
Abstract
Innate lymphoid cells (ILCs) are innate lymphocytes that do not express antigen-specific receptors and largely reside and self-renew in mucosal tissues. ILCs can be categorized into three groups (ILC1–3) based on the transcription factors that direct their functions and the cytokines they produce. Their signature transcription factors and cytokines closely mirror those of their Th1, Th2, and Th17 cell counterparts. Accumulating studies show that ILCs are involved in not only the pathogenesis of mucosal tissue diseases, especially respiratory diseases, and colitis, but also the resolution of such diseases. Here, we discuss recent advances regarding our understanding of the biology of ILCs in mucosal tissue health and disease. In addition, we describe the current research on the immune checkpoints by which other cells regulate ILC activities: for example, checkpoint molecules are potential new targets for therapies that aim to control ILCs in mucosal diseases. In addition, we review approved and clinically- trialed drugs and drugs in clinical trials that can target ILCs and therefore have therapeutic potential in ILC-mediated diseases. Finally, since ILCs also play important roles in mucosal tissue homeostasis, we explore the hitherto sparse research on cell therapy with regulatory ILCs. This review highlights various therapeutic approaches that could be used to treat ILC-mediated mucosal diseases and areas of research that could benefit from further investigation.
Similar content being viewed by others
Introduction
Our bodies are constantly being challenged by pathogens and noxious stimuli, and mucosal tissues serve as the primary defense against these threats. This defense is mediated not only by the barrier function of the mucosal tissues but also by a sophisticated local immune system that connects with the systemic immune system. A key component of the local system is innate lymphoid cells (ILCs): Multiple studies have revealed that ILCs play important roles in tissue homeostasis and inflammation1. They are largely tissue-resident lymphocytes whose precursors settle in the tissues during organ development; once mature, they perform multiple effector functions that are often highly tissue-specific1,2. Although we now know that ILCs are present in almost all organs, the fact that ILCs were first discovered in the mucosa and are abundant in these tissues compared to other organs suggests that they play a particularly essential role in mucosal tissues.
ILCs lack specific antigen receptors such as T-cell receptors (TCRs) or B-cell receptors; rather, they recognize signals emitted by the tissue, particularly cytokines. They also have receptors for microbial products, nutrient components, lipid mediators, and neuronal transmitters1. Interestingly, ILC subsets closely resemble T-cell subsets in terms of the transcription factors that drive their activities and the cytokines they produce. Specifically, natural killer (NK) cells resemble cytotoxic T cells; type-1 helper ILCs (ILC1s) are similar to T-helper (Th)1 cells; group-2 ILCs (ILC2s) correspond to Th2 cells; and group-3 ILCs (ILC3s) are similar to Th17/22 cells. ILC1s and NK cells together form group-1 ILCs. They constitutively express the T-box transcription factor T-bet (encoded by Tbx21), which is essential for NK cell maturation and the early development of ILC1s3. However, the formation of NK cells and their cytotoxic functions also require the expression of Eomesodermin (Eomes)4. NK cells and helper ILC1s bear receptors for IL-12 and IL-18 and produce IFN-γ and TNF-α5. ILC2s express two transcription factors, namely, GATA-binding protein-3 (GATA3) and retinoic acid receptor-related orphan receptor (ROR) α (RORα). They bear the IL-33 receptor (IL-33R)6 and produce large amounts of type 2 cytokines (e.g., IL-5 and IL-13) as well as other cytokines such as amphiregulin, GM-CSF, and IL-9 7,8,9. ILC3s are regulated by the RORγt and aryl hydrocarbon receptor (AHR) transcription factors, express receptors for IL-1 and IL-23, and secrete IL-17 and IL-2210,11. ILC3s are subdivided into three populations depending on their expression of CCR6 and natural cytotoxicity receptor (NCR, i.e., NKp46 in mice and NKp44 in humans). CCR6+ cells are designated lymphoid tissue inducer cells (LTis) and produce lymphotoxins (LTs). CCR6- cells are further divided into NCR+ ILC3s and NCR- ILC3s12.
ILCs are highly sensitive to tissue-intrinsic and -extrinsic signals, which result in transcriptional and epigenetic modifications that permit these cells to exert their wide array of effector functions; these functions include lymphoid organogenesis, regulating innate immune responses against insults to the mucosal tissue, and maintaining metabolic and immunological tissue homeostasis13. Here, we provide an overview of recent studies on the biology of ILCs in mucosal tissues, particularly the lung and intestine. Of particular interest are the tissue-specific characteristics and behaviors of ILCs. We also discuss what is currently known about potential ILC immune checkpoints that could be targeted therapeutically. Finally, we detail the drugs currently used or that could be repurposed to treat ILC-mediated diseases of mucosal tissues and the potential of cell-based therapies for these diseases.
Tissue-specific distribution of ILCs
Most ILCs are tissue-resident lymphocytes: parabiosis experiments have shown that in adulthood, the vast majority of helper ILCs in various organs are derived from self-renewing ILC precursors (ILCps) that develop in the fetal liver during ontogeny or infancy and then migrate to emerging tissue14. These cells are defined as lineage- PLZF+ α4β7+ IL-7Rα (CD127)+ PD-1+ cells15,16. The remaining minority of tissue ILCs in adults are derived from ILCps that originate as hematopoietic stem cells in the bone marrow and then migrate to the tissue during adulthood17; this phenomenon is reflected by the fact that at any given time, 30–40% of the IL-18Rα+ ST2− ILCps in the lung are located in the pulmonary vasculature and express migration-associated genes18. Despite their distinct origins, however, the tissue-resident ILCps (identified as lineage- RORα+ Thy1+ CD127+ IL-18Rα+ ST2− cells) derived from the fetal liver closely resemble those derived from bone marrow in terms of phenotype and differentiation potential16.
The homing of both fetal liver- and bone marrow-derived ILCps from the circulation into mucosal and non-mucosal tissues is partially mediated by integrin α4β7, which is expressed by ILCps19,20, and its binding partners on endothelial cells, namely, MAdCAM-1 (mucosal addressin cell adhesion molecule 1) and VCAM-1 (vascular cell adhesion protein 1)21,22. However, many other factors also shape ILCp homing to tissues, including chemoattractant receptors23,24,25. For example, CXCR6 on ILCps partially determines their retention in both the bone marrow in adults and the fetal liver during embryonic life23. Moreover, some homing-related factors promote tissue-tropic ILCp homing; for example, IL-33, which is secreted by lung epithelial cells when they are damaged (e.g., by Nippostrongylus brasiliensis infection of the lungs), induces the egress of ILC2ps from the adult bone marrow and their trafficking to the lung. The mechanism involves IL-33 downregulating CXCR4 in bone marrow ILCps, which normally enforces retention of these cells in the bone marrow18,26. In addition, the differentiation of tissue ILCps into ILC1s, ILC2s, or ILC3s appears to be largely driven by local signals; for example, although IL-18Rα+ST2− lung-resident ILCps normally differentiate into ILC2s, Mycobacterium tuberculosis infection can induce their differentiation into IFN-γ-producing ILC1-like cells27.
As a result of these local factors, ILC1s are largely concentrated in the intraepithelial layer of the intestine28, while ILC2s predominate in the lung, skin, and white adipose tissue, and ILC3s mainly reside in the lamina propria (LP), cryptic patches, and Peyer’s patches of the intestinal tract29,30. This tissue-specific localization of ILCs in peripheral tissues may play important roles in the homeostasis and inflammation of these tissues and the body.
Pathogenic roles of ILCs in mucosal tissues
While non-mucosal tissues also bear ILCs, they are particularly abundant in mucosal tissues and often demonstrate significant changes in mucosal diseases13. Therefore, this review will focus on the most studied lung and intestinal ILCs.
ILCs in the lung
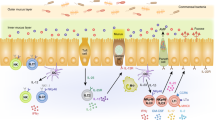
The lung comprises the lower respiratory tract, including the bronchi, bronchioles, and alveoli31. The epithelial lining of the lung forms an essential barrier that not only exchanges gas but also senses and responds to external insults31. In mice, ILC2s are the predominant ILC population in the respiratory tract, whereas ILC3s are the most abundant in humans32. Respiratory diseases such as asthma, chronic obstructive pulmonary disease (COPD), and infection can be associated with dramatic and dynamic changes in the composition and functions of ILC subsets in the lung (Fig. 1). Below, we discuss what is known about the roles of ILCs in lung diseases.

ILCs play a significant role in respiratory diseases, including asthma, COPD, and respiratory infections. A In allergic asthma, alarmins released by allergen-damaged lung epithelium induce activation of ILC2s. ILC2s recruit other immune cells, including eosinophils and alternatively activated M2 macrophages, leading to the induction of AHR and airway Inflammation. In non-allergic asthma caused by obesity and pollutants, IL-1β is induced by lung macrophages and activates lung ILC3s. B In COPD, the roles of ILC1s and ILC3s are better understood than that of ILC2s. IFN-γ-producing ILC1s induce alveolar M1 macrophages to release elastolytic proteases and nitric oxide, resulting in emphysema generation. NK cells have been shown to have enhanced cytotoxic function, which is driven by IL-15 secreted by dendritic cells. C Viral infection and long-term smoking convert lung ILC2s into ILC1-like populations. ILCs can mediate versatile effects in respiratory infection. NK cells and ILC1s contribute to viral clearance. However, ILC2s induce AHR and airway inflammation via type 2 cytokines and epithelium recovery through amphiregulin. ILC3s also play both protective and inflammatory roles in infection mediated by IL-17A and IL-22 secretion.
