significant role of low-abundance bacteria in modulating host immunity. Further studies are needed to establish the causal link between these low-abundance microbes and specific immune responses.
Association between low-abundance bacteria and diseases
Low-abundance bacteria play a crucial role in host disease. An important point is that they play different roles in diseases; sometimes they have a preventive role, but in other situations, they are associated with the disease. Mucispirillum schaedleri, which belongs to the phylum Deferribacteres and is a low-abundance commensal bacteria in humans and rodents, plays both types of roles33. IBD is representative of gut microbiome-associated disease and has been the subject of extensive research concerning host-microbe interactions. Numerous investigations have consistently demonstrated an association between IBD and M. schaedleri. This bacterium was identified as an indicator of dextran sodium sulfate (DSS)-induced colitis in mice34. A20 functions as a potent inhibitor of both the NF-kB and apoptotic signaling pathways, making it a pivotal gene associated with susceptibility to various inflammatory diseases, including IBD. The abundance of M. schaedleri was significantly higher in A20 knockout mice than in wild-type mice35. There is another noteworthy IBD-associated bacterium, E. lenta, a human commensal with known Th17-modulating properties. This bacterium is enriched in IBD patients and contributes to colitis in a Rorc-dependent manner in mice27. Furthermore, previous studies have indicated an observed correlation between E. lenta and both rheumatoid arthritis (RA)36 and multiple sclerosis (MS)37. Moreover, mice with social stress have an increased abundance of M. schaedleri in the intestine38. In contrast, M. schaedleri protects against enteric pathogen infection. In the mouse intestine, M. schaedleri acted as an antagonist of Salmonella enterica serovar Typhimurium virulence. M. schaedleri protected the host against Salmonella colitis by competing for anaerobic respiration substrates and inhibiting the expression of virulence factor, a type 3 secretion system encoded on Salmonella pathogenicity island-1 (T3SS-1)39.
The relationship between commensal microbes and metabolic diseases has been reported. One of the most well-known relationships is between obesity and gut microbes, and low-abundance bacteria are associated with obesity. White et al. observed that the phylum Actinobacteria, which are known as low-abundance bacteria in the gut, is associated with obesity. In this study, among obese subjects, Actinobacteria constituted ~5% of the gut microbiota; meanwhile, these bacteria had a significantly lower prevalence in lean individuals40.
The gut microbiome is closely related to various gastrointestinal tract diseases and is considered both a disease determinant and a potential therapeutic target. In the oral cavity, specific groups of low-abundance bacteria, such as Fretibacterium sp. OT 361 in subgingival plaque (SGP), Prevotella_intermedia in saliva, and Porphyromonas endodontalis in SGP and saliva, were identified as keystone species in pregnant patients with gingivitis41.
Alzheimer’s disease (AD), the most common cause of dementia, is a neurodegenerative disorder that results in the deterioration of cognitive function42. Emerging evidence suggests that the composition of the gut microbiome, alongside the presence of low-abundance bacteria, plays a notable impact on the progression of AD43. Ferreiro et al. recently reported that intestinal commensal bacteria can be an indicator of AD in humans44. They showed that multiple intestinal low-abundance bacteria, such as Dorea formicigenerans, Oscillibacter sp. 57_20, Coprococcus catus, and Ruminococcus lactaris, are associated with a preclinical AD status, whereas Methanosphaera stadtmanae is associated with a healthy status.
Furthermore, low-abundance bacteria are key determinants of a healthy airway microbiome early in human life. Pust and Tümmler reanalyzed public shotgun metagenomic data of healthy children and children with cystic fibrosis (CF)45. They observed that rare taxa are the most important factors in deciding whether a child is healthy or suffering from life-limiting CF disease. These rare members are essential to improving the underdeveloped CF background network. Collectively, these studies detail the robust relationships between low-abundance bacteria and various diseases. However, most findings have primarily focused on establishing associations rather than elucidating causality or underlying mechanisms. Therefore, it is imperative to conduct consistent and comprehensive investigations to further understand these relationships and to explore their potential therapeutic implications.
Impact of low-abundance bacteria on the bacterial community
Low-abundance bacteria play a key role mediators of microbial composition. Low-abundance bacteria are usually not considered core members of the intestine; however, Benjamino et al. revealed that they act as drivers of compositional alteration after diet changes. In this study, they provided six different lignocellulose food sources to termites and performed 16S rRNA gene sequencing and artificial neural network analysis to predict the relative abundances of taxa at random points in the termite hindgut. The study reported that low-abundance taxa, which frequently exist outside of the core community, maintain community-driving correlations in the hindgut microbiome22. The enterotype study of the human gut microbiome is another example underscoring the role of low-abundance bacteria in maintaining community structure. In this study, the genus Ruminococcus was identified as a representative marker for enterotype 3. Notably, the relative abundance of this genus was lower than that of Bacteroides, a marker for enterotype 1, and Prevotella, a marker for enterotype 2, albeit the relative abundance was slightly higher than 1%46. Hildebrand et al. demonstrated the possibility of low-abundance bacteria as keystone species in restoring the gut microbial community after antibiotic intervention47. The authors identified a novel bacterial species, initially named UBorkfalki ceftriaxensis and subsequently revised as Candidatus Borkfalkia ceftriaxoniphila48, from the human intestine. This bacterium bloomed to 92% relative abundance after administration of cephalosporin (ceftriaxone) in the intestine and restored the microbial community into a healthy but shifted community state in the long term. Importantly, most species co-occurred with UB. ceftriaxensis were probiotic species frequently used to treat antibiotic-associated diarrhea (AAD).
