
Abstract
Mitochondria, ubiquitous double-membrane-bound organelles, regulate energy production, support cellular activities, harbor metabolic pathways, and, paradoxically, mediate cell fate. Evidence has shown mitochondria as points of convergence for diverse cell death-inducing pathways that trigger the various mechanisms underlying apoptotic and nonapoptotic programmed cell death. Thus, dysfunctional cellular pathways eventually lead or contribute to various age-related diseases, such as neurodegenerative, cardiovascular and metabolic diseases. Thus, mitochondrion-associated programmed cell death-based treatments show great therapeutic potential, providing novel insights in clinical trials. This review discusses mitochondrial quality control networks with activity triggered by stimuli and that maintain cellular homeostasis via mitohormesis, the mitochondrial unfolded protein response, and mitophagy. The review also presents details on various forms of mitochondria-associated programmed cell death, including apoptosis, necroptosis, ferroptosis, pyroptosis, parthanatos, and paraptosis, and highlights their involvement in age-related disease pathogenesis, collectively suggesting therapeutic directions for further research.
Similar content being viewed by others

Introduction
Mitochondria, at the core of cellular metabolism, play pivotal roles in key metabolic activities, from energy production to cellular signaling1, all of which are critical to organism health. Mitochondrial dysfunction is a profound factor contributing to aging and various age-related diseases, such as neurodegenerative, cardiovascular, and metabolic diseases. Due to their irreplaceable roles and incompletely characterized mechanisms, mitochondria have garnered increased interest in recent decades, especially in the last five years, in which more than 10,000 research papers have been published annually and in which novel protective pathways and therapeutic targets, such as mitochondrial quality control (MQC) networks2 and programmed cell death (PCD), have been described. As accumulated evidence indicates, in an apparent paradox, mitochondria determine cell fate by governing PCD pathways. Since its discovery as a decisive form of cell lifespan termination, PCD has garnered considerable attention. The first PCD described, apoptosis, has been the topic of research for more than 5 decades and ha been described in as many as 30,000 annual publications in a 5-year period, and surprisingly, a significant portion of these publications have characterized and substantiated the unequivocal involvement of mitochondria in apoptosis3. Research describing newly discovered forms of nonapoptotic PCD, such as necroptosis, ferroptosis, and pyroptosis, has been conducted for more than 20 years, and on average, more than 5,000 annual publications in this period (Fig. 1a) implicated mitochondria in non-PCD mechanisms of action4. However, the specific interactions between mitochondrial and PCD factors and the underlying mechanisms remain poorly characterized.

a Publications of typical programmed cell death in the last 50 years. b Mitochondria are vital in regulating cellular metabolism and are involved in the pathogenesis and progression of numerous human diseases, directly or indirectly, through many pathways. Mitochondria are also essential in regulating cell fate via PCD, an evolutionarily conserved process in multicellular organisms that is indispensable for modulating homeostasis, containing diverse patterns, including apoptosis, necroptosis, ferroptosis, and pyroptosis. Mitochondria-associated PCD is extensively involved in the pathological progression of various disorders in different organ systems. In this regard, fresh insights into the interplay between mitochondria and PCD are expected to be inspired in disease treatment development in contexts ranging from in vitro to preclinical and clinical trials. PCD Programmed cell death, EMR Electronic medical record.
Here, we provide an overview of the current state of the field with a specific focus on regulatory mechanisms of MQC systems discovered to date and mitochondrial contributions to various forms of PCD, including the underlying molecular mechanisms and effects on human diseases. We highlight mitochondria as the primary nodes in which PCD is triggered, death signaling is propagated, cell death is realized, and the considerable impacts that cell fate exerts on the incidence of numerous diseases. We also highlight unique insights into the interactions between mitochondrial and PCD factors that have been applied for developing therapies on the basis of preclinical research in vitro and clinical trials (Fig. 1b), which may provide further direction for effective therapy.
Mitochondrial biology
Mitochondrial structure
Mitochondria, cytoplasmic organelles in most eukaryotic cells (Fig. 2a), are enclosed by two unique phospholipid membranes, the inner mitochondrial membrane (IMM) and outer mitochondrial membrane (OMM), which separate specialized and functionally compartmentalized structures, defined as the intermembrane space and matrix. These two membranes show clearly distinct chemical and molecular properties in terms of lipid composition, transmembrane protein functions, and permeability due to the organelle’s endosymbiotic origin. In contrast to the IMM, the OMM permits ions and small molecules to flow through voltage-dependent anion channels that enable only the free transport of water, gaseous substrates, and products of oxidative phosphorylation. This selectivity enables the formation of cross-membrane electrochemical gradients needed for ATP production. The IMM undergoes distinct morphological organizational changes to form densely clustered invaginations into the matrix referred to as cristae, which establish a platform for imported host-derived proteins and form crista junctions5 (Fig. 2b).

a, b Hypothetical depiction of the origin of mitochondria from Aerobic Proteobacterium and mitochondrial simplified structure. c Microstructure and enzyme complexes of mitochondria. d Mitochondrial functions rely on cellular interactions. OMM Outer mitochondrial membrane, IMM Inner mitochondrial membrane, IMS Intermembrane mitochondrial space, mtDNA Mitochondrial DNA, ROS Reactive oxygen species, ER Endoplasmic reticulum, ATP Adenosine triphosphate.
Mitochondria carry a unique genetic code, mitochondrial DNA (mtDNA), with 16,569 base pairs but no introns6. In contrast to two copies of nuclear DNA (nDNA), multiple copies of mtDNA, corresponding to the number of genes related to the energy production needed in different tissues, are present in cells. Surprisingly, during evolution, the majority of mitochondrial genes were lost or were translocated to nuclei, becoming nDNA; 37 genes, however, remained in mtDNA, and they encode 2 ribosomal RNAs and 22 transfer RNAs, which are essential components of the translational apparatus, and mtDNA encodes 13 proteins, which function as core constituents of OXPHOS complexes embedded in the IMM7. An estimated 1500 proteins are expressed in mitochondria, the majority of which are encoded by nDNA and translated outside of mitochondria before being transported into the organelle via specialized import pathways8. In mitochondria, these proteins constitute the components of the electron transport chain, metabolic pathways, etc.
Mitochondrial function
Mitochondria are widespread intracellular organelles in virtually all eukaryotes, where they function as metabolism and signaling hubs for a broad range of cellular activities; their primary roles involve regulating energy production and cell signaling, with additional functions in mitonuclear crosstalk, such as that related to fatty acid oxidation, iron–sulfur cluster biosynthesis, and thermogenesis9,10,11 (Fig. 2c).
Because mitochondria are cell powerhouses, many catabolic pathways converge in mitochondria, particularly pathways involved in ATP production mediated through oxidative phosphorylation. Specifically, the catabolism of metabolites generates acetyl-CoA, which is oxidized in the tricarboxylic acid cycle to produce two reduced electron donors, driving the mitochondrial electron transport chain. Electrons are transferred to and flow through electron transport chain complexes, resulting in proton outflow through the IMM and formation of an electrochemical gradient; this gradient generates the proton-motive force that drives protons back to the matrix via complex V, a rotary turbine that catalyzes the synthesis of ATP1.
Mitochondria regulate cell signaling primarily via the influx of signaling molecules and by forming platforms on which multiple signaling interactions occur. Mitochondria modulate signaling pathways ranging from cytochrome c (cyt c) release to caspase activation, accompanied by signaling complex activation, immune responses, and PCD induction4. Mitochondria regulate the levels of intracellular molecules, including reactive oxygen species (ROS) and Ca2+. When Ca2+ accumulates in mitochondria, the activity of Ca2+-sensitive dehydrogenases and metabolite transporters is modulated, which accelerates oxidative metabolism (Fig. 2d). High-conductance channel-induced pore formation in the IMM, called mitochondrial permeability transition (MPT), is triggered by Ca2+ overload and ROS. It is linked to significant alterations in mitochondrial morphology and functional activities of various enzymes, which modulate mitochondrial respiration and ATP production12. ROS are produced by electron transport from redox donors to molecular oxygen, which is then converted to hydrogen peroxide and dispersed into the cytoplasm; ROS participate directly in intercellular signaling to regulate basal or adaptation responses that maintain organismal homeostasis13.
Mitochondrial quality control and mitohormesis
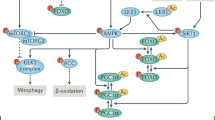
Because mitochondria are hubs of energy production and guardian of multiple metabolic processes and cell signaling pathways, dysfunctional mitochondria cause diverse diseases across species14,15. To maintain functionality and ensure homeostasis, different mechanisms in cells sense and react to aberrant mitochondrial activities; these mechanisms constitute MQC systems2 (Fig. 3). In response to general insults, such as electron transfer chain (ETC) suppression, protein misfolding, and redox imbalances, mitochondrial integrity and metabolism are strictly regulated by DNA repair networks, antioxidants, and proteases or chaperones engaged in the mitochondrial unfolded protein response (UPRmt), which has been extensively described16. Notably, an adaptive response, mitochondria–cytosol–nucleus crosstalk, also known as mitochondrial retrograde signaling or mitohormesis, is triggered nonautonomously to attenuate cellular disorder. The coordinated interplay among crosstalk factors leads to the production of versatile mitokines, which facilitate the transcriptional upregulation of stress response proteins and promote cytoprotective and stress resistance mechanism activation17. However, when mitochondria are not promptly rescued from excessive damage, overexpression of certain proteotoxic signals induces the activity of additional protein quality control (PQC) networks, including the UPRmt and mitophagy, through which malfunctioning mitochondria are engulfed, preserving proteostasis18.

a Activation of the UPRmt pathway is triggered by the accumulation of unfolded proteins in the mitochondrial matrix, expression of mitochondrial protein import components, and upregulation of chaperones and proteases for reestablishing mitochondrial proteostasis. b Molecular chaperones and proteolytic enzymes in the mitochondria perform a major function in refolding or destroying mitochondrial misfolded and unfolded proteins. c Mitochondrial proteins are likely to be decomposed by being transported to lysosomes, where vesicles formed from mitochondrial tubules seize specific mitochondrial cargos for lysis. d Fragmented dysfunctional mitochondria are removed by mitophagy. Bnip3, FUNDC1, and NIX are mitophagy receptors that bind LC3 to tether mitochondria to autophagosomes. OMM proteins and Parkin are ubiquitinated when PINK1 phosphorylates proteins on the surface of depolarized mitochondria. The p62 adaptor can recognize ubiquitinated proteins to initiate mitophagy. UPRmt mitochondrial Unfolded Protein Response, MURE Mitochondrial Unfolded Protein Response Element, ATF Activating Transcription Factor, NRF Nuclear Respiratory Factor, HSP Heat Shock Protein, LC3 microtubule-associated protein Light Chain 3, Bnip3 Bcl-2 interacting protein 3, Ub Ubiquitin, PINK1 PTEN-induced kinases 1.
