
Abstract
Cancer-associated fibroblasts (CAFs), as a central component of the tumor microenvironment in primary and metastatic tumors, profoundly influence the behavior of cancer cells and are involved in cancer progression through extensive interactions with cancer cells and other stromal cells. Furthermore, the innate versatility and plasticity of CAFs allow their education by cancer cells, resulting in dynamic alterations in stromal fibroblast populations in a context-dependent manner, which highlights the importance of precise assessment of CAF phenotypical and functional heterogeneity. In this review, we summarize the proposed origins and heterogeneity of CAFs as well as the molecular mechanisms regulating the diversity of CAF subpopulations. We also discuss current strategies to selectively target tumor-promoting CAFs, providing insights and perspectives for future research and clinical studies involving stromal targeting.
Similar content being viewed by others


Introduction
The concept of the tumor microenvironment (TME), although first proposed by Stephen Paget in the “seed and soil theory” in 1889, has been widely appreciated only in recent years based on mounting evidence showing that the heterotypic signaling among the diverse cell types within a tumor cooperatively creates a supportive niche that favors cancer cell survival, outgrowth and escape from immunosurveillance1. Thus, the behavior of cancers depends not only on cancer cell-autonomous defects but also on cancer cell-extrinsic factors, in particular, the intricate tumor microenvironment (TME). The TME, as a dynamic niche composed of a set of cellular and molecular components, closely interacts with cancer cells to meet their nutrient and metabolic demands2. Diverse cell types contribute to TME formation, including immune cells, cancer-associated fibroblasts (CAFs) and even normal epithelial cells, which together are termed stromal cells3. Soluble factors, such as growth factors and cytokines, as well as capillaries and the extracellular matrix (ECM) surrounding cancer cells, together constitute the complex network of tumor stromal signaling mediated by both tumor and stromal cells3.
CAFs, also known as activated fibroblasts, are central to the reactive stroma within the TME, as they not only interact extensively with cancer cells via secreted molecules or cell‒cell adhesion but also indirectly influence cancer cells via ECM remodeling and immune cell infiltration4. While there is an overwhelming abundance of studies that support a tumor-promoting function for CAFs, some studies have noted that CAFs may restrain cancer progression as a host defense mechanism against neoplasia5,6. This contradiction can be explained by the high heterogeneity and plasticity of CAFs, which are possibly dependent on CAF precursor origin, cancer type and tumor progression stage. Indeed, distinct CAF subtypes have been identified with specific molecular markers, such as myofibroblast-like CAFs (myCAFs)7, inflammatory CAFs (iCAFs)7 and antigen-presenting CAFs (ApCAFs)8, which perform different and, in some cases, even contradictory roles in tumorigenesis. Moreover, recent studies using single-cell RNA-sequencing and proteomic technology have further dissected CAF subpopulations based on their distinct transcriptional profiles and demonstrated dynamic heterogeneous modifications of stromal myofibroblast populations in a context-dependent manner2,9,10. The variations in stromal composition not only shape the intratumoral architecture but also contribute to functional changes in tumor cell behavior, highlighting the importance of precise assessment of CAFs when considering treatment options. In this review, the proposed origins and heterogeneity of CAFs are summarized with a special focus on the therapeutic potential of CAFs. We also present recent advancements in specific targeting of protumorigenic CAFs or interference with their activity as potential strategies in anticancer therapy.
Origins and Heterogeneity of CAFs
Fibroblasts and acute wound healing
Initially identified in a wound healing response, fibroblasts play a central role in tissue repair, in which quiescent local tissue fibroblasts respond to tissue injury and become reversibly activated to initiate regenerative repair11. Activated fibroblasts, also termed myofibroblasts due to their high expression of α‑smooth muscle actin (α-SMA), acquire the capabilities of enhanced contractility, ECM production and inflammatory mediator secretion that together initiate wound healing responses, such as closure of the wound and production of connective tissue12. Interestingly, myofibroblasts also contribute to ECM turnover at the late stage of tissue repair with the synthesis of ECM-degrading proteases, such as matrix metalloproteinases (MMPs) and urokinase-type plasminogen activator (uPA), thus facilitating the restoration of the normal tissue architecture without scarring13. Once the repair process is complete, these transiently activated fibroblasts undergo either apoptosis or reprogramming to the resting state14.
Fibroblasts and chronic wound healing/cancer
Unlike the acute wound healing response, which is a natural physiological reaction to acute tissue injury, chronic or repetitive injury such as toxic, metabolic, or infectious insult often results in continuous activation of fibroblasts and excessive ECM component deposition, ultimately leading to pathological tissue fibrosis with impaired organ function15. One of the key mechanisms underlying tissue repair versus irreversible fibrosis is that in acute wound repair, fibroblasts are transiently activated, while during repetitive damage, fibroblasts become resistant to apoptosis or have a limited ability to reacquire a quiescent phenotype14.
Cancer, especially solid tumors, has long been considered a nonhealing wound16 and shares many features with tissue fibrosis, such as activated fibroblasts and increased stiffness of the ECM17. Fibroblasts at the site of a tumor, specifically referred to as CAFs, remain perpetually activated with a high capacity for ECM synthesis and microenvironmental remodeling, leading to stromal desmoplasia, a phenomenon characterized by increased deposition of ECM components in tumors. In this regard, CAFs share many basic characteristics, such as a secretory phenotype and capacity to synthesize ECM components, with fibroblasts found in nonmalignant tissue fibrosis. Therefore, the classic markers found to be expressed in fibroblasts, including α-SMA, vimentin, desmin, fibroblast-specific protein 1 (FSP1; also known as S100A4) and fibroblast activation protein (FAP), have been conventionally used to distinguish CAFs in recent years3,18. Notably, CAFs, while remodeling the TME and influencing cancer cell behavior, are also directly or indirectly reprogrammed by cancer cells and other stromal cells, ultimately displaying distinct epigenetic and transcriptional profiles correlated with their robust proliferative and invasive properties19. Thus, a set of surface markers, such as platelet-derived growth factor receptor-α/β (PDGFRα)/PDGFRβ20, discoidin domain-containing receptor 2 (DDR2)21 and integrin α11β122, have emerged to identify CAFs in the context of a specific TME. Interestingly, it is suggested that the same marker at different expression levels may define the CAF subsets associated with specific stages of cancer development, as loss of caveolin1 (CAV1) is found in metabolically reprogrammed CAFs that promote tumorigenesis23, while high CAV1-expressing CAFs contribute to invasion and metastasis in breast cancer24. However, none of these markers is exclusively expressed by CAFs; for example, desmin and PDGFRβ are also present in perivascular cells25, while FAP is expressed in a subset of CD45+ immune cells26, suggesting the possibility of diverse cellular origins of CAFs and necessitating in-depth biological deciphering of CAF evolution. Indeed, single-cell RNA-sequencing (scRNA-seq) analysis, which enables profiling of gene expression over the whole transcriptome at single-cell resolution, indicates that no single marker appears capable of discriminating CAFs from all other cell types and or even discriminating CAF subtypes27,28,29. Thus, combinations of two or more biomarkers with high discriminatory capacity are emerging to differentiate and isolate all CAFs across distinct cancer types (Table 1).
