
Abstract
The N6-Methyladenosine (m6A) modification of RNA transcripts is the most prevalent and abundant internal modification in eukaryotic messenger RNAs (mRNAs) and plays diverse and important roles in normal biological processes. Extensive studies have indicated that dysregulated m6A modification and m6A-associated proteins play critical roles in tumorigenesis and cancer progression. However, m6A-mediated physiological consequences often lead to opposite outcomes in a biological context-dependent manner. Therefore, context-related complexity must be meaningfully considered to obtain a comprehensive understanding of RNA methylation. Recently, it has been reported that m6A-modified RNAs are closely related to the regulation of the DNA damage response and genomic integrity maintenance. Here, we present an overview of the current knowledge on the m6A modification and its function in human cancer, particularly in relation to the DNA damage response and genomic instability.
Similar content being viewed by others

Introduction
The RNA m6A modification, that is, the methylation of adenosine at the nitrogen-6 position in an RNA molecule, is considered an epitranscriptomic and posttranscriptional regulatory mark. Among more than 150 posttranscriptional chemical modifications on RNA molecules1,2,3, m6A is the most abundant internal modification of mRNAs and noncoding RNAs. Since the discovery of m6A4,5, development of m6A transcriptome-wide mapping technology based on next-generation sequencing (NGS) methods has been used to extensively study this modification, and many related studies have found that this RNA chemical modification exhibits biological significance6,7. Indeed, the m6A modification is closely associated with almost all aspects of RNA-related biological processes, including transcription, pre-mRNA splicing and processing, pri-microRNA (pri-miRNA) processing, nuclear export, translation, RNA stability and decay7,8,9,10,11,12,13,14,15,16,17,18,19,20. In addition to its role in RNA metabolic processes, the m6A modification is involved in other biological processes, such as transcriptional regulation, signal transduction, and the DNA damage response21,22,23,24,25,26. As the m6A modification and associated factors are significantly dysregulated in cancers, understanding their roles in tumorigenesis and cancer progression will provide in-depth insight into the development of new therapeutic strategies for cancer treatment. In this review, we describe the current understanding of m6A modification and its function in biological processes and cancers, particularly its contribution to the DNA damage response and genomic instability.
The dynamics of the m6A modification and its molecular functions in RNA metabolism
The m6A modification is dynamically deposited and removed by m6A methyltransferase complexes (m6A writers) and demethylases (m6A erasers), respectively (Fig. 1). As the core subunit of the m6A methyltransferase complex, METTL3 and METTL14 form a heterodimer and recognize the consensus sequence motif [G > A](m6A)C[A/C/U], which is preferentially located near stop codons, 3′ untranslated regions (UTRs), and long internal exons6,7,17,18. The m6A methyltransferase activity of the METTL3-METTL14 heterodimeric complex is modulated by regulatory proteins, including WTAP, VIRMA, RBM15/15B, ZC3H13 and HAKAI;19,20,21,27 these factors are required for nuclear localization as well as the recruitment of the m6A methyltransferase complex to target RNA substrates. The expression levels or activities of the components of the m6A methyltransferase complex can alter the overall level of m6A in cells, and this change significantly affects transcriptome-wide landscape and biological functions. Although extensive studies have identified components of the m6A methyltransferase complex, silencing each component of the current m6A methyltransferase complex via RNA interference (RNAi) or gene knockout (KO) partially reduced the level of m6A in cells but did not abrogate it. Therefore, future studies need to be focused on investigating uncharacterized components of the m6A methyltransferase complex or enzymes that regulate the abundance of the m6A modification in cells.

A m6A methyltransferase complex is composed of core m6A writer components (METTL3, METTL14, and WTAP) and regulatory proteins (VIRMA, RBM15/RBM15B, HAKAI, and ZC3H13). The m6A writer adds a methyl group to the N6-position of adenosine within the consensus sequence ([G > A](m6A)C[A/C/U]) of an RNA transcript (mRNA, lncRNA, and so on). Another methyltransferase comprising METTL16, METTL5 and ZCCHC4 specifically methylates snRNA, rRNA and a few noncoding RNAs. WTAP is an adapter in the regulation of the nuclear localization and activity of methyltransferase with regulatory proteins. The m6A erasers are ALKBH5 and FTO. ALKBH5 is a primary m6A demethylase that removes the methyl group from N6 adenosine from target mRNAs. FTO demethylates both internal m6A and N6,2′-O-dimethyladenosine in the 5′ cap (m6Am). This image was created with BioRender (https://biorender.com/).
One of the putative human m6A writer proteins, METTL16, is critical for methylating a few transcripts, such as the U6 small nuclear RNA (snRNA), MALAT1, XIST, and the pre-mRNA MAT2A28,29,30,31,32. However, it has been recently reported that METTL16 preferentially localized to the cytosol and methylated more than 334 mRNA transcripts. In addition, METTL16 KO caused a significant reduction in the rate of m6A deposition on target nascent RNAs compared to poly(A) RNAs. Interestingly, in addition to the previously proposed role involved in RNA splicing, METTL16 interaction with eukaryotic initiation factor 3a/b (eIF3a/b) and ribosomal RNAs (rRNAs) promotes ribosome assembly, resulting in enhanced translation of more than 4000 target mRNAs33. However, this translational regulation by METTL16 is neither methyltransferase activity-dependent nor m6A dependent.
Two other putative m6A writer proteins are METTL5 and ZCCHC4, which are critical for the m6A modification of 18 S rRNA and 28 S rRNA, respectively34,35. METTL5, not ZCCHC4, forms a heterodimeric complex with TRMT112, a methyltransferase activator34 (Fig. 1). Their biological function on 18 S and 28 S rRNAs remains unknown because knocking out Mettl5 or Zcchc4 negligibly affected human colon cancer HCT116 cell growth or mature rRNA processing and production34. However, a study showed that the proliferation of ZCCHC4 KO HepG2 cells, derived from the liver tissue of a patient with hepatocellular carcinoma (HCC), was significantly inhibited, which was consistent with the defective translation of specific mRNAs in regulating tumorigenesis34. This phenotypical discrepancy of ZCCHC4 KO might be cell-type specific or context dependent. Nevertheless, METTL5 and ZCCHC4 clearly methylate 18 S and 28 S rRNAs. Thus, the biological significance of METTL5- or ZCCHC4-mediated rRNA methylation in tumorigenesis should be addressed in different contexts.