Low-abundance bacteria also play important microbial functions in the community. For example, the genus Escherichia includes well-studied bacteria and their abundance is low in the healthy gut; however, the function of this genus should not be ignored. Low-abundance Escherichia contributes over 90% of the amount of two abundant proteins related to bacterial pilus assembly, FimA and PapC, in the human intestine46. Pili help bacteria stay longer in the intestine and are key components for transferring plasmids between bacteria. These findings underscore the fundamental significance of minor members within the bacterial population, highlighting their crucial role not only in promoting community stability but also in contributing to essential functions.
Fungi
Fungi are commensal members of the host and ubiquitously exist in diverse body sites such as the intestine, lungs, and skin, and fungal composition is very different based on their niche49. However, they are usually considered to be minor members and are not well studied compared to bacteria due to their low abundance (~0.1% of total microbes in the gut)46,49. The significance of commensal fungi in host-microbiota interactions has often been underestimated, primarily due to the limited size of fungal databases and the comparatively low abundance of fungal cells within the host when compared to bacteria. However, it is important to note that fungal cells are substantially larger, ~100-fold, than bacterial cells, and they occupy a significant amount of physical space. Moreover, as eukaryotes, fungi contribute distinctive metabolic attributes to both the host and the microbiome49. It is worth noting that even minor members of the microbiome can exert profound effects on the host, including via the immune system (Table 2).
Roles of fungi in the host immune system
Commensal fungi have been recognized for their close association with the host’s immune system, and among them, Candida albicans, a commensal fungus in humans, has emerged as a widely employed model organism for investigating host-fungal interactions. Although C. albicans typically colonizes as a commensal in healthy individuals, under conditions of microbial dysbiosis or in individuals with compromised immune systems, this fungus transitions to a pathogenic state, leading to significant mucosal and systemic infections50,51. The formation of C. albicans hyphae is a pivotal characteristic linked to the transition from a commensal to a pathogenic state, as hyphae exhibit increased invasiveness and virulence. In the commensal state, most C. albicans exists in the yeast form, with only a limited presence of hyphae52. The host epithelium plays a barrier function and separates C. albicans from the host, collaborating with the immune system and commensal bacteria to maintain homeostasis51. The mucus layer and commensal bacteria actively contribute, through both direct and indirect mechanisms, to the physical separation of C. albicans from the epithelium. Consequently, impairment of the epithelial barrier or bacterial dysbiosis can facilitate C. albicans infection. Furthermore, epithelial cells actively modulate the commensal state of C. albicans by initiating immune responses. These immune responses encompass the activation and recruitment of innate immune cells, including neutrophils, monocytes, and macrophages, as well as the production of secretory IgA (sIgA) and β-defensins, which are triggered by the release of IL-22 from Th17 cells or innate lymphoid cells. Notably, these immune reactions exhibit a preferential induction of the hyphal form of C. albicans53,54,55.
Numerous studies have consistently highlighted the capacity of commensal C. albicans to induce certain immune responses in both human and murine hosts. Mono-colonization of bacteria-depleted mice with either C. albicans or Saccharomyces cerevisiae in the intestine positively calibrated the activation of protective CD8+ T cells and protected the host against influenza A virus infection. Moreover, treatment with mannans, a fungal cell wall component, recapitulated the protection of these two commensal fungal species56. Furthermore, the intestinal colonization of C. albicans has been shown to elicit Th17 responses in both humans and mice57,58. C. albicans was chosen as a main inducer of human Th17 responses, and cross-reactive Th17 cells against C. albicans contributed to Aspergillus fumigatus-driven non-intestinal inflammation57. In mice, intestinal colonization by C. albicans induced systemic Th17 responses and protected the host against C. albicans invasive infection as well as Staphylococcus aureus invasive infection58. C. albicans colonization in the gut also enhances granulopoiesis. Oral inoculation of laboratory mice with C. albicans induced persistent expansion of myeloid progenitors in the born marrow dependent on IL-6R signaling. This expansion consequently conferred host protection against systemic methicillin-resistant Staphylococcus aureus (MRSA) infection as well as intraperitoneal or intranasal infection with Streptococcus pneumoniae59.
Human gut commensal mycobiota, primarily C. albicans, induce CARD9+CX3CR1+ macrophage-mediated antifungal immunoglobulin G (IgG) production and protect the host against systemic Candida infection60. Malassezia restricta, a commensal fungus found on the skin, is another example of an immune-stimulating fungus. Intestinal colonization of specific-pathogen-free (SPF) mice with this particular fungal species induced upregulation of IL-17A and IFN-γ-producing CD4+ T cells in the colon61. A distinct fungal community inhabits the intestinal mucosa of humans and mice, whereby these mucosa-associated fungi (MAF) induce Th17-mediated immune responses62. MAF, including C. albicans, S. cerevisiae, and Saccharomycopsis fibuligera, protected the host against DSS-induced colitis and Citrobacter rodentium infection via IL-22-dependent mechanisms. Furthermore, MAF promote social behavior by activating IL-17-mediated signaling in neurons. The gut mycobiome also mediates immune cell migration. Zhang et al. reported that commensal fungi are responsible for inducing the migration of CD45+CD103+RALDH+ dendritic cells (DCs) to the peripheral lymph nodes after birth63. Intestinal colonization of both SPF and germ-free (GF) mice with Candida tropicalis triggered the migration of CD45+CD103+RALDH+ DCs from the lamina propria of the intestine to the inguinal and mesenteric lymph nodes. These DCs utilized retinoic acid signaling to initiate an increase in lymph node cellularity and volume expansion.
