
Abstract
The endoplasmic reticulum (ER) is an essential organelle of eukaryotic cells. Its main functions include protein synthesis, proper protein folding, protein modification, and the transportation of synthesized proteins. Any perturbations in ER function, such as increased demand for protein folding or the accumulation of unfolded or misfolded proteins in the ER lumen, lead to a stress response called the unfolded protein response (UPR). The primary aim of the UPR is to restore cellular homeostasis; however, it triggers apoptotic signaling during prolonged stress. The core mechanisms of the ER stress response, the failure to respond to cellular stress, and the final fate of the cell are not yet clear. Here, we discuss cellular fate during ER stress, cross talk between the ER and mitochondria and its significance, and conditions that can trigger ER stress response failure. We also describe how the redox environment affects the ER stress response, and vice versa, and the aftermath of the ER stress response, integrating a discussion on redox imbalance-induced ER stress response failure progressing to cell death and dynamic pathophysiological changes.
Similar content being viewed by others
Introduction
The endoplasmic reticulum (ER) functions as a tool for synthesizing and folding 30% of the proteome, but its usual function is easily affected by external factors1,2,3. The ER protein quality control system maintains cell homeostasis. The quality control system does not function with at least one-third of the polypeptides transported into the ER. The misfolded or unfolded proteins in the ER are retro-translocated to the cytosol for proteasomal degradation, which involves the ER-associated degradation (ERAD) pathway, whose primary function is protein clearance4,5. When ubiquitination and proteasomal degradation are impaired, the high levels of accumulated misfolded/unfolded proteins in the ER block the ER lumen3,6. In stressful environments, the demand for secreted and membrane proteins increases quickly, leading to increased protein synthesis levels that exceed the cellular protein degradation capability, resulting in protein accumulation6. Genetic, environmental, and nutrition-related insults induce imbalances in the ER quality control system or imbalanced ER proteostasis, leading to ER stress-dependent nonnative protein secretions. Dysregulation or disruption of the oxidation–reduction pattern of equilibration–disruption of redox functions, changes in calcium levels, or posttranslation can exacerbate the imbalance to a critical point, initiating ER stress-induced cell death1,6,7. In addition, defective autophagy, energy deficiency, and inflammatory stimulation contribute to the accumulation of misfolded proteins6. Because of the high protein load on the organelle or impaired ER quality control mechanisms, protein degradation may be disrupted, leading to high protein accumulation in the ER, inducing the unfolded protein response (UPR), and thus initiating a stress response1,2. Here, we explain canonical ER stress, noncanonical ER stress, and ER stress response failure to enhance our understanding of ER stress signaling. This review also discusses how ER stress signaling affects mitochondria and vice versa and cell fate determination.
Endoplasmic reticulum stress and the unfolded protein response
When the ER becomes overloaded, cells experience ER stress disorder, characterized by the accumulation of misfolded proteins within the ER lumen. Various conditions, such as nutrient deprivation, hypoxia, viral infection, exposure to oxidative stress, and calcium depletion, can affect the homeostasis of cellular compartments and cause ER stress. Cells developed an evolutionarily conserved signal transduction mechanism called the UPR to resolve imbalanced ER protein folding, with the primary purpose of restoring ER homeostasis. In summary, the UPR is a signaling mechanism that is activated in cells in response to ER stress8. In general, UPR signaling is controlled by three main ER transmembrane-associated sensor proteins, namely, inositol-requiring enzyme 1 alpha (IRE1α), protein kinase R-like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6)6. The ER-resident chaperone BiP/GRP78 strikes a complex balance between unfolded and intraluminal (to be folded) proteins and the three ER stress sensors. Accumulated unfolded proteins sense disrupted equilibrium, resulting in GRP78 dissociation from the ER stress sensors that cooperate extensively in protein folding. Here, we describe the major signal transduction pathway of the ER stress response (Fig. 1).