Mitokines
Mitokines, a term initially coined in the context of prolonged lifespan after mitochondrial chain disruption16, are currently defined as metabolic cytokines or mitochondrion-derived peptides (MDPs) that are released by cells in response to mitochondrial stresses and that affect systematic metabolism and longevity17. Mitokines are nucleus-encoded proteins and respond to mitochondrial insults, such as growth differentiation factor 15 (GDF15) and fibroblast growth factor 21 (FGF21), and alterations in both of these factors are involved in multiple mitochondrion-related disorders and inherited metabolic diseases19. Moreover, various MDPs generated by short open-reading frames (ORFs) located in the regions encoding 12 S and 16 S rRNA in mtDNA have been discovered; these mitochondrial ORFs encode 12 S rRNA-c (MOTS-c), humanin (HN), and six small HN-like peptides (SHLPs)20, which exhibit systemic effects such as neuroprotection, enhanced metabolism, antiapoptotic functions and increased viability. However, the specific functions underlying their activity are unclear.
GDF15
Several studies have indicated that GDF15, a vital humoral factor secreted from skeletal muscle and adipose tissue cells after mitochondrial stress- and UPRmt-induced activation, may drive mitohormesis17. Circulating GDF15 levels are elevated in individuals with mitochondrial diseases and subjects characterized by mitochondrial impairment or mitoribosomal protein deficiency21, and GDF15 systematically increases metabolism in dozens of diseases22,23. When overexpressed, GDF15 regulates systemic energy homeostasis, attenuates mitochondrial malfunction, and promotes metabolic reorganization and adaptability24. However, some reports have indicated that GDF15 exerts certain detrimental effects under pathophysiological conditions and is partially involved in muscle wasting and cachexia25. However, the systemic mechanisms underlying GDF15 action in mitohormesis are unclear, and further studies are needed.
FGF21
Research indicates that FGF21 functions as a potent mitokine, in addition to an endocrine regulator and longevity hormone, as originally described. It is secreted under mitochondrial stress conditions induced by abnormalities in the ETC or via autophagy. An in-depth study has revealed that FGF21 can also be activated in reaction to various mitochondrial stresses, including mitofusin deletion, mtDNA mutations, and mitochondrial ROS overexpression, encouraging a health-promoting response. A recent study discovered an intriguing feature FGF21: It is induced under mild-to-moderate mitochondrial stress conditions but is dispensable or overridden via whole-body metabolic adaptation under severe disease conditions26. However, despite these findings, which revealed a more nuanced function of FGF21, the health-promoting mitohormetic effects of FGF21 are generally recognized. Similar to GDF15, FGF21 exhibits various beneficial regulatory metabolic functions, conferring protection against hormone resistance and increasing glucolipid metabolism and lipothermogenesis, as thoroughly described in a prior study27. The functions of FGF21 have been confirmed by mechanistic evidence showing that FGF21 suppresses oxidative phosphorylation gene expression and rescues the ETC to activate ATF4, a transcription factor targeting the FGF21 promoter28 and a master regulator of the integrated stress response29, leading to cellular homeostasis restoration.
MDP
Typical MDP functions include predominantly the regulation of metabolic signaling and restoration of mitochondrial homeostasis30. HN was the first MDP proposed to exert cytoprotective effects on different aging diseases, such as cardiovascular diseases (CVDs), neurodegenerative diseases, and hepatic steatosis, owing to its anti-inflammatory and antiapoptotic effects that are triggered via increased Ca2+ flux and activation of extracellular signal-regulated kinase and downstream signal cascades31. MOTS-c may be transported to the nucleus and drive responsive gene expression under stress conditions via interconnections between chromatin and stress-induced transcriptional regulators32. MOTS-c, similar to FGF21, demonstrates dominant metabolome-protective features in multiple types of tissues with metabolic disorders, principally functioning to maintain glucolipid metabolism and regulate lipothermogenesis33. SHLPs, especially SHLP2 and SHLP3, play promising roles in inhibiting apoptosis by inhibiting caspase activation and ROS production and promoting adipocyte differentiation34.
The UPRmt
The UPRmt is an adaptable transcriptional process that maintains mitochondrial homeostasis. It is activated by proteotoxic stress, such as that induced by misfolded mitochondrial precursor protein deposition and protein mistargeting35, to limit cytosolic protein production and degrade mislocated proteins in mitochondria that cannot be transported. Both the ubiquitin‒proteasome system and mitophagy are advantageous for mitochondrial proteostasis and are mediated via the PQC system; these processes are mediated by chaperones and proteases that manage protein folding, assembly, and degradation.
The UPRmt signaling pathway has been most extensively characterized in invertebrates36, in which signaling cascades have, however, proven to be significant different from those in mammals. The UPRmt response has been preserved throughout evolution, as indicated by the levels of specific genetic markers in genetically engineered mouse models and humans. According to recent research and bioinformatics analysis, in mammals, the UPRmt pathway is epigenetically regulated by histone H3 lysine 27 demethylases and characterized by the stimulation of multiple chaperones (CHOP, TRAP1, mtHSP70, and the HSP60-MSP10 multimer) and proteases (ClpP, YME1L1, and LONP1)37. However, the underlying mechanisms of most retrograde messengers or promotors remain poorly understood, with some studies reporting paradoxical results.
Mitochondrial stress promotes the activation of the UPRmt either directly via mtDNA disorder-induced imbalance among OXPHOS complexes and indirectly via ATF4 and ATF538,39 to maintain cell proliferation in an elF2α-dependent manner and as proposed mediators in cardioprotection, respectively40.
Mitophagy
Mitophagy is an evolutionarily conserved mechanism in which identification and clearance of mitochondria targeted for elimination in response to misfolded mitochondrial protein accumulation or mitochondrial depolarization are realized, and it is required for MQC maintenance2. In this process, distinct mitophagy pathways are likely to be involved in diverse mechanisms and coordinate to initiate rapid responses to overload-induced damage with highly specialized functions to preserve mitochondrial homeostasis41.
Parkin and PINK
A pair of well-characterized mitophagy mediators involved in one mechanism is PTEN-induced putative kinase 1 (PINK1) and E3 ubiquitin ligase Parkin41. Recent evidence highlights PINK1 as a sensor or mitochondrial damage and in a surveillance mechanism that maintains mitochondrial fitness; it recruits Parkin to damaged mitochondria, promoting autophagic clearance in a Parkin-dependent ubiquitination process42. This pathway is also involved in regulating mitochondrial dynamics through fission and fusion. Consistent with these functions, Parkin is translocated into mitochondria after mitochondrial membrane depolarization and induces mitofusin polyubiquitination, targeting it for proteasomal degradation to prevent defective mitochondria from fusing with functioning mitochondrial networks; mitofusin polyubiquitination is required for subsequent degradation by mitophagy43.
BNIP3 and BNIP3L
Bnip3, in the B-cell lymphoma 2 (Bcl-2) family of PCD-regulating factors, plays a paradoxical but pivotal role in regulating diverse mitochondrial functions, such as oxidation, calcium overload, ATP shortage, and secondary mitochondrial disorders44. Similar to the conventional hypoxia-responsive regulator, Bnip3 induces mitophagy after autophagy stimulation and is involved in mitochondrial fragmentation, triggering mitochondrial depolarization, the subsequent sequestration of disordered mitochondria into autophagosomes, and ultimately, mitochondrial elimination.
Another mitophagy receptor, Bnip3-like (Bnip3L) or NIX, has recently been shown to exhibit functions in addition to its function as a BH3-only proapoptotic factor45. Bnip3L promotes mitophagy by facilitating autophagosome aggregation via disruption of the Bcl-2-BECN1 complex and, more directly, by recruiting autophagosomes to mitochondria via interplay with proteins in the Atg8 family.
FUNDC1
FUN14 domain-containing 1 (FUNDC1) was originally identified as a hypoxia-induced mitophagy modulator that interacts with and recruits LC3 to mitochondria to induce mitophagy mediated by various upstream phosphorylases or phosphatases that regulate mitophagy46. In-depth studies have demonstrated that FUNDC1 mediates mitophagy and mitochondrial fission or fusion, regulates the coupling of the double mitochondrial membranes to promote MQC, and regulates mitochondrial dynamics by interacting with both DNM1L/dynamin-related protein 1 (DRP1) and optic atrophy 1 (OPA1) in response to stresses such those associated with energy metabolism or oxidation47.
Mitochondria-associated programmed cell death
PCD is an essential feature of multicellular organism development and a major cause of degenerative diseases. As the field expands, pathways that coordinate diverse cell death pathways to preserve cellular homeostasis are being discovered48. PCD manifests as alterations in macroscopic morphotypes. In addition to manifestations of dead cells and their fragments before elimination, morphological characteristics of dying cells have typically been used to identify cell death modalities.
Apoptosis
Apoptosis is an outcome of a rapid response to stimuli, and it is morphologically characterized by cell shrinkage, nuclear pyknosis, and karyorrhexis, culminating in the formation of apoptotic bodies that are eventually engulfed by resident phagocytic cells49. Apoptosis is critical for physiological homeostasis in almost every organ system and is induced by multiple stimuli, such as hypoxic, immune reaction, ischemic, and infectious factors.
The central signaling pathway of apoptosis includes a set of cysteine proteases called cysteinyl aspartate proteinases (caspases), which are activated by proteolysis and processing cascades triggered by proapoptotic signaling50. To date, approximately 14 mammalian caspases have been discovered and classified into three categories: apoptotic effectors (caspase-3, -6, and -7), apoptotic initiators (caspase-2, -8, -9, and -10), and inflammatory caspases50. The former two sets of caspases interact and determine apoptosis, which is mediated by the cleavage of certain substrates by effector caspase-3 and -7 when initiator caspase-8, -9, and -10 are activated in response to cues from upstream adaptor molecules. In contrast, instead of functioning in apoptosis, inflammatory caspases are involved in inflammatory cytokine signaling and other forms of PCD, such as pyroptosis.
Cyt c is a heme protein synthesized as an apoprotein in the cytoplasm. It is then translocated to mitochondrial intermembranous and intercristal regions, where it functions as an important hub in the respiratory chain51. Upon activation, Cyt c is released into the cytoplasm, where it facilitates the allosteric activation and oligomerization of the adaptor molecule apoptosis-protease-activating factor 1 and induces heptameric structure assembly, called an apoptosome. This site promotes caspase-9 mutation to induce subsequent caspase-activation cascades, and then, resident phagocytes degrade apoptotic cell remnants50. A conformational change in the protein caused by the connection of Cyt c with cardiolipin (Cl)-specific phospholipids in the mitochondrion enables it to function as a peroxidase to induce Cl oxidation, which triggers a cascade of events that results in apoptosis.