As mentioned above, C. albicans is the most studied fungal species in the context of commensal-immune interactions. Nevertheless, the use of C. albicans as a model commensal fungus in mice presents a certain limitation. Specifically, C. albicans is not a natural murine commensal, which forces the modulation of intestinal bacterial communities through antibiotic treatment or the use of GF mice to establish successful C. albicans colonization, and this modified, bacteria-free environment is not the natural habitat of commensal fungi. Given the limitations of C. albicans as a murine commensal fungus, many researchers have undertaken efforts to investigate the impact of murine commensal fungi on the immune system in mice. One of the strategies is using host-adapted strains of C. albicans. Rahman et al. isolated C. albicans strain 529 L using a low-estrogen murine model of concurrent oral and vaginal C. albicans colonization64, and this strain exhibited stable colonization in the mouse intestine65. Another strategy is rewilding SPF laboratory mice, wherein they are exposed to natural environments, enabling interactions with diverse microbes, including fungi, that they would not encounter under controlled laboratory conditions. Rewilded mice displayed increases in activated T cells and circulating neutrophils with notable increases in the colonization of intestinal fungi. Notably, colonization of SPF mice with fungal consortium isolated from wild mice—Aspergillus candidus, Aspergillus proliferans, Chaetomium globosum and Dichotomopilus indicus—was sufficient to induce expansion of circulating SSChi granulocytes and neutrophils16. In rewilded mice, intestinal fungal colonization resulted in a notable increase in the quantity and frequency of multipotent progenitors (MPPs), particularly within the myeloid-biased MPP3 subset that resides in the bone marrow59. Collectively, these findings highlight the significance of the fungal microbiome in the context of immune development. Therefore, it is imperative to incorporate the study of the mycobiome when investigating interactions between the immune system and the microbiome.
Association between fungi and diseases
The association between commensal fungi and host diseases has consistently been reported66,67. As we mentioned before, IBD is one of the most actively studied diseases in the context of host-bacteria interactions. Commensal fungi also show significant involvement in the pathogenesis of this disease. In IBD patients, reduced fungal diversity and increased relative abundance of Candida species in the gut were observed68. Furthermore, the analysis revealed an elevated Basidiomycota/Ascomycota ratio, a reduced proportion of S. cerevisiae, and an augmented proportion of C. albicans in comparison to the fungal composition of healthy individuals69. The human intestinal mycobiota, which includes C. albicans, stimulates the production of sIgA, with the induced sIgA exhibiting a preference for binding to hyphae, a fungal morphotype commonly associated with virulence55. In CD patients, the antifungal sIgA titer was lower with increased granular hyphal morphologies when compared to healthy individuals. M. restricta is another IBD-associated fungal species. M. restricta was more abundant in CD patients than in healthy controls. In particular, patients carrying the CD-linked polymorphism in CARD9, CARD9S12N, were strongly associated with M. restricta, and intestinal colonization of this fungus exacerbated DSS-induced colitis in mice61.
Obesity is also related to gut commensal fungi, and the supplementation of antifungals such as amphotericin B or fluconazole can effectively inhibit high-fat-diet-induced obesity in mice70. The commensal gut fungus Candida parapsilosis is a strong candidate for obesity-associated fungal species. This fungus increased free fatty acids in the gut through the production of fungal lipases, and it promoted diet-induced obesity in mice70. On the other hand, Mucor racemosus and M. fuscus were more abundant in nonobese subjects than in their obese counterparts in the human gut71.
Asthma is a chronic airway disease, and evidence of the association between the composition of bacteria in the intestine and lung and childhood asthma has been reported72,73,74. In addition, recent studies have indicated that asthma is linked not only to commensal bacteria but also to commensal fungi75,76,77,78. Arrieta et al. analyzed the fecal microbiome of Ecuadorian babies using 16S and 18S rRNA gene sequencing76. They observed that microbial dysbiosis at 3 months of age is associated with later atopic wheeze development that includes fungal dysbiosis. Despite no differences in the alpha- and beta-diversity of the fungal community between healthy control and atopic wheeze groups, atopic wheeze subjects showed a distinct fungal composition characterized by an overgrowth of Pichia kudriavzevii and a reduction in Saccharomycetales compared to the healthy control infants. The same research group replicated these study findings in Canadian babies77. In comparison to the healthy control group, subjects with asthma symptoms showed an overgrowth of P. kudriavzevii in the intestine at 3 months of age. Furthermore, neonatal intestinal overgrowth of P. kudriavzevii exacerbated the manifestations of type-2 and type-17 inflammation during allergic airway disease in subsequent stages in mice. Candida species are also asthma-associated fungal candidates. In the house dust mite (HDM)-induced mouse asthma model, oral administration of C. albicans led to increased airway inflammation, including increased total white cell and eosinophil counts in the airway and total immunoglobulin E (IgE) concentrations in the serum78. In addition, Candida-colonized mice showed no fungal burden but an increase in the abundance of group 2 innate lymphoid cells (ILC2s) compared to the control in the lungs. In humans, patients experiencing severe asthma exacerbation exhibited an elevated Candida burden in the intestine.
The gut–liver axis refers to the reciprocal interaction between the gastrointestinal tract, its resident microbiome, and the liver, resulting from the integration of signals generated by dietary, genetic, and environmental factors79. Intestinal fungi, a part of the gut microbiome, are also associated with liver diseases80. Yang et al. observed that chronic alcohol consumption increases the intestinal fungal burden and the level of circulating β-glucan, a fungal cell wall component, in mice81. Treatment with amphotericin B, an antifungal agent, reduced intestinal fungal burden and the level of circulating β-glucan and ameliorated the symptoms of alcoholic liver disease (ALD). The authors observed that β-glucan elicits hepatic inflammation through the C-type lectin-like receptor, CLEC7A, present on Kupffer cells, and the subsequent increase in IL-1β, a proinflammatory cytokine, actively contributes to hepatocyte impairment and facilitates the progression of ethanol-induced liver disease. In ALD patients, a less diverse fungal community and overgrowth of Candida were observed compared with healthy individuals. Intriguingly, Saccharomyces boulardii, a probiotic yeast that is not gut commensal, exhibits prominent hepatoprotective effects in D-galactosamine-induced liver injury and nonalcoholic steatohepatitis82,83,84.
