
Abstract
Foxp3+ regulatory T cells (Tregs) are a subset of CD4+ T cells that exert suppressive control over other immune cells. Tregs are critical for preventing systemic autoimmunity and maintaining peripheral tolerance, and yet they also assist in orchestration of immunity to pathogenic insult, wherein they limit collateral immunopathology and assist in facilitating a fine balance between immune tolerance and effector activity. Tregs have been extensively studied in lymphoid tissues, and a growing body of work has characterized phenotypically distinct Tregs localized in various nonlymphoid tissue compartments. These tissue Tregs can perform location-specific, alternative functions, highlighting their dynamic, context-dependent roles. Tregs have also been identified in mucosal tissues where specialized physiological functions are paramount, including helping the host to respond appropriately to pathogenic versus innocuous antigens that are abundant at mucosal portals of antigen entry. As in other tissue Treg compartments, mucosal Tregs in the respiratory, gastrointestinal, and genitourinary tracts are distinct from circulating counterparts and can carry out mucosa-specific functions as well as classic suppressive functions that are the hallmark of Tregs. In this review, we summarize current knowledge regarding mucosal Tregs in both health and disease.
Similar content being viewed by others


Introduction
Regulatory T cells (Tregs) are a subset of CD4+ T cells defined by expression of the transcription factor forkhead box P3 (Foxp3) and their ability to suppress conventional T cells and other immune cells1,2,3. Early on, Tregs were demonstrated to be an essential cell type, as patients with immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome and scurfy mice lacking a functional Foxp3 gene suffered from various autoimmune-like conditions involving multiple tissues and organs, plus dysregulated effector T cell activity4,5,6. Additional studies demonstrated that Foxp3 is the master regulator of Treg development and is indeed required for both their differentiation as well as function as suppressive cells7,8,9,10,11,12. Subsequently, a mouse model wherein Foxp3 is ablated in mature Tregs demonstrated that continuous Foxp3 expression is required to maintain the Treg developmental program and thus for Tregs to sustain suppressive function13.
Several decades of active research has since confirmed these findings and demonstrated Tregs to be critical mediators of the immune system. Immunosuppressive mechanisms utilized by Tregs are diverse and varied. Tregs express several immunosuppressive cytokines, including TGFβ and IL-1014,15, and they have also been shown to express granzyme B, leading to death of immune cells including B cells, conventional T cells, and antigen-presenting cells (APC)16,17,18. More recently, this has been shown to occur under particular inflammatory conditions such as infection19. In addition, Tregs express high levels of CTLA-4 at steady state, which allows them to limit availability of CD80 and CD86 on dendritic cells (DC) to conventional T cells20. Tregs also express the ectoenzymes CD39 and CD73, which facilitate the conversion of extracellular ATP to adenosine, which thus directly limits proliferation of effector T cells, as well as suppressing myeloid cells including DCs21,22,23,24. Finally, through their high expression of CD25, a subunit of the IL-2R, Tregs can serve as an IL-2 sink, effectively depriving effector T cells of IL-2 and thus limiting their proliferative capacity25. Not only do Tregs utilize these immunosuppressive mechanisms, and many others, to limit immune responses to self and other innocuous antigens to prevent autoimmunity at homeostasis2,26,27, but they additionally mediate inflammatory responses to infection28,29.
Since their discovery, Tregs have been primarily studied in the thymus, where they develop, and in peripheral lymphoid organs. However, a growing body of research has shown that Tregs also reside in nonlymphoid organs, where they can have unique phenotypes and execute highly specialized roles outside of the canonical regulatory functions, suggesting that Treg phenotype and activity is highly context dependent. In particular, tissue Tregs have been described in visceral adipose tissue (VAT), skin, and muscle30,31,32. These nonlymphoid tissue Tregs are transcriptionally and phenotypically distinct from Tregs in spleen and lymph node (LN) and can perform highly specialized, location-dependent roles. Tregs in human skin, concentrated near hair follicles, regulate epithelial stem cell differentiation and display an activated memory phenotype, limiting inflammation and pro-fibrotic responses at steady state32,33,34. In the mouse VAT, Tregs express the IL-33 receptor (IL-33R), where IL-33 expressed by mesenchymal stromal cells promotes Treg accumulation and proliferation35,36. These Tregs acquire robust expression of the master regulator of adipose differentiation—peroxisome proliferator-activated receptor (PPAR)-γ—which maintains VAT insulin sensitivity31,35,37. Muscle Tregs respond to IL-33 produced by fibroblast-like fibro/adipogenic progenitor (FAP) cells during muscle damage in mice and in turn produce amphiregulin (Areg), a growth factor that directly acts on satellite cells to facilitate muscle repair30,38.Thus, phenotypically distinct and highly specialized Treg populations reside in various nonlymphoid tissue sites, underscoring phenotypic and functional heterogeneity within Tregs based on tissue location. Furthermore, these studies suggest that dynamic populations likely exist throughout the body in other discrete tissue types.
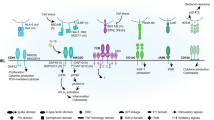
In addition to nonlymphoid organs such as the muscle, VAT, and skin, Tregs have also been characterized in several mucosal tissue sites. The mucosa has critical location-dependent physiologic functions, including air exchange in the respiratory tract, nutrient absorption in the gastrointestinal tract, and reproduction in the genitourinary tract. The mucosal tissues also share a common barrier function in protecting the host from invading pathogenic stimuli, as the mucosa serves as a portal of entry for many pathogens of global health importance. The unique physiological and barrier roles of mucosal sites require a tightly coordinated immune response to allow for necessary tolerance to harmless antigens such as dietary antigens and sperm during reproduction, while also patrolling for and eliminating viral, bacterial, and fungal pathogens at infection entry sites. Given the canonical roles of Tregs as immune response regulators, as well as their more recently discovered location-specific functions, an increasing body of research demonstrates that Treg populations in mucosal tissues are crucial in both homeostasis and disease (Fig. 1).

Mucosal barrier tissues serve as portals of entry to a wide variety of antigens, both harmful and harmless to the host. Thus, the mucosal immune system has the complex job of balancing tolerance to innocuous antigens such as food, allergens, semen, and commensals while at the same time patrolling for pathogens so that an appropriate immune response can be generated to benefit but not harm the host. Tregs, depicted here at the fulcrum of this balance, participate in this process of maintaining a homeostatic state of active immunosurveillance and tolerance rather than persistent mucosal inflammation.
In this review, we summarize a selection of current knowledge of lung, small intestine, colonic, uterine, and vaginal Treg populations during health as well as infection, autoimmunity, and cancer.
Respiratory tract tissue Tregs
The respiratory tract experiences constant exposure to inhaled antigens and pathogens, yet this uniquely delicate mucosal tissue must maintain a state of relative quiescence for the preservation of tissue architecture and oxygen exchange. In this regard, overt immune responses to perturbations such as infection must be tightly regulated, striking a balance between rapid pathogen clearance and tissue preservation (Fig. 1). Tregs employ multiple mechanisms to dictate respiratory tract tissue homeostasis, including promoting effector immune responses mitigating inflammation, and facilitating tissue preservation and healing15,19,29,39,40,41,42,43,44,45,46,47. Other reviews have more comprehensively detailed the multifaceted roles for Tregs during infection28,29,42,48. Here, we mention only a handful of key examples of various functions of Tregs in the respiratory tract with the intention of setting the stage to consider the effects of respiratory tract tissue-specific cues on Treg phenotype and function.
In the context of influenza virus infection in mice, Tregs guide the formation of antigen-specific T follicular helper (Tfh) cell and germinal center (GC) B cell responses in the lymph node through their consumption of excess IL-241. Thus, by controlling IL-2 availability, Tregs promote appropriate effector immune responses for viral clearance41,49. Additionally, respiratory tract Tregs have been demonstrated to mount rapid and robust antigen-specific primary and memory recall responses to influenza virus infection, acting to appropriately limit antiviral T cell responses and ameliorate excess inflammation in mice39,45. Importantly, after secondary influenza challenge, naïve Tregs are insufficient in recapitulating CD8+ T cell regulation39. Similarly, upon Mycobacterium tuberculosis (Mtb) challenge in mice, thymically derived Tregs specific to the immunodominant Mtb epitope expand robustly, exhibit an activated phenotype, and delay the priming and accumulation of effector T cells in the lung. These Mtb-specific Tregs are then specifically culled though Th1 inflammation-derived IL-12, which promotes high Tbet expression in Mtb-specific Tregs50,51. In other examples of respiratory tract infections such as respiratory syncytial virus (RSV) in mice and SARS-CoV-2 in humans, disease severity and pulmonary pathology have been linked to functional differences among Tregs, where a loss in Tregs leads to accelerated viral clearance at the expense of immune-mediated tissue damage and worsened disease outcomes19,52,53,54. Notably, one study identified a decrease in airway Tregs in patients with COVID-19 compared to healthy controls54, thereby raising the possibility that a Treg deficit in the lung could be contributing to disease and the dysregulated immune response in COVID-19, though this requires further study of the mucosal immune responses to natural SARS-CoV-2 infection.