Increasing evidence indicates that the m6A modification is deposited cotranscriptionally on nascent transcripts22,36,37,38,39 (Fig. 2a). In acute myeloid leukemia (AML), CAATT-box binding protein (CEBPZ) directs METTL3 at the transcriptional start site (TSS) in a METTL14-independent manner22. METTL3 recruitment by CEBPZ promotes the m6A modification of target mRNA transcripts and enhances their translation. In mouse embryonic stem cells (mESCs), m6A deposition by Mettl3 occurs cotranscriptionally at Mettl3-bound chromatin regions, as indicated by both the genomic binding of Mettl3 and m6A modification are mainly enriched in the 3’UTR36. Moreover, m6A modification depends upon the transcriptional dynamics of RNA polymerase II38. Attenuated transcriptional activity of RNA polymerase II induced an increase in m6A modification abundance mediated through the physical interaction between RNAPII and METTL3, resulting in inefficient translation. These studies suggest that the m6A modification is, at least, a cotranscriptional event and is one of the factors linking transcription to translation. However, H3 trimethylation at Lys36 (H3K36me3), is associated with transcript elongation, which recruits the m6A methyltransferase complex to chromatin through the physical interaction between H3K36me3 andMETTL14 to deposit m6A cotranscriptionally on nascent transcripts39. Furthermore, several study groups have independently shown that nascent RNAs labeled with 4-thiouridine (4SU) in a short time (5–20 min) were deposited with m6A25, and chromatin-associated regulatory RNAs (carRNAs) were highly modified with m6A marks, revealing it to be among the major substrates for the methyltransferase of m6A modification40,41. Overall, the data obtained to date suggest that m6A deposition occurs cotranscriptionally on nascent- and chromatin-associated RNA molecules via chromatin association with the m6A methyltransferase complex.

a The m6A modification occurs cotranscriptionally via an m6A methyltransferase complex (an m6A writer). b ALKBH5 and FTO dynamically demethylate N6 adenosine in target RNAs. m6A readers (YTHDC1-2, YTHDF1-3, IGF2BP1-3, HNRNPC/G, and HNRNPA2B1) determine the fate of m6A-modified RNAs. c YTHDC1, a nuclear m6A reader, interacts with SRSF3/10 and regulates alternative splicing. d YTHDC1 also controls nuclear export mediated through SRSF3 and NXF1. e YTHDC2, YTHDF1, and YTHDF3 are involved in mRNA stabilization and translation. f YTHDF2 interacts with the mRNA decay machinery and the CCR4-NOT complex, leading to mRNA degradation. g IGF2BP1-3 regulate the stabilization and translation of mRNAs. h HNRNPC and HNRNPG modulate pre-mRNA processing. i HNRNPA2B1 recruits DROSHA-DGCR8 to pri-miRNA for miRNA processing. This image was created with BioRender (https://biorender.com/).
The identification of two m6A demethylases, namely, the m6A erasers α-ketoglutarate-dependent dioxygenase alk B homolog 5 (ALKBH5) and fat mass and obesity-associated protein (FTO), has suggested that m6A deposition is dynamically reversible23,24. ALKBH5 selectively removes the methyl group from N6-adenosine of target mRNAs, whereas FTO demethylates both internal m6A marks and N6,2′-O-dimethyladenosine in the 5′ cap (m6Am)10, suggesting that ALKBH5 is a primary m6A demethylase (Fig. 2b). Interestingly, notable phenotypes in mammalian development have indicated that these two m6A demethylases play a broad regulatory role in developmental processes23,24. Fto-KO mice display partial embryonic lethality, postnatal growth retardation, and increased postnatal lethality41,42,43,44,45. Recently, it has been reported that RNA transcribed from long-interspersed element 1 (LINE1) is a physiological substrate of FTO that regulates the chromatin state in mammalian tissues and during development41. Alkbh5 KO led to impaired fertility through a dysregulated splicing process in sperm development24,46, although Alkbh5-KO mice were viable and reached adulthood. Indeed, ALKBH5 localizes to nuclear speckles (also known as interchromatin granule clusters) and nuclear domains enriched in pre-mRNA splicing factors24. Deficient Alkbh5 leads to high levels of m6A deposition on spermatogenesis-associated mRNAs and induces aberrant RNA splicing in nuclear speckles, resulting in the abrogation of fertility. Taken together, the data show that, along with the m6A methyltransferase complex, the identification of two m6A demethylases indicates the reversible and dynamic m6A modification of mRNAs, which plays critical roles in fundamental biological processes, adding a layer to posttranscriptional regulatory mechanisms.
The m6A modification determines the fate of RNAs via selective m6A-recognizing factors (m6A readers). The YTH domain-containing proteins YTHDC1-2 (YTHDC1 and YTHDC2) and YTHDF1-3 (YTHDF1, YTHDF2, and YTHDF3) are direct m6A readers that modulate the fate of m6A-modified RNAs in cells. First, YTHDC1 binds m6A-modified target RNAs and regulates RNA splicing, nuclear export, exosome-mediated RNA decay and stability of carRNAs in the nucleus14,25,40,47,48,49,50,51 (Fig. 2c, d). YTHDC2 also regulates the stability of m6A-modified RNAs and promotes translation efficiency in the cytoplasm52,53 (Fig. 2e). YTHDF1-3 bind m6A-modified mRNAs and facilitate cytosolic mRNA decay9,54,55 (Fig. 2e, f). In particular, YTHDF1 and YTHDF3 enhance the translation of m6A-modified mRNAs with translation initiation complexes55,56 (Fig. 2e). In addition to YTH domain-containing proteins, insulin-like growth factor 2 mRNA-binding proteins, IGF2BP1-3, recognize the m6A mark on mRNAs and thus regulate their stability and translation57,58 (Fig. 2g). Finally, heterogeneous nuclear ribonucleoproteins (HNRNPs), namely, hnRNPA2B1, hnRNPG, and hnRNPC, are potential m6A readers59,60,61 (Fig. 2h, i). However, since they bind to m6A-modified RNAs and change the structure of target RNAs, they seem to play roles as “m6A switches” not as direct m6A readers60,61 (Fig. 2h). Nevertheless, hnRNPA2B1 modulates the processing of m6A-modified primary miRNAs (pri-miRNAs) through the recruitment of DGCR8, a component of the microprocessor complex, and regulates the alternative splicing process59 (Fig. 2i).
Cellular and molecular functions of the m6A modification
mRNA instability by the m6A modification
One of the best characterized functions of m6A modification causes the destabilization of m6A-modified mRNAs8,9,62. Although all YTHDF1-3 proteins contribute to the destabilization of m6A on target mRNAs, recent studies have implied that YTHDF2 is the major m6A reader involved in the decay of m6A-modified RNAs9,63,64 (Fig. 2e, f). Increasing evidence has revealed that YTHDF2 is required for directing mRNAs to processing bodies (P-bodies), where mRNA decay-associated proteins accumulate9,54,65,66 (Fig. 2f). All three YTHDF1-3 proteins facilitate phase-separation with m6A-modified mRNAs to induce the accumulation of transcripts at P-bodies, stress granules and neuronal RNA granules54. Independent of P-bodies, YTHDF2 directly interacts with CNOT1 and recruits the CCR4/NOT deadenylase complex to m6A-modified mRNAs, leading to the deadenylation and degradation of mRNAs64. Furthermore, YTHDF2 associates with RNase P/MRP, an endoribonuclease, through direct interaction with heat-responsive protein 12 (HRSP12)63. The depletion of any of these three proteins abrogated m6A-mediated degradation of mRNAs, suggesting that YTHDF2 recognition of m6A-modified mRNAs mediates mRNA decay mediated via HRSP12 and RNase P/MRP. A transcriptome-wide analyses of HRSP12-binding sites and cleavage sites of RNase P/MRP further supported the finding that HRSP12 binds upstream of YTHDF2-binding sites and that RNase P/MRP endoribonucleolytically cleaves downstream YTHDF2-binding sites within target m6A-modified mRNAs, demonstrating that the m6A modification promotes the degradation of target mRNAs through the recruitment of RNase P/MRP mediated via HRSP12.