Cellular origins of CAFs
CAFs derived from tissue resident fibroblasts or nonfibroblast lineages
Although generally termed CAFs to describe all activated fibroblasts in the TME of solid cancers, CAFs are actually highly heterogeneous populations that can originate from disparate precursors (Fig. 1). To date, the precise cellular origins of CAFs remain elusive owing to their substantial heterogeneity and a lack of definitive biomarkers for each subset. The most direct source of CAFs is normal resident fibroblasts or quiescent stellate cells. CAFs in the pancreas and liver are traditionally thought to originate from pancreatic stellate cells (PSCs)30 and hepatic stellate cells (HSCs)31, respectively, under the influence of tumor-derived stimuli. For example, cancer cell-derived chemokines, cytokines and microRNAs are found to activate PSCs or HSCs, enabling them to gain myofibroblast-like features and transcriptional signatures associated with CAFs in pancreatic ductal adenocarcinoma (PDAC) or hepatocellular carcinoma32,33,34. Activated PSCs and HSCs further maintain their own activity with enhanced synthetic and secretory capacities via autocrine loops and contribute to desmoplasia35. Although extensively studied in vitro, the contribution of PSCs to PDAC CAFs in vivo in the context of tumorigenesis remains elusive. In a recent work using a lineage-labeling approach to trace the fate of PSCs in vivo, Helms et al. found that, contrary to expectations, PSCs only give rise to a small minority of CAFs in PDAC36, suggesting the existence of diverse CAF progenitors and raising an important question that needs to be addressed regarding the additional cellular origins of PDAC CAFs.

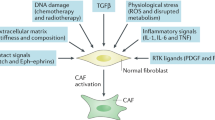
The heterogeneity of fibroblasts could be attributed to the multiple origins of the precursor cells, the phenotypical diversity of subsets and the distinct function of each subset. Potential cellular sources include local tissue resident stellate cells and normal fibroblasts and nonfibroblast lineage or recruited bone marrow-derived mesenchymal stem cells (MSCs) and macrophages. The main subsets of CAFs include myCAFs, iCAFs and apCAFs, which exhibit different biological features and result in phenotypical diversity and functional heterogeneity in cancer progression. However, the distinct subsets of CAFs are not permanent but interconvertible via manipulation of specific signaling, as shown by the conversion between iCAFs and myCAFs via the TGFβ or IL-6 signaling pathway of CAFs. myCAFs: myofibroblast-like CAFs; iCAFs: inflammatory CAFs; apCAFs: antigen-presenting CAFs.
In fact, normal resident fibroblasts, which reside around tumor cells, have been found to be activated via the tumor cell-derived signaling pathway and give rise to a subset of CAFs in PDAC37, gastrointestinal cancer38 and breast cancer20. Furthermore, even local fibroblasts are not homogenous but consist of distinct populations39; therefore, current studies attempt to trace specific lineages of fibroblasts and delineate their contribution to stroma formation. In a recent study using lineage tracing and dual recombinase approaches to follow the fate of 2 normal fibroblast populations marked by the expression of Gli1 and Hoxb6, Garcia et al discovered that Gli1, but not Hoxb6, specifically contributes to a portion of PDAC CAFs40. Similarly, Kobayashi et al. found a CAF subset marked by melanoma cell adhesion molecule (MCAM) derived predominantly from intestinal pericryptal leptin receptor (Lepr) lineage cells38, suggesting that inherent fibroblast heterogeneity may be linked to a specific subpopulation of CAFs.
In addition to the mesenchymal lineage, CAFs have been found to originate from multiple nonfibroblast lineage cells, including epithelial41 and endothelial cells41, through epithelial/endothelial-to-mesenchymal transition (EMT/EndMT). Other suggested CAF precursors, although less common, include adipocytes42, pericytes43, mesothelial cells44, and smooth muscle cells45.
CAFs derived from recruited bone marrow cells
Although predominantly observed to be of local origin38,46, lineage tracing using murine models and human samples has revealed the potential of bone marrow contribution to the CAF pool in several neoplasias, including rectal adenoma47, gastric cancer47, hepatocellular carcinoma48, PDAC49, and breast cancer50. Mesenchymal stem cells (MSCs) recruited from the bone marrow can differentiate into a subpopulation of CAFs under tumor-derived TGF-β, WNT, and IL-6/STAT3 signaling51,52. Furthermore, the finding that BM-derived multilineage hematopoietic cells were engrafted in the tumors of recipient mouse models suggests the possibility that other bone marrow-derived cells may serve as CAF precursors. Indeed, some studies have shown that bone marrow macrophages/monocytes can convert into CAFs in PDAC53,54 and Lewis lung carcinoma (LLC)55 via macrophage–myofibroblast transition (MMT).
Collectively, overwhelming evidence now points toward multiple origins contributing to the CAF pool rather than one28. In fact, a collection of diverse subpopulations of CAFs from different progenitors coexist in distinct tumor types56 and coevolve with epithelial genetic events during the development of cancer, resulting in the temporal and spatial dynamics of CAFs20. Thus, it is tempting but also challenging to investigate the full CAF reservoir and the mechanisms governing the transformation from normal precursors to CAF subtypes during cancer evolution to gain an in-depth understanding of the tumor-associated stroma for targeted anticancer therapy.
CAF heterogeneity and plasticity
While the presence of CAF subtypes based on their distinct expression patterns has long been accepted, functional categorization was first recapitulated in a coculture system of PDAC organoids and murine PSCs, which identified two mutually exclusive subtypes of CAFs7, termed αSMAhigh IL-6low myofibroblasts (myCAFs) and αSMAlow IL-6high inflammatory CAFs (iCAFs). These two phenotypes, confirmed later in patient-derived PDAC specimens9, are dependent on their spatial location and biomedical niche within the TME. For example, myCAFs, activated by direct contact with neoplastic cells, reside adjacent to tumor foci, whereas iCAFs, induced by cancer cell-derived factors such as IL-1α and TNFα7,57, are located more distant from tumor cells. While iCAFs are generally confirmed to be tumor-promoting via the secretion of inflammatory cytokines and growth factors58, which confer proliferation, metastasis and chemoresistance of cancer cells59, myCAFs exhibit dual tumor-restraining and tumor-promoting roles, depending on the stage of the tumor and the complex context of the surrounding TME. One school of thought is that activation of fibroblasts reflects a host defense mechanism acting as a dense barrier limiting tumor spread3. It is also possible that the cues emanating from the TME may contribute to the opposing effects of myCAFs at different stages of tumor development60. Importantly, the distinct subsets of CAFs are not permanent but interconvertible via manipulation of specific signaling, as evidenced by conversion of iCAFs to myCAFs via the TGFβ signaling pathway, supporting the notion that CAF subpopulations have high potential for plasticity rather than terminally differentiated states7 and thereby providing a rationale for the induction of CAF phenotypic switching as a strategy in developing anticancer therapy.