Recent studies employing a combination of epigenetic, single-cell chromatin accessibility, gene expression, and T cell receptor (TCR) fate mapping methods have identified a generalizable tissue-specific signature for Tregs that is conserved among mice and humans55,56,57,58,59,60,61. At homeostasis, roughly 10–20% of the total Tregs found in the lungs of specific pathogen-free (SPF) mice exhibit a tissue-specific profile56. Phenotypically, mouse tissue Tregs, including those found in the lungs, can be identified by their expression of KLRG1 and IL-33R and are regulated by BATF56,57. Interestingly, lung tissue Tregs expressing IL-33R appear to be particularly sensitive to IL-33 signaling, undergoing a 60-fold expansion in response to treatment with rIL-33 in vivo, as compared to other nonlymphoid tissue IL-33R+ Tregs isolated from peripheral tissues such as fat (10-fold) and skin (5-fold)56. Other transcription factors contributing to the core tissue Treg signature in the lung include Nfil3, GATA-3, and Maf, as well as the Delta-Notch signaling pathway, among others55,56,57. Notably, although tissue Tregs isolated from various nonlymphoid tissues share an overlapping “core” signature with lung tissue Tregs, tissue-specific cues also appear to influence their overall phenotype, revealing a possible relationship for site-specific functions.
Studies elucidating a comprehensive ascription of the functional capacities for respiratory tract tissue Tregs are incomplete. However, it is clear that lung tissue Tregs play a major role in respiratory immunity and resolution. One prominent example is the ability of lung tissue Tregs (defined as CD44hiCD62Llo with increased expression of CD103, PD-1, GITR, CTLA-4 and KLRG1) to promote tissue healing through their production of Areg after influenza infection in mice44. Importantly, the selective deficiency of Areg production by Tregs results in profound tissue damage and decreased oxygen exchange but does not impact other Treg suppressive functions, demonstrating a non-redundant requirement for tissue Tregs to produce Areg. Perhaps not surprisingly, IL-18 and IL-33 signaling are pivotal in initiating the tissue repair program, including among non-infectious scenarios44,62. Conversely, when IL-18 signaling is antagonized by Notch4, Tregs lose their ability to produce Areg resulting in severe morbidity, as has been recently demonstrated for COVID-19 disease63. Themes of lung tissue-specific roles for Tregs are further recapitulated by a model of chronic inflammation and fibrosis established by repetitive Aspergillus fumigatus challenge wherein lung tissue Tregs (CD69hiCD103hi) ameliorate Th2-mediated pathology in mice, possibly through immunosuppressive functions and/or the production of amphirgulin47 (Fig. 2). Notably, other mechanisms of tissue healing may also be mediated by Treg expression of the soluble growth factor, keratinocyte growth factor (KGF), which can be induced in Tregs derived from lung tissue in mice and humans64. Characterization of additional functions for respiratory tract tissue Tregs are still forthcoming, but will likely include quintessential Treg functions, possibly with extra guidance from tissue-specific cues during homeostasis and insult.

Foxp3+ Tregs found throughout the body have immunosuppressive roles and can limit inappropriate inflammatory responses to self- and foreign antigens through a variety of mechanisms, including but not restricted to CTLA-4, ectoenzymes CD39 and CD73, IL-2 deprivation, and secretion of soluble regulatory mediators such as IL-10 and TGFβ. In addition, as has been demonstrated in non-mucosal and nonlymphoid tissues such as the visceral adipose tissue, skin, and muscle, Tregs can execute additional specialized functions that are induced by and meet the needs of their unique tissue environment. Here we highlight one example from each of the major mucosal tissue sites. In the respiratory tract, infection or inflammation can induce Tregs to produce amphiregulin, which promotes lung tissue repair. In the gastrointestinal tract, ingestion of dietary antigens early in life induces peripherally generated Tregs that are neuropilin-1 (NRP1) and Helios double-negative, as well as negative for the transcription factor RORγt. These cells have been shown to be critical for tolerance to dietary antigens, and so are major players in the prevention of food allergy. In addition, microbiota induces RORγt+c-Maf+ pTregs that are crucial to maintain tolerance to both food and commensal bacteria. Finally, exposure to sperm and seminal fluid induces an increase in the number of Tregs present in uterine tissues, and Tregs have a demonstrated role in preventing pre-eclampsia and gestational hypertension. Future work to characterize the unique phenotypic and functional traits of mucosal tissue Tregs will surely reveal additional novel roles for these cells in these barrier tissue compartments.
In addition to immune responses to infection, respiratory tract Tregs have been implicated in restraining immune reactions to innocuous antigens to restrict allergic responses including asthma65,66. In humans with loss-of-function mutations in Foxp3 resulting in IPEX, clinical presentation includes not only autoimmune manifestations, but also severe allergic inflammatory conditions67,68, and mice with mutant Foxp3 also spontaneously develop allergic airway inflammation69. Indeed, mouse model studies have demonstrated that Tregs can suppress Th2 responses to allergens through an IL-10-dependent mechanism and that Treg-mediated suppression of DC activation is also involved in restriction of the response70,71. Furthermore, peripherally induced Tregs play a crucial role in restricting Th2-pathologies such as allergic inflammation and asthma in the lungs72. Consistent with this finding, patients with atopic asthma were documented to have decreased abundance of Tregs in bronchoalveolar lavage fluid73.
Finally, whereas tissue-specific Tregs represent only a fraction of the total respiratory tract Treg population in SPF mice56, it remains likely that this frequency would be impacted by antigen experience, such as in the context of “dirty/pet store” mice74. Interestingly, the frequency and distribution of tissue Tregs in humans appears to be related to age, wherein pediatric tissues demonstrate the highest frequencies of Tregs in mucosal and nonlymphoid tissues followed by a shift toward accumulation in lymphoid tissues during adulthood75. In this regard, whereas the total frequency of Tregs among total CD4+ T cells in the lung tissue of pediatric donors has been estimated to be roughly 15%, in adult lung tissue that number is reduced to roughly 5%. Phenotypically, among both children and adults, human lung tissue Tregs are mostly CD45RA- and CD69+, with a smaller fraction dually expressing CD10375. Importantly, each individual’s past history of antigen experience is also likely to impact their current status76,77,78. Studies examining the tissue-specific Treg TCR repertoire in the human respiratory tract will be influential79. Notably, among unmanipulated SPF mice, tissue context significantly impacts the Treg TCR repertoire wherein clonal diversity tends to be more restricted in peripheral tissues as compared to the spleen57. A final consideration, specific to the lung, is the elucidation of recent evidence demonstrating that respiratory tract tissue-resident memory T cells (Trm) may be more transient than has been reported for other peripheral tissues80,81,82. For example, the local draining lymph node may serve as a more permanent reservoir for respiratory Trm in lieu of long-term maintenance within the delicate pulmonary tissue80, thereby raising the notion that respiratory tract Tregs may not persist long-term within the tissue parenchyma. In conclusion, studies further elucidating the functional capabilities, antigen-specificities, and longevity of respiratory tract-specific tissue Tregs among both mice and humans will be informative.
Gastrointestinal tract tissue Tregs
The gastrointestinal (GI) tract, including the small intestine and colon, is the site of food ingestion and metabolite transport in addition to hosting a complex microbiome. Therefore, the mucosal GI immune system has the extraordinary task of tolerating orally ingested dietary antigens (a phenomenon known as oral tolerance)83,84 as well as beneficial commensal bacterial species (spp.), while concurrently providing barrier immunity against ingested and enteric pathogens (Fig. 1). Tregs are crucial mediators of GI tract tolerance84 and are maintained in high numbers in the GI tract, making up 20–30% of total CD4+ cells in the lamina propria of the small intestine and colon85,86. Ablation of Tregs or mutations in Foxp3 leads to systemic autoimmunity in mice and humans, including in the GI tract5,26,87,88. Moreover, Tregs are necessary for prevention of GI tract inflammation and Inflammatory Bowel Diseases (IBD) such as colitis89,90. Due to the increased need for tolerance in the GI tract, Tregs have been widely studied in the small intestine and colon, and more than any other mucosal site, a large quantity of research has been published91. Here we provide a broad rather than exhaustive overview of the vast and nuanced field of GI tract Treg biology in both health and disease.
