
Abstract
The importance of the complement component C1q has been highlighted by its involvement in autoimmunity, infection, inflammatory diseases, and tumors. The unique tulip-like structure of C1q has both a collagen-like stalk (C1q tail) and heterotrimeric globular head (gC1q), each with different binding specificities, and the binding of these components to their respective receptors leads to functional complexities in the body and bridges innate and adaptive immunity. This review describes the fundamental roles of C1q in various microenvironments and focuses on the importance of the interactions of C1q and its receptors with the inhibitory receptor LAIR-1 in maintaining homeostasis. Current therapeutic opportunities modulating LAIR-1 are also discussed.
Similar content being viewed by others
Introduction
C1q, an evolutionarily ancient protein canonically known as the initiator of the classical complement pathway, is increasingly appreciated to have a variety of complement-independent functions in innate and adaptive immunity. The structure of C1q, consisting of its globular head (gC1q) and its collagen-like stalk (C1q tail), enables it to interact with multiple binding partners in circulation and on the cell surface to influence local and systemic immune functions. However, current gaps in knowledge include why C1q deficiency predisposes to development of systemic lupus erythematosus (SLE) and how C1q impacts tolerance by suppressing the immune response. This review provides an overview of the current literature on C1q and its receptors. Understanding the downstream consequences of C1q-targeted therapies will be critical for their success in the clinic. Along with these interests, the present review will focus on the role and therapeutic potential of the inhibitory receptor LAIR-1, which plays a significant role in C1q’s maintenance of homeostasis and prevention of autoimmunity.
Characteristics of C1q
As previously well reviewed1,2,3,4, the first complement component, C1q, is an ~460 kDa macromolecular glycoprotein that has a tulip-like structure and is found circulating in the blood. C1q is composed of 18 subunits (6 C1qa, 6 C1qb, and 6 C1qc subunits) that contain an N-terminal triple-helical collagen-like region (C1q tail) and a C-terminal globular head region (gC1q)5. The C1q tail has the repeating sequence Glycine-X–Y (where X is any amino acid and Y is proline or hydroxyproline), which is also found in collagens1,6,7. The transcription factors IRF8 and PU.1 upregulate the synchronized expression of the three chains of C1q8. In addition, MafB is one of the critical regulators of C1q production9. MafB directly regulates all C1q genes, including the promoter regions of C1qa, C1qb, and C1qc. However, the regulation of C1q production at the molecular level is still incompletely understood.
The hematopoietic system is the primary source of C1q production. Although most components of the complement cascade are produced in the liver, C1q is constitutively synthesized by monocytes, macrophages, dendritic cells (DCs), and microglia10. Transplantation of wild-type bone marrow to C1q-deficient mice results in a complete restoration of circulating serum levels of C1q11. Inappropriate circulating C1q levels are associated with almost all inflammatory or inflammation-related diseases, including cancer, Alzheimer’s disease (AD), and metabolic disease12. Toll-like receptor (TLR) ligands induce C1q production by macrophages or DCs in infectious and inflammatory diseases. Moreover, serum C1q levels are also increased with aging but are similar between men and women13. Age-related increases in C1q may play an active role in inhibiting muscle repair and regeneration, as evidenced by a study that found that the administration of C1q suppressed muscle regeneration14. In the context of tumor biology, C1q is primarily expressed in vascular endothelial and fibroblast cells and in infiltrating monocytes and is associated with tumor invasion15. C1q is also highly expressed in the stroma and vascular endothelium in the tumor microenvironment (TME) and acts to enhance tumor progression by promoting angiogenesis16.
C1q exhibits diverse binding abilities of cell surface receptors via its gC1q and C1q tail regions. The diversity of C1q functions is related to its domains and differs for each receptor, as its interaction with different receptors will result in different downstream signaling events, which can have inflammatory or anti-inflammatory effects. The interactions of C1q with receptor for advanced glycation end products (RAGE), CD91, scavenger receptor class F member 1 (SCARF-1), CD33 (Siglec-3), integrins and dendritic cell-specific intercellular adhesion molecule-3 grabbing nonintegrin (DC-SIGN), calreticulin and leukocyte-associated immunoglobulin-like receptor 1 (LAIR-1, CD305), and discoidin domain receptor (DDR) 1/2 have been thoroughly and critically reviewed4,17,18,19,20. Nonetheless, it is important to further assess the consequences of C1q in hematopoietic and nonhematopoietic cells.
Immune functions of C1q
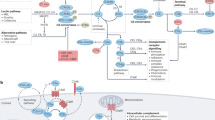
C1q affects the overall state of the immune system to promote tolerance and quiescence. During the steady state, circulating C1q contributes to the clearance of naturally occurring apoptotic cells; regulates immune cell differentiation by suppressing the differentiation of monocytes into professional antigen-presenting cells, which take part in initiating adaptive immune responses;21,22 regulates immune cell polarization, such as by inducing a tolerogenic phenotype in macrophages;23 and suppresses proinflammatory cytokine production in innate immune cells, including type 1 interferon (IFN) production by DCs24. C1q also upregulates the expression of engulfment machinery, including Mer tyrosine kinase and Gas6, in macrophages25,26. C1q enhances macrophage foam cell efferocytosis and cell survival27.