The Bcl-2 family, a set of 25 cytoplasmic proteins, has been identified as another driver of apoptosis. Based on their distinct modes of action, Bcl-2 proteins are classified into 3 groups, proapoptotic three-domain proteins (Bax and Bak), proapoptotic BH3-only proteins (tBid, Bad, NOXA, PUMA, and Bim), and antiapoptotic four-domain proteins (Bcl-2 and Bcl-xL). The anti-apoptotic family members mainly antagonize the effects of proapoptotic members by preventing homo- and heterodimeric interactions to maintain mitochondrial membrane integrity51,52. Normally, Bak is translocated to mitochondria and inactivates Bax in the cytoplasm. However, following activation, Bax is inserted into the OMM, accompanied by Bak, and they are directly bound and activated by BH3-only proteins in the hydrophobic groove, leading to extensive conformational alteration of the OMM50.
Apoptosis is a genetically specified program mediated by a plethora of molecular pathways. Two of these pathways, the extrinsic and intrinsic pathways, have been extensively described as distinct but ultimately convergent processes that culminate in caspase activation53. The extrinsic pathway, or death receptor pathway, drives apoptosis when extracellular ligands bind cognate transmembrane death receptors, including Fas, TNF, or TRAIL (Fig. 4a). Death ligand stimulation causes the receptors to oligomerize and recruit caspase-8 and the Fas-associated death domain, promoting a the formation of death-inducing complex that, in turn, triggers the downstream caspase cascade leading to apoptotic PCD54. In contrast, activation of caspase-8 can drive Bid activity in the intrinsic pathway, also termed the mitochondrial or Bcl-2-regulated pathway, as bid is required for apoptotic signal amplification52. Intrinsic apoptosis is triggered by irreversible mitochondrial outer membrane permeabilization (MOMP), which is caused either through pore formation mediated by the Bcl-2 family, especially proapoptotic Bak and Bax, or after the MPT following the permeability transition pore (mPTP) opening50,52 (Fig. 4a).

a In the extrinsic pathways, death receptor ligands bind to members of the death receptor family to function, for instance, TNF, FAS, and TRAIL receptors. Numerous diverse stressors, such as DNA damage, the removal of growth factors, and mitotic arrest, activate the intrinsic apoptotic pathway, which in turn activates BH3-only members. By activating the effector proapoptotic Bcl-2 proteins Bax and Bak and inhibiting antiapoptotic Bcl-2 proteins, BH3-only proteins cause MOMP. In particular, cyt c is activated by releasing proteins from the mitochondrial intermembrane gap. The heptameric structure known as the apoptosome is created when cyt c interacts with APAF1. As a result, caspase-9 is recruited and activated, which cleaves and activates caspase-3 and -7. The caspase inhibitor XIAP is blocked by proteins such as SMAC, which are released due to MOMP, promoting apoptosis. The extrinsic and intrinsic apoptotic pathways are linked via caspase-8 cleavage and activation of the BH3-only protein Bid. b In normal cases, Bax localizes to the cytoplasm and Bak to the mitochondria. By interacting with BH3-only proteins, Bax and Bak can be directly activated during apoptosis, which leads to their stabilization on the OMM. Their dimer further oligomerizes into a higher-order multimer, contributing to the release of cyt c and other IMS proteins. Over time, Bax and Bak accumulate with macropore formation, which allows the IMM to protrude through the OMM, after which the IMM herniates and ruptures, eventually releasing mtDNA. TNF Tumor Necrosis Factor, FADD Fas-Associated Protein with DD, DISC Death-Inducing Signaling Complex, TRAIL Tumor necrosis factor-Related Apoptosis-Inducing Ligand, SMAC Second Mitochondria-derived Activator of Caspases, tBid truncated Bid, Apaf1 Apoptotic protease-activating Factor 1, XIAP X-linked Inhibitor of Apoptosis Protein, PUMA p53 Upregulated Modulator of Apoptosis, UV Ultraviolet.
An increasing body of evidence suggests that OMM integrity is the determining factor that determines cell survival or death. MOMP is the cause of primed cell apoptosis55 and is classified into caspase-dependent and caspase-independent forms. Generally, antiapoptotic proteins in the Bcl-2 family reside mainly on the OMM, where they protect mitochondria from factors that induce permeability, likely by coupling to and neutralizing proapoptotic factors. However, external stimuli, such as growth factor withdrawal and diverse cytotoxic insults, or internal stimuli, such as ROS, DNA damage, and hypoxia, activate proapoptotic BH3-only proteins, which exert their effects via two disparate mechanisms. Some of these proteins, such as Bad, preferentially engage with antiapoptotic proteins to release them from proapoptotic proteins to which they are bound, a process that is indispensable for morphogenesis because it induces MOMP. Other proteins, such as tBid, directly initiate proapoptotic proteins to induce MOMP by promoting Bax transport to the OMM or exert localized effects on Bak56. Bax and Bak collaboratively induce pore formation in the OMM, and the size of these pores vary depending on the Bax concentration and degree to which Bax assembles into rings and arcs in apoptotic mitochondria, where they support permeabilization via lipidic pore formation. As explained above, cyt c is released from mitochondria following MOMP and triggers caspase responses through apoptosome assembly, culminating in cellular disintegration; this series of events constitutes the caspase-dependent pathway57. Notably, the position of cyt c within the IMM and cristae does not promote its rapid release via MOMP (Fig. 4b). To enable cyt c transport out of mitochondria, cristae are reconfigured in a proapoptotic Bcl-2-dependent manner; specifically, Bim and Bak widen the pores to facilitate the escape of the bulk of mitochondrial cyt c from the intermembrane mitochondrial space (IMS), ensuring caspase activation4.
The activation of another caspase-dependent pathway is mediated by mPTP, which is formed by the fusion of the outer and inner leaflets of protein-stabilized membranes. mPTP is a multimeric complex harboring a voltage-dependent anion channel (VDAC) that resides in the OMM, adenine nucleotide translocase, an IMM integral protein, and the matrix protein cyclophilin D (CyPD). Among these proteins, VDAC is a mitochondrial porin that mediates Ca2+ movement into the IMS, and impaired Ca2+ transport regulation leads to matrix Ca2+ overload and activation of highly conductive mPTP58. This process is enhanced by CyPD, which increases mitochondria sensitivity to Ca2+, or by direct regulation of MPT55. mPTP opening can be enhanced by other factors, including ROS, inorganic phosphate, low pH, and ATP depletion, as well as factors including the membrane lipid environment and interorganelle communications. The deletion of Bax and Bak inhibits mitochondrial swelling and rupture after mPTP opening, indicating that some of the proteins implicated in mitochondrion-associated apoptosis may also affect the mPTP4.
Although numerous studies have been devoted to investigating the caspase-dependent apoptosis pathway, in each case, the overall picture remains hazy but the outline is gradually appearing. The latest research indicates that, in addition to Bcl-2 family proteins, the Bcl-2-related ovarian killer (BOK) functions as a non-Bcl-2 effector of apoptosis and is considered to play roles that overlap with that of Bcl-2 in MOMP induction, even when Bax and Bak are lacking52. BOK, a highly conserved protein with high similarity to Bax and Bak, is ubiquitously expressed. BOK has been shown to exert a proapoptotic effect on mitochondrial fission and morphology and thus affects MOMP. After cyt c release, another component, the second mitochondrion-derived activator of caspase, is also released from mitochondria into the cytoplasm, where it binds to apoptosis-protein inhibitors and leads to caspase activation50.
Although caspases support MOMP induced wave propagation, the inhibition of caspase activation after exposure to mitochondrial apoptosis stimulation exerted no impact on the kinetics or extent of MOMP in cells; this is an alternate mechanism underlying MOMP called the caspase-independent pathway. Underlying the maladaptive response to the irreversible loss of mitochondrial function, some caspase-independent death effectors, including endonuclease G and apoptosis-inducing factor (AIF), are translocated to the cytoplasm and, ultimately, the nucleus, where they contribute to DNA fragmentation and chromatin condensation59.
One thought-provoking caveat regarding cyt c release suggests that most cyt c is segregated within mitochondrial cristae. During MOMP, despite the pores opening on the OMM, the mechanism by which cyt c escapes from the IMS is unclear. Consistent with cyt c escape, cyt c mobilization is controlled following MOMP, and caspase activation is changed, implying an additional secondary mechanism, which is thought to be associated with cristae remodeling, modulating this process60. Mechanically, several BH3-only proteins, including Bid, Bik, and Bim, have been proven to initiate cristae remodeling in a MOMP-independent manner through Bax/Bak-independent or Bax/Bak-dependent pathways; this remodeling requires that the mitochondrial fission protein DRP1 level, as well as the GTPase OPA1 levels to some extent, be regulated4,52. OPA1 regulates IMM fusion and the size of crista junctions, while OPA1 oligomers restrict the size of the junctions, whereas OPA1 oligomer disintegration promotes junction widening. As auxiliary factors, ubiquitin-like proteins modify DRP1, stabilizing the binding sites between mitochondria and the endoplasmic reticulum (ER) membranes, where Ca2+ is pumped back into mitochondria to promote cristae remodeling61.
Nonapoptotic PCD
Recent research on apoptosis has focused on the homeostasis and development of eukaryotic organisms. However, because mitochondria are sites of diverse cell death-promoting signaling convergence, various nonapoptotic PCD forms have been discovered. These modalities, which differ from apoptosis in their specific morphological or mechanical characteristics, are either triggered independently of apoptosis or, more frequently, kill cells as a backup mechanism when apoptosis is inhibited62.
Necroptosis
Inhibiting apoptosis cannot entirely stop PCD, but it leads to a transitions to a caspase-independent process with morphological traits comparable to those of necrosis called necroptosis3. Apoptosis and necroptosis differ in several respects. Morphologically, the cell membranes of cells undergoing apoptosis are preserved. In contrast, the plasma membranes of necroptotic cells are disrupted, which is accompanied by organelle swelling, nuclear condensation, chromatin digestion, DNA hydrolysis, and eventually cell lysis.