Alternative splicing by the m6A modification
Several lines of evidence show the role played by the m6A modification in mRNA splicing. Previous reports showed that the overall levels of m6A on mRNAs were significantly enriched in the early onset of embryogenesis in Drosophila and were rapidly decreased during embryogenesis. Ime4, a Drosophila METTL3 homolog, regulates the female-specific splicing of the Sex-lethal (Sxl) gene67,68. The m6A demethylases FTO and ALKBH5 are also involved in splicing machinery, regulating alternative splicing of long 3’UTRs containing pre-mRNAs or a subset of adipogenesis-associated mRNAs, respectively24,46,69. Transcriptome-wide mapping of RNAs bound by FTO showed significant overlap with previously reported m6A locations within intronic regions of pre-mRNAs, and depletion of FTO led to the inclusion of alternatively spliced exons70. ALKBH5 also regulates the proper splicing of longer 3’UTR transcripts, particularly in mitotic and meiotic male germ cells46. Even though m6A may affect the splicing process in only a subset of genes, these m6A-mediated splicing events might be functionally important. Although the abundance of m6A marks is related to modulated alternative splicing, the m6A reader protein YTHDC1 interacts with splicing regulators, including SAM68, SC35, SRSF1 and SRSF3, suggesting that the m6A reader plays a role in mRNA splicing47,48,71,72 (Fig. 2c). However, it remains unclear whether YTHDC1 activity is coordinated with these splicing regulators in a m6A-dependent or m6A-independent manner.
Nuclear export mediated via the m6A modification
The m6A modification influences the nuclear export of m6A-modified mRNA14,15,24. ALKBH5 loss induced the nuclear accumulation of m6A-modified mRNAs, suggesting that the m6A mark mediated mRNA export24. Another study demonstrated that YTHDC1 was involved in the nuclear export of m6A-modified mRNAs via its interaction with splicing factor SRSF3 and nuclear RNA export factor 1 (NXF1)14 (Fig. 2d). These results are supported by the interaction exhibited between the m6A-methyltransferase complex YTHDC1 and the TREX (TRanscription-EXport) mRNA export complex15. The nuclear export of mRNAs is cotranscriptionally coupled with the capping, splicing and 3′ end processing of primary transcripts73. However, although the m6A modification of pre-mRNAs has been associated with the splicing process, it has been recently reported that the role of m6A modification in pre-mRNA splicing is limited to a small number of pre-mRNA groups37, suggesting that the m6A-mediated pre-mRNA splicing process is not closely related to the nuclear export process. The TREX subunits ALY/REF and THOC5 make contact with NXF1, expose the RNA-binding domain of NXF1, and allow the interaction between NXF1 and mRNA74. However, it remains unclear exactly how the m6A mark regulates nuclear mRNA export between the nucleus and cytoplasm. Nevertheless, these studies suggest a role for m6A in nuclear export.
RNA translation mediated via the m6A modification
Myriad studies have demonstrated that m6A modification regulates the efficient translation of m6A-modified mRNAs. Some YTH domain-containing m6A readers, including YTHDF1, YTHDF3 and YTHDC2, have been reported to enhance the translation of m6A-modified mRNAs55,56,75 (Fig. 2e). In particular, YTHDF1 promotes the translation of m6A-modified mRNAs through its interaction with the eukaryotic translation initiation factor eIF3 complex56. YTHDF1 binds to m6A sites that are located around a stop codon and in the 3′UTR and facilitates translation. However, it remains to be investigated how eIF3 regulates the translation of m6A-modified mRNAs, since eIF3 is recruited to the 5′UTR or upstream of the translation start site for translation initiation76. Another study reported that eIF3 binds directly to 5′UTR-m6A sites, leading to the recruitment of the ribosomal 43 S preinitiation complex12. Translation initiation by eIF3 binding to 5′UTR-m6A sites does not require the cap-binding factor eIF4E, suggesting cap-independent translation. In addition, most recent studies demonstrate that eIF3H at the 5′UTR directly binds to METTL3 at the 3′UTR-m6A sites, promoting mRNA circularization and increasing cap-dependent or cap-independent ribosome translation efficiency77,78. It is still unclear how METTL3 binds to m6A-modified mRNA and contributes to translation efficiency. Future studies should address the molecular details of how METTL3 and readers recognize m6A marks and how the m6A modification contributes to each step of translation.
RNA m6A modifications in cancer
Although the current knowledge of the precise mechanism by which m6A modification regulates diverse biological processes remains to be further explored, an increasing number of studies examined the effects of m6A modification in various types of cancer. In this section, we summarize the recent findings of these studies with respect to the most common types of human cancer.
Acute myeloid leukemia (AML)
AML is the most common hematopoietic malignant leukemia in adults. Recurring chromosomal aberration and genetic mutations as well as aberrant alteration of epigenetic modifications, including DNA methylation and histone modification, contribute to hematopoietic malignancies such as AML79. The m6A methyltransferase METTL3 is more abundant in AML cells than in CD34-positive stem and hematopoietic progenitor cells (HSPCs) and is required for the differentiation of AML cells22,80. METTL3 binds to promoters associated with the differentiation of AML in a METTL14-independent manner, leading to direct transcriptional activation. METTL3 is recruited by CEBPZ at a transcriptional start site, and promoter-bound METTL3 adds the m6A mark to a cognate mRNAs, enhancing its ribosomal translation38. A study revealed that METTL3 deposited the m6A mark to pro-oncogenes such as the MYC proto-oncogene (c-MYC), B-cell lymphoma 2 (BCL2), and phosphatase and tensin homolog (PTEN), activating phosphoinositide 3-kinase (PI3K) and protein kinase B (PKB) signaling pathways, which regulate cell differentiation and self-renewal80. METTL14, a key component of the m6A methyltransferase complex, is highly expressed in HSPCs and AML cells81. Although negatively regulated by SPI1, METTL14 is involved in the m6A modification of target mRNAs of MYB and MYC, promoting translation and inhibiting myeloid differentiation. Interestingly, FTO, a m6A eraser, also plays a critical oncogenic role in AML, promoting leukemic oncogene-mediated transformation and leukemogenesis by regulating target mRNAs, such as ASB2 and RARA mRNAs, through the removal of methyl group from N6-adenosine82. Moreover, YTHDC1 and YTHDF2, m6A readers, play important roles in the survival and differentiation of AML cells51,83. YTHDC1 undergoes liquid‒liquid phase separation with m6A-modified mRNAs and forms nuclear YTHDC1-m6A condensates (nYACs)51. Abundant nYACs in AML cells protect m6A-modified mRNAs (i.e., MYC and others) from the polyA tail exosome targeting complex (PAXT) and exosome-associated RNA degradation51,84. YTHDF2 destabilizes m6A-modified mRNAs (i.e., tumor necrosis factor receptor Tnfrsf2 and others) that are associated with the function of self-renewing leukemic stem cells (LSCs), contributing to the initiation of AML83.