GRP78/BiP, an ER chaperone, is closely associated with three sensors of the UPR, IRE1, PERK, and ATF6, inhibiting them under normal physiological conditions. Upon ER stress or misfolded protein accumulation, GRP78 dissociates from all these UPR transducers and permits stress sensors to activate downstream signaling. A different signal transduction system activates each pathway. The most conserved signal transducer, IRE1 (which contains a serine/threonine kinase and an RNase domain on its cytosolic side), undergoes homodimerization and autophosphorylation, and its activation mediates the activation of its RNase domain to produce spliced XBP1 mRNA, which is the active isoform of XBP1 that is translocated to the nucleus to increase UPR target genes, including chaperones and ERAD. The RNase domain of IRE1 also regulates the RIDD (regulated IRE1-dependent decay) pathway, where IRE1 degrades ER membrane-localized mRNAs through its RNase activity, resulting in a reduction in the amount of protein imported into the ER lumen. Similarly, during ER stress, the cytosolic domain of IRE1 interacts with TRAF2 and activates the downstream kinase ASK1, enhancing the activated JNK pathway and triggering apoptosis. Similarly, activation of PERK increases the phosphorylation of the alpha subunit of the translation protein eIF2, which attenuates protein translation to reduce ER protein overload, while paradoxically upregulating ATF4 mRNA, which targets the activation of proapoptotic CHOP and other UPR target genes. Upon sensing ER stress, a third UPR transducer, ATF6 alpha, is translocated to the Golgi apparatus, where it undergoes cleavage by site-1 and site-2 proteases. The cleaved fragments are then translocated to the nucleus and activate the transcriptional target genes of ATF6, including chaperones and XBP1. Adaptive response failure may not resolve ER stress and may upregulate UPR signaling to induce apoptosis.
ER stress signaling pathway
The three main UPR branches may act synergistically or differentially according to the activation mode with different strength and time courses9. Each branch activation causes b-ZIP transcription factors to function individually or jointly to activate the downstream target genes of the UPR. Activated UPR controls nonspecific transcriptional ER processes, such as mitochondrial function, amino acid metabolism, cellular redox status, and small molecule transport. The mechanisms of the UPR involving transcriptional activation ensure a certain degree of ER stress (acute or prolonged), which can alter several contributing mechanisms and ideally allow enhanced efficiency of the secretory pathway to alleviate the stressful stimulus. If the adaptive response cannot restore protein folding homeostasis, the UPR signals are continuously emitted and eventually are converted to alternative signals called “terminal UPR” signals, which promote apoptotic signaling10.
The most conserved ER stress signaling branch is IRE1α, and its activation mechanism has been extensively investigated. IRE1α is a type 1 transmembrane bifunctional protein kinase with three domain areas: the luminary N-terminal domain, endoribonuclease cytosolic (RNase) domain, and serine/threonine kinase cytosolic domain11. Responding to the accumulation of unfolded proteins during ER stress conditions, IRE1α dimerizes and trans-autophosphorylates, leading to the activation of the cytosolic region RNase domain, which is an activating result of a conformational modification caused by the excision of a 26-nucleotide intron from mRNA that encodes the transcription factor XBP1 (ref. 12). The consequence of this splicing event is a frameshift in the mRNA, and the transcription factor XBP1 thus becomes active and stable. Subsequently, XBP1 is translocated to the nucleus where it upregulates prosurvival target genes13 to generate multiple cell survival factors. XBP1 also increases the protein secretion rate in ER and Golgi compartments. Furthermore, IRE1α activation triggers the degradation of other ER-localized mRNAs in a process called regulated IRE1-dependent decay (RIDD), in addition to producing stable spliced transcription factors, such as XBP1s14. However, IRE1 interacts with TRAF2 to activate the inflammatory response and cellular apoptosis-associated protein kinases, especially apoptosis signal-regulating kinase 1 (ASK1), which leads to JNK activation15. In addition, IRE1–TRAF2 complexes recruit IκB kinase, which results in the degradation and phosphorylation of IκB, and consequently the translocation of nuclear factor-κB (NF-κB) to the nucleus to control inflammatory gene transcription16.
PERK, an ER-resident transmembrane kinase, is the second UPR signaling branch. PERK phosphorylates the downstream substrate of eukaryotic initiator of translation factor 2α (eIF2α) at serine 51 under ER stress conditions and contributes to the inhibition of protein synthesis in the ER lumen. When initiating translation, eIF2 constitutes a Met–tRNAi–GTP ternary complex that binds the 40 S subunit of 43 S PIC17. During eIF2α phosphorylation at Ser51, the binding affinity of eIF2 to its guanine nucleotide exchange factor, called eIF2B, which blocks the conversion of eIF2-GDP to eIF2-GTP, and thus prevents translation, is significantly enhanced18,19. PERK thus helps to reduce protein streaming into the ER, alleviating ER stress. However, when eIF2α is limited, mRNAs with short open reading frames at 5′ untranslated regions are favored. The transcription factor ATF4 is the produce of one of the encoded genes that induces translation. CHOP (transcription factor C/EBP homologous protein) and GADD34 (growth arrest and DNA damage-inducible 34) are two major target genes induced by ATF4. CHOP is a transcription factor that regulates the genes encoding apoptosis components. Thus, at modest signaling levels, the PERK branch is highly protective, but it can also activate cell death pathways. This dualism is likely to occur at the phosphorylated eIF2α stage and is demonstrated by the results of its unique phosphatase modulation. GADD34 encodes the phosphatase PP1c PERK-inducible regulatory subunit that counteracts PERK by dephosphorylating eIF2α. The GADD34–PP1c complex defends cells against ER stress by expanding low-level phosphorylation of eIF2α and is selectively inhibited by either small molecules or GADD34 deletion.