Studies have revealed certain mechanisms by which tumor necrosis factor (TNFα) plays a critical role in driving necroptosis. When caspase-8 is inhibited, TNFα binds with a complementary receptor, forming a transitory membrane signaling complex containing TRADD, an adapter molecule that mediated the translocation of receptor-interacting protein kinase (RIPK) 1 to TNF, which activates RIPK1 and RIPK3, which assemble into a necrosome complex63. The necrosome then phosphorylates and stimulates a mixed-lineage kinase domain-like pseudokinase, which is transported to the plasma membrane and induces permeability to kill cells64. Recent research has linked necroptosis to mitochondria, with convincing evidence showing that progressive mitochondrial dysfunction during aging may predispose cells to undergo necroptosis. Mitochondrial ROS promote the onset of necroptosis by increasing RIPK1 autophosphorylation, resulting in RIPK3 recruitment and necrosome assembly65. Similarly, RIPK3 promotes mitochondrial energy metabolism and activates mitochondrial ROS generation, increasing the necrosome assembly rate and RIPK3 activity66 (Fig. 5a). In addition, RIPK3 can induce mitochondrial damage by upregulating the expression of mitochondrial NADPH oxidase 4 through posttranscriptional modification; other underlying mechanisms have yet to be further explored.

a Necroptosis is formed from necroptosis-driving factors such as TNF α binding with complementary receptors to recruit RIPK1, where the engagement renders RIPK1 and RIPK3 activated, which fuels necrosome configuration. The necrosome thereafter phosphorylates and activates MLKL, which is translocated to and permeabilizes the plasma membrane and permits DAMP release. b Ferroptosis is driven by iron-dependent lipid peroxidation and ROS overexpression. GPX4 converts excessive peroxides into lipid alcohols to mitigate this process under normal conditions; however, once stimuli occur, overexpressed superoxide from OXPHOS complexes reacts with ferrous ions to yield a slew of unstable radicals, leading to excessive ROS and, afterward, lipid peroxidation reboots to induce ferroptosis. Another route to ferroptosis is the buildup of lipid peroxide by circumstances such as cysteine deprivation, which induces the breakdown of certain iron-binding proteins, including FtMt and heme, to free iron and ROS, therefore triggering nearby MLP and hence ferroptosis. c Pyroptosis is initiated by the pathogen component-activated inflammatory caspase response, which not only converts IL‐1β and IL‐18 to mature forms but also cleaves GSDMD to facilitate pore penetration and enable IL-1β and IL-18 release, thus resulting in cell swelling and pyroptosis. Apart from these, inflammatory stimuli also lead to pyroptosome-mediated caspase-3 activation and GSDME cleavage to unleash membrane permeabilization to favor the release of ions and mtDNA to induce pyroptosis. DAMPs Damage-Associated Molecular Patterns, MLKL Mixed-Lineage Kinase Domain-Like pseudokinase, PDH pyruvate dehydrogenase, GPX4 Glutathione Peroxidase 4, TCA Tricarboxylic acid cycle, ETC Electron Transport Chain, GSH Glutathione, MLP Membrane Lipid Peroxidation, FtMt Mitochondrial Ferritin, α-KG Alpha-ketoglutarate, IDH2 Isocitrate Dehydrogenase 2, NADH Nicotinamide Adenine Dinucleotide Dehydrogenase, OXPHOS mitochondrial Oxidative Phosphorylation System, GSDMD Gasdermin D, GSDME Gasdermin E, IL-18 Interleukin-18, IL-1β Interleukin-1β.
Ferroptosis
Ferroptosis is the novel term coined to explain an oxidation-related PCD modality driven by iron-dependent lipid peroxidation that has been proven to promote healthspan when inhibited67. Since mitochondria, the major organelles involved in iron utilization and catalytic and harboring anabolic pathways, play pivotal roles in iron homeostasis as well as substance and energy metabolism, speculation that mitochondria are regulatory hubs of ferroptosis is reasonable. This supposition is supported by the morphological features of ferroptosis, which include increased mitochondrial membrane condensation and density, reduced overall volume and number of mitochondria cristae, and loss of OMM integrity68.
ROS generation is one of the mechanisms governing ferroptosis. Under normal circumstances, cells harbor intrinsic defensive systems for detoxifying lipid peroxides, predominantly via glutathione peroxidase 4 (GPX4), which converts excess peroxides into lipid alcohols to mitigate ferroptosis. However, under conditions of mitochondrial stress or pathology, excess superoxide produced by OXPHOS complexes is converted to hydrogen peroxide, which then reacts with ferrous ions to yield many unstable radicals. Once the antioxidation ability of GPX4 is surpassed, excessive ROS are likely to disrupt cellular redox homeostasis and induce ferroptosis69. Another ferroptosis pathway involves lipid peroxide accumulation after cysteine deprivation, which increases the glutaminolysis rate and, as a result, accelerates mitochondrial respiration by activating the tricarboxylic acid cycle. Notably, this TCA cycle acceleration results in mitochondrial hyperpolarization and ROS overproduction70 (Fig. 5b).
Pyroptosis
Pyroptosis is an inflammatory PCD culminating in the permeabilization of the plasma membrane, causing leakage of cellular contents, including pro‐inflammatory mediators, especially IL‐1β and IL‐18, which are critical to induction of the innate immune response at infection sites, modulating adaptable immunity and amplifying cytolytic effects71. Principally operating as an innate immunological response to pathogens and oxidative stress, pyroptosis is induced after inflammasome formation and caspase-dependent cleavage of gasdermin D (GSDMD). This mechanism is classified into the caspase‐1-dependent and caspase‐independent pathways, with the latter involving caspase‐4 and caspase‐5 (Fig. 5c).
In a caspase-1-dependent manner, inflammasomes are formed when a class of pathogen-associated molecular patterns, such as pyrin domain-containing 3 (NLRP3), is recognized. These pathogen-associated molecular patterns are integrated with apoptosis-associated speck-like protein (ASC), which comprises a caspase activation and recruitment domain (CARD), to recruit pro-caspase-1 and thus drive inflammasome formation72. In quick succession, caspase-1 is activated, forming mature cleaved caspase-1. Then, activated caspase-1 not only converts precursors IL‐1β and IL-18 into mature proteins73 but also cleaves GSDMD to generate N-GSDMD, which initiates MOMP and induces IL-1β and IL-18 release, jointly forcing cell swelling and pyroptosis74. Notably, activation of caspase-1 can inhibit mitophagy and further increase mitochondrial damage75. Additionally, NLRP3 activation requires Ca2+ conduction; however, Ca2+ overload causes mitochondrial damage, which is followed by ROS overproduction and then activation of the NLRP3 inflammasome. In contrast, NLRP3 inhibition can rescue mitochondrial structure by inhibiting substances that induce mitochondrial damage76.
In contrast, caspase-4 and caspase-5 can be independently triggered after interacting with intracellular lipopolysaccharide via its CARD to regulate the maturation and secretion of IL-1β and IL-18 in some cases or to cleave GSDMD directly to induce NLRP3 inflammasome development, eventually leading to pyroptosis77. A recent report revealed that inflammatory stimuli also lead to mitochondrial damage and subsequent activated caspase-3-mediated gasdermin E (GSDME) cleavage, which drives MOMP in parallel with GSDMD cleavage to favor mtDNA release coupled with ROS production, inducing pyroptosis78.
Parthanatos
Parthanatos is a recently characterized PCD that is induced following the hyperactivation of DNA damage-responsive enzymes, especially poly (ADP-ribose) polymerase 1 (PARP1)79. The mechanisms governing this process have been partially identified. In the normal state, the activation of PARP1 forms a line of defense against the effects of mild stimuli such as high ROS and Ca2+ concentrations, enhancing the restoration of cellular homeostasis. However, PARP1 overactivation involves considerable consumption of cellular NAD+, a critical metabolite involved in a majority of cellular metabolism pathways80,81, inducing an energy crisis82. This crisis is exacerbated by the release of a PAR polymer from the nucleus that is toxic to mitochondria and the translocation of AIF to the nucleus, eventually inducing large-scale DNA fragmentation and chromatin condensation.
Parthanatos is similar to necroptosis to certain respects. Although the extent of their similarities is unclear, a cell undergoing parthanatos shows necroptosis-like morphological change, with PARP1 overexpression associated with rare apoptotic body necrosis. This change highlights the possibility that PARP1 is an active effector of RIPK initiators, presumably involved in mediating necroptosis downstream of RIPK1 and RIPK3 when activated; however, further exploration is still needed.
Paraptosis
Paraptosis is another recently described PCD frequently mediated by mitogen-activated protein kinases via an unknown mechanism83. Little is understood about the mechanisms underlying paraptosis. In contrast, several recent studies have indicated that it is characterized by cytoplasm and intracellular organelles vacuolization through the dilatation of mitochondria and the ER. Perturbations of ion homeostasis or cellular proteostasis are critical to paraptosis, possibly due to Ca2+ translocation from the ER to mitochondria, stimulating mitochondrial dilatation84. Although simple mechanisms underlying paraptosis have been outlined, they have yet to be studied at the molecular level.
Diseases associated with mitochondrial cell death
Increasing evidence has highlighted the critical roles of mitochondrion-associated PCD in the pathogenesis of multiple organ systems (Table 1). Multifaced treatments targeting cell death mediators in clinical trials have been beneficial for multiple disorders. Although many PCD diseases are worthy of detailed discussion, herein, we focus on cancer and neurodegenerative, cardiovascular, and metabolic diseases, as they are the most prevalent diseases and the most recently and extensively studied. However, we briefly introduce the underlying associations among different PCD modalities and other typical diseases, including digestive diseases, inflammatory conditions, and infections.
Cancer
Mitochondrion involved in cell death has been implicated in cancer development. Physiologically, apoptosis is a protective defense mechanism that effectively inhibits tumor growth and eliminates neoplastic cells. A major mechanism triggering apoptosis involves elevated ROS levels in cancer cells resulting from heightened metabolic activity. As previously discussed, ROS at excessive levels disrupt mitochondrial functions, resulting in mitochondrial membrane depolarization and mPTP opening, which subsequently activates intrinsic apoptosis pathways. Moreover, membrane depolarization is a crucial event in the activation of TRAIL, a ligand that causes fragmentation of mitochondria in multiple human cancer cell lines, ultimately inducing apoptosis and necroptosis by activating caspases and RIPK1/RIPK3, respectively. Excessive levels of ROS can also promote necroptosis via RIPK1 oxidation and necrosome complex formation, followed by inflammatory and anticancer immune responses85. ROS generation also mediates ferroptosis. Given that they exhibit increased reliance on cellular iron levels, cancer cells are particularly susceptible to ferroptosis86. The ROS-induced ferroptotic cascade leads to oxidative stress within the cytoplasm, culminating in the release of cytokines and inflammatory mediators such as TNF-α and IL-6, which potentiate the immune system to combat cancer. Novel investigations have revealed that CD8+ T cells, the main forces of cancer eradication, can directly induce ferroptosis in cancer cells, highlighting the potential significance of ferroptosis in cancer treatment87. ROS may also trigger ferroptosis and promote NLRP3 inflammasome activation and subsequent pyroptosis. In addition to higher ROS levels, cancer cells may show greater susceptibility to apoptosis than normal cells. Genetic complexity, metabolic regulation, and stress responses drive cancers closer to apoptosis initiation. In response to oncogene activation, in multiple cancer types, such as chronic lymphocytic leukemia, melanoma, and lung cancer, the levels of proapoptotic proteins are increased, stimulating the oligomerization of Bax or Bak and culminating in MOMP and apoptosis88. Moreover, an upstream stimulator of apoptosis, p53, triggers the innate immune system pathway to drive both cell-intrinsic and cell-extrinsic tumor-suppressing activities89.