In inflammation, C1q acts as an opsonin by binding pathogens, foreign organisms, and apoptotic debris. C1q-dependent engulfment of apoptotic cells is essential for preventing autoimmunity28. C1q can interact with various proteins and form protein complexes in particular microenvironments. C1q in human serum binds danger-associated molecular patterns (DAMPs) released from apoptotic and necrotic cells, including phosphatidylserine, nucleic acids, and high mobility group box protein 1 (HMGB1)3,29,30,31,32. C1q also binds pathogen-associated molecular patterns (PAMPs) in response to infection, including lipopolysaccharides. In the presence of HMGB1, C1q diminishes proinflammatory cytokine production and directs macrophage polarization, leading to the generation of proresolving macrophages (M2-like). Human monocytes cultured with a combination of C1q and HMGB1 upregulated Mer and modulated T-cell proliferation (Fig. 1a). C1q opsonizes oxidized or acetylated lipids and promotes lipoprotein clearance through M2 macrophage polarization in atherosclerosis33,34. Moreover, C1q impacts brain inflammation and atherosclerosis35. C1q–apolipoprotein-E (ApoE) complexes have emerged as markers for ongoing complement activity in atherosclerosis in vivo. C1q can also bind to advanced glycation end products (AGEs) and facilitate their removal in atherosclerosis36.

a C1q activates the classical complement pathway and induces phagocytosis, allowing apoptotic debris clearance. C1q also maintains quiescence by inhibiting antigen-presenting DC differentiation, which induces adaptive immunity. In the presence of DAMPs or PAMPs, such as HMGB1, C1q not only inhibits proinflammatory cytokine production but also promotes anti-inflammatory (M2) macrophage polarization that is critical for the resolution of inflammation. Once SLE develops, C1q suppresses DNA-containing immune complex (IC)-mediated pDC activation. b In contrast, in neuroinflammation or tumors, C1q promotes disease progression. In amyloid β plaques in AD or neuroinflammation, C1q promotes A1 astrocytes and activates microglia that degrade neurons. C1q and HMGB1 promote the loss of dendritic complexity and cognitive impairment. C1q inhibits CD8 + T cells and DCs that kill tumor cells.
C1q is of particular interest in systemic lupus erythematosus (SLE), as C1q deficiencies strongly predispose to SLE development3,11,37. SLE is a systemic autoimmune disease, but the multifaceted pathogenic mechanisms leading to inflammation and organ damage are not fully understood. However, dysregulation of several functions of C1q is strongly related to several hallmark features of SLE pathology38,39,40,41. Of particular note are the defective clearance of apoptotic debris42 and IFN-α production by DCs induced by SLE autoantibodies and immune complexes21,43 (Fig. 1a). In addition to SNPs and other mutations that can alter the expression or function of C1q in some patients, one-third of SLE patients have anti‐C1q antibodies, which can contribute to pathology by inhibiting both C1q opsonization abilities and interaction with inhibitory receptors44. Notably, more than 90% of patients have proliferative lupus nephritis45,46.
Wound healing integrates various resident and migratory cells, the extracellular matrix, growth factors, and cytokines involved in inflammation, proliferation, and tissue remodeling47. C1q promotes the regeneration process and favors wound healing by stimulating angiogenesis in a complement-independent manner48. DDR2 is a receptor for C1q that is involved in wound healing and is present on the surface of epithelial cells. C1q and DDR2 binding improved cell migration and induced MMP2 and MMP9 expression in wound healing in vitro18.
In the brain, C1q is required for normal neuronal maturation, as it directs microglia to synapses to be eliminated in the process of synaptic pruning49,50,51. C1q expression is upregulated in neuronal injury and early in neurodegenerative disorders such as AD. Indeed, C1q protein levels dramatically increase in the normal aging mouse and human brain52. Similar to its functions in the periphery, C1q influences cell polarization in the CNS; astrocytes exist in two reactivity states, A1 and A2, analogous to the M1 and M2 states of macrophages53, wherein A1 astrocytes are more inflammatory and A2 astrocytes are generally considered to be neuroprotective. Interestingly, however, increased C1q in the CNS is associated with a more rather than less inflammatory state; microglial secretion of C1q in combination with IL-1 and TNFα induces A1 astrocytes, and mice deficient in C1q have significantly decreased A1 astrocyte reactivity compared to WT controls53. Our group has discovered that C1q is a critical component of long-term neuronal damage due to dendritic loss and the cognitive dysfunction associated with this loss in the context of autoantibody-mediated neuronal damage54. A subset of SLE-associated autoreactive antibodies (termed DNRAbs) bind double-stranded DNA and cross-react with the excitatory N-methyl-D-aspartate receptors in the brain, causing acute excitotoxicity in the neurons followed by sustained impairments in neuronal integrity and spatial memory. This process is dependent on both microglia and C1q, which colocalizes with synaptic proteins on dendrites, likely tagging the synapses for microglial elimination in a maladaptive form of its normal homeostatic function of synaptic pruning (Fig. 1b). Anti-C1q monoclonal antibodies reduced neuronal damage in AD55, and in a tau-induced AD model, microglia-mediated synapse loss was prevented upon administration of a C1q antibody, and synaptic density was recovered. Therapeutics targeting C1q are currently in development, including a nanobody specific for C1q that competitively prevents C1q from binding to IgG and IgM, effectively blocking complement activation by the classical pathway56. However, the side effects of prolonged C1q inhibition and whether continued C1q inhibition may result in an increased risk of infection or autoimmunity are unknown39.