The third ER stress sensor, ATF6, is a transmembrane transcription factor with an N-terminal cytosolic domain and a C-terminus in the ER lumen that is regulated by ER stress. After sensing ER stress, ATF6 can activate the transcription of ER molecular chaperones. Under ER stress conditions, the S1P and S2P endopeptidases transfer ATF6α for cleavage in the Golgi system, thereby releasing the activated ATF6α form20. The cleavage of ATF6 at a juxta-membrane site contributes to its discharge into the cytosol21. The 50-kDa soluble ATF6 cytosolic fragment is then transported into the nucleus, where it associates with ATF/cAMP response elements and endoplasmic reticulum stress response elements (ERSE-I)22. Consequently, UPR target genes, including XBP1, CHOP, and GRP78, are regulated and activated transcriptionally23.
In addition to these abovementioned canonical ER stress pathways, other pathways or specific elements of the UPR have been linked as determinant of cell fate acting independent of the classical UPR, a process known as noncanonical ER stress. Mainly, the integrated stress response (ISR), translocation of proteins into the ER, extracellular-signal-regulated kinase reactivation, ERAD, ERphagy, and other pathways have been linked to the noncanonical ER stress response24. The ISR is an evolutionarily conserved program affecting homeostasis that is activated by different pathological conditions, such as hypoxia, viral infection, amino acid deprivation, and glucose deprivation, or cell-intrinsic factors, including ER stress caused by the accumulation of unfolded/misfolded proteins in the ER25. The ISR-inducing stressors activate PERK, PKR (double-stranded RNA-activated protein kinase), GCN2 (general control nonderepressible 2), and HRI (heme-regulated inhibitor) kinases, which are linked to the phosphorylation of eIF2α at Ser51 to inhibit the translation of new proteins. Simultaneously, it initiates the translation of specific mRNAs of ATF4, the main effector of ISR. Thus, ATF4 is a critical transcription factor inducing the expression of genes involved in autophagy, antioxidant response, amino acid metabolism, and cell death26. Recently, the PERK pathway was identified as an attenuator of IRE1 signaling via protein phosphatase RNA polymerase II-associated protein 2, which suppresses IRE1 oligomerization and RNase activity, inhibiting the production of spliced XBP1 and ERAD27. Other pathways that are linked with noncanonical ER stress have been described in previously published articles24,25. In addition to canonical or noncanonical ER stress responses, accumulating evidence suggests that ER stress response failure contributes to the development of pathogenesis, which we discuss in a separate section.
ER stress leads to disease progression
During ER stress, a decrease in the capacity of cells to recover misfolded or unfolded proteins may induce cellular dysfunction and disease. The diminished capacity of cells to fold secreted or membrane proteins, the decreased ability to identify or respond to misfolded proteins, or the increased load of misfolded proteins in the ER leads to ER stress and causes several diseases. Improper activation of the UPR can be dangerous because it can destroy the cell or protecting the cell against death (e.g., during neoplastic transformation or viral infection). Each of these conditions has been shown to trigger cellular or organ damage in humans or model organisms under pathological conditions. Table 1 addresses the ER stress-related proteins and their associated pathways that lead to the development of different diseases.
UPR regulation from the redox perspective
ROS are normally small, short-lived, and highly reactive molecules28,29. They can be free radicals derived from oxygen, including anionic superoxide (O2•−) or the hydroxyl radical (OH•) or nonradical molecules, e.g., hydrogen peroxide (H2O2). ROS are generated in cells with a variety of antioxidant defenders in equilibrium. In this case, enzyme scavengers such as superoxide dismutase (SOD), glutathione peroxidase, peroxiredoxins, catalase, and nonenzyme scavengers, such as vitamin C and E, glutathione (reduced glutathione (GSH)), lipoic acid, carotenoids, and iron chelators are involved28. ROS are involved in regulating normal physiological functions by activating various cellular signaling pathways and transcription factors, including phosphoinositide 3 kinase (PI3K)/Akt, mitogen-activated protein kinase, nuclear factor erythroid 2-related factor 2 (NRF2)/Kelch-like ECH-associated protein 1, NF-κB, and tumor suppressor p53, supporting cellular survival or death processes30,31. The transduction of redox-controlled signals is often carried out through reversible thiol protein oxidation. However, further research is needed to understand the physiological relevance, and redox signaling mechanisms at the cellular level to determine the ROS threshold and level and the severity of oxidative stress29,31.