The association between commensal fungi and cancer has also been revealed. Pancreatic ductal adenocarcinoma (PDA) tumors in both humans and a mouse model display an approximately 3000-fold elevation in fungal burden when compared to normal pancreatic tissue85. The mycobiome in PDA tumors showed a composition distinct from that of gut or normal pancreatic tissue. Malassezia species demonstrated remarkable enrichment within the tumor microenvironment and promoted oncogenesis by driving the activation of the complement cascade via mannose-binding lectin (MBL) activation. MBL specifically recognizes glycans present on the fungal cell wall, thus contributing to the accelerated progression of malignancy. Gao et al. reported fungal dysbiosis in fecal samples from polyp and colorectal cancer (CRC) patients86. This dysbiosis was characterized by decreased diversity in polyp patients, an increased Ascomycota/Basidiomycota ratio, and an increased proportion of Trichosporon and Malassezia compared to healthy controls. On the other hand, fungal-derived cell wall components can be utilized in cancer therapy. For example, BTH1677 is a fungal-derived, water-soluble, 1,3–1,6 β-glucan, and this particular agent elicits synchronized anticancer immune responses when administered in conjunction with antitumor antibody therapies87. Taken together, these studies provide compelling evidence regarding the association between commensal fungi and various diseases. The identification of these associations may provide valuable insights into the potential utility of commensal fungi as disease markers or therapeutic targets.
Archaea
Archaea are components of the commensal microbiome and are ubiquitously present in our bodies, including in the intestine, vagina and oral cavity88. Although archaea are often overlooked in microbiome research due to their distinct biological characteristics89, their roles in the host immune system and diseases have been consistently reported (Table 3).
Roles of archaea in the host immune system
Methanobrevibacter smithii is the most dominant methanogenic archaea in the human gut, and this methanogen interacts with immune systems90. In MS patients, the relative abundance of M. smithii was increased in patients with elevated breath methane compared with healthy subjects91. Furthermore, M. smithii exhibited positive correlations with several genes, namely, MAPK14, MAPK1, LTBR, STAT5B, CASP1, and HLA-DRB1, which are known to play roles in the maturation of DCs, interferon signaling, and triggering receptor expressed on myeloid cells (TREM) signaling pathways. Bang et al. observed that M. smithii and another commensal methanogenic archaea, Methanosphaera stadtmanae, interact with monocyte-derived DCs (moDCs)92. This interaction led to an increase in the cell-surface expression of CD197 and CD86, which play crucial roles in providing costimulatory signals essential for moDC maturation. The role of archaea in regulating host immunity is a relatively unexplored area. More studies are needed to identify archaea species that consistently colonize mammalian hosts and affect their physiologies.
Association between archaea and diseases
The associations between archaea and diseases have been reported by several researchers, including the aforementioned association between M. smithii and MS88,93. As an example, Methanobrevibacter oralis is highly present in the oral cavity of periodontitis patients94. Another archaea-associated disease is asthma. In a cross-sectional analysis utilizing fecal samples obtained from a subset of children enrolled in the KOALA birth cohort study in The Netherlands, conducted at ages 6 to 10 years, a significant inverse association was observed between childhood asthma and intestinal archaea, particularly M. stadtmanae95. In addition, it has been suggested that intestinal colonization of archaea promotes gastrointestinal and metabolic diseases such as IBD, obesity, and CRC. Lecours et al. reported an association between IBD and M. stadtmanae. In this study, IBD patients showed higher M. stadtmanae abundance in the intestine than controls96. Moreover, stimulation of mononuclear cells with M. stadtmanae increased TNF production. The association between archaea and obesity was reported by Zhang et al.97. The authors compared the gut microbiome of normal weight, morbidly obese, and post-gastric-bypass surgery individuals and observed a significantly higher amount of the order Methanobacteriales, a H2-oxidizing methanogen, in obese individuals than in other groups. Shotgun metagenomic analysis revealed that CRC patients show a distinct intestinal archaea profile compared with healthy individuals98. Fecal samples from CRC patients had significant enrichment of halophilic Natrinema sp. J7-2 and depletion of methanogenic archaea, including Methanosphaera, Methanococcoides, Methanocorpusculum, Methanocaldococcus, and Methanobacterium. The role of archaea in the context of mutualism remains largely unexplored, yet these findings emphasize the imperative need to investigate the associations of archaea with diseases.
Protozoa
In the past, protozoa were primarily considered parasites (e.g., Cryptosporidium spp., Giardia intestinalis, Entamoeba histolytica, Trichomonas vaginalis, and T. tenax)99,100; however, recent research has indicated the existence of commensal protozoa and their role in the host (Table 3).
Roles of protozoa in the host immune system
Tritrichomonas musculis, a murine commensal protozoan, exhibits protective effects against pathogenic infection in mice101,102. Chudnovskiy et al. reported that T. musculis triggers the activation of the host epithelial inflammasome, leading to subsequent induction of IL-18 production101. Inflammasome-driven IL-18 promoted Th1 and Th17 immunity through interferon regulatory factor 8 (IRF8)- and IRF4-dependent DCs, contributing to protection against Salmonella Typhimurium infection. In contrast to the findings of these probiotic effects, T. musculis exacerbated the development of colitis and CRC. Chiaranunt et al. demonstrated that T. musculis-mediated modulation of host immune responses requires the involvement of the NLRP1B and NLRP3 inflammasomes, which collectively confer host protection against Salmonella infections102. Commensal protozoa increase tuft cells in the intestine, consequently leading to an augmentation of ILC2 populations103,104,105. Tuft cells, also known as taste-chemosensory cells, are characterized by their infrequent occurrence within the intestinal epithelium. These specialized cells exhibit a distinctive ability to respond to protozoan and helminthic stimuli. In response to commensal Tritrichomonas species, intestinal tuft cells exhibited an increased abundance, along with the upregulation of IL-25 expression. Notably, this process was contingent upon the cation channel TRPM5-dependent mechanisms and subsequently induces ILC2 responses103. Tuft cells express the succinate receptor GPR91 (also known as SUCNR1), which enables sense and response to succinate. Remarkably, immune sensing of Tritrichomonas colonization by tuft cells required the succinate receptor, and Tririchomonas-derived succinate was sufficient in triggering ILC2 responses104,105. Based on these findings, it becomes evident that commensal protozoa play a distinct role in modulating the immune system. These results highlight the necessity for ongoing and consistent investigations into commensal protozoa-immune interactions.