As a pivotal characteristic of cancer pathophysiology, tumor cell immune evasion is an intrinsic and vital process that facilitates cancer progression and metastasis due to inefficacy of host immune responses90. Among the intricate mechanisms involved in immune responses, mitochondria are core mediators that both inhibit and promote immune evasion91. Mitochondria inhibit immune responses by suppressing metabolism92 by regulating metabolic pathway activation93, affecting immune cell activity. This metabolic limitation, in turn, promotes tumor cell immune evasion, facilitating their unbridled proliferation. In addition, mitochondria also contribute to tumor cell immune evasion by releasing mtDNA and synthesizing antioxidants, which mitigate oxidative stress in cancer cells94. Intriguingly, recent research has revealed that nanotubes, which connect immune cells and cancer cells, enabling mitochondria to be hijacked from immune cells by cancer cells. This mitochondrial transfer promotes cancer cell metabolism while depleting immune cells, exhibiting a distinctive mechanism of immune evasion in cancer cellss95.
Mitochondrial cell death constitutes a robust barrier that complements MQC in defense against cancers. This outcome is attributable not only to the direct clearance of cancer cells but also to PCD-induced immune attack. The induction of tumor-associated antigen release and promotion of immune-related factor expression induces immunogenic cell death in cancers. In response, however, a myriad of mechanisms in cancer cells mitigates these defense systems; in the crucial response, cancer cells escape PCD as well PCD-induced immune responses. This immune evasion is a pivotal characteristic of cancer pathophysiology. Notably, the dysregulation of apoptosis is a hallmark of numerous malignancies, such as follicular lymphoma and osteosarcoma, as indicated by the upregulation of anti-apoptotic Bcl-2 or Bcl-xL protein expression and the loss of pro-apoptotic BH3 protein expression. Similarly, the expression of proapoptotic effectors, including Bax and Bak, is commonly downregulated in colorectal, lung, and breast cancers96. In other cases, insufficient release of cyt c or an imbalanced redox state induced hinders caspase activation97. Moreover, cells in lung and colon cancers release mtDNA into the extracellular milieu, and this mtDNA is then phagocytosed by nearby immune cells. After internalization, mtDNA triggers immune cell apoptosis, facilitating immune evasion94. Furthermore, a cytotoxic T lymphocyte (CTL) mechanism called “additive cytotoxicity” and is induced via sublethal damage, and it involves perforin-dependent membrane pore formation and nuclear envelope rupture, driving cancer cells to undergo apoptosis. Notably, one of the primary mechanisms that thwarts an immune attack involves the upregulation of immune checkpoint molecules such as PD-L1, which significantly inhibits the activity of CTLs and hence indirectly suppresses apoptosis. In addition to apoptosis, cancer cells leverage multifarious mitochondrion-associated nonapoptotic PCD modalities to realize immune evasion. Factors that inhibit the immune response are secreted from cancer cells and facilitate interference with immune recognition and thus promote necroptosis. The absence or epigenetic silencing of RIPK3 expression via hypermethylation has been documented to inhibit necroptosis in various cancers, such as lung, gastric, ovarian, and colorectal cancer98. In addition, decreased intracellular free ferrous ions and lipid peroxidation rates in cancer cells inhibit immunogenic ferroptosis and prevent inflammatory immune responses. Mutations in BAP1, a nuclear deubiquitinating enzyme that is highly active in various human cancers, also facilitates tumor cell escape from ferroptosis99. Similarly, cancer cells modulate the NF-κB signaling cascade via diverse mechanisms, suppressing innate immunity-triggered pyroptosis. Although low GSDME expression is commonly detected in several cancers, including lung and gastric cancers and is acknowledged to be a pivotal therapeutic target for counteracting pyroptosis evasion and advancing cancer management, the mechanisms underlying its reduced levels are unknown100.
Notably, certain type of mitochondrial cell death might, to some degree, contribute to the proliferation and metastasis of cancer cells. For example, lymphoma and glioblastoma cells exhibit remarkable rates of spontaneous apoptosis, which are correlated with poor prognosis101. In contrast, the facilitated release of a minimal level of cyt c has been observed to induce low yet continuous caspase-9 activation, thereby promoting genomic instability in viable cells and facilitating oncogenic transformation102. MOMP-dependent global mRNA decay may lead to preferential transcription of Bcl-2 and Bcl-xL mRNA, particularly under mild or transient stress conditions, to support cancer progression103. Additionally, necroptosis may fuel carcinogenesis by inducing adaptable immunosuppression, as indicated by the upregulation of RIPK1, RIPK3, and MLKL in some aggressive cancers, including lung cancer and glioblastoma.
Collectively, these findings underscore the intricately involved and elusive nature of mitochondrial cell death in cancer eradication and tumor cell immune evasion, necessitating ongoing efforts to characterize its complex role. Moreover the need to tailor cancer treatments targeting mitochondrion-associated PCD based on specific phenotypes in certain circumstances is increasing.
Neurodegenerative diseases
Various neurodegenerative diseases have been linked to different mechanisms underlying mitochondrion-related dysfunction, such as apoptosis, Ca2+ overload, and ROS generation104, since neurons largely rely on mitochondria for energy production and Ca2+ buffering and, therefore, are very vulnerable to mitochondrial abnormalities.
Alzheimer’s disease (AD) is a canonical paradigm that supports the claim that mitochondrial dysfunction is associated with pathology. Amyloid β (Aβ) accumulation, a key initiator of AD, induced mitochondrial Ca2+ overload in conjunction with increasing hyperphosphorylated tau protein levels, triggering sustained mPTP opening and disrupting mitochondrial homeostasis to stimulate the caspase cascade in apoptosis105. In line with these findings, apoptosis has also been associated with acute brain injury. Most neurons in an ischemic region undergo apoptosis characterized by typical apoptotic features, including apoptotic signaling pathway activation and typical morphological alterations. Additionally, MOMP and apoptosis are likely involved in the development of chronic neurodegenerative diseases106. Shifts in the ER stress response or Ca2+ buffering to activate mitochondrial proteases and cause mitochondrial membrane permeability have been widely described in cells and transgenic AD mouse models.
An increasing body of evidence suggests that nonapoptotic PCD may play an important role in the pathophysiology of neurological diseases such as AD and delayed ischemic injury. Notably, RIPK motifs promote the assembly and accumulation of Aβs, which in turn form active signaling complexes that mediate necroptosis107. Multiple studies have established the role of ferroptosis in neurotoxicity and brain injury; in fact, ferroptosis was initially called glutamate-induced neuronal excitotoxic death, which was blocked by a ferroptosis inhibitor96. Given that high levels of extracellular glutamate trigger ferroptosis and that glutamate neurotoxicity contributes to stroke and other neurodegenerative illnesses, ferroptosis likely contributes to the pathophysiology of multiple brain disorders. Genetic studies have supported the idea that the effects of conditional Gpx4 deletion resemble the effects of neurodegeneration, although the mechanism is unclear108.
Cardiovascular diseases
Similar to neurodegeneration, mitochondrion-associated PCD is a key regulator of different CVDs. Although apoptosis is not the sole mode of cell death in CVDs, it is believed to contribute to various disorders, such as heart failure, atherosclerosis, and aneurysm109. Apoptotic cardiomyocyte death tends to be complicated by irrevocable congestive heart failure, consistent with findings showing that inhibiting apoptosis can attenuate contractile dysfunction and ventricular dilation, both of which are hallmarks of heart failure110. Endothelial cells that undergo apoptosis but are not eliminated in a timely manner, contribute to atherosclerosis111. Plaque formation may be facilitated by an increased propensity of apoptotic cells to coagulate and adhere to platelets; as a result, apoptosis may contribute to plaque instability and rupture. Moreover, apoptosis has been thought to accelerate aneurysm development by depleting medial vascular smooth muscle cells, an outcome that caspase inhibitors100 may prevent.
In addition to apoptotic PCD, necroptosis induced by ischemia/reperfusion (I/R) or oxidative stress contributes to heart failure, predominantly through ATP depletion and mPTP opening, as indicated by attenuated injury in CypD-deficient mice and increased levels of RIPK3 in ischemic tissues post-I/R. Notably, pyroptosis has been more comprehensively investigated in CVD than in any other organ system. As a core regulator, NLRP3 is stimulated by oxidized low-density lipoprotein, which induces the inflammation evident in pyroptosis. Similarly, lipids accumulate following arterial endothelial injury, and large quantities of caspase-1 are seen in plaques deposited after the development of atherosclerosis. In diabetic heart disease, high glucose levels can trigger ROS overexpression and accelerate NLRP3 inflammasome formation, indirectly inducing pyroptosis. Recent research has also suggests that some nonapoptotic PCD modalities may be activated during ischemia. NLRP3 inflammasome inhibitors strengthen cardiac tolerance to ischemic insult, providing convincing justification for further targeting ferroptosis in therapeutic trials112. Additionally, inhibiting ferroptosis attenuates acute and chronic ischemia and protects against heart failure induced by iron overload113.
Metabolic diseases
Metabolic diseases are among the most common disorders related to mitochondrial dysfunction. Mitochondrion-associated cell death has been cited as a critical factor in regulating the survival of different cells under pathological conditions.
In persons with obesity and NASH, adipocyte and hepatocyte cell death via apoptosis plays a central role pathogenesis114,115. In the adipose tissues of patients with obesity, the expression of caspase-3, caspase-7, caspase-8, and caspase-9 is increased, and that of Bcl-2is decreased, correlating with insulin-resistance progression116. Excessive accumulation of lipid species results in adipocyte death, triggering an inflammatory macrophage response. The elevation of circulating FFAs also contributes to an accelerated lipid metabolism rate in the liver and subsequent development of NAFLD. During this disease progression, adipose tissue secretes inflammatory cytokines and exacerbates liver inflammation and hepatocyte death117. In this context, the mechanism underlying apoptosis is activated via caspase-dependent extrinsic and intrinsic pathways. Both pathways initiate MOMP and caspase-2 activation, resulting in caspase-3 and caspase-7 activation and apoptosis. In addition to those in the apoptosis pathway, factors in other cell death pathways, such as +pyroptosis and necroptosis pathways, engage in crosstalk with caspases to regulate cell death mechanisms in metabolic diseases118. Pyroptosis is initiated when gasdermin D is cleaved by caspase-1 or caspase-4, caspase-5, and caspase-11. Caspase-8 regulates the activation of the NLRP3 inflammasome, a critical component in pyroptotic cell death. The activation of the NLRP3 inflammasome is also associated with necroptosis induction. Because of this crosstalk, interventions to block one pathway may affect other pathways to mediate disease pathogenesis.