ER and mitochondrial ROS regulate redox signaling mechanisms
Protein is commonly required to implement biological activity in a particular three-dimensional structure32,33. The indigenous conformation of several proteins, particularly secretory and membrane proteins, is stabilized by intramolecular disulfide bonds34. The ER is a reticular organelle that folds and changes the action of nascent proteins translocated by cytoplasmic ribosomes35. In the ER, H2O2 is produced as a byproduct in the protein folding process, and hence, the ER retains relatively high ROS levels36,37. The ER redox state is closely related to ER homeostasis, and proper functioning of the ER is due to disulfide bridge formation during protein folding. The regulation of the redox state by the ER and mitochondrial pathways is described below.
ER pathway
Intramolecular disulfide bonding, oxidative processes, and possibly the most common posttranslational modification are characteristics of oxidative protein folding (OPF)38. Disulfide bond formation is primarily catalyzed by PDI, which has four Trx domains (a, b, b′, and a′) and a KDEL ER retention series c-domain39,40. The redox state of the CGHC motifs in the a-domains in PDI determines whether the functions of oxidases or isomerases are triggered41,42. The noncatalytic b′ domain identifies unfolded and incorrectly folded proteins via exposed hydrophobic patches on a protruding part of the protein43. The oxidation of nascent proteins is carried out for catalytic purposes by reducing PDI via the CGHC active site42. The next reaction is the reoxidation of PDI active sites44. H2O2 is the principal source for oxidation, in addition to a byproduct produced by O2, which ER oxidoreductases use to oxidize PDI. In peroxidases, H2O2 may be used to shape disulfide bonds that can reoxidize PDI or maintain redox homeostasis in the ER.
Similarly, ERO1 is a highly conserved flavoprotein with two cysteine pairs situated on a flexible loop and on a CXXC motif that is proximal to the flavin adenine dinucleotide cofactor45. O2 is used as a sulfhydryl electron accepter by ERO1 to catalyze the PDI disulfide bond formation that produces H2O2. This process is essential to ensure PDI isomerase activity for oxidizing reduced PDI36,37. To avoid ROS overproduction, yeast ERO1p and mammalian ERO1α and ERO1β are closely regulated to maintain ER redox homeostasis or proper protein folding39.
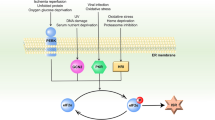
Although ERO1 is important for oxidizing protein dithiols in yeast, ERO1α and ERO1β double knockout results in only a mild ERO1β phenotype that compromises mammalian oxidative folding of proinsulin46,47. In the absence of these flavoproteins, this surprising finding raises the question, What sustains oxidative folding? Similar to ERO1, quiescin sulfhydryl oxidase (QSOX), a flavoprotein containing an ERV/ALR domain fused with a domain, such as Trx, catalyzes the formation of disulfide by coupling disulfide oxidation with oxygen reduction to H2O2. It can also bypass a disulfide exchange reaction catalyzed by PDI due to its unique structure48,49. In vitro, QSOX can transport disulfides to the first thioredoxin domain between the ERV domain and then exchange the disulfide with substrate proteins50. Several NADPH oxidases (NOXs) are positioned on the ER membrane, where ROS generation is catalyzed51,52. For example, Nox4, which has been shown to be in the ER membrane, produces H2O2, which can cause apoptosis after extended ER stress52 (see Fig. 2).

During various pathological conditions, unfolded or misfolded proteins are increased in the ER. During oxidative protein folding in the ER, ROS are generated during electron transfer between PDI and ERO1α. ROS associated with UPR signaling can activate an antioxidant response, such as Nrf2 or can increase ROS generation by activating ERO1 or NOX. Furthermore, ROS are increased in the ER through the association of PDI with ROS, generating Nox4. Although the major site of calcium is in the ER, under stress conditions, calcium may flow to the mitochondrial outer membrane through calcium release channels, such as IP3R or RyR. Increased calcium overload in mitochondria subsequently increases ROS generation. The increased calcium load and ROS in mitochondria may lead to opening of the mitochondrial permeability transition pore, which may cause the release of proapoptotic factors. High oxidative stress during this condition is critical for inducing mitochondrial dysfunction and vice versa. Overall, we suggest that the ER stress response can induce ER or mitochondrial dysfunction, which may increase oxidative stress by dysregulating disulfide bond formation, impairing oxidative protein folding, or the inducing certain UPR genes (e.g., CHOP) where oxidative stress is reversible, which may depend on redox homeostasis (see the text for more details). ER endoplasmic reticulum, ROS reactive oxygen species, PDI protein disulfide isomerase, ERO1α endoplasmic reticulum oxidoreduction 1α, NOX NADPH oxidase, IP3R inositol 1,4,5-trisphosphate receptors, RyR ryanodine receptors.