Impact of protozoa on the bacterial community
Commensal protozoa interact with commensal bacteria and alter bacterial composition in their habitat. In particular, Blastocystis spp. colonization led to distinct bacterial composition in the gut99,106,107. Blastocystis-colonized individuals show increased bacterial diversity with higher abundances of the following taxa: Clostridia, Faecalibacterium, Prevotella 9, Ruminococcaceae UCG-002, Muribaculaceae, Rikenellaceae, Acidaminococcaceae, Phascolarctobacterium, and Ruminococcaceae UCG-005, and lower abundances of the following taxa: Enterobacteriaceae, Enterococcus species, Lactobacillales, and Bacilli compared to Blastocystis-free subjects. Morton et al. also showed that the presence of the gut commensal protozoa Entamoeba is strongly associated with bacterial diversity in rural non-industrialized populations99,108. These findings underscore the critical importance of conducting multi-kingdom interaction studies to better understand host-commensal interactions in a comprehensive manner.
Conclusion
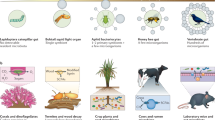
Low-abundance microorganisms, including low-abundance bacteria, fungi, archaea, and protozoa, establish colonization within the animal body and play vital roles in maintaining mutualistic relationships. These microorganisms contribute to the development of the host’s immune system, influence disease status, and play a key role in shaping microbial communities within their niches (Fig. 1). Investigating the roles of low-abundance microbes is imperative; however, researchers must be cautious of potential pitfalls when conducting such studies. First, it is important to acknowledge that the relative abundances of bacteria are inconsistent and variable, both among individuals and even within individuals, particularly in the context of humans and nonlaboratory animals; this fact emphasizes the idea that low-abundance microbes are not always low abundant109. Various factors, including diet, genetic background, and the administration of medications such as antibiotics, significantly influence the composition of microbial communities. Therefore, it is crucial to design robust experiments that incorporate a larger sample size with repeated measurements and diverse backgrounds. This approach is essential for elucidating the significance of low-abundance microbes and identifying potential “biomarkers”. Second, while many studies have highlighted the contribution of individual microorganisms, it is important to recognize that microbial communities are inherently intricate and multifaceted rather than being characterized by simplicity. The interspecies interactions within the same microbial kingdom or between different kingdoms yield different outcomes from those observed in single-species colonization in the context of the roles of microorganisms in the hosts110,111,112. Third, it is crucial to take into account the absolute abundance of microbes within each body site. The intestinal tract, for instance, harbors an abundance exceeding one billion microorganisms, whereas other organs harbor a comparatively lower microbial population. Consequently, the utilization of relative abundances alone may yield disparate numerical representations. In this review, we primarily focus on elucidating the significance of commensal low-abundance microbes within the host. However, to gain a comprehensive understanding of the entirety of host-microbe interactions, it is equally important to explore the reciprocal aspect of this relationship—the role of the host, including the immune system, in shaping a mutualistic association with commensal microbes.

Low-abundance commensals, including bacteria, fungi, archaea, and protozoa, play pivotal roles in various aspects of host physiology. These microorganisms establish mutualistic relationships with the host, exerting profound effects on the host. Notably, they influence diverse phenotypes, including immune activation (e.g., Th17 cells, tuft cells, dendritic cells, MHC class II expression), the occurrence of several diseases (e.g., inflammatory bowel disease, obesity, asthma, cystic fibrosis, Alzheimer’s disease, multiple sclerosis, rheumatoid arthritis, periodontal disease, cancer), social behavior, and protection against pathogenic infections. Moreover, interactions occur among these microorganisms themselves or with other commensal bacteria in their niche. Created with BioRender.com.
References
Yang, J. Y. et al. Gut commensal Bacteroides acidifaciens prevents obesity and improves insulin sensitivity in mice. Mucosal Immunol. 10, 104–116 (2017).
Le Roy, T. et al. Dysosmobacter welbionis is a newly isolated human commensal bacterium preventing diet-induced obesity and metabolic disorders in mice. Gut 71, 534–543 (2022).
Everard, A. et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. P Natl Acad. Sci. USA 110, 9066–9071 (2013).
Hosomi, K. et al. Oral administration of Blautia wexlerae ameliorates obesity and type 2 diabetes via metabolic remodeling of the gut microbiota. Nat. Commun. 13, 4477 (2022).
Turnbaugh, P. J. et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 (2006).
Lu, J. et al. Microbiota influence the development of the brain and behaviors in C57BL/6J mice. PLoS ONE 13, e0201829 (2018).
Foster, J. A. Modulating brain function with microbiota. Science 376, 936–937 (2022).
Needham, B. D. et al. A gut-derived metabolite alters brain activity and anxiety behaviour in mice. Nature 602, 647–653 (2022).
Mangalam, A. et al. Human Gut-derived commensal bacteria suppress CNS inflammatory and demyelinating disease. Cell Rep. 20, 1269–1277 (2017).
Blacher, E. et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature 572, 474 (2019).
Vuong, H. E. & Hsiao, E. Y. Emerging roles for the Gut Microbiome in Autism Spectrum Disorder. Biol. Psychiat. 81, 411–423 (2017).