As we have briefly introduced, Tregs canonically arise in the thymus, where Foxp3 is expressed during thymic differentiation following relatively high avidity interactions of the TCR with self antigens92,93,94. These thymically derived Tregs have thus been termed tTreg95 and are generally defined by the expression of the transcription factor Helios and cell surface receptor neuropilin1 (NRP1)96,97. However, Tregs also arise in the periphery (pTreg) in response to particular signals generated under tolerogenic conditions, including TGFβ98,99,100,101, and can sometimes express Helios despite having extra-thymic origins102. Given the major tolerogenic requirements of the GI tract, the colon and small intestine are a major site of pTreg induction85,95. In the mouse and human colon, ~30–35% of Tregs are NRP1+ and Helios+103,104, suggesting that the majority of GI tract Tregs are induced locally96,97,105. However, some colonic Tregs share TCR repertoires with Tregs in the thymus, suggesting that the colon hosts a combination of both pTregs and tTregs106. Likewise, Helios+ Treg proportions are mouse strain-dependent and can vary greatly107. Numerous studies have identified pTreg to be critical for tolerance to dietary antigens and the microbiota: mice lacking pTregs present with increased type 2 immunity and pathologies as well as dysbiosis in the GI tract72,108, and pTreg have been shown to be important for preventing inappropriate responses to food antigens in the GI tract109.
Given the demonstrated critical roles for pTreg in the GI tract, these cells have been intensely studied in recent years. pTregs in the GI tract can be further classified by expression of transcription factors associated with CD4+ effector T cell lineages. We will first discuss RORγt, the transcription factor associated with T-helper 17 (Th17) lineage differentiation85. In the mouse small intestine and colon, around 40% and 10%, respectively, of Tregs are negative for both RORγt and NRP185,109. In SPF mice weaned onto an antigen-free diet, small intestine NRP1lo Treg numbers are reduced, mostly due to a loss of RORγt- Tregs109. Interestingly, SPF mice treated with antibiotics are depleted of RORγt+, but not RORγT- Tregs, demonstrating that dietary, but not microbial, antigens are crucial for induction of RORγt- Tregs109. Furthermore, SPF mice weaned onto an antigen-free diet followed by immunization and challenge with Ova develop intestinal allergy, implicating RORγt- pTregs in the prevention of food allergy109 (Fig. 2).
Thus, RORγt- Tregs in the small intestine, and, in smaller numbers, in the colon, are induced by dietary antigens and are also necessary for continued tolerance to food-derived antigens. Although preservation of dietary tolerance is mainly attributed to RORγt- Tregs, food allergy in human infants has been associated with a lack of microbiota-induced RORγt+ Tregs, and administration of oral antigen in mice increases RORγt+ Treg numbers, demonstrating a role for RORγt+ Tregs in maintaining tolerance to dietary antigens as well as to commensals110,111,112. Additionally, microbiota-induced pTregs arising around the age of weaning are later required to prevent spontaneous Th2-mediated allergic responses to dietary antigens and protect from dextran sodium sulfate (DSS)-mediated severe colitis in mice113,114,115.
As briefly mentioned above, a large fraction of GI tract pTregs are RORγt positive. RORγt+ Tregs require expression of transcription factor c-Maf for development and function as highly suppressive Tregs that specifically regulate distinct T-helper subsets in different inflammatory contexts, such as limiting pathogenic Th17 inflammatory responses to Helicobacter hepaticus103,112,116,117,118. Many RORγt+ Tregs display a highly activated effector phenotype defined by expression of CD44, ICOS, and CTLA-4111,112,116,119. IL-10, a critical anti-inflammatory cytokine, is constitutively expressed on a large percentage of RORγt+ Tregs in both the small intestine and colon, and a loss of IL-10 results in spontaneous colitis in mice and humans, demonstrating the importance of Treg-derived IL-10 in preventing GI tract inflammation and immunopathology15,120,121,122.
In the mouse colon, 40–60% of Tregs are RORγt+, while RORγt+ Tregs make up a smaller fraction of small intestine Tregs85,103,107,112,116. In humans, IL-17-producing Tregs are found in the circulation and in the microenvironment of chronic ulcerative colitis123, and circulating RORγT+ memory Tregs can be found in humans124. RORγt+ Treg abundance in small intestine and colon is dramatically reduced in germ-free mice or mice treated with antibiotics103,109,112,125, demonstrating that the commensal microbiota is required for the generation of RORγt+ Tregs. A loss of RORγt+ Tregs in mice results in increased Type-2 immune responses and more severe oxazolone-induced colitis, suggesting that commensals regulate intestinal immune responses through induction of RORγt+ Tregs112. Furthermore, local bacterial antigens mediate colonic pTreg selection, especially for RORγt+ Tregs104,109,125,126. In support of Treg induction by commensal antigens, mouse colonic Tregs express TCR specific for Clostridium and commensal spp. that normally colonize the GI mucosal layer and expand in the presence of commensal bacteria126. Other commensals such as lactobacilli and bifidobacteria also increase induction and maturation of GI tract Tregs85. In summary, the small intestine Treg compartment is predominantly RORγt- and responsible for dietary tolerance, while colonic RORγt+ Tregs facilitate tolerance to commensal microbiota, but this distinction is not finite and GI tract Treg phenotype and function overlap.
GI tract pTregs are also heavily influenced by dietary and commensal-derived metabolites. Compared to Tregs in other tissues or lymphoid organs, Tregs in the mouse small intestine and colon express more aryl hydrocarbon receptor (AhR)—a nuclear sensor of dietary, microbiota, and other derivatives127— suggesting an elevated AhR-mediated metabolite dependence in GI tract Tregs91,103,128. Tryptophan, which can be metabolized by commensal microbiota into various AhR ligands127, promotes Treg development through various mechanisms91,129. Retinoic acid (RA), the bioactive derivative of vitamin A, is abundantly found in the GI tract and induces pTregs in mice when in combination with TGFβ91,130,131,132. Likewise, mice given Vitamin A-deficient diets or RA inhibitors display reduced RORγT+ but not Helios+ Treg numbers, suggesting a specific requirement for Vitamin A in the RORγT+ Treg population in the GI tract91,112. Among others, vitamins D3 and B9 promote expression of Foxp3 and anti-apoptotic BCL-2 on colonic Tregs85,133,134. Although some Treg:metabolite processes function independently of the microbiota, others rely heavily on bacteria. Commensals facilitate metabolite production by fermenting dietary fiber to produce short chain fatty acids (SCFA) and other products that promote pTreg induction, expansion, and function, especially in the mouse colon91,112,135,136,137. Bacterial products can also function as Toll-like receptor (TLR) ligands, adjuvanting the induction and function of GI tract pTregs85,91,138,139,140,141. Recently, gut bacteria have been shown transform bile acids into bioactive products that modulate local Treg function and promote pTreg induction in mouse and humans142,143,144.
A third population of GI Tregs expressing GATA3, the master transcription factor for T-helper 2 (Th2) CD4+ T cell differentiation, make up around 20 and 15%, respectively, of mouse small intestine and colon Tregs85,112,145,146. In humans, GATA3+ Tregs have thus far only been identified in blood91. GATA3 directly interacts with Foxp3 to regulate FoxP3 expression and transcriptional programs111,147,148. GATA3+ GI tract Tregs express Helios and are stable under germ-free conditions, suggesting thymic origins103,112. GATA3+ Tregs express IL-33R, which recognizes IL-33, an alarmin produced by intestinal epithelial cells during inflammation145,146. IL-33R ligation, along with TCR activation and IL-2, activates GATA3145, which drives Treg-mediated suppression. A loss of GATA3 expression abrogates accumulation of Tregs in the small intestine, particularly in the context of inflammation, where GATA3+ Tregs are critical to protect mouse GI tissues from collateral immunopathology during enteric infection with pathogenic microbes145. Additionally, small intestinal Tregs maintain epithelial barrier integrity by preserving epithelial cells (IEC) though various IL-10-mediated mechanisms in mice149, where the IEC physically separates lamina propria immune cells and the microbiota in the intestinal lumen111. Thus, GATA3+ Tregs are important for regulation of type-2 immunity and respond to cognate antigen and alarmins during inflammatory events to preserve GI tract tissues111. Further research is warranted to resolve whether GATA3+ Tregs also directly contribute to tissue repair, as is seen in other IL-33R+ Tregs30,38,44,111.