Diabetes mellitus is a common metabolic disorder in which pancreatic β-cell destruction is the main cause of β-cell failure and the onset of diabetes. In response to different stimuli, cells undergo different types of cell death, which modulates the survival of β-cells during disease progression. Apoptosis is the predominant mechanism associated with β-cell loss in Type 1 diabetes and a reduced β-cell population in Type 2 diabetes119,120,121,122. In Type 1 diabetes, β-cell failure is mediated by an immune response characterized by mononuclear and T cells interacting with antigen-presenting cells to generate a high level of inflammatory cytokines, chemokines, and other triggering factors, leading to apoptotic cell death. Three main mechanisms have been reported to be important in diabetes: 1) the expression of Fas on CD8+ T cells and FasL on β-cells, 2) the secretion of proinflammation factors (IL-1β, TNF-α, and IFN-γ) by the immune cells that have infiltrated in islets, and 3) the production of ROS by macrophages123. In Type 2 diabetes, multiple causes, such as glucotoxicity, lipotoxicity, and islet amyloid polypeptide aggregation, contribute to β-cell dysfunction and β-cell death. ER stress pathways (the PERK, IRE1, and ATF6 pathways) are activated in response to these factors. When ER stress is prolonged, increases in the level of the proapoptotic factor CHOP leads to ROS production, mitochondrial dysfunction, and apoptosis124. Apoptotic β-cell death results from an imbalance between proapoptotic Bcl-2 protein (Bad, Bid, Bik, and Bax) and antiapoptotic Bcl family protein (Bcl-2 and Bcl-xL) levels. In addition to apoptosis, other cell death modalities are involved in the pathogenesis of Type 1 and Type 2 diabetes. In Type 1 diabetes, β-cells are susceptible to necrosis through the activation of cytokine signaling. After TNF-α binds to its receptor and forms a necrosome complex, ROS production and DNA fragmentation are initiated, contributing to necroptosis125. Several studies have reported that NLRP3-mediated pyroptosis promotes the pathogenesis and progression of Type 1 diabetes126. Regarding Type 2 diabetes, necroptosis, ferroptosis, and pyroptosis contribute to β-cell death regulation. In particular, ferroptosis may underlie β-cell loss related to Type 2 diabetes, as indicated by an increased risk for metabolic syndrome in individuals with elevated serum ferritin levels. A report showed that necroptosis was the previously unknown mechanism underlying β-cell death in response to islet amyloid formation. NLRP3 inflammasome-mediated pyroptosis also appears to contribute significantly to the development of Type 2 diabetes and insulin resistance122. The activation of the NLRP3 inflammasome can be induced by various factors, such as high glucose, fatty acids, ROS, and IAPP levels127,128.
Mitochondrion-associated cell death is complicated and plays an important pathophysiological role in regulating metabolic diseases. Understanding the relationships between different pathways and the underlying control mechanisms is critical to prevent cell death in the pathogenesis of these diseases.
Other diseases
Almost all PCD modalities have been implicated in the pathogenic progression of digestive diseases to some extent, with some forms critical to the severity and prognosis of diseases. A key mediator of the extrinsic apoptosis pathway mediator for typical apoptotic cascades as well as initiator of signaling cascade reactions of necroptosis, TNF-α is extensively involved in the development of multiple diseases, such as inflammatory bowel disease (IBD), spontaneous colitis129, alcoholic liver disease, and NAFL/NASH130; notably, NAFL/NASH has been proven to be triggered by CyPD-induced mPTP opening, consistent with caspase-3-initiated caspase-6 cleavage and consequent cyt c release. In addition, multiple pathways are clearly involved in necroptosis and ferroptosis, albeit the molecular level details are unclear. Interestingly, the protective effect conferred by caspase-11-dependent pyroptosis on IBD and colitis was proposed to be independent of IL-1β or IL-18 action, indicating unique underlying pathways that need to be further explored.
PCD is intricately linked to inflammation as both a regulator and an endpoint. A pattern recognition receptor member, absent in melanoma 2 (AIM2), has ben reported to activate caspase-3, coupling with NLRP3 to induce caspase signaling in apoptosis. This process is mediated by caspase-1, which drives proinflammatory secretion to induce pyroptosis. The dysregulation of proinflammatory secretion may lead to hereditary autoinflammatory disorders, as evidenced IL-1β prompting chemokine overexpression and leukocyte migration. In addition, IL-18 has been shown to enhance the required for the activation of macrophages and T cells. Additionally, necroptosis is a key initiating event of inflammation, with secreted DAMPs the primary sources by which RIPK3 drives the inflammatory response following MLKL insertion into the plasma membrane. RIPK3 also drives inflammasome assembly to activate caspase-1 and caspase-11. As an RIPK3 regulator, RIPK1 increases circulating the IL-1α level to induce TNF production and the induction of spontaneous inflammatory symptoms via autocrine signaling.
Recent research has highlighted the typical phenotype acquired by cells for which PCD is a defense mechanism against infection. After viral infection of cells, apoptosis can be initiated by granzymes released from activated cytotoxic T lymphocytes and natural killer cells. Necroptotic RIPK3 is activated to limit viral replication. Remarkably, apoptosis and necroptosis seem to be linked to an immune signaling network to confer protection onto cells. Caspase 8 is an innate immune sensor ensconced within the TNF receptor and toll-like receptors in response to viral insults. At the same time, it interacts with RIPK1-RIPK3 to form the triangular guard system to protect each other and eliminate infections. As an auxiliary mechanism, pyroptosis increases viral susceptibility via AIM2 and removes pathogens via caspase-1 activation131.
Clinical trials
As outlined above, mitochondrion-associated PCD is generally associated with the course of multiorgan disorders. In this circumstance, targeted regulatory biomarker or signaling pathway candidates have piqued the interest of scholars, and an increasingly number have been entered into early clinical trials (Table 2). For instance, deferiprone is predicted to attenuate the increase in iron levels in the substantia nigra via ferroptosis inhibition, providing promising therapeutic benefits for several neurodegenerative disorders, such as AD and Parkinson’s disease. Navitoclax, a dual inhibitor of Bcl-2 and Bcl-xL, has been entered into a trial exploring convincing cancer therapies. Similar trials are being planned to evaluate newly developed PCD mediators, with hundreds of registered trials recruiting patients or ongoing. Therefore, it is anticipated that these clinical trials based on extensive PCD research will lead to considerable breakthroughs for disease treatment.
Perspectives
In this review, we have articulated the crucial roles of mitochondria in maintaining homeostasis via quality control networks and regulation of cell fate mediated via mitochondrion-associated PCD. In addition to summarizing the established pathways involved in cell disassembly mediated by apoptotic signaling, we highlighted the involvement of mitochondria in several nonapoptotic PCD modalities, and similar to apoptosis, they associated with considerable disease progression. Notably, it is increasingly evident that cell death modality pathways engage in crosstalk. These interactions involve mitochondria, and although their interplay has been described in broad strokes, the molecular details still need to be identified. In the future, in-depth studies should be focused on identifying novel forms of mitochondrion-associated PCD and characterize the involvement of these death modalities in disease pathogenesis. In addition, PCD-based therapy must be developed and evaluated in future clinical trials.
References
Martinez-Reyes, I. & Chandel, N. S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 11, 102 (2020).
Oh, C. M., Ryu, D., Cho, S. & Jang, Y. Mitochondrial Quality Control in the Heart: New Drug Targets for Cardiovascular Disease. Korean Circ. J. 50, 395–405 (2020).
Weinlich, R., Oberst, A., Beere, H. M. & Green, D. R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 18, 127–136 (2017).
Bock, F. J. & Tait, S. W. G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 21, 85–100 (2020).
Protasoni, M. & Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 22, https://doi.org/10.3390/ijms22020586 (2021).
Chinnery, P. F. & Hudson, G. Mitochondrial genetics. Br. Med Bull. 106, 135–159 (2013).
Rath, S. et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 49, D1541–D1547 (2021).
Hong, H. J. et al. Mitoribosome insufficiency in beta cells is associated with type 2 diabetes-like islet failure. Exp. Mol. Med. 54, 932–945 (2022).
Benador, I. Y., Veliova, M., Liesa, M. & Shirihai, O. S. Mitochondria Bound to Lipid Droplets: Where Mitochondrial Dynamics Regulate Lipid Storage and Utilization. Cell Metab. 29, 827–835 (2019).
Lill, R. & Freibert, S. A. Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis. Annu. Rev. Biochem. 89, 471–499 (2020).
van der Vaart, J. I., Boon, M. R. & Houtkooper, R. H. The Role of AMPK Signaling in Brown Adipose Tissue Activation. Cells-Basel 10, https://doi.org/10.3390/cells10051122 (2021).
Bauer, T. M. & Murphy, E. Role of mitochondrial calcium and the permeability transition pore in regulating cell death. Circ. Res. 126, 280–293 (2020).
Sies, H. & Jones, D. P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 21, 363–383 (2020).
Song, Y. et al. Mitochondrial quality control in intervertebral disc degeneration. Exp. Mol. Med 53, 1124–1133 (2021).
Stoolman, J. S., Porcelli, A. M. & Martinez-Reyes, I. Editorial: Mitochondria as a hub in cellular signaling. Front. Cell Developmental Biol. 10, https://doi.org/10.3389/fcell.2022.981464. eCollection 2022. (2022).
Xin, N. et al. The UPRmt preserves mitochondrial import to extend lifespan. J. Cell Biol. 221, https://doi.org/10.1083/jcb.202201071 (2022).
Klaus, S. & Ost, M. Mitochondrial uncoupling and longevity - A role for mitokines? Exp. Gerontol. 130, https://doi.org/10.1016/j.exger.2019.110796 (2020).
Melber, A. & Haynes, C. M. UPRmt regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res. 28, 281–295 (2018).
Tsygankova, P. G. et al. Plasma FGF-21 and GDF-15 are elevated in different inherited metabolic diseases and are not diagnostic for mitochondrial disorders. J. Inherit. Metab. Dis. 42, 918–933 (2019).