Ansaldo, E. & Belkaidi, Y. How microbiota improve immunotherapy. Science 373, 966–967 (2021).
de Cena, J. A., Zhang, J., Deng, D., Dame-Teixeira, N. & Do, T. Low-abundant microorganisms: the human microbiome’s dark matter, a scoping review. Front. Cell Infect. Microbiol. 11, 689197 (2021).
Beura, L. K. et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532, 512–516 (2016).
Rosshart, S. P. et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 365, 461 (2019).
Yeung, F. et al. Altered immunity of laboratory mice in the natural environment is associated with fungal colonization. Cell Host Microbe 27, 809–822.e6 (2020).
Gause, W. C. & Maizels, R. M. Macrobiota - helminths as active participants and partners of the microbiota in host intestinal homeostasis. Curr. Opin. Microbiol 32, 14–18 (2016).
Ramanan, D. et al. Helminth infection promotes colonization resistance via type 2 immunity. Science 352, 608–612 (2016).
Rapin, A. & Harris, N. L. Helminth-bacterial interactions: cause and consequence. Trends Immunol. 39, 724–733 (2018).
Giacomin, P., Agha, Z. & Loukas, A. Helminths and intestinal flora team up to improve gut health. Trends Parasitol. 32, 664–666 (2016).
Banerjee, S., Schlaeppi, K. & van der Heijden, M. G. A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol 16, 567–576 (2018).
Benjamino, J., Lincoln, S., Srivastava, R. & Graf, J. Low-abundant bacteria drive compositional changes in the gut microbiota after dietary alteration. Microbiome 6, 86 (2018).
Han, G., Luong, H. & Vaishnava, S. Low abundance members of the gut microbiome exhibit high immunogenicity. Gut Microbes 14, 2104086 (2022).
Mazmanian, S. K., Liu, C. H., Tzianabos, A. O. & Kasper, D. L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122, 107–118 (2005).
Ivanov, I. I. et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009).
Wang, Y. et al. Induction of intestinal Th17 cells by flagellins from segmented filamentous bacteria. Front Immunol. 10, 2750 (2019).
Alexander, M. et al. Human gut bacterial metabolism drives Th17 activation and colitis. Cell Host Microbe 30, 17–30.e19 (2022).
Paik, D. et al. Human gut bacteria produce TH17-modulating bile acid metabolites. Nature 603, 907–912 (2022).
Beyaz, S. et al. Dietary suppression of MHC class II expression in intestinal epithelial cells enhances intestinal tumorigenesis. Cell Stem Cell 28, 1922–1935.e1925 (2021).
Kaakoush, N. O. Insights into the role of Erysipelotrichaceae in the human host. Front Cell Infect. Microbiol. 5, 84 (2015).
Palm, N. W. et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 158, 1000–1010 (2014).
Dinh, D. M. et al. Intestinal microbiota, microbial translocation, and systemic inflammation in chronic HIV infection. J. Infect. Dis. 211, 19–27 (2015).
Herp, S., Durai Raj, A. C., Salvado Silva, M., Woelfel, S. & Stecher, B. The human symbiont Mucispirillum schaedleri: causality in health and disease. Med. Microbiol. Immunol. 210, 173–179 (2021).
Berry, D. et al. Phylotype-level 16S rRNA analysis reveals new bacterial indicators of health state in acute murine colitis. ISME J. 6, 2091–2106 (2012).
Vereecke, L. et al. A20 controls intestinal homeostasis through cell-specific activities. Nat. Commun. 5, 5103 (2014).
Zhang, X. et al. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 21, 895–905 (2015).
Cekanaviciute, E. et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl Acad. Sci. USA 114, 10713–10718 (2017).
Werbner, M. et al. Social-stress-responsive microbiota induces stimulation of self-reactive effector T helper cells. mSystems 4, e00292–18 (2019).
Herp, S. et al. Mucispirillum schaedleri antagonizes Salmonella Virulence to protect mice against colitis. Cell Host Microbe 25, 681–694.e688 (2019).
White, J. R., Nagarajan, N. & Pop, M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 5, e1000352 (2009).
Balan, P. et al. Keystone species in pregnancy gingivitis: a snapshot of oral microbiome during pregnancy and postpartum period. Front. Microbiol. 9, 2360 (2018).
Scheltens, P. et al. Alzheimer’s disease. Lancet 388, 505–517 (2016).
Chandra, S., Sisodia, S. S. & Vassar, R. J. The gut microbiome in Alzheimer’s disease: what we know and what remains to be explored. Mol. Neurodegener. 18, 9 (2023).
Ferreiro, A. L. et al. Gut microbiome composition may be an indicator of preclinical Alzheimer’s disease. Sci. Transl. Med. 15, eabo2984 (2023).
Pust, M. M. & Tummler, B. Bacterial low-abundant taxa are key determinants of a healthy airway metagenome in the early years of human life. Comput. Struct. Biotechnol. J. 20, 175–186 (2022).
Arumugam, M. et al. Enterotypes of the human gut microbiome. Nature 473, 174–180 (2011).
Hildebrand, F. et al. Antibiotics-induced monodominance of a novel gut bacterial order. Gut 68, 1781–1790 (2019).
Hildebrand, F., Pallen, M. J. & Bork, P. Towards standardisation of naming novel prokaryotic taxa in the age of high-throughput microbiology. Gut 69, 1358–1359 (2020).
Underhill, D. M. & Lliev, L. D. The mycobiota: interactions between commensal fungi and the host immune system. Nat. Rev. Immunol. 14, 405–416 (2014).
Swidergall, M. & LeibundGut-Landmann, S. Immunosurveillance of Candida albicans commensalism by the adaptive immune system. Mucosal Immunol. 15, 829–836 (2022).