Hepatocellular carcinoma (HCC)
Liver cancer is a highly progressive and the second most life-threatening tumor85,86. It comprises hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (iCCA) in accordance with histological features85,86. Recent reports have shown that METTL3 is upregulated in human HCC, leading to m6A hypermethylation of the tumor suppressor SOCS2 (Suppressor Of Cytokine Signaling 2)87. The m6A reader protein YTHDF2-dependent RNA degradation pathway mediates the degradation of SOCS2 mRNAs, suggesting that METTL3 represses the expression and stability of critical tumor suppressor genes at the posttranscriptional level. In addition, METTL3 regulates the expression of USP7 (Ubiquitin Specific Peptidase 7) through the m6A modification of its target mRNAs88. Upregulated USP7 expression resulted in an increase in oncogenic activities of HCC cells. However, YTHDF2 functioned as a tumor suppressor in HCC via destabilization of epidermal growth factor receptor (EGFR) mRNA89, leading to the inhibition of the ERK/MEK signaling pathway in HCC. These results suggest that m6A modification has a dual role in HCC by promoting HCC tumor progression or inhibiting oncogenic pathways. Thus, the role of m6A modification in HCC remains to be further explored.
Glioblastoma (GBM)
Glioblastoma (GBM) is the most aggressive and common primary brain and central nervous system (CNS) malignancy in adults90,91. GBMs are characterized by heterogeneity; that is, they contain glioblastoma stem-like cell (GSC) populations with stem-like properties, contributing to tumor initiation and therapeutic resistance92. The METTL3 level is highly elevated in GSCs and is required for the maintenance of GSCs and the dedifferentiation of glioma cells through an m6A modification in the 3′ UTR of sex-determining region Y (SRY)-Box 2 (SOX2) mRNA93. The m6A modification of SOX2 mRNAs and recruitment of human antigen R (HuR), which is also highly expressed in GBMs, are essential for SOX2 mRNA stabilization, which leads to the maintenance of GSCs. Most GBM cases are refractory to radiotherapy and the chemotherapy drug temozolomide (TMZ) via rapid DNA repair by O-6-methylguanine-DNA methyltransferase (MGMT)94. Both METTL3 and SOX2 regulate DNA repair genes and partially mediate m6A-dependent radioresistance93. Therefore, further studies are required to examine the potential of SOX2 as a predictor for the outcome of and benefit from TMZ chemotherapy, in addition to MGMT. Interestingly, ALKBH5 is highly expressed in GSCs and demethylates FOXM1 nascent transcripts with a long noncoding RNA antisense strand (FOXM1-AS), leading to enhanced FOXM1 expression, GSC proliferation, and tumorigenesis95. A study showed that m6A modification functions as a tumor suppressor for GSC self-renewal and tumorigenesis96. The abundance of m6A marks after METTL3 or METTL14 knockdown promoted the tumorigenesis of GSCs. In contrast, the inhibition of FTO by the ethyl ester form of meclofenamic acid (MA2) suppressed the progression of GSC-grafted tumors. Taken together, these results suggest that m6A-mediated tumor formation can lead to opposite results, even in the same context. Thus, future studies should address the context- or experimental condition-based comprehensive interpretation to understand the precise role of m6A modification in GBMs.
Breast cancer
Breast cancer is the most frequently cancer diagnosed in women worldwide97. Breast cancer is heterogeneous and classified by the expression of hormone receptors (estrogen receptor and progesterone receptor) and human epidermal growth factor receptor 2 (HER2)98. The molecular subtypes of breast cancer are luminal A, luminal B, HER2-positive, and basal-like triple-negative breast cancer (TNBC), which are considered to show similar clinical behaviors prior to the treatment of breast cancer. In breast cancer, increasing evidence has shown that the m6A modification is critical for tumorigenesis and progression. Silencing of METTL14 and ALKBH5 significantly inhibited breast cancer cell growth and invasive activity99. METTL14 and ALKBH5 regulate m6A-modified mRNAs involved in the cell cycle, the epithelial-mesenchymal transition (EMT), and angiogenesis through the HuR-mediated stabilization of target mRNAs. In particular, a study demonstrated the specific role of m6A modification in regulating the TGFβ signaling pathway in the tumorigenesis of breast cancer. In contrast, another study showed that METTL14 overexpression increased the abundance of m6A marks and inhibited oncogenic activity100. In TNBC, a lower level of METTL3 and a higher level of FTO were associated with poor prognosis, indicating that a lower m6A mark level contributes to the progression of TNBC101. In addition, under hypoxic stress conditions, elevated ALKBH5 reduces the abundance of the m6A mark on pluripotency marker NANOG and KLF4 mRNAs, leading to increased NANOG and KLF4 expression and an enhanced breast cancer stem cell phenotype102.
Lung cancer
Lung cancer is one of the most common cancers in the world, and small-cell lung cancers (SCLCs) and non-small cell lung cancer (NSCLC) are the two major histologic subtypes103,104. Despite our understanding of the biology of this disease and mechanisms of lung tumor progression, the overall cure and survival rates for lung cancer patients remain very low, particularly for patients with metastatic disease. Increasing evidence indicates that METTL3 is highly expressed in NSCLC cells and related to oncogenic activity in lung cancer77,78,105,106,107. Although METTL3 is localized mainly in the nucleus, one study demonstrated that cytosolic METTL3 functioned as a m6A reader that bound to a 3′-UTR near a stop codon. m6A-modified mRNA-bound METTL3 directly interacted with eIF3h in eIF3 complex, facilitating mRNA looping to induce the recycling of polyribosomes77,78. Transcriptome-wide analyses demonstrated that METTL3 regulated a large subset of oncogenic mRNAs through the METTL3-eIF3h axis without affecting mRNA abundance, facilitating oncogenic activity77,78. In addition, several groups supported findings showing that downregulation of METTL3 expression levels through miR-600, miR-33a or RNAi clearly inhibited the oncogenic activities of lung cancer cells105,106,107. miR-600 and miR-33a bound to the 3′-UTR of METTL3 mRNAs, leading to the degradation of METTL3 mRNAs and subsequently inducing apoptosis in lung cancer cells. The level of METTL3 was also controlled posttranslationally. Indeed, METTL3 is SUMOylated by small ubiquitin-related modifier 1 (SUMO1), leading to a reduction in m6A methyltransferase activity. Thus, SUMOylation of METTL3 decreases the global cellular abundance of the m6A mark and subsequently alters the transcriptome of m6A-modified RNAs in cells, facilitating the development of NSCLC.