Recently, a novel subpopulation characterized by the expression of major histocompatibility complex class II molecules was identified and termed antigen-presenting CAFs (apCAFs), suggesting an immunodulatory function of CAFs8. Indeed, the flow cytometry analysis of KPC tumors identified 3 distinct populations of CAFs with MHCII as a unique marker for apCAFs, while Ly6C as an iCAF-specific surface marker and myCAFs are the Ly6C-MHCII- population8. Furthermore, the authors comprehensively evaluated and validated the transcriptomes of each subtype, providing novel marker genes for these cells8 (Table 1). In contrast to other CAF subtypes, apCAFs are considered to be immunosuppressive by inducing T regulatory (Treg) cell formation in breast61 and pancreatic tumors62, whereas recent work points to apCAF-participated T-cell immunity against lung tumors63, suggesting context-dependent tumor-promoting or tumor-suppressive effects of apCAFs.
Furthermore, broad intra- or intertumoral heterogeneity has been more evident by single-cell sequencing and multiomics approaches. New spatially and functionally distinct CAF subpopulations have been increasingly identified in different cancer types, including vascular CAFs (vCAFs), cycling CAFs (cCAFs), and developmental CAFs (dCAFs) in breast cancer, which emerge at different stages of tumor progression and thus play distinct roles in cancer development20. More importantly, based on scRNA-seq analysis of primary intrahepatic cholangiocarcinoma (ICC) tissues and orthotopic murine models, Aoki et al. identified a distinct CAF population that exhibited a higher response to treatment with anti-placental growth factor (PlGF). Blockade of PIGF resulted in an enrichment of a more quiescent subset with a reduced myofibroblast-like phenotype, suggesting that PIGF could be a key regulator of the CAF balance between quiescence and activation states in ICC64. More excitingly, Buechler et al. constructed single-cell and tissue atlases of fibroblast gene expression in health and disease and revealed the existence of a common lineage-wide fibroblast in all organs65. The stem-like ‘universal’ type of fibroblast cell, marked by expression of peptidase inhibitor 16 (Pi16) and Col15a, found in the steady state across tissues, serves as a reservoir that can yield specialized fibroblasts not only in normal tissues but also, more importantly, in the context of disease or injury where they undergo transition into highly activated fibroblasts, as observed by the development of LRRC15+ CAFs in PDAC.
Furthermore, the mechanism regulating the differentiation of universal Pi16+ fibroblasts into LRRC15+ CAFs has been unveiled recently. Krishnamurty et al. found that among 4 CAF clusters in a PDAC murine model, LRRC15+ CAFs, which were absent in normal tissues, emerged as the dominant CAF population under TGF-β signaling during tumor development and resulted in the suppression of antitumor immunity of cytotoxic T cells66. Interestingly, while Pi16 was demonstrated to be a marker of universal fibroblasts with stem-like features65,66, Elyada et al. previously suggested that Pi16 could be specific to iCAFs, as it showed higher expression in Ly6C+ cells, an iCAF marker identified in this study, than in other populations8. The controversy reflects the complexity and incomplete understanding of CAF subpopulations and suggests that the Pi16 subset could be an interesting population that needs further investigation.
In addition, based on cell-surface molecules, a subset of CD10+ GPR77+ CAFs has been defined, which was shown to correlate with chemoresistance by sustaining cancer stemness and may suffice as a prognostic indicator in breast and lung cancer67. Although various subsets of CAFs are emerging, further research is required to understand the full CAF pool in a cancer-dependent manner. A precise understanding of the mechanisms governing CAF heterogeneity and plasticity is a prerequisite for therapeutic interventions that selectively target tumor-supporting CAFs.
The mechanisms regulating CAF heterogeneity
Genetic manipulation
In general, CAF heterogeneity could arise from cancer cell-derived genetic evolution, epigenetic modulation or metabolic reprogramming (Fig. 2). Tumor cells educate CAFs, as evidenced by the difference in signature genes expressed by fibroblasts cocultured with different tumor cells. A successful education process can be manipulated by extrinsic or intrinsic factors. In particular, CAF plasticity can be induced by numerous tumor cell-derived growth factors and chemokines, including TGF-β, epidermal growth factor (EGF), PDGF, fibroblast growth factor (FGF), interleukin 6 (IL-6), and interleukin 1β (IL-1β)68, which skew CAFs toward specific subsets via activation of key regulatory pathways. For example, TGF-β can specify a myofibroblastic phenotype, while IL-6 particularly drives an immune-modulating phenotype in activated PSCs69. More importantly, the genetic heterogeneity of tumor cells profoundly and dynamically defines the CAF phenotype to fulfill their own growth need, highlighting the requirement of personalized medicine. For example, p53, one of the most commonly mutated genes in cancer, has been found to differentially shape the pancreatic stroma based on p53 status. Gain-of-function (GOF) mutant p53 induced a dominant population of CAFs with more permissive invasion and metastasis, while loss of p53 resulted in a more fibrotic stroma compared with wild-type p53 controls70,71. This p53-driven hierarchy in the PDAC stroma could be attributed to downstream paracrine signaling, such as TNF-a, nuclear factor κB (NF-κB)71, or exosomal secretion of certain cargo, such as podocalyxin (PODXL)72, indicating the influence of epithelial genetic events on CAF phenotype and behavior.

The diversity of CAF subtypes is regulated by complex molecular mechanisms, including genetic regulation mediated by tumor cell-derived factors, epigenetic modulation via direct contact between tumor cells and CAFs, and metabolic reprogramming reflecting the reverse Warburg effect. Moreover, these mechanisms can work independently or cooperatively to shape the stromal structure and function, resulting in tumor restraining or tumor supportive effects of CAF subsets in a context-dependent manner. SOCS1: Suppressor of cytokine signaling; STAT3: Signal transducer and activator of transcription 3; LDH: Lactate dehydrogenase; PYCR1: pyrroline-5-carboxylate reductase 1.
Epigenetic modulation
Although rarely harboring genetic aberrations, CAFs have been demonstrated to have highly consistent epigenome changes in the TME, as evidenced by genome-wide DNA methylation profiles of CAFs73,74,75. These differentially methylated regions were found to be particularly enriched at regulatory regions of the genome74 and key transcription factor-binding sites such as androgen receptor in prostate cancer75 and estrogen receptor (ER) in breast cancer74, resulting in local DNA hypermethylation and global DNA hypomethylation76. Alterations in the epigenetic landscape constitutively activate fibroblasts with specialized ECM remodeling capability77, robust autocrine signaling69 and dynamic immunomodulatory functions77. Furthermore, emerging evidence has revealed that the epigenetic switch induced by the crosstalk between cancer cells and CAFs can specifically reprogram the CAF subtype toward a proinvasive state. For example, it has been reported that normal stromal fibroblasts at baseline in the absence of epigenetic changes are naturally hostile to tumorigenesis. Direct contact of MSCs with PDAC cells triggered the induction of MSC DNA methylation in a global panel of genes, including SOCS1. Methylation of the SOCS1 promoter region led to its downregulated expression, resulting in the derepression of STAT3 signaling and subsequent release of procancerous growth factors such as insulin-like growth factor-1 (IGF-1). Moreover, it has been indicated that the tumor-supportive ability of CAFs is, at least in part, associated with the SOCS1 methylation status, as CAFs with SOCS1 methylation are stronger in promoting PDAC growth than CAFs without SOCS1 methylation, highlighting the importance of epigenetic modulation in shaping CAF functional heterogeneity78.