Asthma
Asthma is caused by repeated exposure to specific allergens or host/environmental factors that induce breathing difficulty, coughing, and wheezing. These symptoms are directly caused by airway hyperreactivity (AHR), mucus overproduction, and airway remodeling33. Asthma is a heterogeneous disease composed of many endotypes. Of these, allergic asthma is the most common. It is triggered by inhaled allergens that damage the lung epithelium, which then initiates a cascade of pathogenic events. Specifically, the damaged epithelium releases alarmins, including IL-33, IL-25, and TSLP, which activate ILC2s34,35: these cells then recruit other immune cells, including eosinophils and alternatively activated M2 macrophages, which induce AHR and airway inflammation36. The key roles of alarmins and ILC2s in allergic asthma have been shown by multiple studies. For example, a genome-wide association study showed that asthma is associated with polymorphisms in the genes that encode IL-33 and IL-33 receptor (ST2)37,38. Moreover, the peripheral blood and induced sputum of patients with asthma contain higher IL-33 levels and ILC2 frequencies than those of control subjects39,40,41.
Another common asthma endotype is non-allergic asthma, which affects up to one-third of asthma patients42. One form is induced by obesity, which may be mediated by high-fat diet-induced activation of lung macrophages that produce IL-1β and thereby activate lung ILC3s43. Another form of non-allergic asthma is generated by repeated exposure to environmental pollutants (particulate matter, diesel exhaust particles, ozone, and carbon nanotubes); it may also be associated with ILC-induced airway inflammation since repeated exposure to pollutants increases the numbers of pulmonary ILC2s and their IL-5 and IL-13 production44,45.
ILCs are suspected to be one of the causes of steroid-resistant asthma. For example, one study reported that the levels of circulating ILC2s from steroid-resistant asthma patients increase after steroid treatment; moreover, circulating ILC2s from healthy humans are related to steroid resistance mediated by high expression of the anti-apoptotic genes BCL2 and BCL2L1. The ratio of the proapoptotic genes BAX1 and BAK1 to the anti-apoptotic gene BCL2 was significantly decreased in ILC2s46. Another study reported that asthma patients bear more CD45RO+ ILC2s (which correspond to murine inflammatory ILC2s) in their sputum than controls and that the frequency positively correlates with steroid resistance47. In addition, sputum ILC2s from asthma patients demonstrate steroid resistance, and their numbers are positively correlated with the sputum levels of TSLP48. The outcome of β2-adrenergic receptor (β2AR) agonist treatment also shows a negative correlation with ILC2 frequency and cytokine production, in both murine models and asthma patients49. These results together suggest that ILCs contribute to the development of asthma.
COPD
COPD is an irreversible chronic lung disease characterized by long-term breathing problems and obstructed airflow50. Although the role of ILCs in COPD is less well-researched than that in asthma, a recent study reported that patients with COPD have higher frequencies of ILC1s in the blood than healthy controls and that patients with COPD have higher IFN-γ-producing ILC1 frequencies in the lung than control subjects51. The mechanism by which ILC1s induce COPD may involve their production of IFN-γ: it has been shown that this can induce alterations in the pulmonary protease and protease inhibitor balance which destroy the lung tissue and thereby generate emphysema52,53. In addition, NK cells, which also belong to the group-1 ILCs, acquire a phenotype in COPD that not only heightens the ability of these cells to kill autologous lung cells in vitro but also is correlated with the reduction in lung function and emphysematous destruction in COPD in vivo54. The enhanced killing functions of these cells appear to be driven by trans-presentation of IL-15Rα secreted by lung dendritic cells (DCs)55. Notably, two risk factors for COPD, namely, viral infection and long-term smoking, have been observed to convert lung ILC2s into ILC1-like populations that express IL-12Rβ2, IL-18Rα, T-bet, and IFN-γ both in vitro and in vivo56,57. Moreover, smoke exposure induces lung epithelial cells to secrete IL-33 but also alters lung immune reactions to this cytokine: resident ILC2s demonstrate decreased ST2 expression and type 2 activity, whereas macrophages and NK cells display augmented ST2 expression and greater production of type-1 proinflammatory cytokines58. There is some limited evidence suggesting that ILC3s also promote COPD pathogenesis: compared to those from control subjects, lung tissues from COPD patients have higher NCR- ILC3 frequencies59 and are enriched for the expression of genes related to the LTi subset of ILC3s60.
Thus, risk factors for COPD upregulate ILCs, these cells bear pathological characteristics, and the numbers of these cells correlate with disease severity. These findings suggest that ILCs mediate COPD.
Infection
Respiratory infections are classified as upper and lower respiratory infections61. Studies suggest that while ILCs generally protect the lung from respiratory viral infections, they can also mediate the pathogenic effects of these infections62. With regard to the protective roles of ILCs, NK cells and ILC1s are well known to contribute to immune responses against viruses by secreting IFN-γ. Interestingly, influenza virus infection or vaccination generates memory-like NK cells. Thus, subsequent challenges are associated with a rapid increase in IFN-γ but the downregulation of NKp46-expressing NK cells63. In contrast, ILC2s play both pathogenic and beneficial roles in respiratory viral infections7,64. On the one hand, influenza A virus (H3N1) can rapidly induce AHR by activating ILC2s: these cells produce IL-5 and IL-13, which in turn induce the accumulation of eosinophils in the lung65. On the other hand, ILC2s promote recovery from virus-induced AHR: a recent study reported that the recovery of patients with severe COVID-19 correlates strongly and positively with the frequency of CCR10+ ILC2s in their blood66. Moreover, hospitalized COVID-19 patients have fewer amphiregulin-producing ILC2s in their blood than COVID-19 outpatients and uninfected control subjects67. In addition, when ILC2s are depleted in mice infected with the influenza A virus, the mice demonstrate further reductions in lung function, airway epithelial integrity, and respiratory tissue remodeling; notably, these effects are reversed by intraperitoneal administration of amphiregulin7. Thus, ILC2s may promote lung homeostasis by producing amphiregulin. Finally, lung ILC3s play a protective role mediated by their IL-17 and IL-22 secretion in Streptococcus pneumoniae-infected mice68.
Thus, ILCs generally act to protect the lungs from infection, although they can also promote the damaging effects of respiratory infections in some cases.
ILCs in the intestine
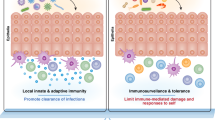
The gastrointestinal (GI) system consists of the stomach, small intestine (i.e., the duodenum, jejunum, and ilium), and large intestine or colon. Moving away from the lumen, the intestine layers consist of the mucosa, submucosa, muscularis propria, and serosa69. The mucosa, in turn, consists of the intestinal epithelium, the LP, and the muscularis mucosa. The mucosa of the small intestine also contains multiple Peyer’s patches and cryptopatches, a unique secondary lymphoid organ. The intestinal epithelium, LP, Peyer’s patches, and cryptopatches are the main sites that contain ILCs; these are also the sites where immune cells act in intestinal immune responses, which are highly sophisticated and enable the GI system to continue absorbing nutrients in an environment constantly exposed to pathogens and commensal microorganisms. Notably, the intestinal epithelium and LP of the murine small and large intestines demonstrate some heterogeneity in terms of ILC composition: specifically, the LP in the lower intestine is occupied by ILC2s, whereas ILC3s predominate in all other intestinal compartments70. As will be detailed below, intestinal ILC3s play crucial homeostatic roles in the intestine but can also contribute to inflammatory bowel diseases (IBDs); intestinal ILC2s are important for expelling worms from the gut; and the intestine also contains regulatory ILCs that may play essential roles in intestinal homeostasis (Fig. 2).

ILCs also play significant roles in maintaining intestinal homeostasis and in disease conditions. A ILC3s contribute to intestinal homeostasis by secreting IL-22, preventing epithelial barrier disruption. Additionally, IL-22 induces mucin production in goblet cells and antimicrobial peptide production in epithelial cells. ILC2s contribute to intestinal homeostasis by stimulating epithelial regeneration through IL-13 production. B During intestinal parasite infection, intestinal ILC2s play important roles in anti-parasitic mechanisms. IL-25 and IL-33 released by parasite-damaged epithelium cause the expansion and activation of local ILC2s, ultimately resulting in parasite expulsion. IL-22 secreted by ILC3s in a parasitic environment promotes intestinal stem cell-mediated epithelial regeneration. C ILCs also play a pathogenic role in intestinal inflammation mediated by the production of IL-17A in ILC3s and IFN-γ in ILC1/3s. Regulatory ILCs (ILCregs) control inflammatory ILC1s and ILC3s through IL-10 and TGF-β1 production in the intestines. Although IL-22 produced by ILC3s acts as a protective factor in maintaining intestinal homeostasis, it can also promote acute colitis by endoplasmic reticulum (ER) stress in epithelial cells.
Role of ILC3s in intestinal homeostasis
ILC3s play essential roles in intestinal homeostasis71,72. In particular, they help prevent the disruption of the epithelial barrier, which can lead to infection and intestinal disorders such as IBDs. This role of ILC3s is mediated by their secretion of IL-22, a key cytokine in intestinal homeostasis, while other cell types can produce IL-22, RORγt+ ILC3s are the main producers of IL-22 in the intestine73,74. IL-22 induces goblet cells to produce mucin proteins that protect the epithelial barrier75. Moreover, IL-22-producing ILC3s (especially NCR+ ILC3s) induce STAT3 phosphorylation in intestinal stem cells, which permits the repair of damaged tissue73,76,77,78. In addition, when the intestine is inflamed, IL-22-producing ILC3s engage in patrolling behavior that directly prevents epithelial cell death79. IL-22 also induces intestinal epithelial cells to produce antimicrobial peptides, including RegIIIβ and RegIIIγ, thus preventing pathogenic bacteria from inducing intestinal lesions74.