C1q has protumorigenic properties in the TME16,57. Tumors developing in WT mice display early deposition of C1q, higher vascular density, and an increase in the number of lung metastases compared with those developing in C1qa-deficient mice. Moreover, C1q inhibits CD8 + T-cell activation, proliferation, and cytotoxic functions, a situation that may occur in the TME via modulation of the mitochondrial metabolism of CD8 + T cells58,59 (Fig. 1b).
C1q may also be detrimental in the context of COVID-19 infection caused by SARS-CoV-260. Clinical studies have shown that defects in type I interferon (IFN) production or antibodies to IFN appear to correlate with severe COVID-19 infection61, and anti-IFN antibodies in critical COVID-19 correlate with a poor interferon signature gene response and upregulation of LAIR-1, an inhibitory C1q receptor in PBMCs62. The genome of SARS-CoV-2 encodes four major structural proteins: the spike (S) protein, nucleocapsid (N) protein, membrane (M) protein, and envelope (E) protein, all of which are required to produce a structurally complete viral particle63. S1, N, M, and E all bind to C1q and activate both the complement pathway and kinin-kallikrein systems64. Viral pneumonia has been associated with complement activation and respiratory failure65. More studies are needed to show whether C1q and/or LAIR-1 are involved in SARS-CoV-2 infection.
Leukocyte-associated Ig-like receptor-1 (LAIR-1)
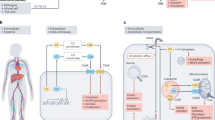
LAIR-1 is expressed on most hematopoietic cells. LAIR-1 has a large availability of ligands in both circulation and tissues, indicating a need for tight regulation of its interactions66. LAIR-1 binds to collagens with high affinity, especially collagen I and III. Additionally, the major LAIR-1-collagen binding site is in the conserved sequence of Gly-Pro-Hyp repeats, present in collagen and C1q67,68. LAIR-1 contains two ITIMs that negatively regulate intracellular signaling through SHP-1 binding associated with various phases of the immune response (reviewed by Meyaard69). The continuous interaction between C1q or collagens and LAIR-1 may control the inhibition capacity of LAIR-1. LAIR-1 is primarily regulated by its level of expression (Fig. 2a adopted from Meyaard69). LAIR-1’s activity is also controlled by secretion of its soluble forms, which include the splice variant LAIR‐2 (CD306) and a shed form of LAIR‐1 (sLAIR‐1), which antagonize LAIR‐1 by acting as decoy receptors69. In vivo, LAIR-2 can be found in urine from pregnant women, in fluids such as synovial fluid from rheumatoid arthritis and osteoarthritis patients, and in the circulation of patients with autoimmune thyroid disease70,71. Soluble forms of LAIR-1 have been utilized as a therapeutic to reverse immune suppression72.

a The levels of expression of the receptor and soluble forms of LAIR-1 and LAIR-2 alter the strength of LAIR-1 inhibition. In addition, partnering of existing ligands with other receptors, such as (b) RAGE or (c) CD33, may also alter the function of LAIR-1.
LAIR-1 is highly expressed on monocytes and plasmacytoid DCs. LAIR-1 is also expressed on immune cells in the skin, mainly on tissue-resident CD14 + cells, macrophages and CD11c + DCs66. LAIR-1 is consistently upregulated on monocytes and DCs during the inflammatory phase of the immune response and tends to return to normal expression levels during the resolution phase. In tumors, high expression of LAIR-1 has been reported in hematopoietic malignancies and kidney, breast, and ovarian cancers73. LAIR-1 is particularly enriched in nonclassical (patrolling) monocytes. LAIR-1-deficient mice have lower Ly6C expression in the steady state lung and enhanced metastatic melanoma in the lung57. Overexpression of LAIR-1 was associated with worse overall survival in patients with hepatocellular carcinoma74. In contrast, LAIR‐1 expression is lower in pDCs and B cells in SLE patients than in healthy donors, and increased IFN-α and antibody secretion result due to the lack of LAIR-1 inhibition75,76.