Since histone methylation is metabolically sensitive to cellular methylation potential79, epigenetic modulation often collaborates with metabolic reprogramming. Recent work identified nicotinamide N-methyltransferase (NNMT) as a master regulator sustaining the protumorigenic phenotype of CAFs via genome-wide DNA and histone hypomethylation. Mechanically, elevated expression of NNMT in the tumor stroma resulted in depletion of SAM (S-adenosyl methionine), a universal methyl donor for histones, which then reduced the global methylation potential of the cell, leading to upregulation of thousands of genes, including protumorigenic cytokines and oncogenic ECM components79. Similarly, in a recently published work, Kay et al. reported a mechanism driven by epigenetic and metabolic interplay in collagen-rich ECM production. Specifically, CAF proline is upregulated via hyperacetylated histone 3 at the promoter sites of pyrroline-5-carboxylate reductase 1 (PYCR1), a key enzyme for proline synthesis. The abundant proline provides CAFs with tumor collagen, resulting in the deposition of pro-tumorigenic extracellular matrix80.
In addition to primary tumors, CAF epigenetic heterogeneity has also been implicated in metastatic niches, where CAFs isolated from liver metastasis and lung metastasis of PDAC displayed distinct DNA methylation patterns, resulting in a more homogenous iCAF phenotype in liver metastasis, whereas CAFs from lung metastasis maintained heterogeneity81. The epigenetic shift toward a more homogeneous iCAF phenotype could, at least in part, explain the aggressive feature of liver metastasis and may provide the rationale for targeting the epigenome in PDAC liver metastasis.
Metabolic reprogramming
Since 1920, the “Warburg effect”, which proposes a model of cancer cell metabolic shift toward aerobic glycolysis, has been the leading principal in understanding the metabolic characteristics of malignant cells. Interestingly, recent investigations have found that the tumor stroma relies on the “reverse Warburg effect” to feed adjacent cancer cells where aerobic glycolysis occurs in CAFs and generates energy-rich metabolites such as lactate that tumor cells then use to perform oxidative phosphorylation82. Thus, metabolic adaptations are another feature of CAF activation toward the glycolytic phenotype and the acquisition of tumor-promoting functions. A number of possible mechanisms have been described to explain the metabolic switch of CAFs from oxidative phosphorylation to aerobic glycolysis. Lactate dehydrogenase (LDH), an enzyme involved in the conversion of pyruvate to lactate, is a key component of the glycolysis pathway, and various studies have therefore focused on the regulatory mechanism of fibroblast LDH expression in the context of tumors.
Intratumoral hypoxia is a typical hallmark of solid tumors, leading to the stabilization of HIF-1α. Sustained elevation of HIF-1α directly induces glycolytic enzymes such as LDH and pyruvate kinase M2 (PKM2), thereby altering CAF metabolism toward a pro-glycolytic phenotype83,84. In addition to HIF-1a, other factors have been shown to directly regulate the transcription of glycolytic enzymes, including oncogenic c-Myc85 and forkhead box protein M1 (FOXM1)86, along with the recently identified POU1F187 and PRRX1 (paired related homeobox1)88. Moreover, chronic hypoxia can lead to hypomethylation of glycolysis-related genes in fibroblasts, suggesting that metabolic coupling with epigenetic events favors the accumulation of pro-glycolytic CAFs. Indeed, chromatin modifiers and transcription factors have been found to act on CAF expression of glycolytic enzymes in a cooperative manner89.
Crosstalk between cancer cells and the TME can also influence the plasticity of stromal cells, as evidenced by the observation that cancer cells with high metastatic potential have a higher capability to metabolically reprogram fibroblasts, which show a more aggressive phenotype, than fibroblasts reprogrammed by low metastatic cancer cells90, supporting the notion that CAFs are the metabolic scar of cancer and represent an important target as cancer cells per se in anticancer therapy.
Development of novel therapeutic strategies against tumor-promoting CAFs
Over the past decade, multiple approaches have attempted to target CAFs and their crosstalk with cancer cells in preclinical models and clinical trials. Direct depletion of CAFs through genetic manipulation, pharmacological targeting or specific antibodies, contrary to previous assumptions, enhanced tumor growth and aggressiveness following ablation of α-SMA+ CAFs in PDAC6. Depletion of FAP+ CAFs, although has been reported to prolong survival in PDAC murine models91, administration of sibrotuzumab, a FAP-specific antibody, failed to improve survival for patients with metastatic colorectal cancer in a Phase II trial92, highlighting the importance of targeted therapy rather than widespread eradication of all CAFs. To date, novel approaches specifically targeting tumor-supportive CAFs and re-engineering the tumor stroma have been explored extensively in preclinical models, some of which have begun to enter clinical trials (Fig. 3).

Current strategies to selectively target tumor-promoting CAFs include (a) blocking the differentiation from precursor cells to tumor-promoting CAFs via inhibition of precursor activation or targeting key signaling pathways in differentiation. b Targeted depletion of tumor-promoting CAFs without affecting tumor-restraining CAFs through genetic manipulation or specific antibodies. c Induction of phenotypic switching from tumor-promoting to tumor-restraining CAFs. d Targeting the crosstalk between cancer cells and tumor-promoting CAFs to inhibit their supportive effect on cancer proliferation, migration and chemoresistance.
Inhibition of progenitor cell differentiation toward a pro-cancerous CAF
To date, elucidation of the molecular mechanisms underlying CAF subset formation has made it possible to inhibit the transition from precursor cells toward tumor-promoting CAFs. It has been reported that during pancreatic cancer progression, tumor paracrine signals, including IL-1 and TGF-β, induce the mesothelial to apCAF transition, which is responsible for immunosuppression62. However, IL-1 and TGF-β may not serve as good targets for specific inhibition of the transition due to their pleiotropic effects. Therefore, a monoclonal antibody (mAb) against mesothelin (MSLN), a specific mesothelial cell marker, was tested and found to effectively block mesothelial cell activation and apCAF generation62.