The production of IL-22 by ILC3s in the intestine appears to be driven by direct and indirect interactions between ILC3s and commensal microbiota76. The direct interactions may be mediated by intrinsic receptors on intestinal ILCs, such as free fatty acid receptor 2 (Ffar2). Ffar2 is a G-protein-coupled receptor that recognizes bacterial metabolites, specifically short-chain fatty acids80. The indirect interactions may involve signals from other cells, such as myeloid cells. When the Toll-like receptors on these cells are engaged by bacterial proteins, they produce IL-23, which activates ILC3s to produce IL-2281,82
Role of ILC3s and ILC1s in intestinal diseases
ILC3s also play a pathogenic role in the intestines and promote IBDs. This role is largely mediated by their production of IL-17 and IFN-γ83,84,85, while IFN-γ production is a hallmark cytokine of ILC1s, ILC3s bear some plasticity that allows them to express IFN-γ when stimulated by combinations of cytokines such as IL-2 + IL-786. In addition, IL-22-expressing ILC3s can be converted into IFN-γ-expressing ILC1s in certain circumstances87. IL-17 induces massive neutrophil transmigration, which disrupts epithelial junctions and increases epithelial permeability, while excessive production of IFN-γ causes epithelial cell damage that exacerbates the inflammatory reaction87. ILC3s may also contribute to IBDs by producing large amounts of GM-CSF, which recruits and maintains inflammatory monocytes; this role is mediated by NCR+ ILC3s88. The inflammatory monocytes induced by NCR+ ILC3s promote tissue damage and dysbiosis and generate a positive feedback loop that amplifies inflammation88. NCR- ILC3s can also promote IBDs: the intestinal inflammatory environment can cause NCR+ ILC3s to convert into NCR- ILC3s that also secrete GM-CSF. This cytokine then recruits neutrophils, which in turn damage the tissue by secreting large amounts of proinflammatory cytokines and matrix metalloproteinases89.
Notably, although IL-22-producing ILC3s play a key protective role in GI homeostasis, they can also promote acute colitis. This may be mediated by IL-22-dependent endoplasmic reticulum stress in colon epithelial cells90. The pleiotropic behavior of intestinal ILC3s is partly explained by their different modes of action following disease progression. Thus, in the context of acute colitis, ILC3s are activated by TNF-like ligand 1 A (TL1A) from CX3CR1+ mononuclear phagocytes to produce IL-22. However, TL1A induces expression of OX40L, a costimulatory molecule, on ILC3s to activate IFN-γ+ CD4+ T cells, which exacerbates colitis during chronic T-cell-mediated colitis91.
Role of ILC2s in the Intestine
Intestinal ILC2s play key roles in eliminating intestinal parasites, including N. brasiliensis92. This is mediated by their production of type 2 cytokines, particularly IL-13, which stimulates mast cells to produce proteases and induces parasite expulsion93,94. Although IL-13 is secreted by various cell types, including Th2 cells, ILC2s are the main producers of IL-13 in parasite infections, especially during the early phase. IL-13 production by ILC2s is induced by the release of IL-25 and IL-33 by parasite-damaged epithelial cells: IL-25 activates and expands local IL-25R (IL-17RB)-expressing ILC2s, termed inflammatory ILC2s (iILC2s), which are abundant in the intestine, while IL-33 activates and expands the small population of IL-33R (ST2)-expressing ILC2s in the intestine, termed natural ILC2s (nILC2s). Both iILC2s and nILC2s participate in the expulsion of N. brasiliensis93,94. The predominance of iILC2s and the small nILC2 population in the intestine under homeostatic conditions is different from the case in other organs, which are dominated by nILC2s95. Notably, IL-25-responsive intestinal iILC2s can migrate to the lungs after N. brasiliensis infection: this migration event depends on sphingosine-1-phosphate (S1P) signaling and contributes to the systemic anti-helminth response96.
Role of regulatory ILCs in the intestine
Along with group-1–3 ILCs, the ILC family also contains a fourth ILC type, namely, regulatory ILCs (ILCregs). They correspond to regulatory T cells (Tregs) in the adaptive immune system. These ILCregs have been found in the intestine97. They produce IL-10 and TGF-β1, which are responsible for controlling various intestinal inflammatory conditions, possibly by regulating inflammatory ILC1s and ILC3s97. Studies on this novel ILC subset are lacking, but regulatory ILCs deserve continued attention since they may serve as critical mediators of the tissue immune response.
Recent advances regarding the regulation of ILCs by immune checkpoint molecules
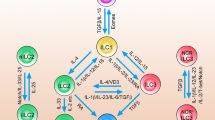
The job of immune cells is to mount appropriate immune responses to immunological threats, but persistent overreactions can lead to tissue damage and autoimmunity. Above, we showed that ILCs can shift from their homeostatic protective roles to become pathogenic. However, due to the relatively recent discovery of ILCs, the mechanisms that could block or mediate these shifts are poorly understood. In contrast, much more is understood about the mechanisms related to the adaptive counterparts of ILCs, namely, T cells. Considerable research on these mechanisms has led to the identification of key immune checkpoint molecules that can be targeted therapeutically to successfully contain T-cell-mediated diseases. These observations have led to the following question: what are the mechanisms that control ILC reactivity? Given that ILCs are innate counterparts of T cells, can the immune checkpoints in T-cell immune responses also play similar roles in ILCs? Since immune checkpoints may play important roles in ILC-mediated tissue homeostasis, inflammatory responses, and antitumor responses, these questions have aroused considerable interest in the last few years. Here, we will review the current understanding regarding the contribution of immune checkpoint molecules to ILC function in mucosal tissues (Fig. 3).

Many different immune checkpoint molecules control both ILCs and interactive cell responses. ICOS-mediated signals regulate ILC function by controlling the expression of Bcl2 and the phosphorylation of STAT5. Both ICOS and ICOSL are expressed in ILC2s, resulting in the production of type 2 cytokines and the expansion of regulatory T cells. GITR enhances ILC2 function by increasing the production of IL-5, IL-13, and IL-9 via the STAT5 pathway. ILCs also express OX40L, which triggers the effector function of OX40+ regulatory T cells. All ILC subsets bear DR3 on their surface, which promotes cytokine production and proliferation. PD-1 acts as a restrictive molecule in ILCs, downregulating inflammatory effects and KLRG1 on ILC2s. The role of KLRG1 in all ILC subtypes remains unclear, but studies have shown that KLRG1 appears to mediate the protective function of ILC2s.
ICOS-ICOSL
Inducible T-cell costimulator (ICOS) is a member of the CD28 receptor family98 and is highly expressed by activated T cells and regulatory T cells99,100. The ICOS ligand (ICOSL) is mainly expressed by DCs, and the binding of ICOSL to ICOS on T cells enhances their differentiation and effector functions101,102. Studies show that ILC2s express high levels of ICOS in the steady state, and ICOS-mediated signals regulate ILC2 functions103,104,105. Specifically, ICOS-deficient mice have reduced ILC2 frequencies and IL-13 production in the lungs and small intestine due to decreased expression of the anti-apoptotic molecule Bcl2103,104. As a result, ICOS-deficient mice do not demonstrate ILC2-dependent lung inflammation in response to intranasal IL-33104 or papain105 administration. Notably, Kamachi et al. showed that while lung ILC2s may interact with ICOSL-expressing DCs in lungs with allergic inflammation105, Mazzi et al. reported that ILC2s express both ICOS and ICOSL and that ILC2-intrinsic ICOS:ICOSL interactions promote ILC2 cytokine production and survival via STAT5 signaling104. Most studies on ICOS in ILCs have focused on ILC2s, but ICOS may affect the functions of ILC3s. Iwanaga et al. showed with single-cell RNA-sequencing analysis that the lungs of Klebsiella pneumoniae-infected Rag2–/– mice contain distinct clusters of Il17a+Il22+Icos+ ILC3s that are critical for host defense106.
Thus, ICOS signaling serves as a key regulator of lung ILC2 survival and asthma-related cytokine production. It may also mediate protective lung ILC3 functions. However, the mechanisms involved and whether these signals participate in diseases other than allergic asthma and infection remain unknown.
GITR-GITRL
The glucocorticoid-induced TNFR-related (GITR) protein is encoded by Tnfrsf18 and is a member of the TNFR superfamily107. It is stimulated by the GITR ligand (GITRL), mainly expressed by antigen-presenting cells and endothelial cells108. Recent studies have reported that ILCs express GITR109,110. Murine ILC1s express moderate levels of GITR, and influenza virus infection upregulates GITR expression on ILC1s, thereby impairing the host antiviral response by reducing their IFN-γ secretion111.
Unlike ILC1s, ILC2s express high levels of GITR109,112. Nonetheless, GITR expression levels on ILC2s are associated with asthma: RNA-sequencing analysis of lung ILC2s from Alternaria-treated mice showed that the expression of GITR in ILC2 populations correlated strongly and positively with their expression of ILC2 signature molecules (Il1rl1 (encoding ST2)) and type 2 cytokines (Il5 and Il13)112. Moreover, when Gitr−/− mice were administered papain or IL-33, they demonstrated fewer lung ILC2s, less eosinophilia, and thus lower inflammation110. Similar results were obtained when GITRL was blocked by GITR-hFc injection before mice were administered papain intranasally. GITR appears to promote ILC2 functions by acting synergistically with ST2: ligation of both receptors causes ILC2s to produce IL-9 via STAT5 phosphorylation. The resulting IL-9 then acts on ILC2s in an autocrine manner to further augment their IL-5 and IL-13 production110. These results show that GITR could serve as an immune checkpoint for activated ILC2s. Thus, targeting GITR may be an attractive new therapeutic strategy for ILC2-mediated allergic and inflammatory diseases.
OX40-OX40L
OX40 is a member of the TNFR superfamily and is expressed by activated CD4+ and CD8+ T cells113, while the OX40 ligand (OX40L, CD252, encoded by Tnfsf4) is expressed by antigen-presenting cells such as DCs and B cells. The engagement of OX40 sustains T-cell proliferation and survival113. Interestingly, ILCs express OX40L rather than OX40. Moreover, multiple studies have shown that OX40L-expressing ILCs can regulate the responses of T cells as antigen-presenting cells do114,115,116.