In addition to maintaining tolerance to commensal and dietary antigens, GI Tregs also contribute to humoral mucosal functions such as control of germinal center reactions to promote secretion of immunoglobulin A (IgA), which blocks invading pathogens from attaching to the mucosal epithelium150. Conversely, a loss of c-Maf+ RORγt+ Tregs leads to excessive, Th17 and IgA responses118, suggesting that GI tract pTregs balance protective vs pathogenic mucosal humoral responses. Mouse Tregs expressing B cell lymphoma 6 (BCL-6) migrate via CXCR5 to Peyer’s patches germinal centers where these Follicular Tregs (Tfr) control Tfh responses, thereby encouraging IgA production151. Small intestine Tregs specific for commensal flagellin antigens dampen mucosal uptake of commensal antigens through supporting production of IgA, in turn limiting local T cell activation and further maintaining tolerance to the microbiota in mice152. Finally, some GI Foxp3+ Tregs that receive environmental cues within Peyer’s patches may convert into Tfh cells, in the process losing Foxp3 expression, to then interact with B cells in mice153.
While Tregs in the small intestine and colon are indispensable for maintaining appropriate immune tolerance during homeostasis, colonic Tregs within the aberrant context of a cancer environment can be counterproductive. Tregs, including those expressing cytotoxic molecule granzyme B, are enriched in colorectal cancer (CRC), colon-draining lymph nodes, and other tumor sites and are associated with poorer disease prognosis in mouse and humans, likely related to the suppression of anti-tumor effector T cells154,155,156,157,158,159. Tumor environment-mediated immune dysbiosis may abnormally increase Treg recruitment to the tumor through chemotactic receptors or by expanding Tregs through TGFβ or IL-10160,161, where they dampen anti-tumor immune responses. To combat tumor-driven accumulation of Tregs in CRC tumors, therapeutic strategies to eliminate Tregs or block their suppressive functions may help to promote anti-tumor effector T cell immunity155. However, the role of Tregs in CRC is nuanced and context-dependent, as Tregs can also protect the mouse and human host from cancer-associated inflammation162,163 and BLIMP-1+ Tregs and Treg-derived IL-10 have been associated with decreased polyps and increased CRC survival in both mice and humans164,165,166. Together, these conflicting results suggest that distinct Treg subsets may perform different functions in intestinal cancers167,168. For example, in mice, Treg-specific loss of transcription factor TCF-1 expression, a suppressor of genes co-bound by FoxP3, increases Treg-mediated T cell suppression and promotes tumor growth in polyposis, demonstrating a role for Treg TCF-1 expression in tumor clearance169. Additionally, tumor-infiltrating Tregs in human CRC express low TCF-1 expression, suggesting that TCF-1 expression levels may predict CRC outcomes169. Therefore, Treg involvement in mucosal tumor immunity warrants further study to understand which Treg subsets may predict improved CRC outcomes and which subsets could be targeted therapeutically to increase survival.
Treg dysfunction has major implications for chronic and debilitating GI tract disease; therefore, additional research is necessary to elucidate potential Treg-targeting therapies to ameliorate IBD and allergy. Furthermore, oral vaccine delivery is an attractive route as a non-invasive vaccine strategy that could induce protective immune responses at the mucosal barrier, as tissue-resident memory T cells (Trm) are known to be indispensable for protection from mucosal infections such as herpes simplex virus 2 (HSV-2)170. However, further studies are needed to understand how to best leverage mucosal immune responses, including Tregs, at the site of infection while overcoming the tolerogenic tendencies of the oral mucosa and GI tract84,171,172,173,174.
Genitourinary tract tissue Tregs
The genitourinary (GU) tract, comprised of the urinary and reproductive organs, requires paradoxical immune responses. The mucosal GU tissues such as the vagina, cervix, and uterus serve as the entry point for both reproductive sperm and sexually transmitted infections (STIs). Therefore, the GU tract must be at once tolerant to select foreign antigens and commensal bacteria, while hostile to invading STI, poising the GU tract as a unique and complex immunological site. Mucosal Tregs are likely to be critical for facilitating the balance between tolerance and immunity.
Uterine Tregs
A healthy mammalian pregnancy requires careful immune tolerance at every stage, from the introduction of male sperm to the end of successful gestation. During sexual reproduction, the GU tract immune system must allow sperm, a foreign antigen, and a fetus— essentially a semi-allogeneic graft—to be tolerated for an extended period. Tregs in the blood and at the maternal-fetal interface in the placenta increase during pregnancy and are known mediators of fetal tolerance, preventing spontaneous abortion, fetal resorption, and preeclampsia in mice and humans175,176,177,178. Given the critical need for Treg-mediated tolerance during pregnancy, it is unsurprising that Tregs resident in the mucosal GU tract also play a role in pregnancy. As early as first exposure to male seminal antigens and before embryonic implantation, Tregs accumulate in the uterus and uterine-draining lymph nodes in mice179,180,181. During pregnancy, CD25+ cells increase two-fold in the mouse iliac and inguinal lymph nodes and make up 30% of uterine CD4+ T cells, and in both allogeneically and syngeneically mated mice, uterine Foxp3 mRNA concentration is 1000-times higher than in age-matched non-pregnant mouse uterine tissue, suggesting a robust increase in uterine Tregs during pregnancy175. A reduction in Tregs in early pregnancy causes uterine artery dysfunction in mice, demonstrating a tissue-specific role for Treg-mediated prevention of gestational hypertension and preeclampsia182 (Fig. 2). Given that immune responses in both mouse and human have been shown to be dampened during the luteal phase of the estrus cycle and in response to seminal extracellular vesicles, it is possible that GU mucosal Tregs in the uterus and vagina have additional immunosuppressive roles to promote pregnancy at the early stages of conception183,184,185,186,187. Treg-focused cellular therapies could be useful in preventing and treating preeclampsia and other pregnancy complications, but further research in humans is needed to design diagnostic tests for Treg function in early pregnancy and to identify the temporal window in which intervention is safe and effective188.
Cervical and vaginal Tregs
Historically, Tregs are understudied in the cervical and vaginal mucosa. However, as a barrier tissue site with constitutive exposure to commensal microbiota, plus the potential for exposures to male seminal antigens and microbial pathogens, the dynamics of the vaginal immune system must be carefully orchestrated, likely in part by Tregs.
In humans, the healthy vaginal mucosa hosts a microbiome dominated by lacotobacillus, and dysbiosis of the vaginal microbiome causes harmful overgrowth of fungal and bacterial pathogens, leading to bacterial vaginosis (BV), vaginal candidiasis, urinary tract infections, and dysregulated vaginal pH, all of which increase susceptibility to STIs and infertility189,190. As in the GI tract, the vaginal immune system must remain tolerogenic to commensal bacteria while allowing for appropriate immune responses to deleterious microbes; therefore, we hypothesize that Tregs in the vagina may also facilitate tolerance to commensal microbiota. In support of this, vaginal isolates of Lactobacillus crispatus, a predominant species of healthy vaginal bacteria, induces Tregs from conventional CD4+ T cells in a human mixed leukocyte reaction191. Vaginal dysbiosis such as BV or abnormal vaginal dominance by anerobic bacterial communities is associated with increased pro-inflammatory cytokines in the vagina and decreased peripheral Treg numbers, suggesting that a breach in normal vaginal commensals triggers a switch from tolerogenic to anti-microbial immune responses190,192,193. Thus, further studies are necessary to determine how Tregs may facilitate the tolerance:inflammation axis based on the presence of either commensals or harmful bacterial spp.
Vaginal and uterine Tregs help maintain a healthy mucosal environment during homeostasis and pregnancy, but they also facilitate appropriate immune responses to harmful pathogens. In vaginal herpes simplex virus 2 (HSV-2) infection in mice, Tregs in the vaginal-draining lymph nodes (dLN) are necessary to promote proper antigen-bearing dendritic cell (DC) migration from the vagina to the dLN, and a loss of these Tregs delays HSV-2-specific CD4+ T cell priming and results in worsened disease194,195. This demonstrates that Tregs in the dLN tune the antiviral CD4 T cell responses in the nearby vaginal tissue. Moreover, Tregs accumulate in the mouse vaginal mucosa early after HSV-2 infection, suggesting an additional need for Treg-mediated immune regulation at the site of infection194,195. Given the multitude of pathogens that may contact the vaginal mucosa, vaginal tissue Tregs likely have important roles in immune response coordination, such as limiting excessive inflammation during infection to prevent collateral immune-mediated tissue damage. In support of this, Treg-related cytokines exert anti-inflammatory effects during infection with Trichomonas vaginalis196. In the human endocervix, Tregs are inversely correlated with inflammatory cytokine concentrations and abundance of CD4+ T cells, suggesting that Tregs in the GU tract prevent genital inflammation and could potentially lower HIV acquisition risk through limiting HIV target cell availability197. Importantly, the roles of vaginal and uterine Tregs must be considered when designing mucosal vaccines, as Tregs promote anti-pathogenic immunity in some contexts, but dampen mucosal Trm responses in others. For example, intra-uterine immunization with non-adjuvanted UV-killed Chlamydia trachomatis in mice induces uterine Tregs that abrogate the effects of local effector T cells elicited by the vaccine198. Additionally, vaginal Tregs may directly contribute to tissue repair through production of Areg after infection or injury, as has been shown in lung and muscle-resident Tregs in mice30,38,44. Further vaginal Treg studies in both mouse and especially in human are necessary to determine how vaginal Tregs modulate local anti-pathogen immune responses, prevent mucosal tissue damage, and potentially execute unique, location-specific functions.