Activation of LAIR-1 inhibits proinflammatory M1-like macrophage differentiation and promotes alternative activation of macrophages66,77. Consequently, LAIR-1 partners with other receptors to improve its own function (Fig. 2b). The C1q-HMGB1 complex polarizes monocytes to anti-inflammatory M2-like macrophages, a pathway mediated through RAGE and LAIR-1 crosslinking, which depends on the relative levels of C1q and HMGB132. CD33 is another inhibitory receptor crosslinked by C1q20. C1q specifically binds to CD33 and gC1q, and the C1q tail engages LAIR-1 improve inhibition. The decreased LAIR-1 and CD33 expression on SLE monocytes, along with the frequent abnormalities related to C1q in SLE, suggest that C1q/CD33/LAIR-1 inhibitory networks are disrupted in SLE (Fig. 2c).
Further studies identified that LAIR-1 is an immune inhibitory receptor for collagen domain-containing proteins, including C1q, surfactant protein D (SP-D) and adiponectin78,79. C1q belongs to the collectin family, including mannose-binding lectin, SP-A, SP-D, and ficolin, which are pattern recognition proteins80. SP‐D also engages LAIR‐1 and inhibits FCαR‐mediated reactive oxygen species production by a human myeloid leukemia cell line81. gC1q has a structure similar to that of tumor necrosis factor (TNF) and belongs to the C1q/TNF superfamily (CTRP), which is involved in inflammation82,83. Among the CTRP family members, adiponectin behaves similarly to C1q by stimulating Mer tyrosine kinase-dependent engulfment of apoptotic cells26. In addition, adiponectin’s globular domain inhibits T-cell activation by interacting with LAIR-179.
In summary, the distribution of collagens, C1q, adiponectin, and SP-D in the body locally regulates the function of LAIR-1 to avoid immune dysfunction. In addition, the interaction of LAIR-1 with C1q (and other ligands) can control immune cells in various stages of the inflammatory response. Thus, LAIR-1 is a targetable receptor that dampens immune reactions.
LAIR-1 modulators as potential therapeutics
Since the LAIR-1 gene was identified in 1997 by the Meyaard group84, studies have demonstrated a critical role for LAIR-1 in the immune imbalance of autoimmune diseases and cancers. Its interactions with C1q alone mediate a major inhibitory pathway for the innate immune response during homeostasis as well as during inflammation and RA and SLE progression75,85. Remarkably, current studies suggest that utilizing C1q and collagen or synthetic peptides to modulate LAIR-1 is a beneficial therapeutic strategy at the molecular level in many diseases under appropriate conditions.
LAIR-1 agonistic antibody (anti-LAIR-1) enhances the activity of LAIR-1. In RA, collagen can suppress the T-cell cytokine response through the action of LAIR-1, and treatment with anti-LAIR-1 ameliorated RA severity85. In the collagen-induced arthritis (CIA) model and DR-1 transgenic mice, CD3-induced cytokine secretion was significantly suppressed in the presence of collagen, whereas LAIR-1-deficient splenocytes showed no attenuation. Treatment with anti-LAIR-1 significantly attenuated CIA in LAIR-1 wild-type mice. Type II collagen-administered B6.DR1/LAIR-1-deficient mice developed more severe arthritis than wild-type mice85.
C1q administration also ameliorates airway inflammation by activating LAIR-186. The findings of Helou et al. suggest that the LAIR-1 pathway is crucial for regulating allergic airway inflammation because it suppresses the activity of type 2 innate lymphoid cells (ILC2s). Crosslinking of LAIR-1 by its known ligand, C1q, decreased type 2 cytokine production by ILC2s in vitro, and IL-33-induced allergic airway inflammation and airway hyperreactivity in humanized mice was significantly reduced86.
Similar to C1q, collagen is beneficial for LAIR-1 activation87. Some bioactive regions play a role in mediating natural immune cell activation and inflammatory responses by engaging LAIR-1. Human collagen III-derived ligand peptide (LAIR1-LP) targets LAIR-188. LAIR1-LP enhances macrophage uptake through interactions with collagen-domain binding surface receptors and inhibits inflammation through interaction with LAIR-189,90. Moreover, LAIR1-LP inhibits the production of proinflammatory cytokines/chemokines upon T-cell activation87. Collagen can stimulate M2 polarization of macrophages in vivo. For instance, implantation of collagen gels into injured muscles of mice resulted in an increased number of M2-like macrophages compared to that seen in control mice91. Furthermore, during skin wound healing in mice, it was also demonstrated that collagen injected into wounds led to the M2 polarization of macrophages92.
LAIR-1 has the capacity to promote tolerogenic immune responses in the context of DAMP release31,93. Our group reported that LAIR-1 can influence macrophage polarization in the presence of HMGB1, which binds the activating receptor RAGE to provoke an inflammatory response but promotes a tolerogenic phenotype when crosslinked by C1q and LAIR-131. In the same study, a fusion protein containing the RAGE-binding fragment of HMGB1 and the LAIR-1-binding fragment of C1q crosslinks the two receptors in the same way as HMGB1 and C1q do. HMGB1 increases leukotriene B4 production in activated monocytes, while HMGB1 plus C1q produces specialized pro-resolving lipid mediators and promotes pro-resolving M2-like macrophage polarization61.