These OX40L-related T-cell regulatory roles include the ability of lung ILC2s to promote local T-cell responses to infections and allergens114,115,116, as shown by the fact that lung ILC2s express high levels of OX40L after intranasal administration of papain or IL-33 and promote the expansion of OX40-expressing Th2 cells. Moreover, specifically deleting OX40L in ILC2s (Il7raCre+/+Tnfsf4fl/fl mice) blocked the effective Th2 and Treg responses induced by N. brasiliensis infection or allergen exposure114. In addition, when mice are infected with respiratory syncytial virus, their lung CD4+ T cells and ILC2s express high levels of OX40 and OX40L, respectively. OX40/OX40L interactions were further shown to promote CD4+ T-cell cytokine production in an ILC-dependent manner115. Similarly, when OX40L+ lung ILC2s and CD4+ T cells from naïve mice were adoptively transferred into naive Il7ra−/− mice (which lack ILC2s and CD4+ T cells) that were then exposed intranasally to OVA and an adjuvant protease (bromelain), robust OVA-specific type 2 cytokine production and airway inflammation ensued. However, the transfer of either cell population alone did not have these effects116. These findings suggest that OX40L expression in ILC2s is critical for inducing OX40-expressing T-helper cells to produce type 2 cytokines.
The OX40L-related T-cell-regulating roles of ILCs are also observed in the intestine. All ILC3 subsets in the intestine express high levels of OX40L, particularly NCR− ILC3s. OX40L expression by ILC3s appears to contribute to intestinal homeostasis91,117. Specifically, ILC3s and Tregs colocalize in the cryptopatches of the intestine, and Tnfsf4−/−Rag1−/− mice (which lack T and B cells and OX40L expression) demonstrate expansion of intestinal Tregs when they are reconstituted with wild-type Tregs and ILC3s. However, when OX40L-deficient ILC3s are transferred along with wild-type Tregs, the Tregs do not demonstrate expansion in the intestine117. Since Tregs play key roles in suppressing inflammation, the crosstalk between Tregs and ILC3s in mucosal tissues via OX40-OX40L signaling is likely to be critical for intestinal homeostasis117. The importance of OX40L in intestinal homeostasis was also demonstrated by the study of Castellanos et al., which was mentioned above91. This study reported that TL1A expressed in CX3CR1+ mononuclear phagocytes could contribute to intestinal homeostasis by acting on OX40L of ILC3s and upregulating IL-22 production. Notably, this study also showed that the phagocyte-TL1A-OX40L axis mediates the ability of MHCII+ ILC3s to activate the antigen-specific T cells that drive chronic T-cell colitis91. Thus, OX40L expression in ILC3s can upregulate both Tregs and pathogenic T cells, thus driving not only intestinal homeostasis but also intestinal inflammation.
The role of OX40L in ILC1s is currently unknown. Nonetheless, the data for ILC2s and ILC3s suggest that OX40L is an important ILC-mediated immune checkpoint molecule, albeit one that can promote both homeostasis and disease depending on the circumstances.
DR3-TL1A
Death receptor 3 (DR3) is encoded by Tnfrsf25 and is a member of the TNFR superfamily. It is expressed by various immune cells, including T cells and macrophages118,119. The TNF family cytokine TL1A (TNF-like cytokine 1 A) appears to be the only DR3 ligand in inflamed tissues, including the lung and gut120. When T cells bind to TL1A via DR3, their cytokine production is stimulated121.
Recent studies have shown that all ILC subsets also bear DR3 on their surface and that this promotes cytokine production and proliferation122,123,124. In particular, TL1A binding to DR3 on human and murine ILC2s promotes expansion, survival, and functions124. Moreover, genetic overexpression of TL1A or exogenous TL1A administration activates lung ILC2s in vivo. In addition, DR3−/− mice cannot elicit the lung ILC2 responses induced by intranasal papain challenge124. A study on patients with allergic asthma also supported the notion that TL1A interactions with DR3 on ILC2s may contribute to this disease: allergen challenges not only increased the TL1A levels in the airways but also elevated DR3 expression in lung ILC2s125. Thus, the TL1A-DR3 axis could contribute to allergen-induced airway inflammation by inducing ILC2s to secrete type 2 cytokines.
Intestinal ILC3s also express DR3 at high levels, and this expression may mediate their pathogenic activities in IBDs126. Colitis is exacerbated by injection of an agonistic anti-DR3 antibody; the colitis-worsening effects of the antibody are not observed in mice that lack ILC3s, which suggests that DR3-expressing ILC3s play a key role in initiating colitis. This effect may be mediated by ILC3 production of GM-CSF, which is upregulated by the agonistic antibody. Notably, this study showed that neutralization of TL1A by soluble DR3 ameliorates colitis, which suggests that DR3-expressing ILC3s may directly recognize TL1A126.
Thus, the limited studies to date suggest that DR3-mediated activation of ILC2s and ILC3s can lead to pulmonary and intestinal inflammation, respectively. DR3 is therefore a candidate ILC checkpoint molecule.
PD-1-PD-L1
Programmed cell death-1 (PD-1) is a member of the immunoglobulin gene superfamily and the most representative inhibitory T-cell checkpoint molecule. It is a receptor that was originally identified in dying activated T cells127, and many tumors have been found to overexpress its ligand (PD-L1), thereby evading cytotoxic T cells128. Moreover, HIV-specific CD8+ and CD4+ T cells overexpress PD-1, which blocks their antiviral functions129. These findings have led to intensive research on the mechanisms by which the binding of PD-1 to PD-L1 regulates T cells in cancer and chronic infection129,130.
PD-1 was recently shown to participate in ILC development and functions as well. Specifically, it plays an important role during the development of all three ILC subsets since the adoptive transfer of PD-1+ ILCps into Rag2−/−Il2rg−/− mice (which lack T and B cells, and ILCs) induces reconstitution of ILC1, ILC2, ILC3 and a small number of conventional NK cells but not T and B cells in the mice131. Moreover, although PD-1 deficiency does not significantly affect ILCp frequencies in the bone marrow or the development of mature ILCs in homeostatic conditions132,133, PD-1 is upregulated on lung ILC2s in lung inflammation, and ILC3s express functional PD-1 in the murine intestine130 human decidua134, and pleural effusions of patients with tumors135 . The role of PD-1-expressing ILCs in antitumor immunity is currently being investigated extensively. Since this topic has been addressed thoroughly by other reviews136,137, we will focus here on the roles that PD-1-expressing ILCs play in mucosal diseases other than cancer.
Lung ILC2s express PD-1 at high levels, and this expression is further increased by influenza infection and asthma-inducing intranasal stimulation with papain131, IL-33, house dust mite extract, or Alternaria extract138. In one study, a PD-1 agonist reduced lung ILC2s and ameliorated ILC2-mediated asthma; in this experiment, Rag2−/−Il2rg−/− mice were reconstituted with ILC2s from human peripheral blood, treated with the agonist, and then challenged intranasally with IL-33 or house dust mite or Alternaria extract138. Moreover, PD-1-deficient Pdcd1−/− mice display exacerbated IL-33-induced AHR and lung inflammation138. Thus, similar to its role in T cells139, PD-1 expression appears to regulate inflammatory lung ILC2s132,138.
The mechanism by which PD-1 restricts ILC2 functions may involve its ability to downregulate killer cell lectin-like receptor G1 (KLRG1) on ILC2s, which (as detailed below) promotes ILC2 effector responses132. This role of PD-1 is reflected by the fact that PD-1-deficient mice expel N. brasiliensis better than wild-type mice, and PD-1 deficiency is associated with high KLRG1 expression by lung ILC2s. Moreover, the adoptive transfer of KLRG1+ lung ILC2s from PD-1-deficient mice into mice lacking T cells and ILCs greatly increases worm expulsion132. PD-1 may also restrict ILC2s by shaping their metabolism: PD-1-deficient ILC2s display a metabolic shift toward glycolysis, glutaminolysis, and methionine catabolism, which is associated with increased effector function and survival138. Moreover, PD-1 may act by limiting STAT5 phosphorylation in ILC2s: blocking PD-1 or its downstream signaling molecule SHP1/2 significantly increases STAT5 phosphorylation in ILC2s132. Thus, PD-1 appears to be an inhibitory ILC2 checkpoint, and PD-1 agonists may be new ILC-specific therapies for asthma and allergy.
As mentioned above, ILC3s in the mouse intestine131 and human decidua134 express functional PD-1. The role of PD-1 in intestinal ILC3s has not been studied. However, Vacca et al. showed that in early pregnancy, ILC3s in the human decidua express PD-1, while invading trophoblasts express PD-L1. The PD-1/PD-L1 interactions regulate the production of cytokines by ILC3s, including IL-22, IL-8, and TNF-α. This suggests that PD-1 regulates the activities of ILC3s at the feto-maternal interface134.
The role of PD-1 expression in ILC1s is unknown. Further studies on the potential of PD-1 to act as an ILC checkpoint protein in ILCs other than ILC2s are warranted.
KLRG1
KLRG1 is a C-type lectin expressed by many immune cell types, including NK cells and ILC2s140,141. However, blood-, tonsil-, and lung-resident ILC2s express significantly higher constitutive levels of KLRG1 than other ILC subsets142. Notably, although KLRG1 deficiency does not affect overall lung ILC2 frequencies or ST2 expression under homeostatic conditions or papain-induced asthma143, KLRG1 appears to be a marker for lung iILC2s. These inflammatory ILC2s are generated by IL-25, as opposed to the IL-33-reactive natural ILC2s (nILC2s). These cell types have already been mentioned above: they are present in the intestine and contribute to N. brasiliensis expulsion. Similarly, Huang et al. showed that local lung iILC2s helped expel N. brasiliensis from the lung; significantly, the iILC2s expressed high levels of KLRG1. KLRG1+ iILC2s also provide partial protection from Candida albicans infection141. Thus, KLRG1 is associated with iILC2-mediated protection from infection. As mentioned above, such KLRG1 expression by iILC2s appears to be downregulated by PD-1 since PD-1 deficiency enhances the ability of iILC2s to expel N. brasiliensis from the lungs132.
It should be noted that Huang et al. performed in vitro experiments that showed that KLRG1+ iILC2s differentiate into nILC2s and ILC3-like cells141. Thus, KLRG1+ iILC2s may be transient ILC progenitors recruited by infection and inflammation into the lung and intestine, where they then develop into nILC2-like cells and ILC3-like cells141. This is supported by a study that showed that KLRG1+ ILC precursors (defined as lineage-CD117+CRTH2– cells) from human blood differentiate into ILC1s, ILC2s, and ILC3s in vitro depending on the input signals144.
