
Abstract
Transcriptional deregulation, a cancer cell hallmark, is driven by epigenetic abnormalities in the majority of brain tumors, including adult glioblastoma and pediatric brain tumors. Epigenetic abnormalities can activate epigenetic regulatory elements to regulate the expression of oncogenes. Superenhancers (SEs), identified as novel epigenetic regulatory elements, are clusters of enhancers with cell-type specificity that can drive the aberrant transcription of oncogenes and promote tumor initiation and progression. As gene regulators, SEs are involved in tumorigenesis in a variety of tumors, including brain tumors. SEs are susceptible to inhibition by their key components, such as bromodomain protein 4 and cyclin-dependent kinase 7, providing new opportunities for antitumor therapy. In this review, we summarized the characteristics and identification, unique organizational structures, and activation mechanisms of SEs in tumors, as well as the clinical applications related to SEs in tumor therapy and prognostication. Based on a review of the literature, we discussed the relationship between SEs and different brain tumors and potential therapeutic targets, focusing on glioblastoma.
Similar content being viewed by others
Introduction
Brain tumors, including glioblastoma, medulloblastoma, diffuse intrinsic pontine glioma, meningioma, ependymoma, and other rare brain tumors, account for approximately 3% of cancer cases worldwide1. While brain tumors are relatively rare, they still deserve research attention because of their significant mortality and morbidity at all ages2. Currently, treatments for brain tumors include surgery, chemotherapy, and radiotherapy. Despite substantial advances in chemotherapy for different brain tumors, chemotherapy resistance and relapse are still challenges. Delivering therapeutic agents to primary brain tumors is particularly challenging because of the unique brain–blood barrier (BBB) and brain–tumor barrier (BTB), resulting in poor response to treatments3,4. Moreover, brain tumor cells breach the BBB and adhere to the BTB, and this diffuse mode of invasion is the fundamental reason for brain metastases and relapse4. Therefore, it is necessary to study pathogenesis further and find more effective targets to improve the prognosis of patients with brain tumors.
Gene transcription is a complex and highly coordinated process. Transcriptional dysregulation mediated by epigenetic modifications in tumors has attracted significant attention. Over the last decades, epigenetic modifications such as DNA methylation and histone modification have been identified as critical drivers of several types of brain cancer5. Enhancers are a class of regulatory DNA sequences that function as cis-regulatory elements to enhance the transcription of target genes occupied by coactivators and transcription factors (TFs)6,7. With the expanding concept of enhancers, “superenhancers (SEs)” have been proposed, which span several kilobases and are enriched with a higher density of TFs, coactivators, and epigenetic modifications8. Compared to typical enhancers, SEs can drive higher gene expression and participate in many biological processes9. It is worth mentioning that oncogenes regulated by SEs in tumor cells are not expressed in normal cells, suggesting that SEs play a critical role in the occurrence and development of tumors10. As tumor-associated variants are significantly enriched in SEs, identifying SEs improves the understanding of the mechanisms of tumorigenesis and provides new insights into the diagnosis, treatment, and prognosis of tumors11. Overall, SE-driven transcriptional disorders are associated with the progression of human cancers, including brain tumors.
This review will summarize the characteristics and identification, unique organizational structures, and activation mechanisms of SEs in tumors, as well as the clinical applications related to SEs in tumor therapy and prognostication. Furthermore, we will discuss SE-associated genes, transcriptional regulatory mechanisms, and therapeutic targets in different brain tumors.
Overview of superenhancers
Characteristics and identification of SEs
In 2013, Young et al. first proposed superenhancers: large clusters of transcriptional enhancers that drive the expression of cell-identity genes10. SEs were initially discovered in embryonic stem cells (ESCs), with ESC-specific master transcription factors Oct4, Sox2, and Nanog binding to enhancer elements and recruiting mediators to activate the gene transcription program12. Subsequently, SEs were identified in other cells, including cancer cells13,14. As novel epigenetic regulatory elements, SEs precisely regulate oncogene transcriptional activation during tumorigenesis and may be potential therapeutic targets15,16.
SEs are bound by many cell-type-specific TFs and recruit a series of mediators, transcriptional coactivators, and chromatin regulators (CBP/p300 and cohesin) as well as RNA polymerase II (RNA pol II), thus forming an SE-promoter loop to initiate downstream transcription17. In addition, histone modifications are important features of SEs. Histone H3 lysine 27 acetylation (H3K27ac) and histone H3 lysine 4 methylation (H3K4me1), which make the chromatin structure looser and provide accessible TF binding sites, are labels of active SEs18,19. H3K27ac and H3K4me1 are required for enhancers to activate target gene transcription, and these active chromatin markers can mediate the recruitment of epigenetic readers, including BET proteins20,21. In conclusion, these key components are integral parts of SE organization and the functional basis of SEs (Fig. 1).

SEs recruit TFs, mediators, RNA pol II, histone modifiers, and other chromatin regulators, activating the expression of downstream genes. The enhancer-promoter loop that is formed with the help of seRNAs contributes to the transcription of target genes. Blocking SEs with BRD4 or CDK7 inhibitors is considered a viable antitumor approach. SE superenhancer, TF transcription factors, BRD4 bromodomain protein 4, CDK7 cyclin-dependent kinase 7.
For the identification of SEs, next-generation sequencing and high-throughput sequencing technologies, including chromatin immunoprecipitation sequencing (ChIP-seq)22, DNase I sequencing (DNase-seq)23, chromosome conformation capture sequencing (3C-seq)24, and assay for transposase-accessible chromatin sequencing (ATAC-seq), are powerful tools25. SEs can be identified based on the strength of their binding with chromatin regulators and histone modification levels. Histone modifications can be used as indicators to identify SEs based on genome-wide factors such as the ChIP-seq-identified level of H3K27ac. Moreover, as an increasing number of SEs are defined in different types of tumors, various databases have been established to explore SE functions and facilitate SE research, such as SEdb (http://www.licpathway.net/sedb) and dbSUPER (http://bioinfo.au.tsinghua.edu.cn/dbsuper/)26,27,28,29,30,31,32,33,34,35,36,37,38,39,40.
The role of SE-derived RNAs
Enhancers regulate the transcription of target genes and are actively transcribed into enhancer RNAs (eRNAs), and this ability to be transcribed is a general feature of functionally active enhancers41. Correspondingly, SEs are also transcribed into seRNAs, including long noncoding RNAs (lncRNAs), microRNAs (miRNAs), and circular RNAs (circRNAs), which play an important role in gene expression and epigenetic regulation42,43,44.
A growing number of studies have revealed the critical roles of seRNAs in regulating target gene transcription. Young’s group proposed an RNA-mediated feedback control model of transcriptional condensates. In the model, low levels of RNAs mediate the formation of transcriptional condensates, while high levels of RNAs cause condensate dissolution, thus affecting the transcriptional output45. The seRNAs act synergistically with SEs to regulate gene expression by maintaining the SE-promoter chromatin loop46. For example, the seRNA HPSE interacts with hnRNPU and p300 and then facilitates chromatin looping between the SE and HPSE promoter to enhance HPSE expression, thus promoting cancer progression47.
Although the role of seRNAs in transcriptional regulation in both cis and trans is clear, the molecular mechanism is still controversial. Three models of seRNA mechanisms have been reported: interacting with TFs or coactivators, driving enhancer-promoter looping, and transferring to the cytoplasm to mediate various cell activities48. While most studies have demonstrated that seRNAs promote enhancer-promoter looping, Panigrahi et al. showed that seRNAs do not affect chromatin looping49. They concluded that mutual costimulation of enhancer and promoter transcription is not dependent on seRNAs49. Further studies are required to explain these conflicting observations regarding the molecular mechanism of seRNAs.
SeRNAs that are generated from genetic alterations or somatic mutations of SEs during tumorigenesis and are termed oncogenic seRNAs46 (Fig. 1). Oncogenic seRNAs are significantly upregulated in several types of tumors and associated with carcinogenic processes, including cell proliferation, apoptosis, autophagy, and epithelial-mesenchymal transition (EMT). In hepatocellular carcinoma (HCC), HCCL5, an SE-driven lncRNA, is significantly overexpressed and promotes cell growth, invasion, and metastasis50. Recently, the SE-lncRNA FASRL has been identified in HCC, and FASRL can increase fatty acid synthesis and lipid accumulation, thereby exacerbating HCC progression51. The novel SE-lncRNA LOC100506178 was found to promote nasopharyngeal carcinoma metastasis by interacting with the TF hnRNPK and controlling the expression of hnRNPK, accelerating the EMT process52. In addition, SEs can promote the transcription of pri-miRNAs and recruitment of the Drosha/Dgcr8 complex, thereby increasing the expression of cell-specific miRNAs43. In HCC, the YY1/p65/p300 complex increases QK1 expression through interaction with the SE-promoter loop, thus promoting HCC malignancy and increasing circRNA formation and EMT53. SEs usually contain discrete loci with seRNA expression peaks, and seRNA signals in cancer samples have been identified to have clinical relevance54. Although some studies have reported a correlation between SE-derived ncRNAs and prognostic indicators, most were meta-analyses, and thus, further clinical validation is required55.
Unique organizational structures of SEs
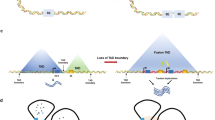
Studies on SEs consider not only the linear structure of the genome but also the three-dimensional (3D) structure of the genome. In the eukaryotic genome, DNA‒protein complexes fold 3D chromatin loops, called topologically associating domains (TADs), which are structural units for transcriptional regulation56. SEs can be enriched in the chromatin loop inside TADs, and SE-containing TADs have more chromatin interactions57. TADs insulate promoters from enhancers and superenhancers, and the disruption of TADs alters regulatory circuits and leads to oncogene activation58,59. The architectural protein CTCF interacting with the cohesin complex defines the boundary for TADs and SEs, and this interaction is specifically required for the formation of chromatin loops, allowing orderly gene regulation58,60,61. In T-cell acute lymphoblastic leukemia, loss of CTCF induces a TAD fusion event, which leads to a direct interaction between the MYC promoter and a distal SE, thus activating MYC62. Especially in the absence of cohesin, SEs tend to form links with each other, resulting in extensive SE fusion63. Therefore, an accurate understanding of the 3D genome chromatin structure is essential for SE-mediated transcriptional regulation.