Cobb, L. J. et al. Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging-Us 8, 796–809 (2016).
Dao, T. et al. Sarcopenia and Muscle Aging: A Brief Overview. Endocrinol. Metab. (Seoul.) 35, 716–732 (2020).
Zeng, F. R. et al. BET inhibitors synergize with sunitinib in melanoma through GDF15 suppression. Experimental and Molecular Medicine, https://doi.org/10.1038/s12276-023-00936-y (2023).
Asrih, M. et al. Overview of growth differentiation factor 15 in metabolic syndrome. J Cell Mol Med, https://doi.org/10.1111/jcmm.17725 (2023).
Chung, H. K. et al. Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. J. Cell Biol. 216, 149–165 (2017).
Moon, J. S. et al. Growth differentiation factor 15 protects against the aging-mediated systemic inflammatory response in humans and mice. Aging Cell 19, e13195 (2020).
Croon, M. et al. FGF21 modulates mitochondrial stress response in cardiomyocytes only under mild mitochondrial dysfunction. Sci. Adv. 8, https://doi.org/10.1126/sciadv.abn7105 (2022).
Barcena, C., Mayoral, P. & Quiros, P. M. Mitohormesis, an Antiaging Paradigm. Int Rev. Cel. Mol. Bio. 340, 35–77 (2018).
Tezze, C., Romanello, V. & Sandri, M. FGF21 as Modulator of Metabolism in Health and Disease. Front Physiol. 10, 419 (2019).
Quiros, P. M. et al. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 216, 2027–2045 (2017).
Wu, Y., Sun, L., Zhuang, Z., Hu, X. & Dong, D. Mitochondrial-Derived Peptides in Diabetes and Its Complications. Front Endocrinol. (Lausanne) 12, 808120 (2021).
Dartora, D. R. et al. Lower Mitochondrial-derived Peptide Humanin in Young Adults Born Preterm vs. Term and Association With Left Ventricular Ejection Fraction. Circulation 142 (2020).
Kim, K. H., Son, J. M., Benayoun, B. A. & Lee, C. The Mitochondrial-Encoded Peptide MOTS-c Translocates to the Nucleus to Regulate Nuclear Gene Expression in Response to Metabolic Stress. Cell Metab. 28, 516 (2018). -+.
Merry, T. L. et al. Mitochondrial-derived peptides in energy metabolism. Am. J. Physiol.-Endoc M 319, E659–E666 (2020).
Nashine, S. & Kenney, M. C. Effects of Mitochondrial-Derived Peptides (MDPs) on Mitochondrial and Cellular Health in AMD. Cells-Basel 9, https://doi.org/10.3390/cells9051102 (2020).
D’Amico, D., Sorrentino, V. & Auwerx, J. Cytosolic Proteostasis Networks of the Mitochondrial Stress Response. Trends Biochem Sci. 42, 712–725 (2017).
Matilainen, O., Quiros, P. M. & Auwerx, J. Mitochondria and Epigenetics - Crosstalk in Homeostasis and Stress. Trends Cell Biol. 27, 453–463 (2017).
Naresh, N. U. & Haynes, C. M. Signaling and Regulation of the Mitochondrial Unfolded Protein Response. Cold Spring Harb Perspect Biol 11, https://doi.org/10.1101/cshperspect.a033944 (2019).
Molenaars, M. et al. A Conserved Mito-Cytosolic Translational Balance Links Two Longevity Pathways. Cell Metab. 31, 549–563.e547 (2020).
Houtkooper, R. H. et al. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457 (2013).
Chamseddine, D. et al. The mitochondrial UPR regulator ATF5 promotes intestinal barrier function via control of the satiety response. Cell Rep. 41, 111789 (2022).
Onishi, M., Yamano, K., Sato, M., Matsuda, N. & Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 40, e104705 (2021).
Vargas, J. N. S., Hamasaki, M., Kawabata, T., Youle, R. J. & Yoshimori, T. The mechanisms and roles of selective autophagy in mammals. Nat. Rev. Mol. Cell Biol. https://doi.org/10.1038/s41580-022-00542-2 (2022).
Zhou, J. et al. Simultaneous treatment with sorafenib and glucose restriction inhibits hepatocellular carcinoma in vitro and in vivo by impairing SIAH1-mediated mitophagy. Exp. Mol. Med. 54, 2007–2021 (2022).
Gao, A. B., Jiang, J. Y., Xie, F. & Chen, L. X. Bnip3 in mitophagy: Novel insights and potential therapeutic target for diseases of secondary mitochondrial dysfunction. Clin. Chim. Acta 506, 72–83 (2020).
Li, Y. et al. BNIP3L/NIX-mediated mitophagy: molecular mechanisms and implications for human disease. Cell Death Dis 13, https://doi.org/10.1038/s41419-021-04469-y (2022).
Li, G. Y., Li, J. L., Shao, R. C., Zhao, J. H. & Chen, M. FUNDC1: A Promising Mitophagy Regulator at the Mitochondria-Associated Membrane for Cardiovascular Diseases. Front. Cell Develop. Biol. 9, https://doi.org/10.3389/fcell.2021.788634 (2021).
Chen, M. et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 12, 689–702 (2016).
Galluzzi, L. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 25, 486–541 (2018).
Chipuk, J. E., Mohammed, J. N., Gelles, J. D. & Chen, Y. Y. Mechanistic connections between mitochondrial biology and regulated cell death. Developmental Cell 56, 1221–1233 (2021).
Van Opdenbosch, N. & Lamkanfi, M. Caspases in cell death, inflammation, and disease. Immunity 50, 1352–1364 (2019).
Giacomello, M., Pyakurel, A., Glytsou, C. & Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 21, 204–224 (2020).
Kist, M. & Vucic, D. Cell death pathways: Intricate connections and disease implications. EMBO J. 40, e106700 (2021).
Spinelli, J. B. & Haigis, M. C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 20, 745–754 (2018).
Vakifahmetoglu-Norberg, H., Ouchida, A. T. & Norberg, E. The role of mitochondria in metabolism and cell death. Biochem Biophys. Res Co. 482, 426–431 (2017).
Amanakis, G. & Murphy, E. Cyclophilin D: An Integrator of Mitochondrial Function. Front. Physiol. 11, https://doi.org/10.3389/fphys.2020.00595 (2020).
Czabotar, P. E. et al. Bax crystal structures reveal how BH3 domains activate bax and nucleate its oligomerization to induce apoptosis. Cell 152, 519–531 (2013).
Ke, F. F. S. et al. Embryogenesis and adult life in the absence of intrinsic apoptosis effectors BAX, BAK, and BOK. Cell 173, 1217 (2018) .
Marchi, S. et al. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 69, 62–72 (2018).
Benitez-Guzman, A., Arriaga-Pizano, L., Moran, J. & Gutierrez-Pabello, J. A. Endonuclease G takes part in AIF-mediated caspase-independent apoptosis in Mycobacterium bovis-infected bovine macrophages. Vet. Res 49, 69 (2018).
Kondadi, A. K. et al. Cristae undergo continuous cycles of membrane remodelling in a MICOS-dependent manner. Embo. Rep. 21, https://doi.org/10.15252/embr.201949776 (2020).
Bhattarai, K. R., Riaz, T. A., Kim, H. R. & Chae, H. J. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp. Mol. Med. 53, 151–167 (2021).
Tang, D., Kang, R., Berghe, T. V., Vandenabeele, P. & Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 29, 347–364 (2019).
Seo, J., Nam, Y. W., Kim, S., Oh, D. B. & Song, J. Necroptosis molecular mechanisms: Recent findings regarding novel necroptosis regulators. Exp. Mol. Med. 53, 1007–1017 (2021).
Galluzzi, L., Kepp, O., Chan, F. K. M. & Kroemer, G. Necroptosis: Mechanisms and Relevance to Disease. Annu Rev. Pathol.-Mech. 12, 103–130 (2017).
Ju, E., Park, K. A., Shen, H. M. & Hur, G. M. The resurrection of RIP kinase 1 as an early cell death checkpoint regulator-a potential target for therapy in the necroptosis era. Exp. Mol. Med 54, 1401–1411 (2022).
Morgan, M. J. & Kim, Y. S. Roles of RIPK3 in necroptosis, cell signaling, and disease. Exp. Mol. Med. 54, 1695–1704 (2022).
Kim, J., Jo, Y., Cho, D. & Ryu, D. L-threonine promotes healthspan by expediting ferritin-dependent ferroptosis inhibition in C. elegans. Nat. Commun. 13, https://doi.org/10.1038/s41467-022-34265-x (2022).
Gan, B. Y. Mitochondrial regulation of ferroptosis. J. Cell Biol. 220, https://doi.org/10.1083/jcb.202105043 (2021).
Han, C. et al. Ferroptosis and Its Potential Role in Human Diseases. Front. Pharmacol. 11, https://doi.org/10.3389/fphar.2020.00239 (2020).
Tang, D., Chen, X., Kang, R. & Kroemer, G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 31, 107–125 (2021).
Kesavardhana, S., Malireddi, R. K. S. & Kanneganti, T. D. Caspases in cell death, inflammation, and pyroptosis. Annu Rev. Immunol. 38, 567–595 (2020).
Yu, P. et al. Pyroptosis: mechanisms and diseases. Signal Transduct Tar. 6, https://doi.org/10.1038/s41392-021-00507-5 (2021).
Liu, X. et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153 (2016).
Bertheloot, D., Latz, E. & Franklin, B. S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell Mol. Immunol. 18, 1106–1121 (2021).
Ding, H. G. et al. Hypercapnia promotes microglial pyroptosis via inhibiting mitophagy in hypoxemic adult rats. CNS Neurosci. Ther. 26, 1134–1146 (2020).
Zhang, W. et al. Cytosolic escape of mitochondrial DNA triggers cGAS-STING-NLRP3 axis-dependent nucleus pulposus cell pyroptosis. Exp. Mol. Med. 54, 129–142 (2022).
Marchi, S., Guilbaud, E., Tait, S. W. G., Yamazaki, T. & Galluzzi, L. Mitochondrial control of inflammation. Nat. Rev. Immunol. https://doi.org/10.1038/s41577-022-00760-x (2022).
Rogers, C. et al. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun. 10, https://doi.org/10.1038/s41467-019-09397-2 (2019).
Robinson, N. et al. Programmed necrotic cell death of macrophages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 26, 101239 (2019).
Ryu, D. et al. NAD(+) repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci. Transl. Med. 8, https://doi.org/10.1126/scitranslmed.aaf5504 (2016).
Zapata-Perez, R., Wanders, R. J. A., van Karnebeek, C. D. M. & Houtkooper, R. H. NAD(+) homeostasis in human health and disease. EMBO Mol. Med. 13, e13943 (2021).