The soluble form of LAIR-1 (LAIR-2) blocks the detrimental LAIR-1-mediated inhibition in tumor treatment. Collagen in the tumor microenvironment can affect the ability of T cells to kill cancer cells by regulating the migration of T cells into the tumor94. The effects of tumor-associated collagen on immune cells could help explain why a high collagen density in tumors is correlated with a poor prognosis95. In mouse models of lung cancer, anti-PD-L1 resistance is associated with enhanced deposition of collagen, as well as fewer and more exhausted tumor-infiltrating CD8 + T cells. Combining anti-PD-1 with blockade of LAIR-1 significantly increases the therapeutic efficacy96. Abrogating LAIR-1 immunosuppression through LAIR-2 overexpression or SHP-1 inhibition sensitizes resistant lung tumors to anti-PD-196. Currently, a dimeric LAIR-2 Fc fusion protein, NC410, which both targets the tumor ECM and promotes T-cell function through blockade of LAIR-1-mediated inhibition, is being tested in a trial as cancer immunotherapy72. In humanized mouse tumor models, NC410 reduces tumor growth dependent on T cells. Immunohistochemical analysis of human tumors shows that NC410 binds to collagen-rich areas where LAIR-1+ immune cells are localized72.
Concluding remarks
Overall, the current review discusses the biological importance of C1q and its partners, mainly LAIR-1, in immunity and their expected therapeutic effects. Although there are still gaps in our understanding of the functions of C1q in complex microenvironments, targeting LAIR-1 will enable the development of new therapeutic strategies for many diseases, including inflammation, SLE, tumors, and hopefully COVID-19.
References
Ghebrehiwet, B., Kandov, E., Kishore, U. & Peerschke, E. I. B. Is the A-Chain the Engine That Drives the Diversity of C1q Functions? Revisiting Its Unique Structure. Front. Immunol. 9, 162 (2018).
Lu, J. H. et al. The classical and regulatory functions of C1q in immunity and autoimmunity. Cell. Mol. Immunol. 5, 9–21 (2008).
Son, M., Diamond, B. & Santiago-Schwarz, F. Fundamental role of C1q in autoimmunity and inflammation. Immunol. Res. 63, 101–106 (2015).
Thielens, N. M., Tedesco, F., Bohlson, S. S., Gaboriaud, C. & Tenner, A. J. C1q: A fresh look upon an old molecule. Mol. Immunol. 89, 73–83 (2017).
Reid, K. B. & Porter, R. R. Subunit composition and structure of subcomponent C1q of the first component of human complement. Biochem. J. 155, 19–23 (1976).
Ghai, R. et al. C1q and its growing family. Immunobiology 212, 253–266 (2007).
Kishore, U. et al. Structural and functional anatomy of the globular domain of complement protein C1q. Immunol. Lett. 95, 113–128 (2004).
Chen, G., Tan, C. S., Teh, B. K. & Lu, J. Molecular mechanisms for synchronized transcription of three complement C1q subunit genes in dendritic cells and macrophages. J. Biol. Chem. 286, 34941–34950 (2011).
Tran, M. T. N. et al. MafB is a critical regulator of complement component C1q. Nat. Commun. 8, 1700 (2017).
Arkwright, P. D., Riley, P., Hughes, S. M., Alachkar, H. & Wynn, R. F. Successful cure of C1q deficiency in human subjects treated with hematopoietic stem cell transplantation. J. Allergy Clin. Immunol. 133, 265–267 (2014).
van de Bovenkamp, F. S., Dijkstra, D. J., van Kooten, C., Gelderman, K. A. & Trouw, L. A. Circulating C1q levels in health and disease, more than just a biomarker. Mol. Immunol. 140, 206–216 (2021).
Veerhuis, R., Nielsen, H. M. & Tenner, A. J. Complement in the brain. Mol. Immunol. 48, 1592–1603 (2011).
Watanabe, S. et al. Serum C1q as a novel biomarker of sarcopenia in older adults. FASEB J. 29, 1003–1010 (2015).
Horii, N. et al. Resistance training prevents muscle fibrosis and atrophy via down-regulation of C1q-induced Wnt signaling in senescent mice. FASEB J. 32, 3547–3559 (2018).
Agostinis, C. et al. Complement component C1q as potential diagnostic but not predictive marker of preeclampsia. Am. J. Reprod. Immunol. 76, 475–481 (2016).
Bulla, R. et al. C1q acts in the tumour microenvironment as a cancer-promoting factor independently of complement activation. Nat. Commun. 7, 10346 (2016).
Abbonante, V. et al. Discoidin domain receptor 1 protein is a novel modulator of megakaryocyte-collagen interactions. J. Biol. Chem. 288, 16738–16746 (2013).
Hayuningtyas, R. A. et al. The collagen structure of C1q induces wound healing by engaging discoidin domain receptor 2. Mol. Med. 27, 125 (2021).
Lee, J. H. et al. Complement C1q stimulates the progression of hepatocellular tumor through the activation of discoidin domain receptor 1. Sci. Rep. 8, 4908 (2018).