Liquid–liquid phase separation (LLPS), a dynamic physicochemical process, forms a membraneless organelle termed a condensate to organize biological processes within cells64. In 2017, the proposed phase separation model limited SE-specific gene regulation to the membraneless organelle65. The intrinsically disordered regions (IDRs) of TFs, the transcriptional coactivators bromodomain protein 4 (BRD4) and mediator complex subunit 1 (MED1), and RNA Pol II can form phase-separated condensates at SE regions, leading to the transcriptional bursting and simultaneous activation of SE-driven genes66,67. The phase-separated condensate can separate SEs from other chromatin regions and concentrate SE-associated transcriptional processes at key regions, which explains why the effect of transcription regulation of SEs is greater than the overall effect of individual typical enhancers66. In leukemias, NUP98–HOXA9, a NUP98 fusion oncoprotein containing IDRs, can form LLPS condensates. LLPS of NUP98–HOXA9 is critical for leukemogenesis because it not only induces CTCF-independent chromatin loops but also leads to the formation of a “superenhancer”-like binding pattern at leukemogenic genes to regulate transcriptional activity68,69. The pharmacological inhibition of LLPS condensates can suppress metastasis and chemoresistance in osteosarcoma, representing a novel therapeutic strategy70. In addition, seRNAs also contribute to the organization of the phase-separated condensate and play a role in transcriptional activation71. Current studies on phase separation mainly focus on the role of biomolecular condensates assembled in tumors. However, such studies have not considered the intrinsic mechanism of the dynamic phase separation process, which still needs further investigation. Therefore, the phase separation model, which can ensure the precise regulation of genes, provides a novel perspective to elucidate the formation and transcriptional regulatory mechanisms of SEs (Fig. 2).

At the superenhancer locus, transcriptional regulators with extensive interactions, including TFs, BRD4, MED1, RNA pol II, and enhancer RNAs, are enriched to form a phase-separated condensate, which is separated from other chromatin domains and can drive transcriptional bursting and produce simultaneous activation of genes.
The gene transcription process requires a variety of transcriptional activators at specific DNA regulatory elements. To coordinate transcription programs in normal and malignant cells, a small group of master TFs forms an interconnected core regulatory circuitry (CRC) by directly co-occupying their SEs and each other’s SEs, allowing close contact between SEs and their target promoters8,72. The activities of SEs and TFs affect each other. On the one hand, SEs coordinate with TFs to regulate the gene expression program, and the activity of SEs is affected by TF enrichment. On the other hand, the expression of TFs is often regulated by the activity of SEs, indicating positive feedback regulation between SEs and TFs8. The interconnected autoregulatory loop containing SEs can regulate specific cell-type transcriptional programs. To date, the CRC model and associated master TFs have been identified in multiple cancer types, including esophageal cancer72, acute myeloid leukemia73, multiple myeloma74, and osteosarcoma70. Because of the important role of CRC in malignant tumors, targeting the core TFs may suppress tumor growth.
Functional activation of SEs during tumorigenesis
Cancer cells proactively construct SEs via mechanisms such as genetic mutation, single-nucleotide polymorphism (SNP), chromosomal rearrangement, and viral infection11 (Fig. 3). During tumorigenesis, the functional activation of SEs leads to the dysregulation of transcriptional programs, making tumors highly dependent on gene expression regulators75.

Genetic mutations, single-nucleotide polymorphisms (SNPs), chromosomal rearrangements, and viral infections lead to SE formation and oncogene activation.
Genetic mutations lead to SE activation by creating new binding sites for TFs, changing SE copy numbers, and changing the spatial structure of the genome. In T-cell acute lymphoblastic leukemia cells, there is a 12 bp insertion upstream of the TAL1 oncogene, which generates an SE region through the binding of MYC and the recruitment of CBP and other core TFs, driving the aberrant expression of key oncogenes76 (Fig. 3). In addition to insertion, focal amplification of SEs is a common mechanism of SE activation. For instance, two focal amplifications of SEs located on the 3’ side of MYC in endometrial carcinoma and lung cancer are close to the MYC promoter region and are associated with aberrant expression of MYC77.
Some specific tumor-related SNPs in regulatory elements are correlated with the activity of SEs. The SNP rs2168101 has been demonstrated to affect neuroblastoma susceptibility; it is located in the LMO1 superenhancer and changes a GATA-binding motif into a TATA motif, thus inhibiting the activity of the SE and decreasing the expression of LMO178 (Fig. 3). Similarly, in diffuse large B-cell lymphoma, two SNPs (rs9831894 and rs6773363) of susceptibility loci have been identified and are a part of a tumor-specific SE79.
It has been shown that structural variations can disrupt 3D genome organization and active SE regions, known as “superenhancer hijacking”, which has been reported in neuroblastoma, leukemia, and colorectal cancer80,81,82,83,84,85. Superenhancer hijacking has been described as a novel mechanism for tumorigenesis in which chromosomal rearrangements cause enhancers to transfer to nearby oncogenes, thereby leading to high expression of oncogenes and the tumor initiation82. In colorectal cancer, IGF2 is a target of enhancer hijacking, which is mediated by the formation of a contact domain comprising IGF2 and a lineage-specific superenhancer82 (Fig. 3). In addition, Wang et al. developed a computational framework called NeoLoopFinder to predict enhancer hijacking by identifying the chromatin interactions induced by structural variations80. They found that enhancer hijacking-driven oncogenes such as MYC, ETV1, PVT1, and CDK12 are significantly upregulated in cancer cells.
Viral infections have also been shown to induce SE formation and drive aberrant transcription of genes involved in the initiation and progression of tumors. Epstein‒Barr virus (EBV), human papilloma virus (HPV), human T-cell leukemia virus (HTLV), and human hepatitis B virus (HBV) are common oncogenic viruses, and their capacity to activate SE-associated genes has been reported in several tumors. HTLV-1 is associated with adult T-cell leukemia/lymphoma (ATLL) and specifically encodes the TF HBZ. HBZ binds to ATLL-specific BATF3 superenhancers, leading to the expression of BATF3 and other downstream genes and promoting the proliferation of ATLL cells86 (Fig. 3). In HPV-infected cervical cancer, integration of the viral genome into the host genome generates a superenhancer that drives the overexpression of viral E6 and E7, promoting the growth of cancer cells87.
Clinical applications related to SEs in cancer
Small-molecule inhibitors targeting SEs for tumor therapy
The high transcriptional activity of superenhancers mediates transcriptional addiction in aggressive tumors, making oncogenes extremely susceptible to transcriptional alterations88. Thus, targeting SE complexes with small-molecule inhibitors is a promising strategy for tumor therapy (Fig. 1). Unlike genetic mutations, epigenetic alterations are reversible, and many drugs targeting epigenetic regulators have exhibited therapeutic potential in clinical trials89. The sensitivity of SEs to emerging small-molecule inhibitors has been confirmed in various tumors, suggesting that SEs might be antitumor targets90. Some drugs have entered clinical trials (Table 1), with the hope that they can be applied clinically.
BRD4, a member of the bromodomain and extraterminal domain protein (BET) family, plays a role in SE organization and oncogene expression regulation91. Mechanistically, BRD4 binds to acetylated lysines in enhancers, SEs, and TFs, bringing them together and mediating transcriptional activation and elongation via RNA pol II and mediators92,93. Inhibition of BRD4 disrupts the communication between SEs and their target promoters, resulting in subsequent repression of oncogenes91. KDM6A, a histone demethylase, is frequently mutated, which promotes tumorigenesis. KDM6A loss was found to regulate aberrant activation of SEs of oncogenes, ultimately leading to pancreatic cancer development. KDM6A-deficient pancreatic cancer is sensitive to BET inhibitors (such as JQ1 and I-BET151), which can decrease the expression of SE-associated genes and suppress tumor growth94. Another BET inhibitor, OTX015, is effective against mouse and human MYCN-driven neuroblastoma in models, as it can selectively disrupt the binding of BRD4 and SEs and lead to the repression of MYCN expression95.
In addition, cyclin-dependent protein kinases (CDKs) are protein-serine/threonine kinases that play an essential role in regulating the cell cycle and transcription96. During transcriptional activation, BRD4 binds to SEs, followed by the recruitment of the TFIIH/CDK7 initiation complex and P-TEFb elongation complex containing CDK975. Cyclin-dependent kinase 7 (CDK7), an important CDK, can phosphorylate the RNA pol II C-terminal domain (CTD) at serine 5 (Ser5) and Ser7, leading to transcriptional initiation97. CDK7 also phosphorylates and activates CDK9/cyclin T, and CDK9 increases the phosphorylation of RNA pol II at Ser2, thus promoting transcriptional elongation98. Therefore, given that CDK7 and CDK9 are directly or indirectly involved in cellular transcriptional regulation, inhibition of CDK7 and CDK9 may interfere with transcription99. Covalent CDK7 inhibitors (such as THZ1 and THZ2) result in the downregulation of oncogene transcription and can serve as SE blockers to inhibit the expression of SE-driven oncogenes100. THZ2 is a newly developed CDK7 inhibitor with a fivefold longer half-life than THZ1, and its antitumor effects have been demonstrated in triple-negative breast cancer, gastric cancer, and osteosarcoma88,101,102. Moreover, the CDK9 inhibitor AZD4573 has shown effective antitumor activity through inhibition of CDK9, and AZD4573 is currently being evaluated in phase I clinical trial for patients with hematological malignancies (NCT03263637)103,104.