The m6A modification and genomic instability
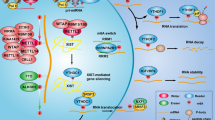
Several studies have found that METTL3-mediated m6A modification plays a critical role in the DNA damage response (DDR) to regulate the DNA repair pathway. RNA m6A modification rapidly occurs in ultraviolet (UV)-irradiated chromatin, indicating that METTL3 is specifically recruited to the UV-damaged chromatin region108 (Fig. 3a). This extensive recruitment of METTL3 depends on ADP-ribose polymerase 1 (PARP1). This DNA repair pathway may be mediated by trans-lesion DNA polymerase κ (Pol κ), which has been implicated in both nucleotide excision repair (NER) and trans-lesion synthesis (TLS)108,109,110. The m6A-mediated recruitment of Pol κ differentially regulates the UV-induced DNA damage response mediated by the canonical NER pathway and the Rad18/PCNA-regulated TLS pathway. However, the precise role of m6A modification and Pol κ in the UV damage response remains to be further investigated. Another study supported these findings by showing that nucleoplasmic fractions of m6A RNAs were immediately concentrated in UV-irradiated DNA lesions without affecting the level of METTL3, METTL14 or FTO111 (Fig. 3a). The authors showed that UV radiation reduced the levels of 2,2,7-methylguanosine (m3G/TMG) and N1-methyladenosine (m1A) in RNA as results of DNA damage. These results suggest that METTL3 rapidly localizes to UV-irradiated genomic regions and methylates RNAs and that these concentrated m6A-modified RNAs regulate the downstream DNA damage repair pathway to promote cell survival.

DNA damage response and repair. a Upon genotoxic stress, ATM phosphorylates METTL3, which binds to a DNA lesion, and m6A-modified RNAs direct PARP1 and Pol K for nucleotide excision repair (NER). b The m6A-modified RNAs are recognized by YTHDC1 or YTHDF2, which recruits RAD51 to the damaged region for homologous recombination repair (HRR). c. Reactive oxygen species (ROS) activate the ERK-JNK pathway, which phosphorylates ALKBH5. UBC9 binds to phosphorylated ALKBH5, inducing its SUMOylation. Inhibition of the demethylase activity of ALKBH5 by SUMOylation increases the level of m6A-modified RNAs that are related to DNA damage repair. Transcriptional regulation of DNA damage response and repair. d METTL3 stabilizes the p53 protein in a m6A-independent manner. METTL3 deposits an m6A mark on p53 target mRNAs for regulating the DNA damage response and tumor suppression. This image was created with BioRender (https://biorender.com/).
METTL3-mediated m6A modification also mediates homologous recombination (HR)-mediated double-strand DNA (dsDNA) break repair112 (Fig. 3b). METTL3 is phosphorylated at Ser43 by ataxia telangiectasia mutated (ATM) in response to double-strand breaks (DSBs). Phosphorylated METTL3 can be localized at DSB regions, leading to the m6A modification of nascent RNAs derived from damaged chromatin regions. These m6A-modified RNAs are recognized by YTHDC1, resulting in the formation of DNA‒RNA hybrids at DSBs. Subsequently, the formation of DNA‒RNA hybrids induces the recruitment of repair-related proteins, including RAD51 and BRCA1, to promote or HR-mediated repair, preventing genomic instability113,114. Depletion of METTL3 significantly enhanced the sensitivity of cancer cells and murine xenograft models to DNA damage-based therapies, such as chemotherapy drugs or radiation. Furthermore, a higher level of METTL3 predicted a poor survival probability for head and neck squamous carcinoma (HNSC) patients who had been treated with cisplatin or radiation. These results suggest that m6A modification in DSB repair is a potential target for cancer therapy. However, since METTL3 can increase the efficiency of DSB repair, it may also contribute to drug resistance in DNA damage-based treatment93,112.
RNA m6A modification also occurs in the majority of DNA‒RNA hybrids (R-loops) in human pluripotent stem cells115 (Fig. 3b). The m6A modification of RNA in R-loops is increased during the G2/M phase and disappears in the G0/G1 phase of the cell cycle, indicating cell cycle-dependent regulation of this modification. YTHDF2 binds to m6A-modified RNAs in R-loops, leading to the degradation of RNAs and the reduction in the number of R-loops. Inhibition of METTL3 or YTHDF2 results in the accumulation of R-loops and γH2AX, a DSB marker, and subsequently induces cell growth retardation. Thus, the regulation of METTL3- and YTHDF2-mediated RNA–DNA hybrids may represent a critical process in preventing genomic instability caused by the accumulation of cotranscriptional R-loops during mitosis. Furthermore, m6A can also resolve R-loops induced by DNA damage via UV or camptothecin (CPT) through tonicity-responsive enhancer-binding protein (TonEBP)116. TonEBP directly binds to R-loops and recruits METTL3, leading to m6A modification on an RNA strand of the R-loop. TonEBP also recruits RNase H1 to resolve R-loops. However, in different studies and cellular contexts, the m6A modification promoted R-loop formation to facilitate transcription termination117, suggesting context-dependent regulation. Nevertheless, the studies demonstrate that the m6A modification plays a critical role in regulating R-loops, the DNA damage response, and genomic stability.
It has been reported that FTO is important for the maintenance of bone mass and functions because it protects osteoblasts from genotoxic damage45. Previously, large-scale genome-wide association studies (GWAS) showed that FTO was closely linked to obesity and body composition in multiple human populations118,119,120. FTO specifically removes a methyl group from the N6-adenoside on the mRNAs of DNA repair genes (e.g. Hspa1, Cdk9, Kdm2a, and Ube2v1), leading to increased mRNA stability45. Subsequently, upregulation of Hspa1a and DNA repair genes protects osteoblasts from genotoxic agent (UV and H2O2)-mediated apoptosis. In addition, FTO protects osteoblasts from genotoxic damage induced by metabolic stress caused by the loss of Fto, which exacerbated osteoblast DNA damage in mice fed a high-fat diet. ALKBH5 also plays a role in the regulation of the DNA damage response and apoptosis mediated via reactive oxygen species (ROS)121 (Fig. 3c). ROS activate the ERK/JNK signaling pathway, which phosphorylates ALKBH5 at Ser87 and Ser321. Phosphorylated ALKBH5 interacts with UBC9, a SUMO E2 conjugating enzyme, leading to SUMOylation of ALKBH5 at Lys86 and Lys321. The enzymatic activity of SUMOylated ALKBH5 was thus inhibited, and then, the abundance of m6A on the mRNA of DNA repair genes increased, protecting cells from ROS-induced DNA damage response.
The m6A mark engages in crosstalk with the transcription factor, p53, and regulates the p53-mediated transcriptomic program induced by DNA damage stimuli122 (Fig. 3d). METTL3 has been identified as a p53-interacting partner after treatment with the DSB inducer doxorubicin. METTL3 stabilizes the p53 protein in a m6A-independent manner. However, METTL3 deposited the m6A mark on p53-targeted mRNAs to regulate the DNA damage response and tumor suppression only in the presence of an intact p53 protein. Therefore, further investigation should address whether RNA is dispensable for the direct interaction between METTL3 and p53 since p53 can bind to other RNA species123,124,125.