Kang, B. E., Choi, J. Y., Stein, S. & Ryu, D. Implications of NAD(+) boosters in translational medicine. Eur. J. Clin. Invest 50, e13334 (2020).
Fontana, F., Raimondi, M., Marzagalli, M., Di Domizio, A. & Limonta, P. The emerging role of paraptosis in tumor cell biology: Perspectives for cancer prevention and therapy with natural compounds. Biochim. Biophys. Acta Rev. Cancer 1873, 188338 (2020).
Lee, D., Kim, I. Y., Saha, S. & Choi, K. S. Paraptosis in the anti-cancer arsenal of natural products. Pharm. Therapeut 162, 120–133 (2016).
Snyder, A. G. et al. Intratumoral activation of the necroptotic pathway components RIPK1 and RIPK3 potentiates antitumor immunity. Sci Immunol 4, https://doi.org/10.1126/sciimmunol.aaw2004 (2019).
Wu, Y. et al. The epigenetic regulators and metabolic changes in ferroptosis-associated cancer progression. Mol. Cancer 19, 39 (2020).
Wang, W. et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274 (2019).
Sharma, A., Boise, L. H. & Shanmugam, M. Cancer Metabolism and the Evasion of Apoptotic Cell Death. Cancers (Basel) 11, https://doi.org/10.3390/cancers11081144 (2019).
Ghosh, M., Saha, S., Li, J., Montrose, D. C. & Martinez, L. A. p53 engages the cGAS/STING cytosolic DNA sensing pathway for tumor suppression. Mol. Cell 83, 266–280.e266 (2023).
Huang, H., Li, S., Tang, Q. & Zhu, G. Metabolic Reprogramming and Immune Evasion in Nasopharyngeal Carcinoma. Front. Immunol. 12, 680955 (2021).
Klein, K. et al. Role of Mitochondria in Cancer Immune Evasion and Potential Therapeutic Approaches. Front Immunol. 11, 573326 (2020).
Sheehan, C. & Muir, A. The requirement for mitochondrial respiration in cancer varies with disease stage. PLoS Biol. 20, e3001800 (2022).
Egan, G. et al. Mitochondrial and metabolic pathways regulate nuclear gene expression to control differentiation, stem cell function, and immune response in leukemia. Cancer Discov. 11, 1052–1066 (2021).
Kopecka, J. et al. Mitochondrial metabolism: Inducer or therapeutic target in tumor immune-resistance? Semin Cell Dev. Biol. 98, 80–89 (2020).
Saha, T. et al. Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat. Nanotechnol. 17, 98–106 (2022).
Low, H. B. et al. DUSP16 promotes cancer chemoresistance through regulation of mitochondria-mediated cell death. Nat. Commun. 12, 2284 (2021).
Abramczyk, H., Brozek-Pluska, B. & Kopec, M. Double face of cytochrome c in cancers by Raman imaging. Sci. Rep. 12, 2120 (2022).
Khan, M. et al. A novel necroptosis-related gene index for predicting prognosis and a cold tumor immune microenvironment in stomach adenocarcinoma. Front Immunol. 13, 968165 (2022).
Carbone, M. et al. Biological mechanisms and clinical significance of BAP1 mutations in human cancer. Cancer Discov. 10, 1103–1120 (2020).
Wei, X. et al. Role of pyroptosis in inflammation and cancer. Cell Mol. Immunol. 19, 971–992 (2022).
Park, W. Y. et al. Apoptosis-induced nuclear expulsion in tumor cells drives S100a4-mediated metastatic outgrowth through the RAGE pathway. Nat. Cancer 4, 419–435 (2023).
Xu, Y. et al. Bile acid-induced “Minority MOMP” promotes esophageal carcinogenesis while maintaining apoptotic resistance via Mcl-1. Oncogene 39, 877–890 (2020).
Koren, E. & Fuchs, Y. Modes of Regulated Cell Death in Cancer. Cancer Discov. 11, 245–265 (2021).
Ghavami, S. et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 112, 24–49 (2014).
Calvo-Rodriguez, M. & Bacskai, B. J. Mitochondria and Calcium in Alzheimer’s Disease: From Cell Signaling to Neuronal Cell Death. Trends Neurosci. 44, 136–151 (2021).
Cui, J. T. et al. Regulated cell death: discovery, features and implications for neurodegenerative diseases. Cell Commun Signal 19, https://doi.org/10.1186/s12964-021-00799-8 (2021).
Richard, R. & Mousa, S. Necroptosis in Alzheimer’s disease: Potential therapeutic target. Biomed. Pharmacother. 152, 113203 (2022).
Jiang, X. J., Stockwell, B. R. & Conrad, M. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 22, 266–282 (2021).
Qi, B. et al. Cardiac-specific overexpression of Ndufs1 ameliorates cardiac dysfunction after myocardial infarction by alleviating mitochondrial dysfunction and apoptosis. Exp. Mol. Med 54, 946–960 (2022).
Teringova, E. & Tousek, P. Apoptosis in ischemic heart disease. J Transl Med 15, https://doi.org/10.1186/s12967-017-1191-y (2017).
Ohsawa, S., Vaughen, J. & Igaki, T. Cell Extrusion: A Stress-Responsive Force for Good or Evil in Epithelial Homeostasis. Developmental Cell 44, 532–532 (2018).
Ren, D. et al. Activated Protein C Strengthens Cardiac Tolerance to Ischemic Insults in Aging (vol 130, pg 252, 2022). Circ. Res. 131, E1 (2022).
Fang, X. X. et al. Ferroptosis as a target for protection against cardiomyopathy. P Natl Acad. Sci. USA 116, 2672–2680 (2019).
Wang, L. et al. YAP and TAZ protect against white adipocyte cell death during obesity. Nat. Commun. 11, 5455 (2020).
Chen, J., Li, X., Ge, C., Min, J. & Wang, F. The multifaceted role of ferroptosis in liver disease. Cell Death Differ. 29, 467–480 (2022).
Guo, H., Callaway, J. B. & Ting, J. P. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med 21, 677–687 (2015).
Wilson, C. H. & Kumar, S. Caspases in metabolic disease and their therapeutic potential. Cell Death Differ. 25, 1010–1024 (2018).
Han, D. et al. Sestrin2 protects against cholestatic liver injury by inhibiting endoplasmic reticulum stress and NLRP3 inflammasome-mediated pyroptosis. Exp. Mol. Med 54, 239–251 (2022).
Lytrivi, M., Castell, A. L., Poitout, V. & Cnop, M. Recent Insights Into Mechanisms of beta-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 432, 1514–1534 (2020).
Yong, J., Johnson, J. D., Arvan, P., Han, J. & Kaufman, R. J. Therapeutic opportunities for pancreatic beta-cell ER stress in diabetes mellitus. Nat. Rev. Endocrinol. 17, 455–467 (2021).
Sha, W., Hu, F., Xi, Y., Chu, Y. & Bu, S. Mechanism of Ferroptosis and Its Role in Type 2 Diabetes Mellitus. J. Diabetes Res. 2021, 9999612 (2021).
Li, X., Xiao, G. Y., Guo, T., Song, Y. J. & Li, Q. M. Potential therapeutic role of pyroptosis mediated by the NLRP3 inflammasome in type 2 diabetes and its complications. Front Endocrinol. (Lausanne) 13, 986565 (2022).
Rojas, J. et al. Pancreatic Beta Cell Death: Novel Potential Mechanisms in Diabetes Therapy. J. Diabetes Res. 2018, 9601801 (2018).
You, S., Zheng, J., Chen, Y. & Huang, H. Research progress on the mechanism of beta-cell apoptosis in type 2 diabetes mellitus. Front. Endocrinol. (Lausanne) 13, 976465 (2022).
Ding, S. et al. Modulatory Mechanisms of the NLRP3 Inflammasomes in Diabetes. Biomolecules 9, https://doi.org/10.3390/biom9120850 (2019).
Durrani, L. et al. Correlation Between High Serum Ferritin Level and Gestational Diabetes: A Systematic Review. Cureus 13, e18990 (2021).
Olona, A., Leishman, S. & Anand, P. K. The NLRP3 inflammasome: regulation by metabolic signals. Trends Immunol. 43, 978–989 (2022).
Swanson, K. V., Deng, M. & Ting, J. P. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19, 477–489 (2019).
Patankar, J. V. & Becker, C. Cell death in the gut epithelium and implications for chronic inflammation. Nat. Rev. Gastroenterol. Hepatol. 17, 543–556 (2020).
Shojaie, L., Iorga, A. & Dara, L. Cell Death in Liver Diseases: A Review. Int. J. Mol. Sci. 21, https://doi.org/10.3390/ijms21249682 (2020).
Jorgensen, I., Rayamajhi, M. & Miao, E. A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 17, 151–164 (2017).
Acknowledgements
We appreciate the Ryu laboratory members, as well as those in all authors’ laboratories, for their insights and suggestions. D.R. and H.H.K. were supported by an SMC-SKKU Future Convergence Research Program Grant. D.R. was supported by grants from the National Research Foundation of Korea (NRF) funded by the Korean government (MSIT, 2023R1A2C3006220 and 2021R1A5A8029876).
Author information
Authors and Affiliations
Contributions
T.T.N. and S.W. contributed equally to this review article. T.T.N., S.W., R.H.H., and D.R. conceived the ideas, designed the structure, and wrote the manuscript together with the coauthors’ inputs. T.H.N., Y.J., Y.Z., W.P., and K.G. contributed to the mitochondria quality control section, and C.M.O., K.S.P., K.T.H., H.H.K., and R.P. contributed to the cell death section. I.K.L., M.S., R.H.H., and D.R. contributed to the paper revision. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nguyen, T.T., Wei, S., Nguyen, T.H. et al. Mitochondria-associated programmed cell death as a therapeutic target for age-related disease. Exp Mol Med 55, 1595–1619 (2023). https://doi.org/10.1038/s12276-023-01046-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-023-01046-5
This article is cited by
-
The pyroptosis mediated biomarker pattern: an emerging diagnostic approach for Parkinson’s disease
Cellular & Molecular Biology Letters (2024)
-
Evaluation of histological and ultrastructural changes provoked by prenatal tramadol on postnatal cortical cerebellar neuronal development in rats: possible implication of Ki67, GFAP and MicroRNA-7/P53 signalling trajectories
Journal of Molecular Histology (2024)
-
Protopanaxadiols Eliminate Behavioral Impairments and Mitochondrial Dysfunction in Parkinson’s Disease Mice Model
Neurochemical Research (2024)
-
From ferroptosis to cuproptosis, and calcicoptosis, to find more novel metals-mediated distinct form of regulated cell death
Apoptosis (2024)