Jacobsen, I. D. The role of host and fungal factors in the commensal-to-pathogen transition of Candida albicans. Curr. Clin. Microbiol. Rep. 10, 55–65 (2023).
Jacobsen, I. D. et al. Candida albicans dimorphism as a therapeutic target. Expert Rev. Anti-Infe 10, 85–93 (2012).
Netea, M. G., Joosten, L. A., van der Meer, J. W., Kullberg, B. J. & van de Veerdonk, F. L. Immune defence against Candida fungal infections. Nat. Rev. Immunol. 15, 630–642 (2015).
Ost, K. S. et al. Adaptive immunity induces mutualism between commensal eukaryotes. Nature 596, 114–118 (2021).
Doron, I. et al. Mycobiota-induced IgA antibodies regulate fungal commensalism in the gut and are dysregulated in Crohn’s disease. Nat. Microbiol. 6, 1493–1504 (2021).
Jiang, T. T. et al. Commensal fungi recapitulate the protective benefits of intestinal bacteria. Cell Host Microbe 22, 809–816.e804 (2017).
Bacher, P. et al. Human anti-fungal Th17 immunity and pathology rely on cross-reactivity against Candida albicans. Cell 176, 1340–1355.e1315 (2019).
Shao, T. Y. et al. Commensal Candida albicans positively calibrates systemic Th17 immunological responses. Cell Host Microbe 25, 404–417.e406 (2019).
Chen, Y. H. et al. Rewilding of laboratory mice enhances granulopoiesis and immunity through intestinal fungal colonization. Sci. Immunol. 8, eadd6910 (2023).
Doron, I. et al. Human gut mycobiota tune immunity via CARD9-dependent induction of anti-fungal IgG antibodies. Cell 184, 1017–1031.e1014 (2021).
Limon, J. J. et al. Malassezia is associated with Crohn’s disease and exacerbates colitis in mouse models. Cell Host Microbe 25, 377–388.e6 (2019).
Leonardi, I. et al. Mucosal fungi promote gut barrier function and social behavior via Type 17 immunity. Cell 185, 831–846.e814 (2022).
Zhang, Z. et al. Peripheral lymphoid volume expansion and maintenance are controlled by gut microbiota via RALDH+ dendritic cells. Immunity 44, 330–342 (2016).
Rahman, D., Mistry, M., Thavaraj, S., Challacombe, S. J. & Naglik, J. R. Murine model of concurrent oral and vaginal Candida albicans colonization to study epithelial host-pathogen interactions. Microbes Infect. 9, 615–622 (2007).
McDonough, L. D. et al. Candida albicans Isolates 529L and CHN1 exhibit stable colonization of the murine gastrointestinal tract. mBio 12, e0287821 (2021).
Limon, J. J., Skalski, J. H. & Underhill, D. M. Commensal fungi in health and disease. Cell Host Microbe 22, 156–165 (2017).
Richard, M. L. & Sokol, H. The gut mycobiota: insights into analysis, environmental interactions and role in gastrointestinal diseases. Nat. Rev. Gastroenterol. Hepatol. 16, 331–345 (2019).
Chehoud, C. et al. Fungal signature in the gut microbiota of pediatric patients with inflammatory bowel disease. Inflamm. Bowel Dis. 21, 1948–1956 (2015).
Sokol, H. et al. Fungal microbiota dysbiosis in IBD. Gut 66, 1039–1048 (2017).
Sun, S. et al. The gut commensal fungus, Candida parapsilosis, promotes high fat-diet induced obesity in mice. Commun. Biol. 4, 1220 (2021).
Mar Rodriguez, M. et al. Obesity changes the human gut mycobiome. Sci. Rep. 5, 14600 (2015).
Hufnagl, K., Pali-Scholl, I., Roth-Walter, F. & Jensen-Jarolim, E. Dysbiosis of the gut and lung microbiome has a role in asthma. Semin Immunopathol. 42, 75–93 (2020).
Liu, C. et al. Microbial dysbiosis and childhood asthma development: Integrated role of the airway and gut microbiome, environmental exposures, and host metabolic and immune response. Front. Immunol. 13, 1028209 (2022).
Chung, K. F. Airway microbial dysbiosis in asthmatic patients: a target for prevention and treatment? J. Allergy Clin. Immunol. 139, 1071–1081 (2017).
van Tilburg Bernardes, E., Gutierrez, M. W. & Arrieta, M. C. The fungal microbiome and asthma. Front. Cell Infect. Microbiol. 10, 583418 (2020).
Arrieta, M. C. et al. Associations between infant fungal and bacterial dysbiosis and childhood atopic wheeze in a nonindustrialized setting. J. Allergy Clin. Immunol. 142, 424–434.e410 (2018).
Boutin, R. C. et al. Bacterial-fungal interactions in the neonatal gut influence asthma outcomes later in life. Elife 10, e67740 (2021).
Kanj, A. N. et al. Dysbiosis of the intestinal fungal microbiota increases lung resident group 2 innate lymphoid cells and is associated with enhanced asthma severity in mice and humans. Respir. Res. 24, 144 (2023).
Albillos, A., de Gottardi, A. & Rescigno, M. The gut-liver axis in liver disease: pathophysiological basis for therapy. J. Hepatol. 72, 558–577 (2020).
Chen, L., Zhu, Y., Hou, X., Yang, L. & Chu, H. The role of gut bacteria and fungi in alcohol-associated liver disease. Front. Med. 9, 840752 (2022).
Yang, A. M. et al. Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Invest. 127, 2829–2841 (2017).
Yu, L. et al. Saccharomyces boulardii administration changes gut microbiota and attenuates D-galactosamine-induced liver injury. Sci. Rep. 7, 1359 (2017).