The m6A RNA modification is involved in the regulation of telomere length and genomic integrity in human cancers26 (Fig. 4). Telomeres are specialized structures at the ends of mammalian linear chromosomes and consist of tandem TTAGGG DNA nucleotide repeats126. Shelterin, a protein complex that binds to single- or double-strand telomeres, protects telomeres from being inappropriately recognized as damaged DNA127,128. In human cancer cells, telomerase, which consists of the catalytic subunit TERT and RNA template TERC, adds TTAGGG repeats during every cell division, preventing gradual telomere shortening due to the end replication problem of semiconservative DNA replication129. In normal somatic cells, TERT is transcriptionally repressed, and therefore, telomere length is gradually shortened with defective telomerase activity and progressive cell divisions, leading to a shortened-telomere-driven crisis point129. A shortened telomere crisis is closely linked to the genomic alterations found in cancer-relevant genomes130. Homeobox-Containing 1 (HMBOX1, also known as HOT1 or TAH1) is a mammalian telomere-binding protein that is involved in the recruitment of active telomerase and is required for telomere maintenance in the alternative lengthening of telomeres (ALT) in cancer cells131,132. Notably, HMBOX1 mRNA has been identified as a de novo target for m6A modification in cancer cells26. The authors of this study found that m6A marks in the 3′ UTR of HMBOX1 mRNA facilitated its degradation through YTHDF2 (Fig. 4a). In line with the function of HMBOX1 in the recruitment of telomerase to telomeres, downregulation of HMBOX1 mediated by the overexpression of METTL3 failed to maintain telomere length, as indicated by the defective recruitment of telomerase to telomeres26,131 (Fig. 4b). A previous report showed that HMBOX1 functions as a transcriptional repressor133. HMBOX1 suppresses the expression of MDM2 and is essential for the competency of p53 signaling26 (Fig. 4c). METTL3 upregulation in human cancer cells leads to shortened telomere-driven telomere dysfunction and inactivation of the p53-dependent DNA damage response pathway through MDM2 derepression26 (Fig. 4b, c). In the cancer-relevant genome, this coordinating environment might contribute to various types of telomere-associated chromosomal aberrations (e.g., translocations, amplifications, and deletions), enhancing the tumorigenicity and aggressiveness of cancer cells (Fig. 4d). Taken together, these results suggest an unexpected regulatory role for m6A marks in telomere biology and genome integrity.

a N6-methyladenosine (m6A) of HMBOX1 mRNA induces YTHDF2-mediated mRNA degradation. b A reduction in HMBOX1 gradually shortens telomeres. c Although shortened telomeres induce a dysfunctional telomere-driven DNA damage response (DDR), derepressed MDM2 inactivates the p53-mediated pathway, which is involved in cell cycle arrest or apoptosis. d Genomic instability mediated through sister chromatid exchange (T-SCE) or an anaphase bridge promotes tumorigenesis and cancer progression. This image was created with BioRender (https://biorender.com/).
Concluding remarks and future directions
The review discusses our current understanding of the multifaceted roles of the m6A modification in regulating RNA metabolic processes and related biological processes, including the DNA damage response and genomic instability. The role of m6A modification and m6A-associated proteins is cell- or disease-context dependent. Because of the significant role played by m6A in a variety of biological and physiological processes, context-dependent coordinated action among m6A-associated proteins determines the outcomes of m6A modification. The molecular details of the crosstalk between m6A modification and m6A-associated proteins and how this crosstalk affects diverse biological processes still need to be investigated. In particular, understanding how m6A modification and its associated proteins modulate DNA damage responses to maintain genomic integrity may lead to a new therapeutic strategy in cancer.
References
Helm, M. & Motorin, Y. Detecting RNA modifications in the epitranscriptome: predict and validate. Nat. Rev. Genet. 18, 275–291 (2017).
Boccaletto, P. et al. MODOMICS: a database of RNA modification pathways. 2021 update. Nucleic Acids Res. 50, D231–D235 (2022).
Nachtergaele, S. & He, C. Chemical modifications in the life of an mRNA transcript. Annu. Rev. Genet. 52, 349–372 (2018).
Desrosiers, R., Friderici, K. & Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl Acad. Sci. USA 71, 3971–3975 (1974).
Perry, R. P. & Kelley, D. E. Existence of methylated messenger RNA in mouse L cells. Cell 1, 37–42 (1974).
Meyer, K. D. et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 149, 1635–1646 (2012).
Dominissini, D. et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 (2012).
Wang, Y. et al. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 16, 191–198 (2014).
Wang, X. et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 (2014).
Mauer, J. et al. Reversible methylation of m(6)Am in the 5′ cap controls mRNA stability. Nature 541, 371–375 (2017).
Yang, Y. et al. Dynamic m6A modification and its emerging regulatory role in mRNA splicing. Sci. Bull. 60, 21–32 (2015).
Meyer, K. D. et al. 5′ UTR m(6)A promotes cap-independent translation. Cell 163, 999–1010 (2015).
Fustin, J. M. et al. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 155, 793–806 (2013).
Roundtree, I. A. et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife 6, e31311 (2017).
Lesbirel, S. et al. The m(6)A-methylase complex recruits TREX and regulates mRNA export. Sci. Rep. 8, 13827 (2018).
Edens, B. M. et al. FMRP modulates neural differentiation through m(6)A-dependent mRNA nuclear export. Cell Rep. 28, 845–854.e845 (2019).
Wang, X. et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature 534, 575–578 (2016).
Liu, J. et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10, 93–95 (2014).
Ping, X. L. et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 24, 177–189 (2014).
Yue, Y. et al. VIRMA mediates preferential m(6)A mRNA methylation in 3’UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 4, 10 (2018).
Patil, D. P. et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537, 369–373 (2016).
Barbieri, I. et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 552, 126–131 (2017).
Jia, G. et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887 (2011).
Zheng, G. et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 49, 18–29 (2013).
Lee, J. H. et al. Enhancer RNA m6A methylation facilitates transcriptional condensate formation and gene activation. Mol. Cell 81, 3368–3385 e3369 (2021).
Lee, J. H. et al. Regulation of telomere homeostasis and genomic stability in cancer by N (6)-adenosine methylation (m(6)A). Sci. Adv. 7, eabg7073 (2021).
Wen, J. et al. Zc3h13 regulates nuclear RNA m(6)A methylation and mouse embryonic stem cell self-renewal. Mol. Cell 69, 1028–1038 e1026 (2018).
Brown, J. A., Kinzig, C. G., DeGregorio, S. J. & Steitz, J. A. Methyltransferase-like protein 16 binds the 3’-terminal triple helix of MALAT1 long noncoding RNA. Proc. Natl Acad. Sci. USA 113, 14013–14018 (2016).
Pendleton, K. E. et al. The U6 snRNA m(6)A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell 169, 824–835 e814 (2017).
Warda, A. S. et al. Human METTL16 is a N(6)-methyladenosine (m(6)A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 18, 2004–2014 (2017).
Mendel, M. et al. Methylation of structured RNA by the m(6)A writer METTL16 is essential for mouse embryonic development. Mol. Cell 71, 986–1000 e1011 (2018).
Doxtader, K. A. et al. Structural basis for regulation of METTL16, an S-adenosylmethionine homeostasis factor. Mol. Cell 71, 1001–1011 e1004 (2018).