More importantly, studies have shown that combination of SE inhibitors with traditional therapy enhances efficacy105,106. The notable antitumor effects of JQ1 and THZ1 have been demonstrated in multiple types of tumors, including triple-negative breast cancer, diffuse large B-cell lymphoma, and pancreatic cancer88,107,108. Simultaneous inhibition of BRD4 and CDK9 with JQ1 and LDC067 suppresses cell growth and migration in medulloblastoma109,110. The coinhibition of these two molecules also shows a similar antiproliferative effect in malignant rhabdoid tumors109,110. Combination therapy with JQ1 and the novel CDK7 inhibitor YKL-5-124 shows a synergistic effect in neuroblastoma and can delay resistance to BRD4 inhibition111. In addition, combination of a histone deacetylase (HDAC) inhibitor and JQ1 or OTX015 results in stronger repression of oncogenes and higher expression of tumor suppressor genes, suggesting that these epigenetic drugs have synergistic antitumor effects112. Similarly, when THZ1 is combined with an HDAC inhibitor, sensitivity notably increases105,113. Given the potential therapeutic effects of small-molecule inhibitors, rational combinational strategies may be more effective for cancer treatment.
Despite significant inhibitory effects in previous studies, some side effects of BRD4 or CDK7 inhibitors have been reported in animal experiments and phase I clinical trials, such as heart toxicity, gastrointestinal toxicity, fatigue, thrombocytopenia, and impairment of muscle function114,115,116. Furthermore, drug resistance is a challenge related to small-molecule inhibitors. In MYCN-driven neuroblastoma cells, upregulation of the multidrug transporters ABCB1 and ABCG2 results in resistance to THZ1117. In tumors with resistance to BRD4 inhibitors, hyperphosphorylation of BRD4 and epigenetic plasticity contribute to decreased sensitivity of JQ1118,119. The safety and efficacy of these small-molecule inhibitors, including potential side effects, off-target effects, and drug resistance, need to be further studied before clinical application.
Utility of SEs in tumor prognostication
SEs can also serve as biomarkers for evaluating the prognosis of tumor patients. In HCC, the SE-associated lncRNA HCCL5 regulates malignant biological behavior and is associated with the prognosis of patients with HCC50. Further analysis revealed that the aberrant SE landscape in HCC is the result of extensive reprogramming. The key components of SEs (BRD4, CDK7, p300, and MED1) are overexpressed in HCC, which is associated with the poor prognosis of patients120. In addition, SEs with broad and high H3K27ac signals have been identified in nasopharyngeal carcinoma and were found to be associated with overexpression of the ETV6 oncogene and poor prognosis121. Xu et al. defined an SE-associated gene risk signature to predict the response to chemotherapy in patients with diffuse large B-cell lymphoma, which may help clinicians make more appropriate treatment decisions122. Moreover, in our previous studies, we constructed prognostic models based on SE-associated genes to predict overall survival for osteosarcoma and multiple myeloma patients, which may be helpful for clinical treatment123,124. Therefore, oncogenic SEs may promote the malignancy of tumors and have utility in predicting the clinical outcome of tumor patients.
The oncogenic roles and regulatory mechanisms of SEs in brain tumors
SEs in glioblastoma
Glioblastoma (GBM), a high-grade glioma (World Health Organization grade IV), is the most common malignant brain tumor and has a 5-year survival rate of 5.6%125. Despite multimodal treatment, including surgical resection, radiation, and standard therapy with temozolomide (TMZ), the prognosis is universally poor due to therapy resistance and recurrence126,127,128. Therefore, it is urgent to explore the molecular mechanisms underlying GBM progression and develop novel therapeutic strategies to improve the prognosis of GBM patients.
Identification of SEs in GBM
In the past few years, several novel epigenetic markers contributing to the pathogenesis of GBM have been reported. In 2014, a population-based single-cell whole-genome sequencing methodology was applied to characterize genomic heterogeneity in EGFR-amplified GBM129. Researchers identified a translocation of a superenhancer to the 5’ promoter region of TERT, which can activate TERT129. Moreover, another study described 3D genome information through Hi-C sequencing in glioblastoma stem cells (GSCs)130. In GSCs, genomic structural variants lead to SE-promoter interactions, such as physical interactions between the two SEs (SE1 and SE2) and JAK1130. In addition, integration of Hi-C and chromatin data revealed strong H3K27ac signals in stemness genes and identified a region overlapping SOX2 as an SE locus130. Currently, the majority of biopsies are preserved as archived formalin-fixed paraffin-embedded (FFPE) samples. Zhao et al. assessed FFPE tissue with antibody-guided chromatin tagmentation with sequencing (FACT-seq), the first highly sensitive method to describe histone modifications in FFPE tissues131. Using FACT-seq of H3K27ac in FFPE human GBM samples, the researchers identified 492 disease-specific SEs (in genes such as EGFR, ETV1, and CDK6)131. In summary, the development of novel technologies allows the identification of complete SE landscapes and histone modifications in the whole genome, which aids the investigation of the epigenetic regulation and heterogeneity of GBM.
SE-mediated aberrant transcriptional programs
Research focused on SE-driven aberrant transcriptional programs in GBM is gradually increasing. Since CDK7 can phosphorylate RNA Pol II to initiate transcription, CDK7 expression is significantly increased in GBM and is associated with poor prognosis98,132. The CDK7 inhibitor THZ1 can disrupt global gene transcription and preferentially target SE-associated genes in GBM cells. For example, five highly expressed SE-associated genes (WNT7B, FOSL1, FOXL1, ZMIZ1, and PHC2) were found to be associated with sensitivity to THZ1, and their knockdown inhibited the proliferation of GBM cells (Fig. 4a)132. In addition, GBM cells harbor a superenhancer in the Mcl-1 locus, leading to an increased level of Mcl-1, a member of the antiapoptotic Bcl-2 family of proteins133. Pharmacological inhibition of the SE by THZ1 decreased Mcl-1 mRNA and protein levels. A similar study identified the SE-derived lncRNA TMEM44-AS1, which was upregulated and correlated with malignant phenotypes in GBM134. When Myc directly binds to the promoter and SE of TMEM44-AS1 and colocalizes with MED1, H3K27ac, and RNA pol II, TMEM44-AS1 is activated134. The Myc inhibitor Myci975 can inhibit the growth of GBM cells by inhibiting the Myc/TMEM44-AS1 feedback loop (Fig. 4b)134. Together, these findings suggest that SE-mediated aberrant transcriptional programs are associated with GBM progression, and targeting these oncogenic SEs may be an effective therapeutic strategy for GBM.

a In GBM cells, five highly expressed SE-associated genes are associated with sensitivity to the CDK7 inhibitor THZ1. b Myc and MED1 mediate the epigenetic activation of TMEM44-AS1, which is directly bound to SerpinB3, and sequentially activate Myc and EGR1/IL-6 signaling; Myc induces transcription of TMEM44-AS1 and binds to the SE region, forming a positive feedback loop with TMEM44-AS1, thus aggravating tumor progression c The RFP-HDAC1 complex contributes to TMZ resistance via aberrant deacetylation of H3K27ac, and the disruption of the complex leads to an increase in TMZ efficacy by changing core histone modifications in GBM. d The core transcriptional regulatory circuitry (CRC) in GBM: SE-driven TFs are highly enriched in SE regions and regulate the expression of SE-associated lncRNAs. The BET degrader dBET6 can effectively disrupt the expression of core TFs.
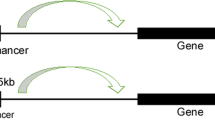
SE-driven mechanisms of chemoresistance
Resistance to TMZ occurs in almost all patients during chemotherapy, leading to a poor prognosis. High expression of O6-methylguanine-DNA methyltransferase (MGMT) is one of the leading causes of TMZ resistance because MGMT is a DNA repair factor that can reverse DNA damage caused by TMZ135. MGMT expression is negatively associated with methylation modification in the MGMT promoter region136. Thus, the higher the methylation degree of the MGMT gene promoter region in GBM patients is, the better the effect of TMZ. Apart from methylation, MGMT expression can be regulated by other factors. It was found that the enhancer (K-M enhancer) located between the Ki67 and MGMT genes can increase MGMT expression and is activated in TMZ-resistant GBM tissues137. Deleting the K-M enhancer reduces MGMT and Ki67 expression, thus increasing sensitivity to TMZ137. Moreover, another study demonstrated that aberrant transcriptional regulation of SEs is a cause of resistance to TMZ138. The H3K27ac status regulates cis-regulatory elements such as SEs. However, the RET finger protein-histone deacetylase 1 (RFP-HDAC1) complex deacetylates H3K27ac marks on core histones, decreasing gene transcription levels138. Disruption of the HDAC complex can increase TMZ sensitivity in GBM cells by affecting the H3K27ac status (Fig. 4c)138,139. In the future, more in-depth studies are needed to investigate whether the epigenetic regulatory mechanism underlying TMZ resistance is mediated by histone acetylation and which SEs drive transcriptional reprogramming. GBM resistance to TMZ may be related to abnormal SE activity, providing new ideas for overcoming GBM drug resistance.
HDACs regulate SE activity
Multiple lines of evidence have reported that the malignant phenotype of GBM is regulated by SEs and HDACs. HDAC1, together with BRD4, RNA pol II, and other key components, is enriched in SEs10. SLC30A3 is significantly reduced in GBM tissues and can inhibit the growth and metastasis of GBM cells140. Overexpression of HDAC1 decreases the H3K27ac level in the SE region of SLC30A3, which inhibits the expression of SLC30A3140. HDAC1 is predominantly associated with transcriptional repression, while the transcriptional regulation mechanism of HDAC7 is complex and related to both activation and repression. In stem-like breast cancer cells, HDAC1 and HDAC3 regulate the expression of HDAC7141. In turn, HDAC7 maintains H3K27ac levels by binding to SEs, thereby promoting the expression of SE-associated oncogenes141. HDAC1/3 knockdown increases the overall H3K27ac level, but HDAC7 knockdown decreases the H3K27ac level, and these changes are only observed in cancer stem cells and not in other cells141. These results suggest functional differences between HDAC1/3 and HDAC7. Inhibition of HDAC1/2/3 disrupts SE activity by increasing the accessibility of TF sites and disrupting chromatin loops, an indirect effect105,142,143,144. In GBM models, the HDAC inhibitors panobinostat and romidepsin have been shown to target SEs and elicit metabolic reprogramming through H3K27ac modification, thereby suppressing GBM progression143,145. Overall, different types of HDACs have different regulatory effects on SE activity.