Epigenetic reprogramming has been revealed to provide dynamic and reversible modulation of stromal cells. Therefore, targeting the epigenome via regulation of DNA methylation or histone modification has been investigated in preclinical models, and several promising molecules are now active in clinical trials. Decitabine (DAC), a DNA methyltransferase (DNMT) inhibitor, was first applied in the treatment of a PDAC xenograft model in which the tumor-free survival with DAC-pretreated MSCs was significantly longer than that of mice with untreated MSCs78. However, it is notable that inhibition of DNA methylation only attenuated but could not fully prevent MSC reprogramming into protumorigenic CAFs, suggesting that multiple mechanisms cooperate in driving CAF differentiation. Following this, NNMT-mediated histone methylation was reported to be a master regulator in defining the protumorigenic stroma. Strikingly, inhibition of NNMT activity led to a reversion of the CAF phenotype in orthotopic high-grade serous ovarian cancer (HGSC), resembling normal omental fibroblasts morphologically and transcriptionally79. This finding implicates that the metastatic stroma can be targeted and even revert back to the normalized state via epigenetic remodeling, is of high translational value and should be further explored as a new therapeutic approach for advanced cancer.
Targeted ablation of tumor-promoting CAFs
Selective targeting of tumor-promoting CAFs relies on the identification of specific and convenient markers. It is proposed that CD10 and GPR77 can define a human CAF subset that sustains cancer stemness and chemoresistance. Depletion of the CD10+GPR77+ subset abrogated tumor growth and reversed chemosensitivity, and the effect also replicated with administration of GPR77 neutralizing mAb in a patient-derived xenograft model of breast cancer. Mechanistically, GPR77 not only serves as a surface marker but also functions as an essential signaling molecule for the maintenance of NF-κB activities in the subset67.
Chemotherapy resistance in PDAC is often associated with desmoplasia93,94, one of which was recently shown to occur through PlGF/VEGF-mediated activation of CAFs in an orthotopic PDAC mouse model treated with gemcitabine95. Therefore, the authors delicately designed and synthesized a multiparatopic VEGF decoy receptor (Ate-Grab), a fusion protein consisting of single-chain Fv of atezolizumab for targeting PD-L1-rich tumor tissues, fused to VEGF-Grab, which was previously developed to target PIGF/VEGF96, resulting in PD-L1-directed PlGF/VEGF blockade. Indeed, Ate-Grab treatment relieved chemotherapy-induced desmoplasia in a PDAC model by sequestering PlGF/VEGF within the TME and thus inhibiting CAF activation, especially the CD141+ population, a key subset activated by gemcitabine treatment responsible for tumor fibrosis95.
However, specific markers to precisely define a unique human CAF subpopulation in vivo are still lacking in many tumor types; therefore, targeting the potential cellular sources of CAFs may provide another way for precision treatment. PSCs are known to give rise to PDAC CAFs, although recent studies have demonstrated multiple cells of origin in addition to PSCs. To functionally dissect the role of PSC-derived CAFs in PDAC progression, PSC CAFs were specifically ablated via Cre-mediated diphtheria toxin (DT) treatment, which resulted in attenuated tumor stiffness and a desmoplastic milieu with lower levels of matricellular proteins36. This finding is worthwhile for further investigation since PDAC patients with abundant matricellular fibrosis have been observed to have shortened survival97. Similarly, in the liver, HSCs, as the main source of CAFs, have been targeted using the same strategy, leading to a reduced tumor burden in ICC60. Notably, HSCs were found to give rise to both myCAFs and iCAFs in this study; therefore, targeted ablation of HSCs not only blocks iCAF but also myCAF formation. Further analysis demonstrated that myCAFs in ICCs act as tumor promoters via the interaction of hyaluronan (HA) receptors and hyaluronan synthase 2 (Has2)60, highlighting a context-dependent myCAF function and the necessity of a thorough understanding of CAF subset function in vivo.
Induction of phenotypic switching toward tumor-restrictive CAFs
Compared with targeted ablation of CAF subsets, fine-tuning of specific CAF subpopulations based on their inherent plasticity seems to be more feasible and less challenging for therapeutic interventions. Mounting evidence has pointed to TGF-β1 signaling as one of the key regulators governing the fate of CAF subpopulations, especially between myCAF and iCAF conversion in PDAC, which is under preclinical investigation by transforming tumor-promoting iCAFs into tumor-restrictive myCAFs through activation of TGF-β signaling69. In addition to the myCAF and iCAF transition, TGF-β signaling has also been found to regulate CAF functional heterogeneity in other tumors. In a recent study, Hu et al. established a living biobank of CAFs from non-small lung cancer (NSCLC) patients, which displayed distinctive functional subsets with subtypes I and II as cancer cell protectors, while subtype III correlated with higher sensitivity to chemotherapy. Furthermore, these subsets were interconvertible by intrinsic TGF-β1 signaling, where loss of TGF-β signaling increased tumor-protection subsets, whereas addition of TGF-β1 downregulated these subsets and thereby attenuated their capacity to confer EGFRi resistance98.
Intrinsic or acquired trastuzumab resistance has been found in some HER2+ breast cancer patients and results in the failure of standard trastuzumab therapy99. Recently, it was demonstrated that the abundance of CD16+ fibroblasts in HER2+ breast cancer patients is correlated with poor response to trastuzumab through severe desmoplasia and inefficient drug delivery. Mechanistically, activated CD16+ fibroblasts enhanced matrix production and stiffness through a VAV2-dependent pathway100. Targeting VAV2 not only blocked the activation of CD16+ fibroblasts but also reversed desmoplasia and decreased trastuzumab resistance100.
The discovery of the IL1α/JAK/STAT signaling pathway in promoting iCAF formation has provided novel pharmacological targets. JAK inhibitors are reported to skew the iCAF subtype toward the myCAF subtype in PDAC69. Furthermore, preclinical studies have confirmed the effectiveness of the IL-1 receptor antagonist anakinra on PDAC progression101, which has now entered a phase 1 clinical trial in combination with standard chemotherapy in PDAC (NCT02021422). Indeed, a series of clinical trials targeting the specific pathway essential for procancerous CAF formation or maintenance, including TGF-β, VEGF and FGF, have shown promising antitumor efficacy and acceptable safety in combination with chemotherapy102,103,104,105,106 (Table 2). However, CAF-targeted clinical trials may not always recapitulate the advantageous effect in preclinical models, as observed by the diminished survival of pancreatic cancer patients treated with the hedgehog inhibitor IP-926 (NCT01130142) or vismodegib (NCT01383538) in combination with gemcitabine. The disappointing outcomes highlight the importance of a careful, thorough and long-term preclinical investigation before translating optimism in the CAF field into the clinic.