Everard, A., Matamoros, S., Geurts, L., Delzenne, N. M. & Cani, P. D. Saccharomyces boulardii administration changes gut microbiota and reduces hepatic steatosis, low-grade inflammation, and fat mass in obese and type 2 diabetic db/db mice. mBio 5, e01011–e01014 (2014).
Yang, A. M. et al. Saccharomyces Boulardii ameliorates non-alcoholic steatohepatitis in mice induced by a methionine-choline-deficient diet through Gut-liver axis. Front. Microbiol. 13, 887728 (2022).
Aykut, B. et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 574, 264–267 (2019).
Gao, R. et al. Dysbiosis signature of mycobiota in colon polyp and colorectal cancer. Eur. J. Clin. Microbiol. Infect. Dis. 36, 2457–2468 (2017).
Thomas, M. et al. A randomized, open-label, multicenter, phase II study evaluating the efficacy and safety of BTH1677 (1,3-1,6 beta glucan; Imprime PGG) in combination with cetuximab and chemotherapy in patients with advanced non-small cell lung cancer. Invest. N. Drugs 35, 345–358 (2017).
Bang, C. & Schmitz, R. A. Archaea associated with human surfaces: not to be underestimated. FEMS Microbiol. Rev. 39, 631–648 (2015).
Pausan, M. R. et al. Exploring the Archaeome: detection of archaeal signatures in the human body. Front. Microbiol. 10, 2796 (2019).
Mohammadzadeh, R., Mahnert, A., Duller, S. & Moissl-Eichinger, C. Archaeal key-residents within the human microbiome: characteristics, interactions and involvement in health and disease. Curr. Opin. Microbiol. 67, 102146 (2022).
Jangi, S. et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 7, 12015 (2016).
Bang, C., Weidenbach, K., Gutsmann, T., Heine, H. & Schmitz, R. A. The intestinal Archaea Methanosphaera stadtmanae and Methanobrevibacter smithii activate human dendritic cells. PLoS ONE 9, e99411 (2014).
Sereme, Y. et al. Methanogenic Archaea: emerging partners in the field of allergic diseases. Clin. Rev. Allerg. Immunol. 57, 456–466 (2019).
Li, C. L. et al. Prevalence and molecular diversity of Archaea in subgingival pockets of periodontitis patients. Oral. Microbiol. Immunol. 24, 343–346 (2009).
Barnett, D. J. M., Mommers, M., Penders, J., Arts, I. C. W. & Thijs, C. Intestinal archaea inversely associated with childhood asthma. J. Allergy Clin. Immunol. 143, 2305–2307 (2019).
Lecours, P. B. et al. Increased prevalence of Methanosphaera stadtmanae in inflammatory bowel diseases. PLoS ONE 9, e87734 (2014).
Zhang, H. et al. Human gut microbiota in obesity and after gastric bypass. Proc. Natl Acad. Sci. USA 106, 2365–2370 (2009).
Coker, O. O., Wu, W. K. K., Wong, S. H., Sung, J. J. Y. & Yu, J. Altered gut archaea composition and interaction with bacteria are associated with colorectal cancer. Gastroenterology 159, 1459–1470.e1455 (2020).
Chabe, M., Lokmer, A. & Segurel, L. Gut Protozoa: friends or foes of the human gut microbiota? Trends Parasitol. 33, 925–934 (2017).
Ribeiro, L. C., Santos, C. & Benchimol, M. Is Trichomonas tenax a parasite or a commensal? Protist 166, 196–210 (2015).
Chudnovskiy, A. et al. Host-protozoan interactions protect from mucosal infections through activation of the inflammasome. Cell 167, 444–456.e14 (2016).
Chiaranunt, P. et al. NLRP1B and NLRP3 control the host response following colonization with the commensal protist Tritrichomonas musculis. J. Immunol. 208, 1782–1789 (2022).
Howitt, M. R. et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 351, 1329–1333 (2016).
Schneider, C. et al. A metabolite-triggered tuft cell-ILC2 circuit drives small intestinal remodeling. Cell 174, 271–284.e214 (2018).
Nadjsombati, M. S. et al. Detection of succinate by intestinal tuft cells triggers a type 2 innate immune circuit. Immunity 49, 33–41.e37 (2018).
Audebert, C. et al. Colonization with the enteric protozoa Blastocystis is associated with increased diversity of human gut bacterial microbiota. Sci. Rep. 6, 25255 (2016).
Kim, M. J., Lee, Y. J., Kim, T. J. & Won, E. J. Gut microbiome profiles in colonizations with the enteric protozoa blastocystis in Korean populations. Microorganisms 10, 34 (2022).
Morton, E. R. et al. Variation in rural African gut microbiota is strongly correlated with colonization by entamoeba and subsistence. PLoS Genet. 11, e1005658 (2015).
Olsson, L. M. et al. Dynamics of the normal gut microbiota: a longitudinal one-year population study in Sweden. Cell Host Microbe 30, 726–739.e723 (2022).
Liu, N. N. et al. Multi-kingdom microbiota analyses identify bacterial-fungal interactions and biomarkers of colorectal cancer across cohorts. Nat. Microbiol. 7, 238–250 (2022).
Patnode, M. L. et al. Interspecies competition impacts targeted manipulation of human gut bacteria by fiber-derived glycans. Cell 179, 59–73.e13 (2019).
Rice, T. A. et al. Interspecies commensal interactions have nonlinear impacts on host immunity. Cell Host Microbe 30, 988–1002.e1006 (2022).
Acknowledgements
This research was supported by grants from the NIH (R21AI168772 and R01DK113265) and AAI Careers in Immunology Fellowship.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Han, G., Vaishnava, S. Microbial underdogs: exploring the significance of low-abundance commensals in host-microbe interactions. Exp Mol Med 55, 2498–2507 (2023). https://doi.org/10.1038/s12276-023-01120-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-023-01120-y