The molecular mechanisms underlying HDAC inhibitor-mediated SE activity are not yet fully understood. Some studies have investigated the regulatory mechanisms of HDAC inhibitors: (1) reshaping epigenetic markers by promoting H3K27 acetylation144,146,147; (2) decreasing the binding of RNA pol II at SE regions143,144,145; (3) inducing SE looping defects by reducing contact with SE components and promoters142; and (4) reducing the expression of core regulatory circuitry TFs to repress SE activity72. These epigenetic alterations cause the global disruption of SEs and thus inhibit SE-associated transcript and gene expression143,144. Based on their antitumor effects, HDAC inhibitors should be able to exert synergistic effects in combination with SE inhibitors (such as THZ1 and JQ1) and chemotherapies in neuroblastoma, glioblastoma, and other tumors72,105,112,113,146.
SE-mediated core regulatory circuitry
SE-mediated core regulatory circuitry plays a crucial role in human cancers, including medulloblastoma, neuroblastoma, and glioblastoma8,148,149,150. An active chromatin landscape was mapped in GSCs, identifying SE-associated genes and core TFs that establish SEs and maintain GSC identity151. The study found that many genes defining GSC identity are highly expressed and regulated by SEs, such as SOX2, SEPT9, CXXC5, CDK6, SALL3, and EGFR, which are required for tumor cell proliferation151. In addition, another study depicted the core TFs and their interconnected regulatory circuitry in GBMs, showing that many motifs of SE-driven TFs (SOX2, JUN, and RFX2) are enriched in SE regions (Fig. 4d)152. The stem cell TF SOX2 can bind to the ELOVL2 superenhancer in GSCs to drive the expression of ELOVL2, which is critical for the maintenance of cancer stem cells153. Furthermore, nine SE-associated lncRNAs were identified in GBM tissues, and LINC01094 is one of the lncRNAs whose disease-specific expression is regulated by tumor-associated core TFs and SEs152. SEs and the core regulatory circuitry in GBM cooperatively regulate transcriptional programs, which is significant for GBM progression.
Roles of seRNAs in gliomas
Some seRNA-regulated immune-related genes (IRGs) relevant in the tumor microenvironment were identified in glioma154, and they were used to construct a prognostic model that can predict the survival outcomes of patients with glioma. These glioma-specific seRNAs that regulate IRGs are located in adjacent sites and coexpressed in tumors, suggesting that they have potential as therapeutic targets154,155.
Promising antitumor effects of targeting BET proteins
The BET inhibitor I-BET151 inhibits the proliferation and eliminates the tumorigenicity of GBM cells156,157,158. The antiproliferative activity is equivalent to that of TMZ, suggesting that I-BET151 may be beneficial for GBM patients with TMZ resistance156. In addition, another BET inhibitor, OTX015, was found to cross the BBB and selectively penetrate tumor tissues in a preclinical study, indicating its potential efficacy in GBM159. In addition, dBET6, a chemical degrader of BET proteins, can inhibit the proliferation and self-renewal ability of GBM cells by significantly reducing BET protein occupancy, RNA pol II activity, and active histone markers, and dBET6 is superior to first-generation BRD4 inhibitors such as JQ1160,161. Interestingly, GBM cells exhibit sex-specific responses to the BRD4 inhibitor JQ1, which is correlated with sex-specific gene expression patterns. The sensitivity of male patient-derived GBM cells to BET inhibitors is higher than that of female patient-derived GBM cells162.
As newly recognized epigenetic elements, numerous SEs have been identified in GBM and have been shown to be related to tumor progression (Table 2). As described above, novel epigenetic mechanisms that regulate the malignant biological behaviors of GBM cells have been revealed, providing potential treatments. In the future, targeting SEs may be a promising complement to traditional therapies for GBM treatment.
SEs in pediatric brain tumors
SEs in medulloblastoma
Medulloblastoma (MB) is the most common malignant brain tumor in children and adolescents younger than 15 years old163. Previous genomic studies have revealed four molecular subgroups of MB (WNT, sonic-hedgehog (SHH), Group 3, and Group 4) with different biological and clinical behaviors164,165. Among these subgroups, Group 3 and Group 4 MBs account for the majority of cases and have the poorest outcomes166.
A study of MB illustrated that oncogenic drivers were restricted to Group 3 and Group 4167. The growth factor independent 1 family oncogenes GFI1 and GFI1B are oncogenes of Group 3 and Group 4 MBs that can cooperate with MYC to promote MB formation in vivo167. Genomic structural variants occur in the GFI1 or GFI1B coding sequences proximal to enhancers and superenhancers, leading to oncogenic activation of GFI1 and GFI1B167. In addition, scientists have identified some subgroup-specific SEs in MBs that can activate oncogenes, including ALK, MYC, SMO, and ETV4148. In Group 3 MB, an SE-driven transcriptional regulatory network consisting of 14 SE-associated genes exists168. A BET inhibitor (JQ1) and a CDK7 inhibitor (THZ1) showed synergistic inhibitory effects, with therapeutic potential for Group 3 MB168. Moreover, some adult SHH MB patients present with truncating mutations in the chromatin reader BRPF1, which are absent or rare in pediatric patients169. Mutated BRPF1 increases the accessibility of a subset of SEs associated with key genes involved in cerebellum development and chromatin remodeling, promoting tumorigenesis in adult SHH MB170.
Superenhancers not only activate oncogene expression but are also associated with tumor suppressor gene activation. Brain-specific knockout of the H3K4 methyltransferase MLL4 in mice can induce MB171. Mechanistically, MLL4 loss leads to MB by reducing broad H3K4me3 and SE signals, downregulating tumor suppressors (Dnmt3a and Bcl6), and extensively damaging epigenomic signatures171. Therefore, MLL4-established superenhancers play an important role in tumor suppression in normal cells. The antitumor role of SEs has also been reported in other types of tumors, such as chronic myelogenous leukemia and breast cancer172. Superenhancers can drive either oncogene expression or tumor suppressor expression, playing dual roles in tumor progression. SE-associated genes and possible mechanisms in MB are shown in Table 2.
SEs in diffuse intrinsic pontine glioma
Diffuse intrinsic pontine glioma (DIPG) is a fatal pediatric brain tumor with a median survival time of 11 months173. Because of its diffuse growth and location, DIPG is inoperable, and the standard therapy is radiation therapy, but its effectiveness is extremely limited174. The characterization and development of drugs are challenging due to the scarcity of DIPG samples.
Approximately 85% of DIPGs are characterized by mutation of lysine 27 to methionine in histone 3 (H3K27M), which leads to oncogenic transcription dysregulation and increased stem-like potential and proliferation105,173,174,175. H3K27M glioma cells exhibit greater proliferation potential due to the aberrant oncogenic program176. Together with RNA pol II and H3K27ac, H3K27M localizes to transcriptionally active regions and can drive SE formation177,178. H3K27M inhibits the enzymatic activity of PCR2 through its interaction with the E2H2 subunit, resulting in the loss of H3K27me3 at SE regions179,180. H3K27M leads to a more accessible chromatin configuration in key regulatory regions, which can expose binding site motifs for key TFs, including ASCL1 and NEUROD1181. These TFs then bind to SEs to activate neurogenesis and NOTCH pathway-related genes, ultimately contributing to glioma formation180,181. Recent studies of H3K27M DIPG in vitro and in vivo have identified some important druggable targets and found some effective small molecules to reverse epigenetic alterations, including the HDAC inhibitor panobinostat182, the H3K27me2/3 inhibitor GSKJ4182, the BET inhibitor JQ1178, and the selective E2H2 inhibitor EPZ6438183.
The analysis of SEs in DIPG has revealed potential cell-identity genes, supporting the idea that DIPG originates from a precursor cell of the oligodendroglial lineage. These SE-associated genes include genes classically associated with oligodendrocyte precursor cells (such as SOX10) and genes expressed by oligodendroglial lineage cells during differentiation (such as OLIG1, MYRF, MYT1, and MBP)105. DIPG is vulnerable to BRD4 and CDK7 blockade, which can impair DIPG cell growth105. For example, the BRD4 inhibitor JQ1 can induce neuron-like differentiation and delay tumor growth in a mouse model of DIPG, and the CDK7 inhibitor THZ1 can disrupt transcription and inhibit DIPG growth105,184. Hyperacetylation in DIPG favors the action of BDR4 and leads to enhancer landscape reprogramming, activating SE-driven oncogenes in DIPG174. In addition, CBP is related to transcription activation, and its activity can be enhanced by BRD4185. The combination of JQ1 and a CBP inhibitor (ICG-001) can reverse the detrimental SE programs activated by BET or CBP174. Collectively, these findings suggest that SE-driven oncogenes in DIPG can be targeted with SE blockers. SE-associated genes and possible mechanisms in DIPG are shown in Table 2.
SEs in other rare pediatric brain tumors
DNA 5-hydroxymethylcytosine (5hmC), one of the molecular alterations in GBM, can recruit DNA-binding proteins and is essential for GBM tumorigenesis186. 5hmC preferentially localizes to the enhancers and superenhancers of tumor-specific genes in glioblastoma, activating disease-specific gene expression programs187. 5hmC alteration is observed not only in adult brain tumors but also in pediatric brain tumors according to a recent study; 5hmC is located in TF binding sites and SE regions and is crucial to cell identity187,188. Thus, the epigenetic alterations of SEs in adult and pediatric brain tumors are somewhat similar, and 5hmC can be used to identify these aberrant regulatory elements.