Inhibiting activity of mature CAFs
Due to the existence of high intra- and intertumor heterogeneity as well as a lack of CAF-specific markers, researchers are more interested in elucidating the crosstalk between cancer cells and CAFs, especially how CAFs foster excessive proliferation and chemoresistance, to inhibit the tumor-promoting activity of CAFs by disrupting signaling pathways. It has been found that paracrine communication between cancer cells expressing PDGF ligands and CAFs expressing cognate receptors leads to the specification of basal-like breast tumors, a hormone receptor-negative subtype that cannot benefit from tamoxifen treatment in the clinic107. Surprisingly, neutralization of the PDGF pathway with administration of an inhibitory antibody resulted in conversion from the basal-like subtype into the luminal subtype, which is susceptible to anti-estrogen therapies107. In addition, it has been reported that pancreatic tumors harboring p53 mutations promote CAF reprogramming toward a prometastatic subset with the secretion of heparin sulfate proteoglycan 2 (HSPG2) in the stroma. HSPG2 deposition creates a permissive environment for invasion and metastasis, and depletion of HSPG2 not only impairs metastasis but also improves chemotherapy efficacy in pancreatic tumors harboring a GOF p53 mutation71, providing a potential avenue toward targeting the stromal feedback of aggressive subsets rather than manipulation of the CAF phenotype.
Conclusions
Although CAFs have long been investigated as a crucial player in cancer development and therefore represent an attractive therapeutic target, clinical trials targeting CAFs have mostly ended in failure and even in some cases, accelerated cancer progression1, demonstrating that the dynamic complexity of CAF identity and function is far beyond our current understanding. Therefore, dissecting the heterogeneous subpopulations and diverse functions of CAFs in a context-dependent manner is of high importance.
Single-cell analysis techniques such as single-cell transcriptomic and proteomic technologies provide a powerful tool for deciphering cellular heterogeneity and identifying new precise biomarkers for targeted therapy. More importantly, functional assessment of each subset regarding their crosstalk not only with cancer cells but also with other stromal components and even between distinct CAF subpopulations should be given sufficient attention to classify CAF subsets into functional categories. Moreover, given the diversity of CAF reservoirs and the plasticity of CAF changes over time with tumor progression, it would be interesting to illustrate the relationship between each CAF subpopulation, whether they are hierarchical, parallel, or overlapping. For this purpose, reliable in vivo tumor models combined with genetic manipulation systems such as the Cre–lox system offer an elegant way to interrogate CAF function at different stages of tumor development.
While current studies focus on the identification of novel tumor-promoting CAF subsets and strategies to target them specifically, it is noteworthy that tumor-suppressive CAF populations and their homeostatic maintenance will also be worthwhile to identify, as enhancement of these functions as a barrier restricting tumor spread or reprogramming of the so-called bad CAF into good CAF should be a goal of future stroma-targeted therapies. Therefore, there is a growing appreciation that therapeutic approaches to normalize or re-engineer the tumor stroma into a quiescent state or even tumor-suppressive phenotypes would offer the potential for translational impact in improving patient survival.
References
Chen, Y., McAndrews, K. M. & Kalluri, R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 18, 792–804 (2021).
Raghavan, S. et al. Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell 184, 6119–6137 e26 (2021).
Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 16, 582–598 (2016).
Yoshida, G. J. Regulation of heterogeneous cancer-associated fibroblasts: the molecular pathology of activated signaling pathways. J. Exp. Clin. Cancer Res. 39, 112 (2020).
Rhim, A. D. et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25, 735–747 (2014).
Ozdemir, B. C. et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734 (2014).
Ohlund, D. et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 214, 579–596 (2017).
Elyada, E. et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 9, 1102–1123 (2019).
Bernard, V. et al. Single-cell transcriptomics of pancreatic cancer precursors demonstrates epithelial and microenvironmental heterogeneity as an early event in neoplastic progression. Clin. Cancer Res. 25, 2194–2205 (2019).
Ligorio, M. et al. Stromal microenvironment shapes the intratumoral architecture of pancreatic cancer. Cell 178, 160–175 e27 (2019).
Gabbiani, G., Ryan, G. B. & Majne, G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 27, 549–550 (1971).
Micallef, L. et al. The myofibroblast, multiple origins for major roles in normal and pathological tissue repair. Fibrogenes. Tissue Repair 5, S5 (2012).
Simian, M. et al. The interplay of matrix metalloproteinases, morphogens and growth factors is necessary for branching of mammary epithelial cells. Development 128, 3117–3131 (2001).
Tomasek, J. J., Gabbiani, G., Hinz, B., Chaponnier, C. & Brown, R. A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 3, 349–363 (2002).
Koliaraki, V., Prados, A., Armaka, M. & Kollias, G. The mesenchymal context in inflammation, immunity and cancer. Nat. Immunol. 21, 974–982 (2020).
Dvorak, H. F. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 315, 1650–1659 (1986).
Braidotti, N., Chen, S. N., Long, C. S., Cojoc, D. & Sbaizero, O. Piezo1 channel as a potential target for hindering cardiac fibrotic remodeling. Int. J. Mol. Sci. 23, 8065 (2022).
Kahounova, Z. et al. The fibroblast surface markers FAP, anti-fibroblast, and FSP are expressed by cells of epithelial origin and may be altered during epithelial-to-mesenchymal transition. Cytometry A 93, 941–951 (2018).
Sahai, E. et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 20, 174–186 (2020).
Bartoschek, M. et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 9, 5150 (2018).
Nguyen, E. V. et al. Proteomic profiling of human prostate cancer-associated fibroblasts (CAF) reveals LOXL2-dependent regulation of the tumor microenvironment. Mol. Cell Proteom. 18, 1410–1427 (2019).
Zeltz, C. et al. alpha11beta1 integrin is induced in a subset of cancer-associated fibroblasts in desmoplastic tumor stroma and mediates in vitro cell migration. Cancers (Basel) 11, 765 (2019).
Guido, C. et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-beta drives tumor growth: connecting TGF-beta signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle 11, 3019–3035 (2012).
Goetz, J. G. et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell 146, 148–163 (2011).
Armulik, A., Genove, G. & Betsholtz, C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 21, 193–215 (2011).
Arnold, J. N., Magiera, L., Kraman, M. & Fearon, D. T. Tumoral immune suppression by macrophages expressing fibroblast activation protein-alpha and heme oxygenase-1. Cancer Immunol. Res. 2, 121–126 (2014).
Kim, D. et al. Identification and characterization of cancer-associated fibroblast subpopulations in lung adenocarcinoma. Cancers (Basel) 14, 3486 (2022).
Venning, F. A. et al. Deciphering the temporal heterogeneity of cancer-associated fibroblast subpopulations in breast cancer. J. Exp. Clin. Cancer Res. 40, 175 (2021).
Sebastian, A. et al. Single-cell transcriptomic analysis of tumor-derived fibroblasts and normal tissue-resident fibroblasts reveals fibroblast heterogeneity in breast cancer. Cancers (Basel) 12, 1307 (2020).
Vonlaufen, A. et al. Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res. 68, 2085–2093 (2008).
Ezhilarasan, D. Hepatic stellate cells in the injured liver: perspectives beyond hepatic fibrosis. J. Cell Physiol. 237, 436–449 (2022).
Heinemann, V. et al. Tumour-stroma interactions in pancreatic ductal adenocarcinoma: rationale and current evidence for new therapeutic strategies. Cancer Treat. Rev. 40, 118–128 (2014).