Son, M. et al. Evidence for C1q-mediated crosslinking of CD33/LAIR-1 inhibitory immunoreceptors and biological control of CD33/LAIR-1 expression. Sci. Rep. 7, 270 (2017).
Son, M., Santiago-Schwarz, F., Al-Abed, Y. & Diamond, B. C1q limits dendritic cell differentiation and activation by engaging LAIR-1. Proc. Natl Acad. Sci. USA 109, E3160–E3167 (2012).
Hosszu, K. K., Santiago-Schwarz, F., Peerschke, E. I. & Ghebrehiwet, B. Evidence that a C1q/C1qR system regulates monocyte-derived dendritic cell differentiation at the interface of innate and acquired immunity. Innate Immun. 16, 115–127 (2010).
Benoit, M. E., Clarke, E. V., Morgado, P., Fraser, D. A. & Tenner, A. J. Complement protein C1q directs macrophage polarization and limits inflammasome activity during the uptake of apoptotic cells. J. Immunol. 188, 5682–5693 (2012).
Fraser, D. A. et al. C1q and MBL, components of the innate immune system, influence monocyte cytokine expression. J. Leukoc. Biol. 80, 107–116 (2006).
Galvan, M. D., Foreman, D. B., Zeng, E., Tan, J. C. & Bohlson, S. S. Complement component C1q regulates macrophage expression of Mer tyrosine kinase to promote clearance of apoptotic cells. J. Immunol. 188, 3716–3723 (2012).
Galvan, M. D., Hulsebus, H., Heitker, T., Zeng, E. & Bohlson, S. S. Complement protein C1q and adiponectin stimulate Mer tyrosine kinase-dependent engulfment of apoptotic cells through a shared pathway. J. Innate Immun. 6, 780–792 (2014).
Pulanco, M. C. et al. Complement Protein C1q Enhances Macrophage Foam Cell Survival and Efferocytosis. J. Immunol. 198, 472–480 (2017).
Galvan, M. D., Greenlee-Wacker, M. C. & Bohlson, S. S. C1q and phagocytosis: the perfect complement to a good meal. J. Leukoc. Biol. 92, 489–497 (2012).
Brencicova, E. & Diebold, S. S. Nucleic acids and endosomal pattern recognition: how to tell friend from foe? Front. Cell Infect. Microbiol. 3, 37 (2013).
Kim, S. Y. et al. High-Mobility Group Box 1-Induced Complement Activation Causes Sterile Inflammation. Front. Immunol. 9, 705 (2018).
Liu, T. et al. HMGB1-C1q complexes regulate macrophage function by switching between leukotriene and specialized proresolving mediator biosynthesis. Proc. Natl Acad. Sci. USA 116, 23254–23263 (2019).
Son, M. et al. C1q and HMGB1 reciprocally regulate human macrophage polarization. Blood 128, 2218–2228 (2016).
Ho, M. M. & Fraser, D. A. Transcriptome data and gene ontology analysis in human macrophages ingesting modified lipoproteins in the presence or absence of complement protein C1q. Data Brief. 9, 362–367 (2016).
Spivia, W., Magno, P. S., Le, P. & Fraser, D. A. Complement protein C1q promotes macrophage anti-inflammatory M2-like polarization during the clearance of atherogenic lipoproteins. Inflamm. Res. 63, 885–893 (2014).
Vogt, L. M. et al. Apolipoprotein E Triggers Complement Activation in Joint Synovial Fluid of Rheumatoid Arthritis Patients by Binding C1q. J. Immunol. 204, 2779–2790 (2020).
Chikazawa, M. et al. Identification of C1q as a Binding Protein for Advanced Glycation End Products. Biochemistry 55, 435–446 (2016).
Ballanti, E. et al. Complement and autoimmunity. Immunol. Res. 56, 477–491 (2013).
Bock, M., Heijnen, I. & Trendelenburg, M. Anti-C1q antibodies as a follow-up marker in SLE patients. PLoS One 10, e0123572 (2015).
Garred, P., Tenner, A. J. & Mollnes, T. E. Therapeutic Targeting of the Complement System: From Rare Diseases to Pandemics. Pharmacol. Rev. 73, 792–827 (2021).
Potlukova, E. & Kralikova, P. Complement component c1q and anti-c1q antibodies in theory and in clinical practice. Scand. J. Immunol. 67, 423–430 (2008).
Trendelenburg, M. Autoantibodies against complement component C1q in systemic lupus erythematosus. Clin. Transl. Immunol. 10, e1279 (2021).
Baumann, I. et al. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 46, 191–201 (2002).
Franchin, G. et al. Anti-DNA antibodies cross-react with C1q. J. Autoimmun. 44, 34–39 (2013).
Son, M., Kim, S. J. & Diamond, B. SLE-associated risk factors affect DC function. Immunol. Rev. 269, 100–117 (2016).