Atypical rhabdoid/teratoid tumors (AT/RTs) are aggressive and lethal cancers that can be diagnosed at a young age189. The SWItch/Sucrose Non-Fermentable (SWI/SNF) chromatin-remodeling complex is an essential regulator of pluripotency in human embryonic stem cells. Nevertheless, mutation of its core subunit SMARCB1 can lead to AT/RTs due to the disruption of enhancer accessibility190. With noticeable H3K27ac features in enhancer regions, SMARCB1 is required for the integrity of the SWI/SNF complex191, which is inactivated abnormally in most rhabdoid tumors192. Loss of SMARCB1 reduces genome-wide targeting at enhancers, thus impairing the functions of the SWI/SNF complex. However, the small amount of residual SWI/SNF complexes preferentially bind to SEs, including some shared by all subtypes, such as SPRY1, and other lineage-specific SEs, such as SOX2, in brain-derived rhabdoid tumors, which is crucial for maintaining aberrant cell identity. Then, these SEs drive oncogenic transformation by locking cells into a poorly differentiated and highly proliferative state191. Further research found that knockdown of SMARCB1 prevents silencing in SE regions, thus leading to transcriptional upregulation in human embryonic stem cells193. Therefore, SMARCB1 and its regulatory effects on SEs in AT/RT are part of a novel AT/RT tumorigenesis mechanism.
Embryonal tumor with multilayered rosettes (ETMR) is a sporadic and difficult-to-treat brain tumor in infants and young children, with rapid progression and only 10–20% overall survival194,195. Because there are few research models, mechanistic and therapeutic studies of this rare disease are extremely limited. Recently, an oncogenic circuit driven by hijacked superenhancers in ETMRs was revealed. The C19MC-TTYH1 gene fusion and MYCN DNA interactions create superenhancers. Then, the interaction between C19MC-TTYH1 superenhancers and MYCN enhancers fortifies the C19MC-LIN28A-MYCN circuitry, driving the expression of embryonically restricted DNMT3B6 to promote a primitive malignant epigenetic state in ETMRs196. Interestingly, the BET inhibitor JQ1 can downregulate key components of the C19MC-LIN28A-MYCN circuit, including MYCN, LIN28A, and DNMT3B6, disrupting the circuit and inducing ETMR cell death196. The unique SE-dependent oncogenic circuit protects the ETMR and is vulnerable to BET inhibition. Therefore, inhibition of BET may be a promising therapeutic strategy for this orphan disease. SE-associated genes and possible mechanisms in these pediatric brain tumors are shown in Table 2.
SEs in other brain tumors
Meningioma is one of the most common intracranial tumors. Most meningioma patients can be cured by surgical resection, but approximately 20% of patients experience an aggressive clinical course with tumor recurrence or progression197. A comprehensive investigation of the genomic landscape has revealed the overall genomic instability in aggressive meningioma198. Upregulation of the SE-associated HOXD gene is associated with meningioma aggressiveness198 (Table 2).
Ependymoma is a rare disease that can arise throughout neuraxis199. In children, ependymomas mainly occur intracranially, while in adults, the spine is the most common location of ependymomas199. The challenges of ependymomas are resistance to chemotherapy and lack of effective molecular targets. More than 60% of ependymomas harbor a ZFTA-RELA (ZFTAfus) gene fusion200. ZFTAfus contributes to an oncogenic transcriptional program because it binds TF motifs and recruits transcriptional coactivators (BRD4, EP300, CBP, RNA pol II), thus driving SE gene expression in ependymoma201. To identify SE-associated genes that ependymoma cells depend on, Mack et al. analyzed the chromatin landscapes of ependymomas202. In two cohorts of ependymoma specimens, they identified that the vast majority of SEs were tumor-specific and enriched in oncogenes. Among the genes, EPHB2 and CCND1 have been previously proven to be ependymoma-related oncogenes203,204. In addition, ependymoma is sensitive to the BET inhibitor JQ1, which can inhibit the proliferation of ependymoma cells202 (Table 2).
Concluding remarks
The discovery of SEs is a novel breakthrough in the field of epigenetics. SEs are core regulatory elements that maintain the identity of cancer cells and drive cancer cells to become highly addicted to oncogene transcription. SEs regulate the expression of oncogenes that facilitate proliferation, migration, invasion, and even drug resistance, thus promoting tumor malignancy. With the accumulation of relevant research, the role of SEs in brain tumors is becoming increasingly clear. However, the composition of tumor-specific SEs and their potential molecular mechanisms remain to be further investigated.
Transcriptional activators (such as BRD4 and CDK7) are highly enriched in oncogenic SE regions, and their inhibition preferentially affects SE-associated genes in tumor cells. Some inhibitors targeting transcriptional activators have been evaluated in clinical trials. Moreover, combining SE inhibitors with other chemotherapeutic drugs can suppress tumor growth, providing a new strategy for cancer treatment. Since transcription is a basic biological process common to all nucleated cells, targeting SE-associated transcription may lead to general toxicity. Although inhibitors targeting SEs, including JQ1, THZ1, and THZ2, have been studied in many tumors, their potential side effects and off-target effects need further study.
As described above, the development of various brain tumors is closely associated with SEs and downstream oncogenes, and SEs may serve as master gene regulators and novel therapeutic targets. Further studies on the underlying mechanisms of SE activation will shed light on the complex pathogenesis of brain tumors. With persistent SE research efforts, breakthroughs in the treatment of malignant brain tumors are expected.
References
Miranda-Filho, A., Piñeros, M., Soerjomataram, I., Deltour, I. & Bray, F. Cancers of the brain and CNS: global patterns and trends in incidence. Neuro. Oncol. 19, 270–280 (2017).
Ostrom, Q. T., Francis, S. S. & Barnholtz-Sloan, J. S. Epidemiology of brain and other CNS tumors. Curr. Neurol. Neurosci. Rep. 21, 68 (2021).
Mo, F., Pellerino, A., Soffietti, R. & Rudà, R. Blood-brain barrier in brain tumors: biology and clinical relevance. Int. J. Mol. Sci. 22, 12654 (2021).
Steeg, P. S. The blood-tumour barrier in cancer biology and therapy. Nat. Rev. Clin. Oncol. 18, 696–714 (2021).
Gusyatiner, O. & Hegi, M. E. Glioma epigenetics: from subclassification to novel treatment options. Semin. Cancer Biol. 51, 50–58 (2018).
Ko, J. Y., Oh, S. & Yoo, K. H. Functional enhancers as master regulators of tissue-specific gene regulation and cancer development. Mol. Cells 40, 169–177 (2017).
Grosveld, F., van Staalduinen, J. & Stadhouders, R. Transcriptional regulation by (super)enhancers: from discovery to mechanisms. Annu. Rev. Genomics Hum. Genet. 22, 127–146 (2021).
Jiang, Y., Jiang, Y. Y. & Lin, D. C. Super-enhancer-mediated core regulatory circuitry in human cancer. Comput. Struct. Biotechnol. J. 19, 2790–2795 (2021).
Zheng, C., Liu, M. & Fan, H. Targeting complexes of super-enhancers is a promising strategy for cancer therapy. Oncol. Lett. 20, 2557–2566 (2020).
Hnisz, D. et al. Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013).
Jia, Q., Chen, S., Tan, Y., Li, Y. & Tang, F. Oncogenic super-enhancer formation in tumorigenesis and its molecular mechanisms. Exp. Mol. Med. 52, 713–723 (2020).
Whyte, W. A. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013).
Ying, Y. et al. Oncogenic HOXB8 is driven by MYC-regulated super-enhancer and potentiates colorectal cancer invasiveness via BACH1. Oncogene 39, 1004–1017 (2020).
Yu, D. et al. Super-enhancer induced IL-20RA promotes proliferation/metastasis and immune evasion in colorectal cancer. Front. Oncol. 11, 724655 (2021).
Niederriter, A. R., Varshney, A., Parker, S. C. & Martin, D. M. Super enhancers in cancers, complex disease, and developmental disorders. Genes (Basel) 6, 1183–1200 (2015).
Thandapani, P. Super-enhancers in cancer. Pharmacol. Ther. 199, 129–138 (2019).
Levine, M., Cattoglio, C. & Tjian, R. Looping back to leap forward: transcription enters a new era. Cell 157, 13–25 (2014).
Lu, B. et al. Epigenetic profiling identifies LIF as a super-enhancer-controlled regulator of stem cell-like properties in osteosarcoma. Mol. Cancer Res. 18, 57–67 (2020).
Li, K. et al. Comprehensive epigenetic analyses reveal master regulators driving lung metastasis of breast cancer. J. Cell Mol. Med. 23, 5415–5431 (2019).
Sengupta, D. et al. Disruption of BRD4 at H3K27Ac-enriched enhancer region correlates with decreased c-Myc expression in Merkel cell carcinoma. Epigenetics 10, 460–466 (2015).
Kang, Y., Kim, Y. W., Kang, J. & Kim, A. Histone H3K4me1 and H3K27ac play roles in nucleosome eviction and eRNA transcription, respectively, at enhancers. FASEB. J. 35, e21781 (2021).
Visel, A. et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457, 854–858 (2009).
Boyle, A. P. et al. High-resolution mapping and characterization of open chromatin across the genome. Cell 132, 311–322 (2008).
Jin, F. et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 503, 290–294 (2013).
Buenrostro, J. D., Wu, B., Chang, H. Y. & Greenleaf, W. J. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 109, 21.29.21–21.29.29 (2015).
Li, Y. et al. TRlnc: a comprehensive database for human transcriptional regulatory information of lncRNAs. Brief. Bioinform. 22, 1929–1939 (2021).
Chen, C. et al. SEA version 3.0: a comprehensive extension and update of the super-enhancer archive. Nucleic Acids Res. 48, D198–D203 (2020).