Another study suggested that KLRG1+ ILC2s may help block allergies via ILCreg-like functions145. Thus, Golebski et al. showed that KLRG1+ ILC2s from human blood produced IL-10 when they were stimulated in vitro with IL-33 and retinoic acid, whereas KLRG1- ILC2s did not exhibit this activity145. Moreover, when cultured with nasal epithelium, KLRG1+ ILC2s (but not KLRG1- ILC2s) protected the epithelium from the destructive effects of grass-pollen allergen in an IL-10-dependent manner. Similarly, when KLRG1+ ILC2s were co-cultured with CD4+ T cells, they reduced T-cell activation and proliferation in an IL-10-dependent fashion. Moreover, patients with grass-pollen allergy had lower IL-10+KLRG1+ ILC2 frequencies in the blood than healthy subjects, but these frequencies were restored by immunotherapy145. Thus, KLRG1+ ILC2s may help to protect against allergies.
The roles of KLRG1 in ILC1s and ILC3s are unclear. Nonetheless, KLRG1 appears to mediate protective functions by ILC2s against infections and allergies. Further studies on the effect of KLRG1 on ILC cytokine production, proliferation, and roles in mucosal diseases are warranted.
ILC-targeting therapeutic applications: current methodologies and perspectives
Given that ILCs play important roles in mucosal tissue homeostasis and inflammation, many studies have asked whether therapies that aim to downregulate pathogenic ILCs or restore ILC-mediated homeostasis can improve various inflammatory diseases146. Therapeutic approaches can be broadly divided according to whether they block inflammatory ILCs or elicit protective ILC activities. Inflammation-blocking approaches, which have received the most research attention, generally employ monoclonal antibodies to block specific cytokines. Below, we will discuss approaches to block ILC-mediated inflammation: the focus will be primarily on therapies that are either in use currently or have been/are being studied in clinical trials and either do or potentially could target ILCs (Fig. 4).

ILCs have the potential to be targeted by therapies to block their inflammatory effects or induce their protective activities. Many biological drugs target alarmins related to the ILC-related inflammatory response. Itepekimab (astegolimab) and tozorakimab (ecleralimab) target the IL-33/ST2 pathway, reducing type 2 inflammation by inhibiting the actions of alarmins. Tezepelumab and ecleralimab, monoclonal antibodies that block the TSLP pathway, are being tested in clinical trials. However, IL-25-targeted drugs have not yet been approved for clinical trials. Mepolizumab and reslizumab are monoclonal antibodies against IL-5, a cytokine produced by ILC2s, which activates IgE production and type 2 inflammation. Dupilumab is a monoclonal antibody that blocks IL-13, a cytokine that promotes eosinophil recruitment. Guselkumab and risankizumab inhibit IL-12, while ustekinumab and briakinumab inhibit IL-23. All of these drugs stop ILC1s and ILC3s from being activated. Some drugs aim to downregulate lipid mediators excreted by ILCs; an example is montelukast, which downregulates CysLT1R. β2AR agonists can reduce lung ILC2 frequencies and cytokine production and ameliorate asthma in a CD4+ T-cell-independent manner. To inhibit ILCs directly, some drugs that target the migration and signaling pathways of ILCs have been used. Intestinal ILC3s express high levels of integrin α4β7, a homing receptor necessary for ILC3 accumulation in the intestine. Thus, the anti-α4β7 monoclonal antibody vedolizumab or the anti-β7 monoclonal antibody etrolizumab can be used as inflammation-reducing drugs. JAK/STAT signaling has been shown to play a role in the development and effector functions of every subset of ILCs. Thus, JAK inhibitors such as tofacitinib, baricitinib, and upadacitinib can alleviate ILC-derived inflammation.
Drugs that target alarmins
The most common strategy for modulating ILC-dependent pathogenesis is to target (i) the cytokines that act upstream or downstream of ILCs or (ii) the receptors on ILCs that recognize the upstream cytokines. Candidate ILC2 upstream cytokines include the alarmins IL-33, TSLP, and IL-25. These molecules promote the type 2 cytokine production of ILC2s and play a well-established and crucial role in allergic diseases147. As a result, most clinical trials on IL-33, TSLP, and IL-25 blockers focus on allergic diseases. The drugs that target the IL-33/ST2 pathway include astegolimab148,149, itepekimab150, and tozorakimab (formerly MEDI3506)35. While these IL-33/ST2-targeting biologics have not yet received FDA approval, some are now in phase 3 clinical trials. The drugs that block the TSLP pathway include tezepelumab, which recently received FDA approval for asthma, and ecleralimab (formerly CSJ117), which is an inhaled drug that is in phase 2 clinical trials (ClinicalTrials.gov Identifier: NCT04882124)151. Clinical trials on drugs against IL-25 have not yet been conducted. Moreover, there are also IL-12/23p40 inhibitors, which target a cytokine subunit shared by IL-23 and IL-23 and include ustekinumab152 and briakinumab153; and IL-23 inhibitors154, such as guselkumab and risankizumab155.
Drugs that target ILC-producing cytokines
Candidates target downstream molecules of pathogenic ILC2s, including the type 2 cytokines IL-5 and IL-13. Although type 2 cytokines are produced by pathogenic Th2 cells as well as ILC2s, ILC2s rapidly secrete large amounts of these cytokines soon after the pathogenic trigger1. Therefore, several biologics target IL-5 or IL-13 and are already in clinical use and thus could be used to regulate ILC2-mediated pathologies in asthma or allergy. Indeed, dupilumab, an anti-IL-4Rα monoclonal antibody that inhibits the binding of IL-4 and IL-13 to the IL-4Rα receptor, significantly reduced ILC2s in a clinical study on patients with atopic dermatitis156. This suggests that anti-IL-5 monoclonal antibodies such as mepolizumab and reslizumab157,158, approved for refractory asthma, may also downregulate ILC2s. However, studies on this issue have not yet been conducted. Similarly, the ability of potential inhibitors of ILC1s and/or ILC3s to downregulate the pathogenic activities of these cells is unknown. Candidate drugs are IL-17 inhibitors, which include secukinumab159 and ixekizumab160.
Drugs that target lipid mediators
There are also a number of molecules other than alarmins that act upstream of ILC2s and could be targeted to reduce pathogenic ILC2 activity. They include lipid mediators, which activate these cells and thereby promote allergic inflammation; examples are prostaglandin D2 (PGD2) and cysteinyl leukotrienes (cysLTs)161. These molecules could be targeted directly or indirectly. An indirect way of targeting PGD2 is to downregulate CRTH2 (chemoattractant receptor-homologous molecule expressed on Th2 cells), which is a PGD2 receptor that is expressed on ILC2s and mediates the PGD2-dependent trafficking of ILC2s that promotes allergies162. While strategies targeting the CRTH2-PGD2 axis using the PGD2 antagonist fevipiprant are still under investigation, clinical trials have shown unsatisfactory results to date162,163,164. In contrast, montelukast, a cysLT1 receptor antagonist, is a widely used anti-asthmatic drug165 that could potentially act, at least in part, by downregulating the potent ability of cysLTs to activate ILC2s166. Studies are needed to verify this idea.
Drugs that target migration of ILCs
Murine ILC2s express β2AR, and treating IL-33- or allergen-induced murine models of asthma with a β2AR agonist reduces their lung ILC2 frequencies and cytokine production and ameliorates asthma in a CD4+ T-cell-independent manner167. Thus, β2AR agonists could improve asthma by downregulating ILC2s.
Intestinal ILC3s express high levels of integrin α4β7, and this homing receptor is required for ILC3 accumulation in the intestine24. Thus, the anti-α4β7 monoclonal antibody vedolizumab or anti-β7 monoclonal antibody etrolizumab, which are currently being used to treat IBD patients168,169,170, could potentially exert beneficial effects by downregulating ILC3 recruitment to the intestines. This possibility is disputed by a report showing that vedolizumab treatment does not alter the frequencies of ILCs in the blood of patients with Crohn’s disease171. However, further research is warranted.
Drugs that target ILC signaling
ILC activity can be modulated by blocking the signaling pathways that drive ILC activation. These include the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway, which is activated by various cytokines and growth factors in inflammatory diseases172. In fact, JAK/STAT signaling contributes to the development and effector functions of all ILC subsets48,173,174. Thus, JAK inhibitors could be repurposed to target inflammatory ILCs in mucosal diseases. These inhibitors include (i) tofacitinib, a pan-JAK inhibitor that is approved for IBD175; (ii) baricitinib, a JAK1/2 inhibitor that is approved for atopic dermatitis176; and (iii) upadacitinib, a JAK1-selective inhibitor that is also approved for atopic dermatitis177.
Concluding remarks
Mucosal organs, including the lungs and digestive tract, are constantly challenged by various stimuli. ILCs are multifunctional immune cells that are permanent residents in these organs; they ceaselessly patrol these tissues and are poised to react instantaneously and strongly when the tissue is compromised. This role means that despite their relatively low frequencies in mucosal tissues, ILCs play important roles in the homeostasis of mucosal organs and the development and progression of mucosal inflammatory diseases. This suggests that ILCs may be an effective therapeutic target for mucosal diseases. Indeed, many approved therapeutic agents and drugs currently in clinical trials have the potential to modulate the function of ILCs in mucosal diseases. Additional drugs for mucosal disorders may be discovered by identifying key interactions (i.e., immune checkpoint interactions) between ILCs and other immune cells and non-immune cells that dictate the immunological outcomes in mucosal tissues. The relatively sparse research to date suggests that DR3-TL1A, PD-1-PD-L1, and KLRG1 may be useful therapeutic targets. Finally, given that ILCs can also play beneficial roles, another potentially effective therapeutic approach is cell therapy. Although this approach is still impeded by technical issues, a few candidate regulatory ILCs have been identified. Further research in this field, and all the other topics mentioned in this review, is warranted.