Mitochondrial pathway
Mitochondria are among the key sources of ROS production, where the process of oxidative phosphorylation produces unpaired electrons that interact with O2 and increase the production of highly reactive free radicals. While ROS are produced as byproducts of oxidative phosphorylation, a widely discussed long-standing issue has been the specific site(s) in the electron transmission chain critical for ROS generation. Under physiological conditions, complex I is usually considered the key site for the development of mitochondrial ROS, where O2•− on the side of the mitochondrial matrix is formed with rapid dismutation to H2O2 by SOD53,54.
Complex III is also classified as O2− producing machinery15,16. Under ischemic and apoptotic conditions, the development of O2•− in complex III is shown to be induced by the inhibition and reduction of the electron transport system55. Cellular ROS production is largely supported by NOX in certain cells, such as phagocytic neutrophils, nonphagocytic fibroblasts, smooth muscle, and vascular endothelial cells56,57,58. Recently, Nox4, a Nox isoform, was found to be expressed in the mitochondria of chronic myeloid leukemia cells that overexpress the nonapoptotic protein BCL-2 (CEM/BCL-2)59 in the renal cortex of rats60 and of cardiomyocytes, and in membrane fractions enriched with mitochondria. However, to date, Nox4 activity in mitochondria has not been evaluated explicitly, but cytoplasmic Nox4 can participate in promoting the modulation of PKCε, MitoKATP, and thioredoxin-2 activity, leading to the upregulation of mitochondrial ROS production by the electron transport chain, which is redox sensitive61. Other mitochondrial proteins, including pyruvate dehydrogenase, α-ketoglutarate dehydrogenase62, and glycerol-3-phosphate dehydrogenase, and fatty acid β-oxidation56 were identified as being produced by the electron transport chain and the NOX family.
In addition to ROS generation, mitochondrial-specific nitric oxide synthase (NOS) can lead to •NO development. The effect of nitric oxide (NO) production on mitochondrial functions is debated, particularly the amount of NO produced and the conditions in which it is generated. Mitochondrial respiration, even with moderate levels of •NO, can be partially inhibited, causing an increase in mitochondrial ROS output. The effect is depolarization of mitochondria, resulting in the reduced mitochondrial uptake of calcium63. The inhibitory effect of •NO is mainly caused by the inactivation of cytochrome c oxidase (COX; complex IV)64 and rate-limiting component of the electron transport chain. As a double-edged sword, •NO has been shown to play a role in mitochondrial biogenesis, in which it is stimulated by •NO generation through the upregulation of cGMP-dependent expression of PGC1α, which in turn increases the expression of mitochondrial transcription factor A and NRF1, and induces increased mitochondrial biosynthesis in adipocytes and liver cells65.
Furthermore, treatment with inorganic nitrates, nitrites, or S-nitroso-2-mercaptopropionyl glycine ensures the heart mounts a defense with different cellular proteins and is protected against injury by temporary S-nitrosylation (SNO)66. In contrast, excessive generation of •NO may lead to severe tissue damage upon certain pathological stimuli. Since a reaction of nitroxyl anion (NO−) and molecular oxygen is also possible, peroxynitrite can be formed through an alternative route. Peroxynitrite (ONOO−) spreads across mitochondrial membranes and can cause oxidative damage to critical components in mitochondria by oxygenation, nitration, and nitrosation. ONOO− has significant impacts on mitochondrial metabolism, calcium homeostasis, and mitochondrial permeability transition67. ONOO− also uncouples eNOS, thereby increasing ROS-producing enzymes where mitochondrial ROS levels are also increased67,68.
Redox-induced cross talk between the ER and mitochondria
The main sources of ROS are mitochondria, but accumulating evidence suggests that the ER also plays a critical role in regulating redox reactions. Therefore, it is important to study the elusive redox interaction between these two ROS sources. Through the emerging role of redox in calcium homeostasis, the relationship between different cellular ROS sources is suitably illustrated. The ER is a large calcium reservoir. The sarco/endoplasmic reticulum calcium ATPase (SERCA) continuously pumps calcium into the ER from the cytoplasm. Calcium release from the ER occurs via the ryanodine receptor and the 1,4,5-triphosphate receptor (IP3R) under normal and pathological conditions. Oxidation of these calcium transporting agents (due to elevated ROS levels during cellular dysfunction or pathological condition) causes net calcium efflux from the ER into the cytoplasm69, particularly the oxidation of luminal cysteine by ROS.