Qian, F. C. et al. SEanalysis: a web tool for super-enhancer associated regulatory analysis. Nucleic Acids Res 47, w248–w255 (2019).
Guo, Z. W. et al. SELER: a database of super-enhancer-associated lncRNA-directed transcriptional regulation in human cancers. Database (Oxford) 2019, baz027 (2019).
Jiang, Y. et al. SEdb: a comprehensive human super-enhancer database. Nucleic Acids Res. 47, d235–d243 (2019).
Tang, Z. et al. TRCirc: a resource for transcriptional regulation information of circRNAs. Brief. Bioinform. 20, 2327–2333 (2019).
Yuan, J., Zhou, J., Wang, H. & Sun, H. SKmDB: an integrated database of next generation sequencing information in skeletal muscle. Bioinformatics 35, 847–855 (2019).
Huang, M. et al. dbCoRC: a database of core transcriptional regulatory circuitries modeled by H3K27ac ChIP-seq signals. Nucleic Acids Res. 46, D71–D77 (2018).
Khan, A. & Zhang, X. dbSUPER: a database of super-enhancers in mouse and human genome. Nucleic Acids Res. 44, D164–D171 (2016).
Feng, C. et al. KnockTF: a comprehensive human gene expression profile database with knockdown/knockout of transcription factors. Nucleic Acids Res. 48, D93–D100 (2020).
Mei, S. et al. Cistrome cancer: a web resource for integrative gene regulation modeling in cancer. Cancer Res. 77, e19–e22 (2017).
Bu, H., Hao, J., Gan, Y., Zhou, S. & Guan, J. DEEPSEN: a convolutional neural network based method for super-enhancer prediction. BMC Bioinform. 20, 598 (2019).
Wang, F. et al. ATACdb: a comprehensive human chromatin accessibility database. Nucleic Acids Res. 49, d55–d64 (2021).
Pan, Q. et al. VARAdb: a comprehensive variation annotation database for human. Nucleic Acids Res. 49, d1431–d1444 (2021).
Ascensión, A. M., Arrospide-Elgarresta, M., Izeta, A. & Araúzo-Bravo, M. J. NaviSE: superenhancer navigator integrating epigenomics signal algebra. BMC Bioinform. 18, 296 (2017).
Mao, R. et al. Enhancer RNAs: a missing regulatory layer in gene transcription. Sci. China Life Sci. 62, 905–912 (2019).
Soibam, B. Super-lncRNAs: identification of lncRNAs that target super-enhancers via RNA:DNA:DNA triplex formation. RNA 23, 1729–1742 (2017).
Suzuki, H. I., Young, R. A. & Sharp, P. A. Super-enhancer-mediated RNA processing revealed by integrative microRNA network analysis. Cell 168, 1000–1014.e1015 (2017).
Huang, S. et al. Loss of super-enhancer-regulated circRNA Nfix induces cardiac regeneration after myocardial infarction in adult mice. Circulation 139, 2857–2876 (2019).
Henninger, J. E. et al. RNA-mediated feedback control of transcriptional condensates. Cell 184, 207–225.e224 (2021).
Tan, Y., Li, Y. & Tang, F. Oncogenic seRNA functional activation: a novel mechanism of tumorigenesis. Mol. Cancer 19, 74 (2020).
Jiao, W. et al. HPSE enhancer RNA promotes cancer progression through driving chromatin looping and regulating hnRNPU/p300/EGR1/HPSE axis. Oncogene 37, 2728–2745 (2018).
Xiao, S., Huang, Q., Ren, H. & Yang, M. The mechanism and function of super enhancer RNA. Genesis 59, e23422 (2021).
Panigrahi, A. K. et al. SRC-3 coactivator governs dynamic estrogen-induced chromatin looping interactions during transcription. Mol. Cell 70, 679–694.e677 (2018).
Peng, L. et al. Super-enhancer-associated long noncoding RNA HCCL5 is activated by ZEB1 and promotes the malignancy of hepatocellular carcinoma. Cancer Res. 79, 572–584 (2019).
Peng, J. Y. et al. Upregulation of superenhancer-driven LncRNA FASRL by USF1 promotes de novo fatty acid biosynthesis to exacerbate hepatocellular carcinoma. Adv. Sci. (Weinh.) 10, e2204711 (2022).
Tan, Y. et al. A novel oncogenic seRNA promotes nasopharyngeal carcinoma metastasis. Cell Death Dis. 13, 401 (2022).
Han, J. et al. YY1 complex promotes quaking expression via super-enhancer binding during EMT of hepatocellular carcinoma. Cancer Res. 79, 1451–1464 (2019).
Chen, H. & Liang, H. A high-resolution map of human enhancer RNA loci characterizes super-enhancer activities in cancer. Cancer Cell 38, 701–715.e705 (2020).
Wang, Y. et al. The emerging role of super enhancer-derived noncoding RNAs in human cancer. Theranostics 10, 11049–11062 (2020).
Dekker, J. & Mirny, L. The 3D genome as moderator of chromosomal communication. Cell 164, 1110–1121 (2016).
Marchal, C. et al. High-resolution genome topology of human retina uncovers super enhancer-promoter interactions at tissue-specific and multifactorial disease loci. Nat. Commun. 13, 5827 (2022).
Zhang, J. et al. Super enhancers-functional cores under the 3D genome. Cell Prolif. 54, e12970 (2021).
Bing, X. Y., Batut, P. J., Levo, M., Levine, M. & Raimundo, J. SnapShot: the regulatory genome. Cell 182, 1674–1674.e1671 (2020).
Dowen, J. M. et al. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 159, 374–387 (2014).
Li, Y. et al. The structural basis for cohesin-CTCF-anchored loops. Nature 578, 472–476 (2020).
Kloetgen, A. et al. Three-dimensional chromatin landscapes in T cell acute lymphoblastic leukemia. Nat. Genet. 52, 388–400 (2020).
Rao, S. S. P. et al. Cohesin loss eliminates all loop domains. Cell 171, 305–320.e324 (2017).
Cai, D., Liu, Z. & Lippincott-Schwartz, J. Biomolecular condensates and their links to cancer progression. Trends Biochem. Sci. 46, 535–549 (2021).
Hnisz, D., Shrinivas, K., Young, R. A., Chakraborty, A. K. & Sharp, P. A. A phase separation model for transcriptional control. Cell 169, 13–23 (2017).
Sabari, B. R. et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958 (2018).
Cho, W. K. et al. Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415 (2018).
Ahn, J. H. et al. Phase separation drives aberrant chromatin looping and cancer development. Nature 595, 591–595 (2021).
Chandra, B. et al. Phase separation mediates NUP98 fusion oncoprotein leukemic transformation. Cancer Discov. 12, 1152–1169 (2022).
Lu, B. et al. Pharmacological inhibition of core regulatory circuitry liquid-liquid phase separation suppresses metastasis and chemoresistance in osteosarcoma. Adv. Sci. (Weinh.) 8, e2101895 (2021).
Corrigendum Referring to: Phase separation and transcription regulation: are super-enhancers and locus control regions primary sites of transcription complex assembly? Bioessays 41, e1970014 (2019).
Jiang, Y. Y. et al. TP63, SOX2, and KLF5 establish a core regulatory circuitry that controls epigenetic and transcription patterns in esophageal squamous cell carcinoma cell lines. Gastroenterology 159, 1311–1327.e1319 (2020).
Assi, S. A. et al. Subtype-specific regulatory network rewiring in acute myeloid leukemia. Nat. Genet. 51, 151–162 (2019).
Jin, Y. et al. Active enhancer and chromatin accessibility landscapes chart the regulatory network of primary multiple myeloma. Blood 131, 2138–2150 (2018).
Bradner, J. E., Hnisz, D. & Young, R. A. Transcriptional addiction in cancer. Cell 168, 629–643 (2017).
Mansour, M. R. et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science 346, 1373–1377 (2014).
Zhang, X. et al. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat. Genet. 48, 176–182 (2016).
Oldridge, D. A. et al. Genetic predisposition to neuroblastoma mediated by a LMO1 super-enhancer polymorphism. Nature 528, 418–421 (2015).
Kleinstern, G. et al. Inherited variants at 3q13.33 and 3p24.1 are associated with risk of diffuse large B-cell lymphoma and implicate immune pathways. Hum. Mol. Genet 29, 70–79 (2020).
Wang, X. et al. Genome-wide detection of enhancer-hijacking events from chromatin interaction data in rearranged genomes. Nat. Methods 18, 661–668 (2021).
Valentijn, L. J. et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat. Genet. 47, 1411–1414 (2015).
Weischenfeldt, J. et al. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat. Genet. 49, 65–74 (2017).
Gröschel, S. et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157, 369–381 (2014).
Zimmerman, M. W. et al. MYC drives a subset of high-risk pediatric neuroblastomas and is activated through mechanisms including enhancer hijacking and focal enhancer amplification. Cancer Discov. 8, 320–335 (2018).
Montefiori, L. E. et al. Enhancer hijacking drives oncogenic BCL11B expression in lineage-ambiguous stem cell leukemia. Cancer Discov. 11, 2846–2867 (2021).
Nakagawa, M. et al. Targeting the HTLV-I-regulated BATF3/IRF4 transcriptional network in adult T cell leukemia/lymphoma. Cancer Cell 34, 286–297.e210 (2018).
Dooley, K. E., Warburton, A. & McBride, A. A. Tandemly integrated HPV16 can form a Brd4-dependent super-enhancer-like element that drives transcription of viral oncogenes. MBio 7, e01446–16 (2016).
Wang, Y. et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 163, 174–186 (2015).
Cheng, Y. et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 4, 62 (2019).
Wang, L. & Hu, G. Remodeling super-enhancers and oncogenic transcription. Cell Cycle 15, 3157–3158 (2016).
Donati, B., Lorenzini, E. & Ciarrocchi, A. BRD4 and cancer: going beyond transcriptional regulation. Mol. Cancer 17, 164 (2018).