References
Vivier, E. et al. Innate lymphoid cells: 10 years on. Cell 174, 1054–1066 (2018).
Kim, J., Ryu, S. & Kim, H. Y. Innate lymphoid cells in tissue homeostasis and disease pathogenesis. Mol. Cells 44, 301–309 (2021).
Zhang, J. et al. T-bet and Eomes govern differentiation and function of mouse and human NK cells and ILC1. Eur. J. Immunol. 48, 738–750 (2018).
Wagner, J. A. et al. Stage-specific requirement for eomes in mature NK cell homeostasis and cytotoxicity. Cell Rep. 31, 107720 (2020).
Chiossone, L., Dumas, P. Y., Vienne, M. & Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 18, 671–688 (2018).
Entwistle, L. J. et al. Pulmonary group 2 innate lymphoid cell phenotype is context specific: determining the effect of strain, location, and stimuli. Front. Immunol. 10, 3314 (2020).
Monticelli, L. A. et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 12, 1045–1054 (2011).
Turner, J-E. et al. IL-9–mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J. Exp. Med. 210, 2951–2965 (2013).
Gschwend, J. et al. Alveolar macrophages rely on GM-CSF from alveolar epithelial type 2 cells before and after birth. J. Exp. Med. 218, e20210745 (2021).
Meininger, I. et al. Tissue-specific features of innate lymphoid cells. Trends Immunol. 41, 902–917 (2020).
Schroeder, J-H., Howard, J. K. & Lord, G. M. Transcription factor-driven regulation of ILC1 and ILC3. Trends Immunol. 43, 564–579 (2022).
Montaldo, E., Juelke, K. & Romagnani, C. Group 3 innate lymphoid cells (ILC3s): origin, differentiation, and plasticity in humans and mice. Eur. J. Immunol. 45, 2171–2182 (2015).
Kim, M. & Kim, C. H. Colonization and effector functions of innate lymphoid cells in mucosal tissues. Microbes Infect. 18, 604–614 (2016).
Gasteiger, G., Fan, X., Dikiy, S., Lee, S. Y. & Rudensky, A. Y. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 350, 981–985 (2015).
Zhong, C. et al. Differential expression of the transcription factor GATA3 specifies lineage and functions of innate lymphoid cells. Immunity 52, 83–95.e84 (2020).
Ghaedi, M. et al. Single-cell analysis of RORalpha tracer mouse lung reveals ILC progenitors and effector ILC2 subsets. J. Exp. Med. 217 https://doi.org/10.1084/jem.20182293 (2020).
Orimo, K. et al. Characteristics of tissue-resident ILCs and their potential as therapeutic targets in mucosal and skin inflammatory diseases. Allergy 76, 3332–3348 (2021).
Zeis, P. et al. In situ maturation and tissue adaptation of type 2 innate lymphoid cell progenitors. Immunity 53, 775–792.e779 (2020).
Possot, C. et al. Notch signaling is necessary for adult, but not fetal, development of RORgammat(+) innate lymphoid cells. Nat. Immunol. 12, 949–958 (2011).
Constantinides, M. G., McDonald, B. D., Verhoef, P. A. & Bendelac, A. A committed precursor to innate lymphoid cells. Nature 508, 397–401 (2014).
Erle, D. J. et al. Expression and function of the MAdCAM-1 receptor, integrin alpha 4 beta 7, on human leukocytes. J. Immunol. 153, 517–528 (1994).
Deem, T. L. & Cook-Mills, J. M. Vascular cell adhesion molecule 1 (VCAM-1) activation of endothelial cell matrix metalloproteinases: role of reactive oxygen species. Blood 104, 2385–2393 (2004).
Chea, S. et al. CXCR6 expression is important for retention and circulation of ILC precursors. Mediators Inflamm. 2015, 368427 (2015).
Kim, M. H., Taparowsky, E. J. & Kim, C. H. Retinoic acid differentially regulates the migration of innate lymphoid cell subsets to the gut. Immunity 43, 107–119 (2015).
Patman, G. Immunology: gut migration of innate lymphoid cells. Nat. Rev. Gastroenterol. Hepatol. 12, 430–430 (2015).
Stier, M. T. et al. IL-33 promotes the egress of group 2 innate lymphoid cells from the bone marrow. J. Exp. Med. 215, 263–281 (2018).
Corral, D. et al. ILC precursors differentiate into metabolically distinct ILC1-like cells during Mycobacterium tuberculosis infection. Cell Rep. 39, 110715 (2022).
Fuchs, A. et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity 38, 769–781 (2013).
Luci, C. et al. Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat. Immunol. 10, 75–82 (2009).
Klose, C. S. et al. A T-bet gradient controls the fate and function of CCR6-RORγt+ innate lymphoid cells. Nature 494, 261–265 (2013).
Hewitt, R. J. & Lloyd, C. M. Regulation of immune responses by the airway epithelial cell landscape. Nat. Rev. Immunol. 21, 347–362 (2021).
Mjosberg, J. & Spits, H. Human innate lymphoid cells. J. Allergy Clin. Immunol. 138, 1265–1276 (2016).
Stephen T. et al. Asthma. Nat. Rev. Dis. Primers 1 https://doi.org/10.1038/nrdp.2015.25 (2015).
Ham, J., Lim, M., Kim, D. & Kim, H. Y. Memory-like innate lymphoid cells in the pathogenesis of asthma. Front. Immunol. 13 https://doi.org/10.3389/fimmu.2022.1005517 (2022).
Ham, J., Shin, J. W., Ko, B. C. & Kim, H. Y. Targeting the epithelium-derived innate cytokines: from bench to bedside. Immune Netw. 22, e11 (2022).
Halim, T. Y. et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 40, 425–435 (2014).
Grotenboer, N. S., Ketelaar, M. E., Koppelman, G. H. & Nawijn, M. C. Decoding asthma: translating genetic variation in IL33 and IL1RL1 into disease pathophysiology. J. Allergy Clin. Immunol. 131, 856–865 (2013).
Tamari, M., Tanaka, S. & Hirota, T. Genome-wide association studies of allergic diseases. Allergol. Int. 62, 21–28 (2013).
Liu, T. et al. Type 2 innate lymphoid cells: a novel biomarker of eosinophilic airway inflammation in patients with mild to moderate asthma. Respir. Med. 109, 1391–1396 (2015).
Bartemes, K. R., Kephart, G. M., Fox, S. J. & Kita, H. Enhanced innate type 2 immune response in peripheral blood from patients with asthma. J. Allergy. Clin. Immunol. 134, 671–678.e674 (2014).
Kim, J. et al. Innate immune crosstalk in asthmatic airways: Innate lymphoid cells coordinate polarization of lung macrophages. J. Allergy Clin. Immunol. 143, 1769–1782.e1711 (2019).
Akar-Ghibril, N., Casale, T., Custovic, A. & Phipatanakul, W. Allergic endotypes and phenotypes of asthma. J. Allergy Clin. Immunol. Pract. 8, 429–440 (2020).
Kim, H. Y. et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat. Med. 20, 54–61 (2014).
Estrella, B. et al. Effects of air pollution on lung innate lymphoid cells: review of in vitro and in vivo experimental studies. Int. J. Environ. Res. Public Health 16 https://doi.org/10.3390/ijerph16132347 (2019).
Kim, J. et al. The effect of air pollutants on airway innate immune cells in patients with asthma. Allergy 75, 2372–2376 (2020).
Luo, J. et al. Resistance to apoptosis underpins the corticosteroid insensitivity of group 2 innate lymphoid cells. J. Allergy Clin. Immunol. 144, 1722–1726.e1710 (2019).
Ploeg, E. K. V. D. et al. Steroid-resistant human inflammatory ILC2s are marked by CD45RO and elevated in type 2 respiratory diseases. Sci. Immunol. 6, eabd3489 (2021).
Liu, S. et al. Steroid resistance of airway type 2 innate lymphoid cells from patients with severe asthma: the role of thymic stromal lymphopoietin. J. Allergy Clin. Immunol. 141, 257–268.e256 (2018).
Moriyama, S. et al. β2-adrenergic receptor-mediated negative regulation of group 2 innate lymphoid cell responses. Science 359, 1056–1061 (2018).
Rabe, K. F. & Watz, H. Chronic obstructive pulmonary disease. Lancet 389, 1931–1940 (2017).
Blomme, E. E. et al. Quantification and role of innate lymphoid cell subsets in Chronic Obstructive Pulmonary Disease. Clin. Transl. Immunology 10, e1287 (2021).
Freeman, C. M. et al. Cytotoxic potential of lung CD8( + ) T cells increases with chronic obstructive pulmonary disease severity and with in vitro stimulation by IL-18 or IL-15. J. Immunol. 184, 6504–6513 (2010).
Wang, Z. et al. Interferon γ Induction of Pulmonary Emphysema in the Adult Murine Lung. J. Exp. Med. 192, 1587–1600 (2000).
Freeman, C. M. et al. Human CD56+ cytotoxic lung lymphocytes kill autologous lung cells in chronic obstructive pulmonary disease. PLoS One 9, e103840 (2014).
Finch, D. K. et al. Lung dendritic cells drive natural killer cytotoxicity in chronic obstructive pulmonary disease via IL-15 Ralpha. Am. J. Respir. Crit. Care Med. 198, 1140–1150 (2018).
Silver, J. S. et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat. Immunol. 17, 626–635 (2016).
Bal, S. M. et al. IL-1beta, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat. Immunol. 17, 636–645 (2016).
Kearley, J. et al. Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin-33-dependent response to infection. Immunity 42, 566–579 (2015).
De Grove, K. C. et al. Characterization and quantification of innate lymphoid cell subsets in human lung. PLoS One 11, e0145961 (2016).
Suzuki, M. et al. The cellular and molecular determinants of emphysematous destruction in COPD. Sci. Rep. 7, 9562 (2017).
Torres, A. et al. Pneumonia. Nat. Rev. Dis. Primers 7, 25 (2021).