Conclusions
Peripheral Tregs are key mediators of systemic immune tolerance and immune orchestration, and mucosal tissue Tregs are no exception. In fact, Tregs at mucosal sites are charged with the paradoxical and highly nuanced role of facilitating protective immune responses to invading pathogens, while also allowing for immune quiescence in the context of sexual reproduction, inhaled or ingested harmless antigens, and commensal microbiota (Fig. 1). This immune balance is especially critical within the very delicate and highly specialized mucosal tissues. Although Tregs found in various mucosal tissues share the responsibility of balancing inflammatory immune responses, residency in distinct tissues is associated with previously unappreciated location-specific functions that lie beyond canonical Treg roles (Fig. 2). These specialized mucosal Tregs have implications for conditions such as allergy, autoimmunity, and cancer, where aberrant mucosal immune responses drive disease progression. Furthermore, the role of mucosal Tregs in anti-microbial immunity—especially respiratory infections and STIs—should not be underestimated. Importantly, immunity elicited by mucosal vaccination, depending on the context, may be either hindered or helped by Tregs. Therefore, further studies are necessary to elucidate how to best leverage Tregs to overcome oral tolerance and elicit protective resident T cells responses at the sites of bacterial and viral infection. Based on this growing body of mucosal Treg data, there is precedent to continue to characterize Tregs in other mucosal and nonlymphoid tissue sites, where they may perform distinct, yet-unknown tissue-specific functions.
References
Josefowicz, S. Z. & Rudensky, A. Control of regulatory T cell lineage commitment and maintenance. Immunity 30, 616–625 (2009).
Smigiel, K. S., Srivastava, S., Stolley, J. M. & Campbell, D. J. Regulatory T-cell homeostasis: steady-state maintenance and modulation during inflammation. Immunol. Rev. 259, 40–59 (2014).
Sakaguchi, S., Yamaguchi, T., Nomura, T. & Ono, M. Regulatory T cells and immune tolerance. Cell 133, 775–787 (2008).
Bennett, C. L. et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet 27, 20–21 (2001).
Brunkow, M. E. et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet 27, 68–73 (2001).
Wildin, R. S. et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet 27, 18–20 (2001).
Fontenot, J. D., Gavin, M. A. & Rudensky, A. Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4, 330–336 (2003).
Hori, S., Nomura, T. & Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061 (2003).
Khattri, R., Cox, T., Yasayko, S. A. & Ramsdell, F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 4, 337–342 (2003).
Gavin, M. A. et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature 445, 771–775 (2007).
Lin, W. et al. Regulatory T cell development in the absence of functional Foxp3. Nat. Immunol. 8, 359–368 (2007).
Hu, W. et al. Regulatory T cells function in established systemic inflammation and reverse fatal autoimmunity. Nat. Immunol. 22, 1163–1174 (2021).
Williams, L. M. & Rudensky, A. Y. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat. Immunol. 8, 277–284 (2007).
Li, M. O., Wan, Y. Y. & Flavell, R. A. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity 26, 579–591 (2007).
Rubtsov, Y. P. et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28, 546–558 (2008).
Gondek, D. C., Lu, L. F., Quezada, S. A., Sakaguchi, S. & Noelle, R. J. Cutting edge: contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J. Immunol. 174, 1783–1786 (2005).
Grossman, W. J. et al. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 21, 589–601 (2004).
Zhao, D. M., Thornton, A. M., DiPaolo, R. J. & Shevach, E. M. Activated CD4+CD25+ T cells selectively kill B lymphocytes. Blood 107, 3925–3932 (2006).
Loebbermann, J. et al. Regulatory T cells expressing granzyme B play a critical role in controlling lung inflammation during acute viral infection. Mucosal Immunol. 5, 161–172 (2012).
Wing, K. et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275 (2008).
Bopp, T. et al. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J. Exp. Med. 204, 1303–1310 (2007).
Borsellino, G. et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood 110, 1225–1232 (2007).
Deaglio, S. et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 204, 1257–1265 (2007).
Kobie, J. J. et al. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5’-adenosine monophosphate to adenosine. J. Immunol. 177, 6780–6786 (2006).
Pandiyan, P., Zheng, L., Ishihara, S., Reed, J. & Lenardo, M. J. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat. Immunol. 8, 1353–1362 (2007).
Kim, J. M., Rasmussen, J. P. & Rudensky, A. Y. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol. 8, 191–197 (2007).
Josefowicz, S. Z., Lu, L. F. & Rudensky, A. Y. Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol. 30, 531–564 (2012).
Belkaid, Y. & Tarbell, K. Regulatory T cells in the control of host-microorganism interactions (*).Annu Rev Immunol 27, 551–589 (2009).
Richert-Spuhler, L. E. & Lund, J. M. The immune fulcrum: regulatory t cells tip the balance between pro- and anti-inflammatory outcomes upon infection. Prog. Mol. Biol. Transl. Sci. 136, 217–243 (2015).
Burzyn, D. et al. A special population of regulatory T cells potentiates muscle repair. Cell 155, 1282–1295 (2013).
Cipolletta, D. et al. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 486, 549–553 (2012).
Sanchez Rodriguez, R. et al. Memory regulatory T cells reside in human skin. J. Clin. Invest 124, 1027–1036 (2014).
Kalekar L. A., et al. Regulatory T cells in skin are uniquely poised to suppress profibrotic immune responses. Sci. Immunol. 2019; 4, eaaw2910 (2019).
Ali, N. et al. Regulatory T cells in skin facilitate epithelial stem cell differentiation. Cell 169, 1119–1129 (2017).
Li, C. et al. TCR transgenic mice reveal stepwise, multi-site acquisition of the distinctive fat-treg phenotype. Cell 174, 285–299 (2018).
Vasanthakumar, A. et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat. Immunol. 16, 276–285 (2015).
Feuerer, M. et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 15, 930–939 (2009).
Kuswanto, W. et al. Poor repair of skeletal muscle in aging mice reflects a defect in local, interleukin-33-dependent accumulation of regulatory T cells. Immunity 44, 355–367 (2016).
Brincks, E. L. et al. Antigen-specific memory regulatory CD4+Foxp3+ T cells control memory responses to influenza virus infection. J. Immunol. 190, 3438–3446 (2013).
Durant, L. R. et al. Regulatory T cells prevent Th2 immune responses and pulmonary eosinophilia during respiratory syncytial virus infection in mice. J. Virol. 87, 10946–10954 (2013).
Leon, B., Bradley, J. E., Lund, F. E., Randall, T. D. & Ballesteros-Tato, A. FoxP3+ regulatory T cells promote influenza-specific Tfh responses by controlling IL-2 availability. Nat. Commun. 5, 3495 (2014).
Belkaid, Y. Regulatory T cells and infection: a dangerous necessity. Nat. Rev. Immunol. 7, 875–888 (2007).
Sell, S., McKinstry, K. K. & Strutt, T. M. Mouse models reveal role of T-cytotoxic and T-reg cells in immune response to influenza: implications for vaccine design. Viruses 11, 1 (2019).
Arpaia, N. et al. A distinct function of regulatory T. Cells Tissue Prot. Cell 162, 1078–1089 (2015).
Betts, R. J. et al. Influenza A virus infection results in a robust, antigen-responsive, and widely disseminated Foxp3+ regulatory T cell response. J. Virol. 86, 2817–2825 (2012).
D’Alessio, F. R. et al. CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J. Clin. Invest. 119, 2898–2913 (2009).
Ichikawa, T. et al. CD103(hi) Treg cells constrain lung fibrosis induced by CD103(lo) tissue-resident pathogenic CD4 T cells. Nat. Immunol. 20, 1469–1480 (2019).
Veiga-Parga, T., Sehrawat, S. & Rouse, B. T. Role of regulatory T cells during virus infection. Immunol. Rev. 255, 182–196 (2013).
Botta, D. et al. Dynamic regulation of T follicular regulatory cell responses by interleukin 2 during influenza infection. Nat. Immunol. 18, 1249–1260 (2017).
Shafiani, S. et al. Pathogen-specific Treg cells expand early during mycobacterium tuberculosis infection but are later eliminated in response to Interleukin-12. Immunity 38, 1261–1270 (2013).