Valkenburg, K. C., de Groot, A. E. & Pienta, K. J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 15, 366–381 (2018).
Sperb, N., Tsesmelis, M. & Wirth, T. Crosstalk between tumor and stromal cells in pancreatic ductal adenocarcinoma. Int. J. Mol. Sci. 21, 5486 (2020).
Schoepp, M., Strose, A. J. & Haier, J. Dysregulation of miRNA expression in cancer associated fibroblasts (CAFs) and its consequences on the tumor microenvironment. Cancers (Basel) 9, 54 (2017).
Helms, E. J. et al. Mesenchymal lineage heterogeneity underlies nonredundant functions of pancreatic cancer-associated fibroblasts. Cancer Discov. 12, 484–501 (2022).
Dominguez, C. X. et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15(+) myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov. 10, 232–253 (2020).
Kobayashi, H. et al. The origin and contribution of cancer-associated fibroblasts in colorectal carcinogenesis. Gastroenterology 162, 890–906 (2022).
Plikus, M. V. et al. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 184, 3852–3872 (2021).
Garcia, P. E. et al. Differential contribution of pancreatic fibroblast subsets to the pancreatic cancer stroma. Cell Mol. Gastroenterol. Hepatol. 10, 581–599 (2020).
Kalluri, R. & Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 6, 392–401 (2006).
Bochet, L. et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 73, 5657–5668 (2013).
Hosaka, K. et al. Pericyte-fibroblast transition promotes tumor growth and metastasis. Proc. Natl Acad. Sci. USA 113, E5618–E5627 (2016).
Rynne-Vidal, A., Jimenez-Heffernan, J. A., Fernandez-Chacon, C., Lopez-Cabrera, M. & Sandoval, P. The mesothelial origin of carcinoma associated-fibroblasts in peritoneal metastasis. Cancers (Basel) 7, 1994–2011 (2015).
McAnulty, R. J. Fibroblasts and myofibroblasts: their source, function and role in disease. Int. J. Biochem. Cell Biol. 39, 666–671 (2007).
Arina, A. et al. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proc. Natl Acad. Sci. USA 113, 7551–7556 (2016).
Worthley, D. L. et al. Human gastrointestinal neoplasia-associated myofibroblasts can develop from bone marrow-derived cells following allogeneic stem cell transplantation. Stem Cells 27, 1463–1468 (2009).
Kurashige, M. et al. Origin of cancer-associated fibroblasts and tumor-associated macrophages in humans after sex-mismatched bone marrow transplantation. Commun. Biol. 1, 131 (2018).
Direkze, N. C. et al. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res 64, 8492–8495 (2004).
Raz, Y. et al. Bone marrow-derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J. Exp. Med 215, 3075–3093 (2018).
Barcellos-de-Souza, P. et al. Mesenchymal stem cells are recruited and activated into carcinoma-associated fibroblasts by prostate cancer microenvironment-derived TGF-beta1. Stem Cells 34, 2536–2547 (2016).
Weber, C. E. et al. Osteopontin mediates an MZF1-TGF-beta1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene 34, 4821–4833 (2015).
Iwamoto, C. et al. Bone marrow-derived macrophages converted into cancer-associated fibroblast-like cells promote pancreatic cancer progression. Cancer Lett. 512, 15–27 (2021).
Vierhout, M. et al. Monocyte and macrophage derived myofibroblasts: Is it fate? A review of the current evidence. Wound Repair Regen. 29, 548–562 (2021).
Tang, P. C. et al. Smad3 promotes cancer-associated fibroblasts generation via macrophage-myofibroblast transition. Adv. Sci. (Weinh.) 9, e2101235 (2022).
Neuzillet, C. et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 248, 51–65 (2019).
Tran, L. L., Dang, T., Thomas, R. & Rowley, D. R. ELF3 mediates IL-1alpha induced differentiation of mesenchymal stem cells to inflammatory iCAFs. Stem Cells 39, 1766–1777 (2021).
Bhattacharjee, S. et al. Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts. J. Clin. Invest. 131 (2021).
Zhang, X. et al. Cancer-associated fibroblast-induced lncRNA UPK1A-AS1 confers platinum resistance in pancreatic cancer via efficient double-strand break repair. Oncogene 41, 2372–2389 (2022).
Affo, S. et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell 39, 866–882 e11 (2021).
Costa, A. et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 33, 463–479 e10 (2018).
Huang, H. et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell 40, 656–673 e7 (2022).
Kerdidani, D. et al. Lung tumor MHCII immunity depends on in situ antigen presentation by fibroblasts. J. Exp. Med. 219, e20210815 (2022).
Aoki, S. et al. Placental growth factor promotes tumour desmoplasia and treatment resistance in intrahepatic cholangiocarcinoma. Gut 71, 185–193 (2022).
Buechler, M. B. et al. Cross-tissue organization of the fibroblast lineage. Nature 593, 575–579 (2021).
Krishnamurty, A. T. et al. LRRC15(+) myofibroblasts dictate the stromal setpoint to suppress tumour immunity. Nature 611, 148–154 (2022).
Su, S. et al. CD10(+)GPR77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell 172, 841–856 e16 (2018).
Santi, A., Kugeratski, F. G. & Zanivan, S. Cancer associated fibroblasts: the architects of stroma remodeling. Proteomics 18, e1700167 (2018).
Biffi, G. et al. IL1-induced JAK/STAT signaling is antagonized by TGFbeta to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 9, 282–301 (2019).
Wormann, S. M. et al. Loss of P53 function activates JAK2-STAT3 signaling to promote pancreatic tumor growth, stroma modification, and gemcitabine resistance in mice and is associated with patient survival. Gastroenterology 151, 180–193 e12 (2016).
Vennin, C. et al. CAF hierarchy driven by pancreatic cancer cell p53-status creates a pro-metastatic and chemoresistant environment via perlecan. Nat. Commun. 10, 3637 (2019).
Novo, D. et al. Mutant p53s generate pro-invasive niches by influencing exosome podocalyxin levels. Nat. Commun. 9, 5069 (2018).
Lawrence, M. G. et al. Alterations in the methylome of the stromal tumour microenvironment signal the presence and severity of prostate cancer. Clin. Epigenetics 12, 48 (2020).
Pidsley, R. et al. Enduring epigenetic landmarks define the cancer microenvironment. Genome Res. 28, 625–638 (2018).
Nash, C. et al. Genome-wide analysis of AR binding and comparison with transcript expression in primary human fetal prostate fibroblasts and cancer associated fibroblasts. Mol. Cell Endocrinol. 471, 1–14 (2018).
Lee, Y. T., Tan, Y. J., Falasca, M. & Oon, C. E. Cancer-associated fibroblasts: epigenetic regulation and therapeutic intervention in breast cancer. Cancers (Basel) 12, 2949 (2020).
Ippolito, L. et al. Lactate rewires lipid metabolism and sustains a metabolic-epigenetic axis in prostate cancer. Cancer Res. 82, 1267–1282 (2022).