Stehle, C., Hernandez, D. C. & Romagnani, C. Innate lymphoid cells in lung infection and immunity. Immunol. Rev. 286, 102–119 (2018).
Dou, Y. et al. Influenza vaccine induces intracellular immune memory of human NK cells. PLoS One 10, e0121258 (2015).
Wills-Karp, M. & Finkelman, F. D. Innate lymphoid cells wield a double-edged sword. Nat. Immunol. 12, 1025–1027 (2011).
Gorski, S. A., Hahn, Y. S. & Braciale, T. J. Group 2 innate lymphoid cell production of IL-5 is regulated by NKT cells during influenza virus infection. PLoS Pathog. 9, e1003615 (2013).
Gomes, A. M. C. et al. SARS-CoV2 pneumonia recovery is linked to expansion of innate lymphoid cells type 2 expressing CCR10. Eur. J. Immunol. 51, 3194–3201 (2021).
Silverstein, N. J. et al. Innate lymphoid cells and COVID-19 severity in SARS-CoV-2 infection. Elife 11, e74681 (2022).
Van Maele, L. et al. Activation of Type 3 innate lymphoid cells and interleukin 22 secretion in the lungs during Streptococcus pneumoniae infection. J. Infect. Dis. 210, 493–503 (2014).
Mowat, A. M. & Agace, W. W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 14, 667–685 (2014).
Yudanin, N. A. et al. Spatial and temporal mapping of human innate lymphoid cells reveals elements of tissue specificity. Immunity 50, 505–519.e504 (2019).
Diefenbach, A., Gnafakis, S. & Shomrat, O. Innate lymphoid cell-epithelial cell modules sustain intestinal homeostasis. Immunity 52, 452–463 (2020).
Sonnenberg, G. F., Fouser, L. A. & Artis, D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol. 12, 383–390 (2011).
Sawa, S. et al. RORgammat+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat. Immunol. 12, 320–326 (2011).
Zheng, Y. et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 14, 282–289 (2008).
Sugimoto, K. et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest. 118, 534–544 (2008).
Satoh-Takayama, N. et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 29, 958–970 (2008).
Sonnenberg, G. F., Monticelli, L. A., Elloso, M. M., Fouser, L. A. & Artis, D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity 34, 122–134 (2011).
Lindemans, C. A. et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 528, 560–564 (2015).
Jarade, A. et al. Inflammation triggers ILC3 patrolling of the intestinal barrier. Nat. Immunol. 23, 1317–1323 (2022).
Chun, E. et al. Metabolite-sensing receptor Ffar2 regulates colonic Group 3 innate lymphoid cells and gut. Immunity 51, 871–884.e876 (2019).
Kinnebrew, M. A. et al. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity 36, 276–287 (2012).
Longman, R. S. et al. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J. Exp. Med. 211, 1571–1583 (2014).
Takatori, H. et al. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J. Exp. Med. 206, 35–41 (2009).
Powell, N. et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity 37, 674–684 (2012).
Bhatt, B. et al. Gpr109a limits microbiota-induced IL-23 production to constrain ILC3-mediated colonic inflammation. J. Immunol. 200, 2905–2914 (2018).
Cella, M., Otero, K. & Colonna, M. Expansion of human NK-22 cells with IL-7, IL-2, and IL-1β reveals intrinsic functional plasticity. Proc. Natl. Acad. Sci. USA 107, 10961–10966 (2010).
Zeng, B. et al. ILC3 function as a double-edged sword in inflammatory bowel diseases. Cell Death Dis. 10, 315 (2019).
Song, C. et al. Unique and redundant functions of NKp46 + ILC3s in models of intestinal inflammation. J. Exp. Med. 212, 1869–1882 (2015).
Chang, Y. et al. Increased GM-CSF-producing NCR(-) ILC3s and neutrophils in the intestinal mucosa exacerbate inflammatory bowel disease. Clin. Transl. Immunol. 10, e1311 (2021).
Eken, A., Singh, A. K., Treuting, P. M. & Oukka, M. IL-23R+ innate lymphoid cells induce colitis via interleukin-22-dependent mechanism. Mucosal Immunol. 7, 143–154 (2014).
Castellanos, J. G. et al. Microbiota-Induced TNF-like ligand 1 A drives group 3 innate lymphoid cell-mediated barrier protection and intestinal T cell activation during colitis. Immunity 49, 1077–1089.e1075 (2018).
Bouchery, T., Le Gros, G. & Harris, N. ILC2s-trailblazers in the host response against intestinal helminths. Front. Immunol. 10, 623 (2019).
Neill, D. R. et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464, 1367–1370 (2010).
Price, A. E. et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc. Natl. Acad. Sci. USA 107, 11489–11494 (2010).
Ricardo-Gonzalez, R. R. et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat. Immunol. 19, 1093–1099 (2018).
Huang, Y. et al. S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science 359, 114–119 (2018).
Wang, S. et al. Regulatory innate lymphoid cells control innate intestinal inflammation. Cell 171, 201–216.e218 (2017).
Hutloff, A. et al. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature 397, 263–266 (1999).
Coyle, A. J. et al. The CD28-related molecule ICOS is required for effective T cell-dependent immune responses. Immunity 13, 95–105 (2000).
Toker, A. et al. Regulatory T cells in ovarian cancer are characterized by a highly activated phenotype distinct from that in melanoma. Clin. Cancer Res. 24, 5685–5696 (2018).
Dong, C. et al. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature 409, 97–101 (2001).
Van, D. V., Bauer, L., Kroczek, R. A. & Hutloff, A. ICOS costimulation differentially affects T cells in secondary lymphoid organs and inflamed tissues. Am. J. Respir. Cell Mol. Biol. 59, 437–447 (2018).
Paclik, D., Stehle, C., Lahmann, A., Hutloff, A. & Romagnani, C. ICOS regulates the pool of group 2 innate lymphoid cells under homeostatic and inflammatory conditions in mice. Eur. J. Immunol. 45, 2766–2772 (2015).
Maazi, H. et al. ICOS:ICOS-ligand interaction is required for type 2 innate lymphoid cell function, homeostasis, and induction of airway hyperreactivity. Immunity 42, 538–551 (2015).
Kamachi, F., Isshiki, T., Harada, N., Akiba, H. & Miyake, S. ICOS promotes group 2 innate lymphoid cell activation in lungs. Biochem. Biophys. Res. Commun. 463, 739–745 (2015).
Iwanaga, N. et al. Host immunology and rational immunotherapy for carbapenem-resistant Klebsiella pneumoniae infection. JCI Insight 5, e135591 (2020).
Ward-Kavanagh, L. K., Lin, W. W., Sedy, J. R. & Ware, C. F. The TNF receptor superfamily in co-stimulating and co-inhibitory responses. Immunity 44, 1005–1019 (2016).
Tian, J., Zhang, B., Rui, K. & Wang, S. The role of GITR/GITRL interaction in autoimmune diseases. Front. Immunol. 11, 588682 (2020).
Galle-Treger, L. et al. Costimulation of type-2 innate lymphoid cells by GITR promotes effector function and ameliorates type 2 diabetes. Nat. Commun. 10, 713 (2019).
Nagashima, H. et al. GITR cosignal in ILC2s controls allergic lung inflammation. J. Allergy Clin. Immunol. 141, 1939–1943.e1938 (2018).
Vashist, N. et al. Influenza-activated ILC1s contribute to antiviral immunity partially influenced by differential GITR expression. Front. Immunol. 9, 505 (2018).
Cavagnero, K. J. et al. Unconventional ST2- and CD127-negative lung ILC2 populations are induced by the fungal allergen Alternaria alternata. J. Allergy Clin. Immunol. 144, 1432–1435.e1439 (2019).
Sugamura, K., Ishii, N. & Weinberg, A. D. Therapeutic targeting of the effector T-cell co-stimulatory molecule OX40. Nat. Rev. Immunol. 4, 420–431 (2004).
Halim, T. Y. F. et al. Tissue-restricted adaptive type 2 immunity is orchestrated by expression of the costimulatory molecule OX40L on group 2 innate lymphoid cells. Immunity 48, 1195–1207.e1196 (2018).
Wu, J. et al. Critical role of OX40/OX40L in ILC2-mediated activation of CD4( + )T cells during respiratory syncytial virus infection in mice. Int. Immunopharmacol. 76, 105784 (2019).
Drake, L. Y., Iijima, K. & Kita, H. Group 2 innate lymphoid cells and CD4 + T cells cooperate to mediate type 2 immune response in mice. Allergy 69, 1300–1307 (2014).
Deng, T. et al. ILC3-derived OX40L is essential for homeostasis of intestinal Tregs in immunodeficient mice. Cell Mol. Immunol. 17, 163–177 (2020).
McLaren, J. E. et al. The TNF-like protein 1A-death receptor 3 pathway promotes macrophage foam cell formation in vitro. J. Immunol. 184, 5827–5834 (2010).
Twohig, J. P. et al. The death receptor 3/TL1A pathway is essential for efficient development of antiviral CD4(+) and CD8( + ) T-cell immunity. FASEB J. 26, 3575–3586 (2012).
Valatas, V., Kolios, G. & Bamias, G. TL1A (TNFSF15) and DR3 (TNFRSF25): a co-stimulatory system of cytokines with diverse functions in gut mucosal immunity. Front. Immunol. 10, 583 (2019).
Prehn, J. L. et al. The T cell costimulator TL1A is induced by FcgammaR signaling in human monocytes and dendritic cells. J. Immunol. 178, 4033–4038 (2007).
Ahn, Y. O. et al. Human group3 innate lymphoid cells express DR3 and respond to TL1A with enhanced IL-22 production and IL-2-dependent proliferation. Eur. J. Immunol. 45, 2335–2342 (2015).
Meylan, F. et al. The TNF-family cytokine TL1A promotes allergic immunopathology through group 2 innate lymphoid cells. Mucosal Immunol. 7, 958–968 (2014).