Shafiani, S., Tucker-Heard, G., Kariyone, A., Takatsu, K. & Urdahl, K. B. Pathogen-specific regulatory T cells delay the arrival of effector T cells in the lung during early tuberculosis. J. Exp. Med. 207, 1409–1420 (2010).
Vick S. C., et al. A differential regulatory T cell signature distinguishes the immune landscape of COVID-19 hospitalized patients from those hospitalized with other respiratory viral infections. medRxiv https://pubmed.ncbi.nlm.nih.gov/33791720/ (2021).
Sadeghi, A. et al. Th17 and Treg cells function in SARS-CoV2 patients compared with healthy controls. J. Cell Physiol. 236, 2829–2839 (2021).
Szabo, P. A. et al. Longitudinal profiling of respiratory and systemic immune responses reveals myeloid cell-driven lung inflammation in severe COVID-19. Immunity 54, 797–814 (2021). e796.
Delacher, M. et al. Single-cell chromatin accessibility landscape identifies tissue repair program in human regulatory T cells. Immunity 54, 702–720 (2021). e717.
Delacher, M. et al. Genome-wide DNA-methylation landscape defines specialization of regulatory T cells in tissues. Nat. Immunol. 18, 1160–1172 (2017).
Delacher, M. et al. Precursors for nonlymphoid-tissue treg cells reside in secondary lymphoid organs and are programmed by the transcription factor BATF. Immunity 52, 295–312 (2020). e211.
DiSpirito J. R., et al. Molecular diversification of regulatory T cells in nonlymphoid tissues. Sci. Immunol. 3, eaat5861 (2018).
Miragaia, R. J. et al. Single-cell transcriptomics of regulatory T cells reveals trajectories of tissue adaptation. Immunity 50, 493–504 (2019). e497.
Szabo, P. A. et al. Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat. Commun. 10, 4706 (2019).
Munoz-Rojas A. R., Mathis D. Tissue regulatory T cells: regulatory chameleons. Nat. Rev. Immunol. 21, 597–611 (2021).
Liu Q., et al. IL-33-mediated IL-13 secretion by ST2+ Tregs controls inflammation after lung injury. JCI Insight 4, e123919 (2019).
Harb, H. et al. Notch4 signaling limits regulatory T-cell-mediated tissue repair and promotes severe lung inflammation in viral infections. Immunity 54, 1186–1199 (2021). e1187.
Dial, C. F., Tune, M. K., Doerschuk, C. M. & Mock, J. R. Foxp3(+) regulatory T cell expression of keratinocyte growth factor enhances lung epithelial proliferation. Am. J. Respir. Cell Mol. Biol. 57, 162–173 (2017).
Noval Rivas, M. & Chatila, T. A. Regulatory T cells in allergic diseases. J. Allergy Clin. Immunol. 138, 639–652 (2016).
Singh, R. et al. Regulatory T cells in respiratory health and diseases. Pulm. Med. 2019, 1907807 (2019).
Chatila, T. A. et al. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J. Clin. Invest 106, R75–R81 (2000).
Verbsky, J. W. & Chatila, T. A. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-related disorders: an evolving web of heritable autoimmune diseases. Curr. Opin. Pediatr. 25, 708–714 (2013).
Lin, W. et al. Allergic dysregulation and hyperimmunoglobulinemia E in Foxp3 mutant mice. J. Allergy Clin. Immunol. 116, 1106–1115 (2005).
Kearley, J., Barker, J. E., Robinson, D. S. & Lloyd, C. M. Resolution of airway inflammation and hyperreactivity after in vivo transfer of CD4+CD25+ regulatory T cells is interleukin 10 dependent. J. Exp. Med. 202, 1539–1547 (2005).
Lewkowich, I. P. et al. CD4+CD25+ T cells protect against experimentally induced asthma and alter pulmonary dendritic cell phenotype and function. J. Exp. Med. 202, 1549–1561 (2005).
Josefowicz, S. Z. et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 482, 395–399 (2012).
Hartl, D. et al. Quantitative and functional impairment of pulmonary CD4+CD25hi regulatory T cells in pediatric asthma. J. Allergy Clin. Immunol. 119, 1258–1266 (2007).
Beura, L. K. et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532, 512–516 (2016).
Thome, J. J. et al. Early-life compartmentalization of human T cell differentiation and regulatory function in mucosal and lymphoid tissues. Nat. Med. 22, 72–77 (2016).
Goulding, J. et al. Respiratory infections: do we ever recover? Proc. Am. Thorac. Soc. 4, 618–625 (2007).
Wissinger, E., Goulding, J. & Hussell, T. Immune homeostasis in the respiratory tract and its impact on heterologous infection. Semin Immunol. 21, 147–155 (2009).
Snyder, M. E. & Farber, D. L. Human lung tissue resident memory T cells in health and disease. Curr. Opin. Immunol. 59, 101–108 (2019).
Shevyrev, D. & Tereshchenko, V. Treg heterogeneity, function, and homeostasis. Front Immunol. 10, 3100 (2019).
Stolley J. M., et al. Retrograde migration supplies resident memory T cells to lung-draining LN after influenza infection. J. Exp. Med. 217, e20192197 (2020).
Takamura, S. Persistence in temporary lung niches: a survival strategy of lung-resident memory CD8(+) T cells. Viral Immunol. 30, 438–450 (2017).
Masopust, D. & Soerens, A. G. Tissue-resident t cells and other resident leukocytes. Annu. Rev. Immunol. 37, 521–546 (2019).
Pabst, O. & Mowat, A. M. Oral tolerance to food protein. Mucosal Immunol. 5, 232–239 (2012).
Hadis, U. et al. Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity 34, 237–246 (2011).
Tanoue, T., Atarashi, K. & Honda, K. Development and maintenance of intestinal regulatory T cells. Nat. Rev. Immunol. 16, 295–309 (2016).
Atarashi, K. et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331, 337–341 (2011).
Sakaguchi, S., Sakaguchi, N., Asano, M., Itoh, M. & Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 155, 1151–1164 (1995).
Sharma, R., Sung, S. S., Fu, S. M. & Ju, S. T. Regulation of multi-organ inflammation in the regulatory T cell-deficient scurfy mice. J. Biomed. Sci. 16, 20 (2009).
Mayer, C. T. et al. Few Foxp3(+) regulatory T cells are sufficient to protect adult mice from lethal autoimmunity. Eur. J. Immunol. 44, 2990–3002 (2014).
Mottet, C., Uhlig, H. H. & Powrie, F. Cutting edge: cure of colitis by CD4+CD25+ regulatory T cells. J. Immunol. 170, 3939–3943 (2003).
Whibley, N., Tucci, A. & Powrie, F. Regulatory T cell adaptation in the intestine and skin. Nat. Immunol. 20, 386–396 (2019).
Picca, C. C. et al. Role of TCR specificity in CD4+ CD25+ regulatory T-cell selection. Immunol. Rev. 212, 74–85 (2006).
Hsieh, C. S., Lee, H. M. & Lio, C. W. Selection of regulatory T cells in the thymus. Nat. Rev. Immunol. 12, 157–167 (2012).
Lee, H. M., Bautista, J. L., Scott-Browne, J., Mohan, J. F. & Hsieh, C. S. A broad range of self-reactivity drives thymic regulatory T cell selection to limit responses to self. Immunity 37, 475–486 (2012).
Savage, P. A., Klawon, D. E. J. & Miller, C. H. Regulatory T cell development. Annu. Rev. Immunol. 38, 421–453 (2020).
Thornton, A. M. et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J. Immunol. 184, 3433–3441 (2010).
Singh, K., Hjort, M., Thorvaldson, L. & Sandler, S. Concomitant analysis of Helios and Neuropilin-1 as a marker to detect thymic derived regulatory T cells in naive mice. Sci. Rep. 5, 7767 (2015).
Chen, W. et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 198, 1875–1886 (2003).
Kretschmer, K. et al. Inducing and expanding regulatory T cell populations by foreign antigen. Nat. Immunol. 6, 1219–1227 (2005).
Selvaraj, R. K. & Geiger, T. L. A kinetic and dynamic analysis of Foxp3 induced in T cells by TGF-beta. J. Immunol. 179, 11 (2007). p following 1390.
Zheng, S. G., Wang, J. H., Gray, J. D., Soucier, H. & Horwitz, D. A. Natural and induced CD4+CD25+ cells educate CD4+CD25- cells to develop suppressive activity: the role of IL-2, TGF-beta, and IL-10. J. Immunol. 172, 5213–5221 (2004).
Pratama A., Schnell A., Mathis D., Benoist C. Developmental and cellular age direct conversion of CD4+ T cells into RORgamma+ or Helios+ colon Treg cells. J. Exp. Med. 217, e20190428 (2020).