Su, R. et al. METTL16 exerts an m(6)A-independent function to facilitate translation and tumorigenesis. Nat. Cell Biol. 24, 205–216 (2022).
van Tran, N. et al. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 47, 7719–7733 (2019).
Ma, H. et al. N(6-)Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat. Chem. Biol. 15, 88–94 (2019).
Knuckles, P. et al. RNA fate determination through cotranscriptional adenosine methylation and microprocessor binding. Nat. Struct. Mol. Biol. 24, 561–569 (2017).
Ke, S. et al. m(6)A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 31, 990–1006 (2017).
Slobodin, B. et al. Transcription impacts the efficiency of mRNA translation via Co-transcriptional N6-adenosine methylation. Cell 169, 326–337 e312 (2017).
Huang, H. et al. Histone H3 trimethylation at lysine 36 guides m(6)A RNA modification co-transcriptionally. Nature 567, 414–419 (2019).
Liu, J. et al. N (6)-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science 367, 580–586 (2020).
Wei, J. et al. FTO mediates LINE1 m(6)A demethylation and chromatin regulation in mESCs and mouse development. Science 376, 968–973 (2022).
van der Hoeven, F. et al. Programmed cell death is affected in the novel mouse mutant Fused toes (Ft). Development 120, 2601–2607 (1994).
Fischer, J. et al. Inactivation of the Fto gene protects from obesity. Nature 458, 894–898 (2009).
Gao, X. et al. The fat mass and obesity associated gene FTO functions in the brain to regulate postnatal growth in mice. PLoS ONE 5, e14005 (2010).
Zhang, Q. et al. The RNA demethylase FTO is required for maintenance of bone mass and functions to protect osteoblasts from genotoxic damage. Proc. Natl Acad. Sci. USA 116, 17980–17989 (2019).
Tang, C. et al. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3’-UTR mRNAs in male germ cells. Proc. Natl Acad. Sci. USA 115, E325–E333 (2018).
Xiao, W. et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol. Cell 61, 507–519 (2016).
Kasowitz, S. D. et al. Nuclear m6A reader YTHDC1 regulates alternative polyadenylation and splicing during mouse oocyte development. PLoS Genet. 14, e1007412 (2018).
Shima, H. et al. S-adenosylmethionine synthesis is regulated by selective N(6)-adenosine methylation and mRNA degradation involving METTL16 and YTHDC1. Cell Rep. 21, 3354–3363 (2017).
Li, Y. et al. N(6)-Methyladenosine co-transcriptionally directs the demethylation of histone H3K9me2. Nat. Genet. 52, 870–877 (2020).
Cheng, Y. et al. N(6)-Methyladenosine on mRNA facilitates a phase-separated nuclear body that suppresses myeloid leukemic differentiation. Cancer Cell 39, 958–972.e958 (2021).
Wojtas, M. N. et al. Regulation of m(6)A transcripts by the 3′→5′ RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol. Cell 68, 374–387.e312 (2017).
Mao, Y. et al. m(6)A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nat. Commun. 10, 5332 (2019).
Ries, R. J. et al. m(6)A enhances the phase separation potential of mRNA. Nature 571, 424–428 (2019).
Shi, H. et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 27, 315–328 (2017).
Wang, X. et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399 (2015).
Huang, H. et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 20, 285–295 (2018).
Hu, X. et al. IGF2BP2 regulates DANCR by serving as an N6-methyladenosine reader. Cell Death Differ. 27, 1782–1794 (2020).
Alarcón, C. R. et al. HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell 162, 1299–1308 (2015).
Liu, N. et al. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518, 560–564 (2015).
Liu, N. et al. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 45, 6051–6063 (2017).
Lee, Y., Choe, J., Park, O. H. & Kim, Y. K. Molecular mechanisms driving mRNA degradation by m(6)A modification. Trends Genet. 36, 177–188 (2020).
Park, O. H. et al. Endoribonucleolytic cleavage of m(6)A-containing RNAs by RNase P/MRP complex. Mol. Cell 74, 494–507.e498 (2019).
Du, H. et al. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat. Commun. 7, 12626 (2016).
Luo, Y., Na, Z. & Slavoff, S. A. P-bodies: composition, properties, and functions. Biochemistry 57, 2424–2431 (2018).
Sheth, U. & Parker, R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300, 805–808 (2003).
Lence, T. et al. m(6)A modulates neuronal functions and sex determination in Drosophila. Nature 540, 242–247 (2016).
Haussmann, I. U. et al. m(6)A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature 540, 301–304 (2016).
Zhao, X. et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 24, 1403–1419 (2014).
Bartosovic, M. et al. N6-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3’-end processing. Nucleic Acids Res. 45, 11356–11370 (2017).
Wilkinson, F. L. et al. Emerin interacts in vitro with the splicing-associated factor, YT521-B. Eur. J. Biochem. 270, 2459–2466 (2003).
Hartmann, A. M., Nayler, O., Schwaiger, F. W., Obermeier, A. & Stamm, S. The interaction and colocalization of Sam68 with the splicing-associated factor YT521-B in nuclear dots is regulated by the Src family kinase p59(fyn). Mol. Biol. Cell 10, 3909–3926 (1999).
Heath, C. G., Viphakone, N. & Wilson, S. A. The role of TREX in gene expression and disease. Biochem. J. 473, 2911–2935 (2016).
Viphakone, N. et al. TREX exposes the RNA-binding domain of Nxf1 to enable mRNA export. Nat. Commun. 3, 1006 (2012).
Hsu, P. J. et al. Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 27, 1115–1127 (2017).
Jackson, R. J., Hellen, C. U. & Pestova, T. V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 11, 113–127 (2010).
Lin, S., Choe, J., Du, P., Triboulet, R. & Gregory, R. I. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 62, 335–345 (2016).
Choe, J. et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature 561, 556–560 (2018).
Chen, J., Odenike, O. & Rowley, J. D. Leukaemogenesis: more than mutant genes. Nat. Rev. Cancer 10, 23–36 (2010).
Vu, L. P. et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 23, 1369–1376 (2017).
Weng, H. et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell 22, 191–205 e199 (2018).
Li, Z. et al. FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-methyladenosine RNA demethylase. Cancer Cell 31, 127–141 (2017).
Paris, J. et al. Targeting the RNA m(6)A reader YTHDF2 selectively compromises cancer stem cells in acute myeloid leukemia. Cell Stem Cell 25, 137–148 e136 (2019).
Meola, N. et al. Identification of a nuclear exosome decay pathway for processed transcripts. Mol. Cell 64, 520–533 (2016).
Marquardt, J. U., Andersen, J. B. & Thorgeirsson, S. S. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat. Rev. Cancer 15, 653–667 (2015).
Sia, D., Villanueva, A., Friedman, S. L. & Llovet, J. M. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology 152, 745–761 (2017).
Chen, M. et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology 67, 2254–2270 (2018).
Li, Y. et al. METTL3 facilitates the progression of hepatocellular carcinoma by modulating the m6A level of USP7. Am. J. Transl. Res. 13, 13423–13437 (2021).