Golan, M. D., Burger, R. & Loos, M. Conformational changes in C1q after binding to immune complexes: detection of neoantigens with monoclonal antibodies. J. Immunol. 129, 445–447 (1982).
Siegert, C., Daha, M., Westedt, M. L., van der Voort, E. & Breedveld, F. IgG autoantibodies against C1q are correlated with nephritis, hypocomplementemia, and dsDNA antibodies in systemic lupus erythematosus. J. Rheumatol. 18, 230–234 (1991).
Velnar, T., Bailey, T. & Smrkolj, V. The wound healing process: an overview of the cellular and molecular mechanisms. J. Int. Med. Res. 37, 1528–1542 (2009).
Bossi, F. et al. C1q as a unique player in angiogenesis with therapeutic implication in wound healing. Proc. Natl Acad. Sci. USA 111, 4209–4214 (2014).
Berkowitz, S. et al. Complement and Coagulation System Crosstalk in Synaptic and Neural Conduction in the Central and Peripheral Nervous Systems. Biomedicines 9, 1950 (2021).
Stevens, B. et al. The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178 (2007).
Boche, D., Perry, V. H. & Nicoll, J. A. Review: activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 39, 3–18 (2013).
Stephan, A. H. et al. A dramatic increase of C1q protein in the CNS during normal aging. J. Neurosci. 33, 13460–13474 (2013).
Liddelow, S. A. et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017).
Nestor, J. et al. Lupus antibodies induce behavioral changes mediated by microglia and blocked by ACE inhibitors. J. Exp. Med. 215, 2554–2566 (2018).
Dejanovic, B. et al. Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron 100, 1322–1336 (2018).
Laursen, N. S. et al. Functional and Structural Characterization of a Potent C1q Inhibitor Targeting the Classical Pathway of the Complement System. Front. Immunol. 11, 1504 (2020).
Ain, D., Shaikh, T., Manimala, S. & Ghebrehiwet, B. The role of complement in the tumor microenvironment. Fac. Rev. 10, 80 (2021).
Ling, G. S. et al. C1q restrains autoimmunity and viral infection by regulating CD8(+) T cell metabolism. Science 360, 558–563 (2018).
Roumenina, L. T. et al. Tumor Cells Hijack Macrophage-Produced Complement C1q to Promote Tumor Growth. Cancer Immunol. Res. 7, 1091–1105 (2019).
Knight, J. S. et al. The intersection of COVID-19 and autoimmunity. J. Clin. Invest. 131, e154886 (2021).
Peng, T., Du, S. Y., Son, M. & Diamond, B. HIF-1alpha is a negative regulator of interferon regulatory factors: Implications for interferon production by hypoxic monocytes. Proc. Natl Acad. Sci. U. S. A. 118, e2106017118 (2021).
van der Wijst, M. G. P. et al. Longitudinal single-cell epitope and RNA-sequencing reveals the immunological impact of type 1 interferon autoantibodies in critical COVID-19. bioRxiv https://doi.org/10.1101/2021.03.09.434529 (2021).
Masters, P. S. The molecular biology of coronaviruses. Adv. Virus Res. 66, 193–292 (2006).
Savitt, A. G. et al. SARS-CoV-2 Exacerbates COVID-19 Pathology Through Activation of the Complement and Kinin Systems. Front. Immunol. 12, 767347 (2021).
Labarrere, C. A. & Kassab, G. S. Pattern Recognition Proteins: First Line of Defense Against Coronaviruses. Front. Immunol. 12, 652252 (2021).
Carvalheiro, T. et al. Leukocyte Associated Immunoglobulin Like Receptor 1 Regulation and Function on Monocytes and Dendritic Cells During Inflammation. Front. Immunol. 11, 1793 (2020).
Lebbink, R. J. et al. Collagens are functional, high affinity ligands for the inhibitory immune receptor LAIR-1. J. Exp. Med. 203, 1419–1425 (2006).
Lebbink, R. J. et al. Identification of multiple potent binding sites for human leukocyte associated Ig-like receptor LAIR on collagens II and III. Matrix Biol. 28, 202–210 (2009).
Meyaard, L. The inhibitory collagen receptor LAIR-1 (CD305). J. Leukoc. Biol. 83, 799–803 (2008).
Olde Nordkamp, M. J. et al. Enhanced secretion of leukocyte-associated immunoglobulin-like receptor 2 (LAIR-2) and soluble LAIR-1 in rheumatoid arthritis: LAIR-2 is a more efficient antagonist of the LAIR-1-collagen inhibitory interaction than is soluble LAIR-1. Arthritis Rheum. 63, 3749–3757 (2011).
Simone, R. et al. Serum LAIR-2 is increased in autoimmune thyroid diseases. PLoS One 8, e63282 (2013).
Ramos, M. I. P. et al. Cancer immunotherapy by NC410, a LAIR-2 Fc protein blocking human LAIR-collagen interaction. Elife 10, e62927 (2021).
Joseph, C. et al. The ITIM-Containing Receptor: Leukocyte-Associated Immunoglobulin-Like Receptor-1 (LAIR-1) Modulates Immune Response and Confers Poor Prognosis in Invasive Breast Carcinoma. Cancers (Basel) 13, 80 (2020).