Sefik, E. et al. MUCOSAL IMMUNOLOGY. Individual intestinal symbionts induce a distinct population of RORgamma(+) regulatory T cells. Science 349, 993–997 (2015).
Nutsch, K. et al. Rapid and efficient generation of regulatory T cells to commensal antigens in the periphery. Cell Rep. 17, 206–220 (2016).
Weiss, J. M. et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J. Exp. Med. 209, 1723–1742 (2012). S1721.
Cebula, A. et al. Thymus-derived regulatory T cells contribute to tolerance to commensal microbiota. Nature 497, 258–262 (2013).
Ramanan, D. et al. An immunologic mode of multigenerational transmission governs a gut treg setpoint. Cell 181, 1276–1290 (2020). e1213.
Campbell, C. et al. Extrathymically generated regulatory T cells establish a niche for intestinal border-dwelling bacteria and affect physiologic metabolite balance. Immunity 48, 1245–1257 (2018). e1249.
Kim, K. S. et al. Dietary antigens limit mucosal immunity by inducing regulatory T cells in the small intestine. Science 351, 858–863 (2016).
Abdel-Gadir, A. et al. Microbiota therapy acts via a regulatory T cell MyD88/RORgammat pathway to suppress food allergy. Nat. Med. 25, 1164–1174 (2019).
Cosovanu, C. & Neumann, C. The many functions of Foxp3(+) regulatory T cells in the intestine. Front. Immunol. 11, 600973 (2020).
Ohnmacht, C. et al. MUCOSAL IMMUNOLOGY. The microbiota regulates type 2 immunity through RORgammat(+) T cells. Science 349, 989–993 (2015).
Knoop K. A., et al. Synchronization of mothers and offspring promotes tolerance and limits allergy. JCI Insight 5 e137943 (2020).
Knoop, K. A., McDonald, K. G., Hsieh, C. S., Tarr, P. I. & Newberry, R. D. Regulatory T cells developing peri-weaning are continually required to restrain Th2 systemic responses later in life. Front Immunol. 11, 603059 (2020).
Al Nabhani, Z. et al. A weaning reaction to microbiota is required for resistance to immunopathologies in the adult. Immunity 50, 1276–1288 (2019). e1275.
Yang, B. H. et al. Foxp3(+) T cells expressing RORgammat represent a stable regulatory T-cell effector lineage with enhanced suppressive capacity during intestinal inflammation. Mucosal Immunol. 9, 444–457 (2016).
Xu, M. et al. c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 554, 373–377 (2018).
Neumann, C. et al. c-Maf-dependent Treg cell control of intestinal TH17 cells and IgA establishes host-microbiota homeostasis. Nat. Immunol. 20, 471–481 (2019).
Lochner, M. et al. In vivo equilibrium of proinflammatory IL-17+ and regulatory IL-10+ Foxp3+ RORgamma t+ T cells. J. Exp. Med. 205, 1381–1393 (2008).
Glocker, E. O. et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 361, 2033–2045 (2009).
Glocker, E. O., Kotlarz, D., Klein, C., Shah, N. & Grimbacher, B. IL-10 and IL-10 receptor defects in humans. Ann. N. Y. Acad. Sci. 1246, 102–107 (2011).
Kamanaka, M. et al. Expression of interleukin-10 in intestinal lymphocytes detected by an interleukin-10 reporter knockin tiger mouse. Immunity 25, 941–952 (2006).
Kryczek, I. et al. IL-17+ regulatory T cells in the microenvironments of chronic inflammation and cancer. J. Immunol. 186, 4388–4395 (2011).
Ayyoub, M. et al. Human memory FOXP3+ Tregs secrete IL-17 ex vivo and constitutively express the T(H)17 lineage-specific transcription factor RORgamma t. Proc. Natl Acad. Sci. USA 106, 8635–8640 (2009).
Lochner, M. et al. Restricted microbiota and absence of cognate TCR antigen leads to an unbalanced generation of Th17 cells. J. Immunol. 186, 1531–1537 (2011).
Lathrop, S. K. et al. Peripheral education of the immune system by colonic commensal microbiota. Nature 478, 250–254 (2011).
Shinde, R. & McGaha, T. L. The aryl hydrocarbon receptor: connecting immunity to the microenvironment. Trends Immunol. 39, 1005–1020 (2018).
Ye, J. et al. The aryl hydrocarbon receptor preferentially marks and promotes gut regulatory T cells. Cell Rep. 21, 2277–2290 (2017).
Mezrich, J. D. et al. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 185, 3190–3198 (2010).
Coombes, J. L. et al. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J. Exp. Med. 204, 1757–1764 (2007).
Mucida, D. et al. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317, 256–260 (2007).
Sun, C. M. et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 204, 1775–1785 (2007).
Kang, S. W. et al. 1,25-Dihyroxyvitamin D3 promotes FOXP3 expression via binding to vitamin D response elements in its conserved noncoding sequence region. J. Immunol. 188, 5276–5282 (2012).
Yamaguchi, T. et al. Control of immune responses by antigen-specific regulatory T cells expressing the folate receptor. Immunity 27, 145–159 (2007).
Arpaia, N. et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455 (2013).
Furusawa, Y. et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450 (2013).
Smith, P. M. et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573 (2013).
Danne, C. et al. A large polysaccharide produced by helicobacter hepaticus induces an anti-inflammatory gene signature in macrophages. Cell Host Microbe 22, 733–745 (2017). e735.
Round, J. L. & Mazmanian, S. K. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl Acad. Sci. USA 107, 12204–12209 (2010).
Shen, Y. et al. Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell Host Microbe 12, 509–520 (2012).
Verma R., et al. Cell surface polysaccharides of Bifidobacterium bifidum induce the generation of Foxp3(+) regulatory T cells. Sci. Immunol. 3, eaat6975 (2018).
Campbell, C. et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 581, 475–479 (2020).
Hang, S. et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 576, 143–148 (2019).
Li, W. et al. A bacterial bile acid metabolite modulates Treg activity through the nuclear hormone receptor NR4A1. Cell Host Microbe 29, 1366–1377 (2021). e1369.
Wohlfert, E. A. et al. GATA3 controls Foxp3(+) regulatory T cell fate during inflammation in mice. J. Clin. Invest. 121, 4503–4515 (2011).
Schiering, C. et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 513, 564–568 (2014).
Rudra, D. et al. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat. Immunol. 13, 1010–1019 (2012).
Wang, Y., Su, M. A. & Wan, Y. Y. An essential role of the transcription factor GATA-3 for the function of regulatory T cells. Immunity 35, 337–348 (2011).
Biton, M. et al. T helper cell cytokines modulate intestinal stem cell renewal and differentiation. Cell 175, 1307–1320 (2018). e1322.
Corthesy, B. Multi-faceted functions of secretory IgA at mucosal surfaces. Front. Immunol. 4, 185 (2013).
Kawamoto, S. et al. Foxp3(+) T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity 41, 152–165 (2014).
Cong, Y., Feng, T., Fujihashi, K., Schoeb, T. R. & Elson, C. O. A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc. Natl Acad. Sci. USA 106, 19256–19261 (2009).
Tsuji, M. et al. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer’s patches. Science 323, 1488–1492 (2009).
Sun, B., Liu, M., Cui, M. & Li, T. Granzyme B-expressing treg cells are enriched in colorectal cancer and present the potential to eliminate autologous T conventional cells. Immunol. Lett. 217, 7–14 (2020).
Zhang, X., Kelaria, S., Kerstetter, J. & Wang, J. The functional and prognostic implications of regulatory T cells in colorectal carcinoma. J. Gastrointest. Oncol. 6, 307–313 (2015).
Chang, L. Y. et al. Tumor-derived chemokine CCL5 enhances TGF-beta-mediated killing of CD8(+) T cells in colon cancer by T-regulatory cells. Cancer Res. 72, 1092–1102 (2012).
Betts, G. et al. Suppression of tumour-specific CD4(+) T cells by regulatory T cells is associated with progression of human colorectal cancer. Gut 61, 1163–1171 (2012).
Colombo, M. P. & Piconese, S. Regulatory-T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nat. Rev. Cancer 7, 880–887 (2007).
Zou, W. Regulatory T cells, tumour immunity and immunotherapy. Nat. Rev. Immunol. 6, 295–307 (2006).
Tokuno, K., Hazama, S., Yoshino, S., Yoshida, S. & Oka, M. Increased prevalence of regulatory T-cells in the peripheral blood of patients with gastrointestinal cancer. Anticancer Res. 29, 1527–1532 (2009).
Terme, M. et al. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 73, 539–549 (2013).