The work of Booth and colleagues70 highlights the complex relationship between ROS, calcium, the ER, and mitochondria. These authors demonstrated that mitochondria uptake of the calcium released from the ER, stimulating the mitochondrial ETC and accumulation of mitochondrial peroxide. Redox reactions involving the ER and mitochondria were suggested to culminate in cell death by apoptosis to clear cells with excessive ROS. Using a yeast model, Leadsham and colleagues revealed a clear interplay between mitochondrial dysfunction and the production of peroxide by ER-localized NOX71. This study gives rise to possible conflicts regarding the sources of ROS and the importance of the ER. The authors found that a reduction in COX activity contributes to increased cellular ROS levels. These observations suggest that the mitochondrial ETC is critical for the increase in superoxide. However, this increase was traced to ER-localized NOX.
There is an unusual redox cycle capable of combining oxidative folding with ROS accumulation in the ER. ER protein folding ability is regulated by the activity of the UPR: cells show increased chaperone levels when ER folding ability is affected72. Paradoxically, when the UPR is generated in the ER, ROS levels seem to increase, but ER stress is expected to be minimized by the UPR69,72. The reestablishment of ER homeostasis appears to have an inadvertent result on ROS production at face value; the increase in ER ROS levels is a possible byproduct of higher ERO1-PDI levels and UPR targeting of NOX enzymes. However, increased ROS may not be the only products of UPR induction, and ROS may signal and modify the cellular stress response (see Fig. 2). For example, cysteine residues in UPR sensors can increase or diminish UPR signals emitted through ROS-mediated oxidation69. Cells that demonstrate prolonged UPR activation (failure to recover ER homeostasis) may evoke mitochondrial activity to activate ER calcium release and thus induce apoptosis.
Cellular fate during ER stress and redox imbalance
OPF, which is characterized by the production of intermolecular or intramolecular disulfide bonds, is the leading source of H2O2 in the ER32,33,73. In conjunction with PDIs, which play significant roles in OPF, ERO1 proteins, for instance, respond to how the ER can generate oxidative power36,37. To preserve redox homeostasis, H2O2 is used as a common oxidant in some oxidation disorders, such as peroxiredoxin 4, glutathione peroxidase 7/8, and ascorbate peroxidase, to ensure OPF74,75. Furthermore, GSH, the most common reducing agent in cells, contributes to the elimination of excessive ROS76. ROS homeostasis is important in the ER. Although OPF friendly, over accumulated ROS (referred to as oxidative stress) may disrupt the redox homeostasis of the ER, leading to the accumulation of malfunctioning proteins and causing ER stress73. Upon disruption of ER homeostasis (protein folding homeostasis or redox homeostasis), the UPR is stimulated to restore stress77. To restore protein folding ability, UPR sensors, including IRE1α, PERK, and ATF6α, are activated to induce complex pathway networks, including prosurvival autophagy mechanisms, antioxidant reactions, ERAD, and ER biogenesis78,79 and apoptosis and ferroptosis prodeath mechanisms35. Growing evidence suggests that ROS and redox signaling are profoundly involved in deciding cellular fate.
The most critical link between ER stress and redox regulation is composed of many sources of ROS (from the ER or mitochondria). These sources can interfere with ER protein folding and cause ER stress, which may stimulate the UPR to induce apoptosis69. Misfolded proteins in the ER, important sources of ROS, may lead to oxidative stress, given the role played by the formation of disulfide bonds in the ER. During ER stress, dysregulated disulfide bond formation or breakage may induce ROS generation and cause oxidative stress by depleting ER GSH, which may eventually lead to apoptosis80. Some UPR elements, such as CHOP, are also critical for inducing oxidative stress. The overactivation of ERO1α by CHOP during ER stress increases the ROS production. ERO1α also induces ER IP3R-mediated Ca2+ leakage, activating Ca2+ sensor kinase and Ca2+/calmodulin-dependent protein kinase II (CaMKII) in the cytosol, which results in the activation of proapoptotic pathways, including the mitochondrial membrane permeability transition and Fas81,82,83. ERO2α also causes Ca2+/CaMKII sensing kinase activation in the cytosol. CaMKII also induces NOX subunit Nox2, a positive feed-forward loop during ER stress84, and thus triggers the ROS oxidative stress. Since the process of protein folding depends on redox homeostasis, increased oxidative stress may compromise protein folding machinery, leading to the production of misfolded proteins and exaggerating ER stress85 (see Fig. 2). Overall, redox signaling mediators have important roles in generating ROS during ER stress, and mitochondria greatly contribute to the ROS generation86,87. Further research is required to study the in-depth mechanism by which ER and mitochondrial associations play roles in regulating redox reactions, which will help with treating diseases caused by protein misfolding.