Zhong, L. et al. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett. 442, 252–261 (2019).
Louis, D. N. et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 114, 97–109 (2007).
Thakkar, J. P. et al. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 23, 1985–1996 (2014).
Lathia, J. D., Mack, S. C., Mulkearns-Hubert, E. E., Valentim, C. L. & Rich, J. N. Cancer stem cells in glioblastoma. Genes Dev. 29, 1203–1217 (2015).
Visvanathan, A. et al. Essential role of METTL3-mediated m(6)A modification in glioma stem-like cells maintenance and radioresistance. Oncogene 37, 522–533 (2018).
Stupp, R. et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466 (2009).
Zhang, S. et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell 31, 591–606 e596 (2017).
Cui, Q. et al. m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 18, 2622–2634 (2017).
Harbeck, N. et al. Breast cancer. Nat. Rev. Dis. Prim. 5, 66 (2019).
Abramowitz, M. C. et al. Dermal lymphatic invasion and inflammatory breast cancer are independent predictors of outcome after postmastectomy radiation. Am. J. Clin. Oncol. 32, 30–33 (2009).
Panneerdoss, S. et al. Cross-talk among writers, readers, and erasers of m(6)A regulates cancer growth and progression. Sci. Adv. 4, eaar8263 (2018).
Wu, L., Wu, D., Ning, J., Liu, W. & Zhang, D. Changes of N6-methyladenosine modulators promote breast cancer progression. BMC Cancer 19, 326 (2019).
Shi, Y. et al. Reduced expression of METTL3 promotes metastasis of triple-negative breast cancer by m6A methylation-mediated COL3A1 up-regulation. Front. Oncol. 10, 1126 (2020).
Zhang, C. et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc. Natl Acad. Sci. USA 113, E2047–E2056 (2016).
Siegel, R., Naishadham, D. & Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 63, 11–30 (2013).
Chen, Z., Fillmore, C. M., Hammerman, P. S., Kim, C. F. & Wong, K. K. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat. Rev. Cancer 14, 535–546 (2014).
Wei, W., Huo, B. & Shi, X. miR-600 inhibits lung cancer via downregulating the expression of METTL3. Cancer Manag. Res. 11, 1177–1187 (2019).
Du, M. et al. MiR-33a suppresses proliferation of NSCLC cells via targeting METTL3 mRNA. Biochem. Biophys. Res. Commun. 482, 582–589 (2017).
Du, Y. et al. SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res. 46, 5195–5208 (2018).
Xiang, Y. et al. RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature 543, 573–576 (2017).
Ogi, T. & Lehmann, A. R. The Y-family DNA polymerase kappa (pol kappa) functions in mammalian nucleotide-excision repair. Nat. Cell Biol. 8, 640–642 (2006).
Yoon, J. H., Prakash, L. & Prakash, S. Highly error-free role of DNA polymerase eta in the replicative bypass of UV-induced pyrimidine dimers in mouse and human cells. Proc. Natl Acad. Sci. USA 106, 18219–18224 (2009).
Svobodová Kovaříková, A. et al. N(6)-adenosine methylation in RNA and a reduced m(3)G/TMG level in non-coding RNAs appear at microirradiation-induced DNA lesions. Cells 9, 360 (2020).
Zhang, C. et al. METTL3 and N6-methyladenosine promote homologous recombination-mediated repair of DSBs by modulating DNA-RNA hybrid accumulation. Mol. Cell 79, 425–442 e427 (2020).
Ohle, C. et al. Transient RNA-DNA hybrids are required for efficient double-strand break repair. Cell 167, 1001–1013 e1007 (2016).
D’Alessandro, G. et al. BRCA2 controls DNA:RNA hybrid level at DSBs by mediating RNase H2 recruitment. Nat. Commun. 9, 5376 (2018).
Abakir, A. et al. N(6)-methyladenosine regulates the stability of RNA:DNA hybrids in human cells. Nat. Genet. 52, 48–55 (2020).
Kang, H. J. et al. TonEBP recognizes R-loops and initiates m6A RNA methylation for R-loop resolution. Nucleic Acids Res. 49, 269–284 (2021).
Yang, X. et al. m(6)A promotes R-loop formation to facilitate transcription termination. Cell Res. 29, 1035–1038 (2019).
Scuteri, A. et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 3, e115 (2007).
Frayling, T. M. et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316, 889–894 (2007).
Dina, C. et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat. Genet. 39, 724–726 (2007).
Yu, F. et al. Post-translational modification of RNA m6A demethylase ALKBH5 regulates ROS-induced DNA damage response. Nucleic Acids Res. 49, 5779–5797 (2021).
Raj, N. et al. The Mettl3 epitranscriptomic writer amplifies p53 stress responses. Mol. Cell 82, 2370–2384 e2310 (2022).
Riley, K. J. & Maher, L. J. 3rd p53 RNA interactions: new clues in an old mystery. RNA 13, 1825–1833 (2007).
Tournillon, A. S. et al. p53 binds the mdmx mRNA and controls its translation. Oncogene 36, 723–730 (2017).
Yoshida, Y. et al. Binding of RNA to p53 regulates its oligomerization and DNA-binding activity. Oncogene 23, 4371–4379 (2004).
Blackburn, E. H. The molecular structure of centromeres and telomeres. Annu. Rev. Biochem. 53, 163–194 (1984).
Palm, W. & de Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 42, 301–334 (2008).
de Lange, T. How telomeres solve the end-protection problem. Science 326, 948–952 (2009).
Shay, J. W. & Wright, W. E. Hayflick, his limit, and cellular ageing. Nat. Rev. Mol. Cell Biol. 1, 72–76 (2000).
Maciejowski, J. & de Lange, T. Telomeres in cancer: tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 18, 175–186 (2017).
Kappei, D. et al. HOT1 is a mammalian direct telomere repeat-binding protein contributing to telomerase recruitment. EMBO J. 32, 1681–1701 (2013).
Feng, X. et al. The telomere-associated homeobox-containing protein TAH1/HMBOX1 participates in telomere maintenance in ALT cells. J. Cell Sci. 126, 3982–3989 (2013).
Chen, S. et al. Isolation and functional analysis of human HMBOX1, a homeobox containing protein with transcriptional repressor activity. Cytogenet. Genome Res. 114, 131–136 (2006).
Acknowledgements
This work was supported by 5R01GM140084 (National Institute of General Medical Sciences) of K.X.
Author information
Authors and Affiliations
Contributions
J.H., K.X., and J.L. designed and wrote the manuscript. J.L. revised and supervised manuscript preparation.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hong, J., Xu, K. & Lee, J.H. Biological roles of the RNA m6A modification and its implications in cancer. Exp Mol Med 54, 1822–1832 (2022). https://doi.org/10.1038/s12276-022-00897-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-022-00897-8
This article is cited by
-
Transcriptome-wide profiling identifies colon cancer-associated m6A transcripts and potential RNA methyl modifiers
Molecular Biology Reports (2024)
-
dTrmt10A impacts Hsp70 chaperone m6A levels and the stress response in the Drosophila brain
Scientific Reports (2023)