Wu, X. et al. Clinicopathologic significance of LAIR-1 expression in hepatocellular carcinoma. Curr. Probl. Cancer 43, 18–26 (2019).
Bonaccorsi, I. et al. The immune inhibitory receptor LAIR-1 is highly expressed by plasmacytoid dendritic cells and acts complementary with NKp44 to control IFNalpha production. PLoS One 5, e15080 (2010).
Colombo, B. M. et al. Defective expression and function of the leukocyte associated Ig-like receptor 1 in B lymphocytes from systemic lupus erythematosus patients. PLoS One 7, e31903 (2012).
Jin, J. et al. LAIR-1 activation inhibits inflammatory macrophage phenotype in vitro. Cell Immunol. 331, 78–84 (2018).
Fouet, G. et al. Headless C1q: a new molecular tool to decipher its collagen-like functions. FEBS J. 288, 2030–2041 (2021).
Zhang, Y. et al. Adiponectin’s globular domain inhibits T cell activation by interacting with LAIR-1. Biochem. Biophys. Res. Commun. 573, 117–124 (2021).
Teixeira, J. E., Heron, B. T. & Huston, C. D. C1q- and collectin-dependent phagocytosis of apoptotic host cells by the intestinal protozoan Entamoeba histolytica. J. Infect. Dis. 198, 1062–1070 (2008).
Olde Nordkamp, M. J. et al. Leukocyte-associated Ig-like receptor-1 is a novel inhibitory receptor for surfactant protein D. J. Leukoc. Biol. 96, 105–111 (2014).
Schaffler, A. & Buechler, C. CTRP family: linking immunity to metabolism. Trends Endocrinol. Metab. 23, 194–204 (2012).
Shapiro, L. & Scherer, P. E. The crystal structure of a complement-1q family protein suggests an evolutionary link to tumor necrosis factor. Curr. Bio. l 8, 335–338 (1998).
Meyaard, L. et al. LAIR-1, a novel inhibitory receptor expressed on human mononuclear leukocytes. Immunity 7, 283–290 (1997).
Kim, S. et al. The Role of Leukocyte-Associated Ig-like Receptor-1 in Suppressing Collagen-Induced Arthritis. J. Immunol. 199, 2692–2700 (2017).
Helou, D. G. et al. LAIR-1 acts as an immune checkpoint on activated ILC2s and regulates the induction of airway hyperreactivity. J. Allergy Clin. Immunol. 149, 223–236 e226 (2022).
Rowley, A. T., Nagalla, R. R., Wang, S. W. & Liu, W. F. Extracellular Matrix-Based Strategies for Immunomodulatory Biomaterials Engineering. Adv. Healthc. Mater. 8, e1801578 (2019).
Rowley, A. T. et al. Effects of Surface-Bound Collagen-Mimetic Peptides on Macrophage Uptake and Immunomodulation. Front. Bioeng. Biotechnol. 8, 747 (2020).
Dervan, A. et al. Biomaterial and Therapeutic Approaches for the Manipulation of Macrophage Phenotype in Peripheral and Central Nerve Repair. Pharmaceutics 13, 2161 (2021).
Kim, Y. K. et al. Incorporation of a Ligand Peptide for Immune Inhibitory Receptor LAIR-1 on Biomaterial Surfaces Inhibits Macrophage Inflammatory Responses. Adv. Healthc. Mater. https://doi.org/10.1002/adhm.201700707 (2017).
Madsen, D. H. et al. M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J. Cell Bio. l 202, 951–966 (2013).
Larsen, A. M. H. et al. Collagen Density Modulates the Immunosuppressive Functions of Macrophages. J. Immunol. 205, 1461–1472 (2020).
Andersson, U. & Tracey, K. J. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 29, 139–162 (2011).
Kuczek, D. E. et al. Collagen density regulates the activity of tumor-infiltrating T cells. J. Immunother. Cancer 7, 68 (2019).
Romer, A. M. A., Thorseth, M. L. & Madsen, D. H. Immune Modulatory Properties of Collagen in Cancer. Front. Immunol. 12, 791453 (2021).
Peng, D. H. et al. Collagen promotes anti-PD-1/PD-L1 resistance in cancer through LAIR1-dependent CD8(+) T cell exhaustion. Nat. Commun. 11, 4520 (2020).
Acknowledgements
This work was supported by grant from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health [R01AI135063 (MS)]. The author wishes to thank all contributors cited. The author also appreciates Dr. Kaitlin Carroll for helpful editing and BioRender.com for creating graphics.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Son, M. Understanding the contextual functions of C1q and LAIR-1 and their applications. Exp Mol Med 54, 567–572 (2022). https://doi.org/10.1038/s12276-022-00774-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-022-00774-4
This article is cited by
-
Collagen in the central nervous system: contributions to neurodegeneration and promise as a therapeutic target
Molecular Neurodegeneration (2024)