ER stress response failure
In addition to the ER stress response, ER stress response failure is of considerable interest to researchers in this field. ER stress coping mechanisms in cellular health have been established, but cell fate after ER stress response failure remains unclear. Our recent paper suggests that the ER stress response and ER stress response failure play divergent roles in cells88. Upon ER stress, the UPR, primarily functions to achieve cellular adaptation by maintaining ER homeostasis. However, the UPR and ER stress response may fail to alleviate cellular stress under severe stress or pathological conditions. During this condition, ER stress sensors may not be fully activated or aberrantly expressed, further downregulating the downstream signaling. This downregulation may diminish protein quality control and drive cells toward demise. We previously described that aging and its associated diseases, such as obesity and diabetes, may induce ER stress response failure, most likely under severe ER stress conditions. We speculate that various degrees of cellular stress can determine the degree of adaptive UPR, ER stress, and ER stress response or response failure, leading to either cell survival or cell death.
How does ER stress response failure contribute to disease progression?
Although ER stress response failure has not been studied extensively, our recent paper suggests that this mechanism is associated with several diseases and is more specific to metabolic diseases, including obesity and diabetes88. Aging and its related metabolic disturbances can also induce ER stress response failure, where various complex mechanisms are involved. Several previous reports have revealed that ER stress is highly induced in an obesity context, such as in high-fat diet (HFD)-fed rodents or palmitate-exposed cells23,89. However, some contradictory data from studies of muscle show that while body fat and glucose intolerance are increased during HFD treatment, the UPR is not activated. The major UPR sensors, such as IRE1 and PERK, or other UPR elements, such as GRP78, calnexin, CHOP, ATF4, or XBP1s mRNA, are not changed after HFD exposure90. These data suggest that ER stress response failure can contribute to obesity or glucose intolerance. Whether the ER stress response is dependent on tissue type needs to be further explored. However, another study was performed in liver tissue where ER stress response failure was observed in obese and diabetic mice and patients. In a nonalcoholic fatty liver disease (NAFLD) or insulin resistance context, ER stress response failure is a causal factor contributing to the progression of obesity-associated diabetes or nonalcoholic steatohepatitis (NASH)91.
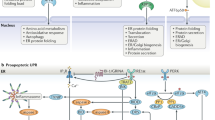
Mainly, physiological disturbances in the ER cause ER stress response dysfunction in which various factors are involved. The exact cause of ER stress response failure is undetermined; however, the contributing factors are postulated to be involved under specific pathological conditions. For example, chronic stress increases ER dysfunction and leads to protein homeostasis diseases, such as aging-associated Alzheimer’s or Parkinson’s disease, metabolic diseases, amyotrophic lateral sclerosis, etc. Chaperones and foldases, such as GRP78 and PDI, play protective roles by reducing protein aggregation, increasing ER function, maintaining proteostasis, or reducing ER stress. Posttranslational modifications such as SNO, for instance, SNO modification of PDI, inactivate protein function and increase ER stress. The addition of a NO group to a cysteine residue within a protein is called a SNO modification, which has diverse functions, such as regulation of metabolism, cellular apoptosis, and transcription factors92,93,94. The excessive uncontrolled production of NO can induce cell death by several mechanisms. ROS-mediated oxidative stress generated from different sources, such as mitochondria or the ER, also leads to ER stress and cell death (discussed in the previous section). We can easily suggest that the nitro-oxidative stress induced by excessive ROS or reactive nitrogen species (RNS) may contribute to ER stress response failure-mediated cell death. However, it is surprising that the overproduction of NO-derived RNS (NO-RNS) can increase SNO-IRE1, which inhibits the endoribonuclease activity of IRE1 to inhibit XBP1 splicing. Since XBP1s acts as a transcription factor, the inhibition of XBP1s or its nuclear translocation may not sufficiently induce the expression of its target genes, such as ER chaperones or ERAD target genes (Fig. 3). ER chaperones or ERAD target genes are critical for maintaining ER homeostasis.

During acute or short-term ER stress, the cell follows its natural adaptive pathway (as explained in Fig. 1) to maintain cellular homeostasis. However, during prolonged ER stress or under certain conditions, such as aging or metabolic diseases (e.g., obesity or diabetes), the activated UPR sensors may not activate downstream signaling (here, we focus on IRE1 signaling). For example, failure of XBP1s to translocate to the nucleus to activate its target genes leads to decreased activation of XBP1s target genes, such as chaperones or ERAD. This diminished effect is called ER stress response failure, which may trigger apoptotic signaling rather than adaptive responses. Evidence of ER response failure in metabolic diseases suggests that the impaired interaction of XBP1s with the insulin receptor or the regulatory subunits of PI3K p85α and p85β blocks XBP1s translocation to the nucleus. Similarly, excessive reactive oxygen species and/or reactive nitrogen species-induced nitro-oxidative stress production induces sulfonation (SO3H) of IRE1 or SNO of IRE1α, which can decrease IRE1α ribonuclease activity, thereby inhibiting the production of XBP1s88,108,160. This impaired signaling may disrupt the ER chaperones, ERAD, or their functions, which may negatively affect cell survival and trigger apoptosis, leading to the subsequent disease progression.