Hajmirza, A. et al. BET family protein BRD4: an emerging actor in NFκB signaling in inflammation and cancer. Biomedicines 6, 16 (2018).
Sengupta, S. & George, R. E. Super-enhancer-driven transcriptional dependencies in cancer. Trends Cancer 3, 269–281 (2017).
Andricovich, J. et al. Loss of KDM6A activates super-enhancers to induce gender-specific squamous-like pancreatic cancer and confers sensitivity to BET inhibitors. Cancer Cell 33, 512–526.e518 (2018).
Henssen, A. et al. Targeting MYCN-driven transcription by BET-bromodomain inhibition. Clin. Cancer Res. 22, 2470–2481 (2016).
Cheng, W. et al. Recent development of CDK inhibitors: an overview of CDK/inhibitor co-crystal structures. Eur. J. Med. Chem. 164, 615–639 (2019).
Zhou, Q., Li, T. & Price, D. H. RNA polymerase II elongation control. Annu. Rev. Biochem. 81, 119–143 (2012).
Larochelle, S. et al. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat. Struct. Mol. Biol. 19, 1108–1115 (2012).
Diab, S., Yu, M. & Wang, S. CDK7 inhibitors in cancer therapy: the sweet smell of success? J. Med. Chem. 63, 7458–7474 (2020).
Rasool, R. U. et al. CDK7 inhibition suppresses castration-resistant prostate cancer through MED1 inactivation. Cancer Discov. 9, 1538–1555 (2019).
Huang, J. R. et al. Cyclin-dependent kinase 7 inhibitor THZ2 inhibits the growth of human gastric cancer in vitro and in vivo. Am. J. Transl. Res. 10, 3664–3676 (2018).
Zhang, J. et al. Targeting super-enhancer-associated oncogenes in osteosarcoma with THZ2, a covalent CDK7 inhibitor. Clin. Cancer Res. 26, 2681–2692 (2020).
Cidado, J. et al. AZD4573 is a highly selective CDK9 inhibitor that suppresses MCL-1 and induces apoptosis in hematologic cancer cells. Clin. Cancer Res. 26, 922–934 (2020).
Barlaam, B. et al. Discovery of AZD4573, a potent and selective inhibitor of CDK9 that enables short duration of target engagement for the treatment of hematological malignancies. J. Med. Chem. 63, 15564–15590 (2020).
Nagaraja, S. et al. Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell 31, 635–652.e6 (2017).
Wang, J. et al. CDK7 inhibitor THZ1 enhances antiPD-1 therapy efficacy via the p38α/MYC/PD-L1 signaling in non-small cell lung cancer. J. Hematol. Oncol. 13, 99 (2020).
Chapuy, B. et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 24, 777–790 (2013).
Lu, P. et al. THZ1 reveals CDK7-dependent transcriptional addictions in pancreatic cancer. Oncogene 38, 3932–3945 (2019).
Song, H. et al. Targeting cyclin-dependent kinase 9 sensitizes medulloblastoma cells to chemotherapy. Biochem. Biophys. Res. Commun. 520, 250–256 (2019).
Moreno, N. et al. Combined BRD4 and CDK9 inhibition as a new therapeutic approach in malignant rhabdoid tumors. Oncotarget 8, 84986–84995 (2017).
Gao, Y. et al. Synergistic anti-tumor effect of combining selective CDK7 and BRD4 inhibition in neuroblastoma. Front. Oncol. 11, 773186 (2021).
Meng, W. et al. Enhanced efficacy of histone deacetylase inhibitor combined with bromodomain inhibitor in glioblastoma. J. Exp. Clin. Cancer Res. 37, 241 (2018).
Wong, M. et al. JMJD6 is a tumorigenic factor and therapeutic target in neuroblastoma. Nat. Commun. 10, 3319 (2019).
Piquereau, J. et al. The BET bromodomain inhibitor I-BET-151 induces structural and functional alterations of the heart mitochondria in healthy male mice and rats. Int. J. Mol. Sci. 20, 1527 (2019).
Stathis, A. et al. Clinical response of carcinomas harboring the BRD4-NUT oncoprotein to the targeted bromodomain inhibitor OTX015/MK-8628. Cancer Discov. 6, 492–500 (2016).
Ma, X. et al. Covalent CDK7 inhibitor THZ1 inhibits myogenic differentiation. J. Cancer 9, 3149–3155 (2018).
Gao, Y. et al. Overcoming resistance to the THZ series of covalent transcriptional CDK inhibitors. Cell Chem. Biol. 25, 135–142.e135 (2018).
Shu, S. et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 529, 413–417 (2016).
Rathert, P. et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 525, 543–547 (2015).
Tsang, F. H. et al. Aberrant super-enhancer landscape in human hepatocellular carcinoma. Hepatology 69, 2502–2517 (2019).
Ke, L. et al. Nasopharyngeal carcinoma super-enhancer-driven ETV6 correlates with prognosis. Proc. Natl Acad. Sci. USA. 114, 9683–9688 (2017).
Xu, H. et al. A novel defined super-enhancer associated gene signature to predict prognosis in patients with diffuse large B-cell lymphoma. Front. Genet. 13, 827840 (2022).
Ouyang, Z. et al. Construction of a five-super-enhancer-associated-genes prognostic model for osteosarcoma patients. Front. Cell Dev. Biol. 8, 598660 (2020).
Qi, T. et al. Super-enhancer associated five-gene risk score model predicts overall survival in multiple myeloma patients. Front. Cell Dev. Biol. 8, 596777 (2020).
Ostrom, Q. T. et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro. Oncol. 20, iv1–iv86 (2018).
Campos, B., Olsen, L. R., Urup, T. & Poulsen, H. S. A comprehensive profile of recurrent glioblastoma. Oncogene 35, 5819–5825 (2016).
Carlsson, S. K., Brothers, S. P. & Wahlestedt, C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol. Med. 6, 1359–1370 (2014).
Stupp, R. et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: a randomized clinical trial. JAMA 318, 2306–2316 (2017).
Francis, J. M. et al. EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov. 4, 956–971 (2014).
Johnston, M. J. et al. High-resolution structural genomics reveals new therapeutic vulnerabilities in glioblastoma. Genome Res. 29, 1211–1222 (2019).
Zhao, L. et al. FACT-seq: profiling histone modifications in formalin-fixed paraffin-embedded samples with low cell numbers. Nucleic Acids Res. 49, e125 (2021).
Meng, W. et al. CDK7 inhibition is a novel therapeutic strategy against GBM both in vitro and in vivo. Cancer Manag. Res. 10, 5747–5758 (2018).
Shang, E. et al. Epigenetic targeting of Mcl-1 is synthetically lethal with Bcl-xL/Bcl-2 inhibition in model systems of glioblastoma. Cancers (Basel) 12, 2137 (2020).
Bian, E. et al. Super-enhancer-associated TMEM44-AS1 aggravated glioma progression by forming a positive feedback loop with Myc. J. Exp. Clin. Cancer Res. 40, 337 (2021).
Jiapaer, S., Furuta, T., Tanaka, S., Kitabayashi, T. & Nakada, M. Potential strategies overcoming the temozolomide resistance for glioblastoma. Neurol. Med. Chir. (Tokyo) 58, 405–421 (2018).
Everhard, S. et al. Identification of regions correlating MGMT promoter methylation and gene expression in glioblastomas. Neuro. Oncol. 11, 348–356 (2009).
Chen, X. et al. A novel enhancer regulates MGMT expression and promotes temozolomide resistance in glioblastoma. Nat. Commun. 9, 2949 (2018).
Natsume, A., Hirano, M., Ranjit, M., Aoki, K. & Wakabayashi, T. Aberrant transcriptional regulation of super-enhancers by RET finger protein-histone deacetylase 1 complex in glioblastoma: chemoresistance to temozolomide. Neurol. Med. Chir. (Tokyo) 59, 293–298 (2019).
Ranjit, M. et al. Aberrant active cis-regulatory elements associated with downregulation of RET finger protein overcome chemoresistance in glioblastoma. Cell Rep. 26, 2274–2281.e2275 (2019).
Zhang, L., Liu, Z., Dong, Y. & Kong, L. Epigenetic targeting of SLC30A3 by HDAC1 is related to the malignant phenotype of glioblastoma. IUBMB Life 73, 784–799 (2021).
Caslini, C., Hong, S., Ban, Y. J., Chen, X. S. & Ince, T. A. HDAC7 regulates histone 3 lysine 27 acetylation and transcriptional activity at super-enhancer-associated genes in breast cancer stem cells. Oncogene 38, 6599–6614 (2019).
Gryder, B. E. et al. Chemical genomics reveals histone deacetylases are required for core regulatory transcription. Nat. Commun. 10, 3004 (2019).
Nguyen, T. T. T. et al. HDAC inhibitors elicit metabolic reprogramming by targeting super-enhancers in glioblastoma models. J. Clin. Invest. 130, 3699–3716 (2020).
Sanchez, G. J. et al. Genome-wide dose-dependent inhibition of histone deacetylases studies reveal their roles in enhancer remodeling and suppression of oncogenic super-enhancers. Nucleic Acids Res. 46, 1756–1776 (2018).
Nguyen, T. T. T., Westhoff, M. A., Karpel-Massler, G. & Siegelin, M. D. Targeting super-enhancers reprograms glioblastoma central carbon metabolism. Oncotarget 12, 1309–1313 (2021).
Shi, K. et al. PAX8 regulon in human ovarian cancer links lineage dependency with epigenetic vulnerability to HDAC inhibitors. Elife 8, e44306 (2019).
Feng, Q. et al. Lactate increases stemness of CD8+ T cells to augment anti-tumor immunity. Nat. Commun. 13, 4981 (2022).
Lin, C. Y. et al. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature 530, 57–62 (2016).
Zeid, R. et al. Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma. Nat. Genet. 50, 515–523 (2018).