Khazaie, K., Bonertz, A. & Beckhove, P. Current developments with peptide-based human tumor vaccines. Curr. Opin. Oncol. 21, 524–530 (2009).
Curiel, T. J. Regulatory T cells and treatment of cancer. Curr. Opin. Immunol. 20, 241–246 (2008).
Ward-Hartstonge, K. A. et al. Inclusion of BLIMP-1(+) effector regulatory T cells improves the Immunoscore in a cohort of New Zealand colorectal cancer patients: a pilot study. Cancer Immunol. Immunother. 66, 515–522 (2017).
Chung, A. Y. et al. Oral interleukin-10 alleviates polyposis via neutralization of pathogenic T-regulatory cells. Cancer Res. 74, 5377–5385 (2014).
Dennis, K. L. et al. Adenomatous polyps are driven by microbe-instigated focal inflammation and are controlled by IL-10-producing T cells. Cancer Res. 73, 5905–5913 (2013).
Olguin J. E., Medina-Andrade I., Rodriguez T., Rodriguez-Sosa M., Terrazas L. I. Relevance of regulatory T cells during colorectal cancer development. Cancers 12, 1888 (2020).
Szeponik, L. et al. Intratumoral regulatory T cells from colon cancer patients comprise several activated effector populations. BMC Immunol. 22, 58 (2021).
Osman, A. et al. TCF-1 controls Treg cell functions that regulate inflammation, CD8(+) T cell cytotoxicity and severity of colon cancer. Nat. Immunol. 22, 1152–1162 (2021).
Iwasaki, A. Exploiting mucosal immunity for antiviral vaccines. Annu. Rev. Immunol. 34, 575–608 (2016).
Park, J. Y., Chung, H., DiPalma, D. T., Tai, X. & Park, J. H. Immune quiescence in the oral mucosa is maintained by a uniquely large population of highly activated Foxp3(+) regulatory T cells. Mucosal Immunol. 11, 1092–1102 (2018).
Bhattacharjee, A. & Hand, T. W. Role of nutrition, infection, and the microbiota in the efficacy of oral vaccines. Clin. Sci. 132, 1169–1177 (2018).
Williams, W. B. et al. HIV-1 VACCINES. Diversion of HIV-1 vaccine-induced immunity by gp41-microbiota cross-reactive antibodies. Science 349, aab1253 (2015).
Price, D. N., Kusewitt, D. F., Lino, C. A., McBride, A. A. & Muttil, P. Oral tolerance to environmental mycobacteria interferes with intradermal, but not pulmonary, immunization against tuberculosis. PLoS Pathog. 12, e1005614 (2016).
Aluvihare, V. R., Kallikourdis, M. & Betz, A. G. Regulatory T cells mediate maternal tolerance to the fetus. Nat. Immunol. 5, 266–271 (2004).
Quinn, K. H., Lacoursiere, D. Y., Cui, L., Bui, J. & Parast, M. M. The unique pathophysiology of early-onset severe preeclampsia: role of decidual T regulatory cells. J. Reprod. Immunol. 91, 76–82 (2011).
Santner-Nanan, B. et al. et al. Systemic increase in the ratio between Foxp3+ and IL-17-producing CD4+ T cells in healthy pregnancy but not in preeclampsia. J. Immunol. 183, 7023–7030 (2009).
Sasaki, Y. et al. Proportion of peripheral blood and decidual CD4(+) CD25(bright) regulatory T cells in pre-eclampsia. Clin. Exp. Immunol. 149, 139–145 (2007).
Guerin, L. R. et al. Seminal fluid regulates accumulation of FOXP3+ regulatory T cells in the preimplantation mouse uterus through expanding the FOXP3+ cell pool and CCL19-mediated recruitment. Biol. Reprod. 85, 397–408 (2011).
Moldenhauer, L. M. et al. Cross-presentation of male seminal fluid antigens elicits T cell activation to initiate the female immune response to pregnancy. J. Immunol. 182, 8080–8093 (2009).
Robertson, S. A. et al. Seminal fluid drives expansion of the CD4+CD25+ T regulatory cell pool and induces tolerance to paternal alloantigens in mice. Biol. Reprod. 80, 1036–1045 (2009).
Care, A. S. et al. Reduction in regulatory T cells in early pregnancy causes uterine artery dysfunction in mice. Hypertension 72, 177–187 (2018).
Vojtech, L. et al. Extracellular vesicles in human semen modulate antigen-presenting cell function and decrease downstream antiviral T cell responses. PLoS One 14, e0223901 (2019).
Wira, C. R. & Fahey, J. V. A new strategy to understand how HIV infects women: identification of a window of vulnerability during the menstrual cycle. AIDS 22, 1909–1917 (2008).
Wira, C. R. et al. Sex hormone regulation of innate immunity in the female reproductive tract: the role of epithelial cells in balancing reproductive potential with protection against sexually transmitted pathogens. Am. J. Reprod. Immunol. 63, 544–565 (2010).
Wira, C. R., Fahey, J. V., Rodriguez-Garcia, M., Shen, Z. & Patel, M. V. Regulation of mucosal immunity in the female reproductive tract: the role of sex hormones in immune protection against sexually transmitted pathogens. Am. J. Reprod. Immunol. 72, 236–258 (2014).
Hughes S. M., et al. Impact of the menstrual cycle and ethinyl estradiol/etonogestrel contraceptive vaginal ring on granulysin and other mucosal immune mediators. Am. J. Reprod. Immunol. 86, e13412 (2021).
Robertson, S. A. et al. Therapeutic potential of regulatory T cells in preeclampsia-opportunities and challenges. Front Immunol. 10, 478 (2019).
Ma, B., Forney, L. J. & Ravel, J. Vaginal microbiome: rethinking health and disease. Annu Rev. Microbiol 66, 371–389 (2012).
Campisciano, G., Zanotta, N., Licastro, D., De Seta, F. & Comar, M. In vivo microbiome and associated immune markers: new insights into the pathogenesis of vaginal dysbiosis. Sci. Rep. 8, 2307 (2018).
Eslami, S. et al. Lactobacillus crispatus strain SJ-3C-US induces human dendritic cells (DCs) maturation and confers an anti-inflammatory phenotype to DCs. APMIS 124, 697–710 (2016).
Schellenberg, J. J. et al. Bacterial vaginosis, HIV serostatus and T-cell subset distribution in a cohort of East African commercial sex workers: retrospective analysis. AIDS 26, 387–393 (2012).
Amabebe, E. & Anumba, D. O. C. The vaginal microenvironment: the physiologic role of lactobacilli. Front. Med. 5, 181 (2018).
Soerens, A. G., Da Costa, A. & Lund, J. M. Regulatory T cells are essential to promote proper CD4 T-cell priming upon mucosal infection. Mucosal Immunol. 9, 1395–1406 (2016).
Lund, J. M., Hsing, L., Pham, T. T. & Rudensky, A. Y. Coordination of early protective immunity to viral infection by regulatory T cells. Science 320, 1220–1224 (2008).
Nemati M., Malla N., Yadav M., Khorramdelazad H., Jafarzadeh A. Humoral and T cell-mediated immune response against trichomoniasis. Parasite Immunol. 40, https://doi.org/10.1111/pim.12510 (2018).
Ssemaganda A., et al. Endocervical regulatory T cells are associated with decreased genital inflammation and lower HIV target cell abundance. Front. Immunol. 12, 726472 (2021).
Stary, G. et al. VACCINES. A mucosal vaccine against Chlamydia trachomatis generates two waves of protective memory T cells. Science 348, aaa8205 (2015).
Acknowledgements
This work was supported by R01 AI141435 and R01 AI131914 from the National Institutes of Allergy and Infectious Diseases (NIAID) (to J.M.L.). B.R.T. was funded by the Viral Pathogenesis Training Grant (T32 AI083203). L.E.R. is a 2021 recipient of an AAI Career Reentry Fellowship.
Author information
Authors and Affiliations
Contributions
B.R.T., L.E.R. and J.M.L. conceived of and wrote the manuscript jointly.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Traxinger, B.R., Richert-Spuhler, L.E. & Lund, J.M. Mucosal tissue regulatory T cells are integral in balancing immunity and tolerance at portals of antigen entry. Mucosal Immunol 15, 398–407 (2022). https://doi.org/10.1038/s41385-021-00471-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-021-00471-x
This article is cited by
-
Regulatory T cells in the face of the intestinal microbiota
Nature Reviews Immunology (2023)
-
Mucosal viral infection induces a regulatory T cell activation phenotype distinct from tissue residency in mouse and human tissues
Mucosal Immunology (2022)
-
A spotlight on heightened T cell complexity and relevance in mucosal tissues
Mucosal Immunology (2022)