Yu, X. et al. TNF superfamily member TL1A elicits type 2 innate lymphoid cells at mucosal barriers. Mucosal Immunol. 7, 730–740 (2014).
Machida, K. et al. Type 2 innate lymphoid cells expressing death receptor 3 are increased in airway of mild atopic asthmatic subject following allergen inhalation challenge. J. Allergy Clin. Immunol. 141 https://doi.org/10.1016/j.jaci.2017.12.908 (2018).
Li, J. et al. Activation of DR3 signaling causes loss of ILC3s and exacerbates intestinal inflammation. Nat. Commun. 10, 3371 (2019).
Ishida, Y., Agata, Y., Shibahara, K. & Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 11, 3887–3895 (1992).
Andrews, L. P., Yano, H. & Vignali, D. A. A. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: breakthroughs or backups. Nat. Immunol. 20, 1425–1434 (2019).
Day, C. L. et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443, 350–354 (2006).
Pardoll, D. M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012).
Yu, Y. et al. Single-cell RNA-seq identifies a PD-1(hi) ILC progenitor and defines its development pathway. Nature 539, 102–106 (2016).
Taylor, S. et al. PD-1 regulates KLRG1(+) group 2 innate lymphoid cells. J. Exp. Med. 214, 1663–1678 (2017).
Seillet, C. et al. Deciphering the innate lymphoid cell transcriptional program. Cell Rep. 17, 436–447 (2016).
Vacca, P. et al. PD-1 is expressed by and regulates human group 3 innate lymphoid cells in human decidua. Mucosal Immunol. 12, 624–631 (2019).
Tumino, N. et al. Presence of innate lymphoid cells in pleural effusions of primary and metastatic tumors: Functional analysis and expression of PD-1 receptor. Int. J. Cancer 145, 1660–1668 (2019).
Mariotti, F. R., Quatrini, L., Munari, E., Vacca, P. & Moretta, L. Innate lymphoid cells: expression of PD-1 and other checkpoints in normal and pathological conditions. Front. Immunol. 10, 910 (2019).
Mallett, G., Laurence, A. & Amarnath, S. Programmed cell death-1 receptor (PD-1)-mediated regulation of innate lymphoid cells. Int. J. Mol. Sci. 20, 2836 (2019).
Helou, D. G. et al. PD-1 pathway regulates ILC2 metabolism and PD-1 agonist treatment ameliorates airway hyperreactivity. Nat. Commun. 11, 3998 (2020).
Myklebust, J. H. et al. High PD-1 expression and suppressed cytokine signaling distinguish T cells infiltrating follicular lymphoma tumors from peripheral T cells. Blood 121, 1367–1376 (2013).
Huntington, N. D. et al. NK cell maturation and peripheral homeostasis is associated with KLRG1 up-regulation. J. Immunol. 178, 4764–4770 (2007).
Huang, Y. et al. IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential ‘inflammatory’ type 2 innate lymphoid cells. Nat. Immunol. 16, 161–169 (2015).
Mazzurana, L. et al. Tissue-specific transcriptional imprinting and heterogeneity in human innate lymphoid cells revealed by full-length single-cell RNA-sequencing. Cell Res. 31, 554–568 (2021).
Blanquart, E. et al. Targeting androgen signaling in ILC2s protects from IL-33-driven lung inflammation, independently of KLRG1. J. Allergy Clin. Immunol. 149, 237–251.e212 (2022).
Nagasawa, M. et al. KLRG1 and NKp46 discriminate subpopulations of human CD117( + )CRTH2(-) ILCs biased toward ILC2 or ILC3. J. Exp. Med. 216, 1762–1776 (2019).
Golebski, K. et al. Induction of IL-10-producing type 2 innate lymphoid cells by allergen immunotherapy is associated with clinical response. Immunity 54, 291–307.e297 (2021).
Cobb, L. M. & Verneris, M. R. Therapeutic manipulation of innate lymphoid cells. JCI Insight. 6, e146006 (2021).
Matsuyama, T. et al. The therapeutic potential for targeting group 2 innate lymphoid cells in asthma. Front. Immunol. 13, 930862 (2022).
Kelsen, S. G. et al. Astegolimab (anti-ST2) efficacy and safety in adults with severe asthma: a randomized clinical trial. J Allergy Clin. Immunol. 148, 790–798 (2021).
Yousuf, A. J. et al. Astegolimab, an anti-ST2, in chronic obstructive pulmonary disease (COPD-ST2OP): a phase 2a, placebo-controlled trial. Lancet Respir. Med. 10, 469–477 (2022).
Wechsler, M. E. et al. Efficacy and safety of itepekimab in patients with moderate-to-severe asthma. N. Engl. J. Med. 385, 1656–1668 (2021).
O’Byrne, P. M. et al. Development of an inhaled anti-TSLP therapy for asthma. Pulm. Pharmacol. Ther. 78, 102184 (2022).
Sands, B. E. et al. Ustekinumab as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 381, 1201–1214 (2019).
Reich, K. et al. A 52-week trial comparing briakinumab with methotrexate in patients with psoriasis. N. Engl. J. Med. 365, 1586–1596 (2011).
Reich, K. et al. Guselkumab versus secukinumab for the treatment of moderate-to-severe psoriasis (ECLIPSE): results from a phase 3, randomised controlled trial. Lancet 394, 831–839 (2019).
Gordon, K. B. et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet 392, 650–661 (2018).
Imai, Y., Kusakabe, M., Nagai, M., Yasuda, K. & Yamanishi, K. Dupilumab effects on innate lymphoid cell and helper T cell populations in patients with atopic dermatitis. JID Innov. 1, 100003 (2021).
Ortega, H. G. et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N. Engl. J. Med. 371, 1198–1207 (2014).
Castro, M. et al. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir. Med. 3, 355–366 (2015).
Langley, R. G. et al. Secukinumab in plaque psoriasis–results of two phase 3 trials. N. Engl. J. Med. 371, 326–338 (2014).
Gordon, K. B. et al. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N. Engl. J. Med. 375, 345–356 (2016).
Orimo, K., Saito, H., Matsumoto, K. & Morita, H. Innate lymphoid cells in the airways: their functions and regulators. Allergy Asthma Immunol. Res. 12, 381–398 (2020).
Xue, L. et al. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J. Allergy Clin. Immunol. 133, 1184–1194 (2014).
Farne, H. et al. Effect of CRTH2 antagonism on the response to experimental rhinovirus infection in asthma: a pilot randomised controlled trial. Thorax 77, 950–959 (2021).
Brightling, C. E. et al. Effectiveness of fevipiprant in reducing exacerbations in patients with severe asthma (LUSTER-1 and LUSTER-2): two phase 3 randomised controlled trials. Lancet Respir. Med. 9, 43–56 (2021).
Leff, J. A. et al. Montelukast, a leukotriene-receptor antagonist, for the treatment of mild asthma and exercise-induced bronchoconstriction. N. Engl. J. Med. 339, 147–152 (1998).
Salimi, M. et al. Cysteinyl leukotriene E(4) activates human group 2 innate lymphoid cells and enhances the effect of prostaglandin D(2) and epithelial cytokines. J. Allergy Clin. Immunol. 140, 1090–1100.e1011 (2017).
Moriyama, S. et al. beta(2)-adrenergic receptor-mediated negative regulation of group 2 innate lymphoid cell responses. Science 359, 1056–1061 (2018).
Feagan, B. G. et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 369, 699–710 (2013).
Dai, B. et al. Dual targeting of lymphocyte homing and retention through alpha4beta7 and alphaEbeta7 inhibition in inflammatory bowel disease. Cell Rep. Med. 2, 100381 (2021).
Peyrin-Biroulet, L. et al. Etrolizumab as induction and maintenance therapy for ulcerative colitis in patients previously treated with tumour necrosis factor inhibitors (HICKORY): a phase 3, randomised, controlled trial. Lancet Gastroenterol. Hepatol. 7, 128–140 (2022).
Forkel, M. et al. Distinct alterations in the composition of mucosal innate lymphoid cells in newly diagnosed and established crohn’s disease and ulcerative colitis. J. Crohns Colitis 13, 67–78 (2019).
Georas, S. N., Donohue, P., Connolly, M. & Wechsler, M. E. JAK inhibitors for asthma. J. Allergy Clin. Immunol. 148, 953–963 (2021).
Stabile, H. et al. JAK/STAT signaling in regulation of innate lymphoid cells: the gods before the guardians. Immunol. Rev. 286, 148–159 (2018).
Robinette, M. L. et al. Jak3 deficiency blocks innate lymphoid cell development. Mucosal Immunol. 11, 50–60 (2018).
Sandborn, W. J. et al. Tofacitinib as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 376, 1723–1736 (2017).
Simpson, E. L. et al. Baricitinib in patients with moderate-to-severe atopic dermatitis and inadequate response to topical corticosteroids: results from two randomized monotherapy phase III trials. Br. J. Dermatol. 183, 242–255 (2020).
Guttman-Yassky, E. et al. Upadacitinib in adults with moderate to severe atopic dermatitis: 16-week results from a randomized, placebo-controlled trial. J. Allergy Clin. Immunol. 145, 877–884 (2020).
Acknowledgements
All the illustrations in this article were created with BioRender.com. This work was supported by the National Research Foundation of Korea (2022R1A2C3007730, 2021R1C1C2010051, and SRC 2017R1A5A1014560).
Author information
Authors and Affiliations
Contributions
Conceptualization, writing–review & editing: H.Y.K.; investigation: S.R., M.Y.L., J.K.; Writing–original draft: S.R., M.Y.L., J.K., and H.Y.K.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ryu, S., Lim, M., Kim, J. et al. Versatile roles of innate lymphoid cells at the mucosal barrier: from homeostasis to pathological inflammation. Exp Mol Med 55, 1845–1857 (2023). https://doi.org/10.1038/s12276-023-01022-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-023-01022-z
This article is cited by
-
Amorphous silica nanoparticles and the human gut microbiota: a relationship with multiple implications
Journal of Nanobiotechnology (2024)