Wang, X. et al. Reciprocal signaling between glioblastoma stem cells and differentiated tumor cells promotes malignant progression. Cell Stem Cell 22, 514–528.e515 (2018).
Mack, S. C. et al. Chromatin landscapes reveal developmentally encoded transcriptional states that define human glioblastoma. J. Exp. Med. 216, 1071–1090 (2019).
Xu, L. et al. Topography of transcriptionally active chromatin in glioblastoma. Sci. Adv. 7, eabd4676 (2021).
Gimple, R. C. et al. Glioma stem cell-specific superenhancer promotes polyunsaturated fatty-acid synthesis to support EGFR signaling. Cancer Discov. 9, 1248–1267 (2019).
Tian, W. et al. A novel prognostic tool for glioma based on enhancer RNA-regulated immune genes. Front. Cell Dev. Biol. 9, 798445 (2021).
Tian, W. et al. Development and validation of a novel prognostic model for lower-grade glioma based on enhancer RNA-regulated prognostic genes. Front. Oncol. 12, 714338 (2022).
Pastori, C. et al. BET bromodomain proteins are required for glioblastoma cell proliferation. Epigenetics 9, 611–620 (2014).
Pastori, C. et al. The Bromodomain protein BRD4 controls HOTAIR, a long noncoding RNA essential for glioblastoma proliferation. Proc. Natl Acad. Sci. USA. 112, 8326–8331 (2015).
Tao, Z. et al. BRD4 regulates self-renewal ability and tumorigenicity of glioma-initiating cells by enrichment in the Notch1 promoter region. Clin. Transl. Med. 10, e181 (2020).
Berenguer-Daizé, C. et al. OTX015 (MK-8628), a novel BET inhibitor, displays in vitro and in vivo antitumor effects alone and in combination with conventional therapies in glioblastoma models. Int. J. Cancer 139, 2047–2055 (2016).
Xu, L. et al. Targetable BET proteins- and E2F1-dependent transcriptional program maintains the malignancy of glioblastoma. Proc. Natl Acad. Sci. USA. 115, e5086–e5095 (2018).
Bauer, K. et al. Degradation of BRD4 – a promising treatment approach not only for hematologic but also for solid cancer. Am. J. Cancer Res. 11, 530–545 (2021).
Kfoury, N. et al. Brd4-bound enhancers drive cell-intrinsic sex differences in glioblastoma. Proc. Natl Acad. Sci. USA. 118, e2017148118 (2021).
Khatua, S., Song, A., Citla Sridhar, D. & Mack, S. C. Childhood medulloblastoma: current therapies, emerging molecular landscape and newer therapeutic insights. Curr. Neuropharmacol. 16, 1045–1058 (2018).
Archer, T. C., Mahoney, E. L. & Pomeroy, S. L. Medulloblastoma: molecular classification-based personal therapeutics. Neurotherapeutics 14, 265–273 (2017).
Taylor, M. D. et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 123, 465–472 (2012).
Northcott, P. A. et al. Medulloblastomics: the end of the beginning. Nat. Rev. Cancer 12, 818–834 (2012).
Northcott, P. A. et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 511, 428–434 (2014).
Li, M. et al. Dissecting super-enhancer driven transcriptional dependencies reveals novel therapeutic strategies and targets for group 3 subtype medulloblastoma. J. Exp. Clin. Cancer Res. 41, 311 (2022).
Kool, M. et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 25, 393–405 (2014).
Aiello, G. et al. Truncated BRPF1 cooperates with smoothened to promote adult Shh medulloblastoma. Cell Rep. 29, 4036–4052.e4010 (2019).
Dhar, S. S. et al. MLL4 is required to maintain broad H3K4me3 peaks and super-enhancers at tumor suppressor genes. Mol. Cell 70, 825–841.e826 (2018).
Cao, F. et al. Super-enhancers and broad H3K4me3 domains form complex gene regulatory circuits involving chromatin interactions. Sci. Rep. 7, 2186 (2017).
Hoffman, L. M. et al. Clinical, radiologic, pathologic, and molecular characteristics of long-term survivors of diffuse intrinsic pontine glioma (DIPG): a collaborative report from the International and European Society for Pediatric Oncology DIPG Registries. J. Clin. Oncol. 36, 1963–1972 (2018).
Wiese, M. et al. Combined treatment with CBP and BET inhibitors reverses inadvertent activation of detrimental super enhancer programs in DIPG cells. Cell Death Dis. 11, 673 (2020).
Iorgulescu, J. B. et al. Molecular biomarker-defined brain tumors: epidemiology, validity, and completeness in the United States. Neuro. Oncol. 24, 1989–2000 (2022).
Filbin, M. G. et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360, 331–335 (2018).
El-Hashash, A. H. K. Histone H3K27M mutation in brain tumors. Adv. Exp. Med. Biol. 1283, 43–52 (2021).
Piunti, A. et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 23, 493–500 (2017).
Lewis, P. W. et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861 (2013).
Chen, K. Y. et al. Reciprocal H3.3 gene editing identifies K27M and G34R mechanisms in pediatric glioma including NOTCH signaling. Commun. Biol. 3, 363 (2020).
Lewis, N. A., Klein, R. H., Kelly, C., Yee, J. & Knoepfler, P. S. Histone H3.3 K27M chromatin functions implicate a network of neurodevelopmental factors including ASCL1 and NEUROD1 in DIPG. Epigenetics Chromatin 15, 18 (2022).
Grasso, C. S. et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 21, 555–559 (2015).
Mohammad, F. et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 23, 483–492 (2017).
Mendez, F. M. et al. Epigenetic reprogramming and chromatin accessibility in pediatric diffuse intrinsic pontine gliomas: a neural developmental disease. Neuro. Oncol. 22, 195–206 (2020).
Wiese, M. et al. The β-catenin/CBP-antagonist ICG-001 inhibits pediatric glioma tumorigenicity in a Wnt-independent manner. Oncotarget 8, 27300–27313 (2017).
Takai, H. et al. 5-Hydroxymethylcytosine plays a critical role in glioblastomagenesis by recruiting the CHTOP-methylosome complex. Cell Rep. 9, 48–60 (2014).
Johnson, K. C. et al. 5-Hydroxymethylcytosine localizes to enhancer elements and is associated with survival in glioblastoma patients. Nat. Commun. 7, 13177 (2016).
Azizgolshani, N. et al. DNA 5-hydroxymethylcytosine in pediatric central nervous system tumors may impact tumor classification and is a positive prognostic marker. Clin. Epigenetics 13, 176 (2021).
Pawel, B. R. SMARCB1-deficient tumors of childhood: a practical guide. Pediatr. Dev. Pathol. 21, 6–28 (2018).
Hodges, H. C. et al. Dominant-negative SMARCA4 mutants alter the accessibility landscape of tissue-unrestricted enhancers. Nat. Struct. Mol. Biol. 25, 61–72 (2018).
Wang, X. et al. SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat. Genet. 49, 289–295 (2017).
Eaton, K. W., Tooke, L. S., Wainwright, L. M., Judkins, A. R. & Biegel, J. A. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr. Blood Cancer 56, 7–15 (2011).
Langer, L. F., Ward, J. M. & Archer, T. K. Tumor suppressor SMARCB1 suppresses super-enhancers to govern hESC lineage determination. Elife 8, e45672 (2019).
Kumar, N. et al. Embryonal tumors with multilayered rosettes: a tertiary care centre experience. Clin. Neurol. Neurosurg. 202, 106508 (2021).
Korshunov, A. et al. Embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, and medulloepithelioma share molecular similarity and comprise a single clinicopathological entity. Acta Neuropathol. 128, 279–289 (2014).
Sin-Chan, P. et al. A C19MC-LIN28A-MYCN oncogenic circuit driven by hijacked super-enhancers is a distinct therapeutic vulnerability in ETMRs: a lethal brain tumor. Cancer Cell 36, 51–67.e57 (2019).
Preusser, M., Brastianos, P. K. & Mawrin, C. Advances in meningioma genetics: novel therapeutic opportunities. Nat. Rev. Neurol. 14, 106–115 (2018).
Paramasivam, N. et al. Mutational patterns and regulatory networks in epigenetic subgroups of meningioma. Acta Neuropathol. 138, 295–308 (2019).
Gerstner, E. R. & Pajtler, K. W. Ependymoma. Semin. Neurol. 38, 104–111 (2018).
Pajtler, K. W. et al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell 27, 728–743 (2015).
Arabzade, A. et al. ZFTA-RELA dictates oncogenic transcriptional programs to drive aggressive supratentorial ependymoma. Cancer Discov. 11, 2200–2215 (2021).
Mack, S. C. et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature 553, 101–105 (2018).
Johnson, R. A. et al. Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature 466, 632–636 (2010).
Mohankumar, K. M. et al. An in vivo screen identifies ependymoma oncogenes and tumor-suppressor genes. Nat. Genet. 47, 878–887 (2015).
Funding
This work was supported by the National Natural Science Foundation of China (No. 82073944), Scientific Research Project of Hunan Health Commission (No. 202113010170), Hunan Provincial Natural Science Foundation of China (No. 2022JJ30834 and 2022JJ80119), and Natural Science Foundation of Changsha (No. kq2202386).
Author information
Authors and Affiliations
Contributions
J.Q. and Q.Q. conceived of the article. H.-H.Z., Y.-H.D. and J.Q. performed the literature search and data collection. H.H.-Z., J.Q., Q.Q. and X.-Q.T. drafted the manuscript. J.Q. and Q.Q. critically revised the work. All authors modified and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhuang, HH., Qu, Q., Teng, XQ. et al. Superenhancers as master gene regulators and novel therapeutic targets in brain tumors. Exp Mol Med 55, 290–303 (2023). https://doi.org/10.1038/s12276-023-00934-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-023-00934-0
This article is cited by
-
3C methods in cancer research: recent advances and future prospects
Experimental & Molecular Medicine (2024)