Xiao, Q. et al. Cancer-associated fibroblasts in pancreatic cancer are reprogrammed by tumor-induced alterations in genomic DNA methylation. Cancer Res. 76, 5395–5404 (2016).
Eckert, M. A. et al. Proteomics reveals NNMT as a master metabolic regulator of cancer-associated fibroblasts. Nature 569, 723–728 (2019).
Kay, E. J. et al. Cancer-associated fibroblasts require proline synthesis by PYCR1 for the deposition of pro-tumorigenic extracellular matrix. Nat. Metab. 4, 693–710 (2022).
Pan, X. et al. Cancer-associated fibroblast heterogeneity is associated with organ-specific metastasis in pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 14, 184 (2021).
Pavlides, S. et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 8, 3984–4001 (2009).
Becker, L. M. et al. Epigenetic reprogramming of cancer-associated fibroblasts deregulates glucose metabolism and facilitates progression of breast cancer. Cell Rep. 31, 107701 (2020).
Zhang, D. et al. Metabolic reprogramming of cancer-associated fibroblasts by IDH3alpha downregulation. Cell Rep. 10, 1335–1348 (2015).
Shim, H. et al. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc. Natl Acad. Sci. USA 94, 6658–6663 (1997).
Cui, J. et al. FOXM1 promotes the warburg effect and pancreatic cancer progression via transactivation of LDHA expression. Clin. Cancer Res. 20, 2595–2606 (2014).
Martinez-Ordonez, A. et al. POU1F1 transcription factor induces metabolic reprogramming and breast cancer progression via LDHA regulation. Oncogene 40, 2725–2740 (2021).
Lee, K. W. et al. PRRX1 is a master transcription factor of stromal fibroblasts for myofibroblastic lineage progression. Nat. Commun. 13, 2793 (2022).
Linares, J. F. et al. The lactate-NAD(+) axis activates cancer-associated fibroblasts by downregulating p62. Cell Rep. 39, 110792 (2022).
Kogure, A. et al. Cancer cells with high-metastatic potential promote a glycolytic shift in activated fibroblasts. PLoS One 15, e0234613 (2020).
McAndrews, K. M. et al. Identification of functional heterogeneity of carcinoma-associated fibroblasts with distinct IL6-mediated therapy resistance in pancreatic cancer. Cancer Disco. 12, 1580–1597 (2022).
Hofheinz, R. D. et al. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie 26, 44–48 (2003).
Peran, I. et al. Cadherin 11 promotes immunosuppression and extracellular matrix deposition to support growth of pancreatic tumors and resistance to gemcitabine in mice. Gastroenterology 160, 1359–1372 e13 (2021).
Incio, J. et al. Obesity-induced inflammation and desmoplasia promote pancreatic cancer progression and resistance to chemotherapy. Cancer Discov. 6, 852–869 (2016).
Kim, D. K. et al. PD-L1-directed PlGF/VEGF blockade synergizes with chemotherapy by targeting CD141(+) cancer-associated fibroblasts in pancreatic cancer. Nat. Commun. 13, 6292 (2022).
Lee, D. H. et al. Multi-paratopic VEGF decoy receptor have superior anti-tumor effects through anti-EGFRs and targeted anti-angiogenic activities. Biomaterials 171, 34–45 (2018).
Laklai, H. et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat. Med. 22, 497–505 (2016).
Hu, H. et al. Three subtypes of lung cancer fibroblasts define distinct therapeutic paradigms. Cancer Cell 39, 1531–1547 e10 (2021).
Arteaga, C. L. et al. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat. Rev. Clin. Oncol. 9, 16–32 (2011).
Liu, X. et al. CD16(+) fibroblasts foster a trastuzumab-refractory microenvironment that is reversed by VAV2 inhibition. Cancer Cell 40, 1341–1357 e13 (2022).
Zhuang, Z. et al. IL1 receptor antagonist inhibits pancreatic cancer growth by abrogating NF-kappaB activation. Clin. Cancer Res. 22, 1432–1444 (2016).
Gaspar, N. et al. Phase I/II study of single-agent lenvatinib in children and adolescents with refractory or relapsed solid malignancies and young adults with osteosarcoma (ITCC-050)(☆). ESMO Open 6, 100250 (2021).
Feng, J. et al. SHR-1701, a bifunctional fusion protein targeting PD-L1 and TGFbeta, for recurrent or metastatic cervical cancer: a clinical expansion cohort of a phase I study. Clin. Cancer Res. 28, 5297–5305 (2022).
Strauss, J. et al. Phase I Trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGFbeta, in advanced solid tumors. Clin. Cancer Res. 24, 1287–1295 (2018).
Bibeau, K., Feliz, L., Lihou, C. F., Ren, H. & Abou-Alfa, G. K. Progression-free survival in patients with cholangiocarcinoma with or without FGF/FGFR alterations: a FIGHT-202 post hoc analysis of prior systemic therapy response. JCO Precis. Oncol. 6, e2100414 (2022).
Dorff, T. B. et al. Clinical and correlative results of SWOG S0354: a phase II trial of CNTO328 (siltuximab), a monoclonal antibody against interleukin-6, in chemotherapy-pretreated patients with castration-resistant prostate cancer. Clin. Cancer Res. 16, 3028–3034 (2010).
Roswall, P. et al. Microenvironmental control of breast cancer subtype elicited through paracrine platelet-derived growth factor-CC signaling. Nat. Med. 24, 463–473 (2018).
Funding
This work was supported by grants from the Natural Science Foundation of China (82100666), the National Natural Science Foundation for Young Scientists of Jiangsu Province (BK20200906), and the Young Scientists Initiative Foundation of Jiangsu University.
Author information
Authors and Affiliations
Contributions
D.Y. conceived the idea and wrote the manuscript. J.L., H.Q., and Q.Z. contributed to the conceptual evaluation of the manuscript.
Corresponding authors
Ethics declarations
COMPETING INTERESTS
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, D., Liu, J., Qian, H. et al. Cancer-associated fibroblasts: from basic science to anticancer therapy. Exp Mol Med 55, 1322–1332 (2023). https://doi.org/10.1038/s12276-023-01013-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-023-01013-0
This article is cited by
-
Understanding the matrix: collagen modifications in tumors and their implications for immunotherapy
Journal of Translational Medicine (2024)
-
Identification of BGN positive fibroblasts as a driving factor for colorectal cancer and development of its related prognostic model combined with machine learning
BMC Cancer (2024)
-
Crosstalk between cancer-associated fibroblasts and regulated cell death in tumors: insights into apoptosis, autophagy, ferroptosis, and pyroptosis
Cell Death Discovery (2024)
-
Heterogeneity and interplay: the multifaceted role of cancer-associated fibroblasts in the tumor and therapeutic strategies
Clinical and Translational Oncology (2024)
-
Modulation of tumor microenvironment by targeting histone acetylation in bladder cancer
Cell Death Discovery (2024)