ROS play major roles in inducing the ER stress response and vice versa. Mitochondria were previously shown to be involved in inducing ROS production; however, accumulating evidence suggests that ROS originating from the ER have roles similar to those of mitochondria-derived ROS, disturbing ER protein folding, and generating ER stress95. ROS and RNS can stimulate oxidative stress and ER stress, potentially driving cellular apoptosis. Excessive ROS and RNS production from H2O2 or NO can cause nitro-oxidative stress and lead to ER stress-induced cell death93,95,96. Under this condition, severe tissue damage may be caused, which may lead to pathogenesis. Furthermore, the loss of antioxidants such as GSH, SOD, or catalase, or the disruption of disulfide bond formation in the ER can increase ROS generation and protein unfolding or misfolding in the ER, contributing to oxidative stress. Some of the contributors to the ER stress response pathway, such as the high activation of CHOP or ERO1α, can also induce ROS generation96. ROS produced by metabolic stress can trigger phospholipase C activation and ER calcium release, and can increase ER stress and mitochondrial dysfunction97. It is suggested that protein folding is highly redox reaction-dependent. Therefore, oxidative stress and ER stress are considered integral UPR components, where ROS generation can be either upstream or downstream targets of the UPR. We have already discussed the impact of ROS on the ER stress response; however, ER stress response failure is currently the focus of understanding the post-ER stress response and its interplay with redox signaling.
Mouse model investigations revealed high oxidative stress and dysregulated UPR responses (response failure) in the kidneys of aged mice after a high dose of tunicamycin injection. This failure was indicated by the loss of p-PERK and XBP1 splicing. The use of antioxidants prevented renal function failure by reducing oxidative stress and ER stress. Oxidative stress inhibition largely corrected the altered UPR in the aged kidneys and protected the old mice from a renal injury caused by a high dose of tunicamycin98,99. Similarly, in NAFLD, NASH, and pathological conditions of obesity and aging, the dysregulated expression of ER stress proteins and dysfunctional autophagy and apoptosis were observed. The expression of ER chaperones, such as GRP78, GRP94, or calnexin and foldases, such as PDI, ERp44, and ERp72 was reduced under NASH conditions. The transcription factors associated with ER stress, such as cleaved ATF6, spliced XBP1, and CHOP, were highly increased in NASH tissues. Dysregulated expression of BCL-2 family proteins was observed, where BCL-2 was highly upregulated, and BIM and MCL-1 were reduced significantly in NASH patients compared with control patients. These phenomena were correlated with the dysregulated expression of autophagy-related proteins, with high levels of Atg16L1 and LC3B found, and no significant differences were observed in the expression levels of Beclin 1, the Atg5–Atg12 conjugate, LC3A, or p62 in NASH tissues. These findings support the notion that ER stress response impairment/failure or autophagy dysregulation can induce severe pathologies, such as NASH, where the protein quality control system is highly compromised100. Several research groups also believe that UPR dysfunction is one of the major factors that contributes to the accumulation of disease-related proteins leading to pathogenesis, such as amyotrophic lateral sclerosis, Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease94,101,102. Since the main sensors of the UPR are localized in the ER, the role of the ER is identified as the hub for regulating the UPR102.
The ER has a role in fundamental functions such as posttranslational modifications that affect proper folding, and assembly of individual subunits and oligomerization. The interaction with chaperone proteins plays a very important role at each cotranslational and posttranslational step. In addition, it plays a crucial role in catalyzing isomerization reactions, balancing the proteins in a folding-competent state and in degradation pathways103,104. Notably, several chaperones or foldases, such as GRP78 or PDI, reduce the accumulation of misfolded proteins. The best example is observed in neurodegenerative diseases, where the chaperones have a critical role in ameliorating the oxidative/nitrosative stress-induced misfolded proteins, representing an adaptive response of cells93. In Alzheimer’s or Parkinson’s disease, PDI is S-nitrosylated, which is induced by the excessive production of NO and NO-RNS. This modification leads to the inhibition of PDI enzymatic activity and the accumulation of polyubiquitinated proteins, thereby activating the UPR. Here, NO blocks the defensive effect of PDI, suggesting that the overproduction of NO-mediated SNO of PDI can be deleterious to the cell by promoting prolonged UPR activation and cell death93,105,106. In addition to PDI SNO, other ER stress pathway proteins also become S-nitrosylated, affecting the UPR in Parkinson’s disease. Excessive NO stimulates the SNO modification of ER stress sensors, such as IRE1α and PERK. When the ER stress sensors are S-nitrosylated, downstream elements are affected. For example, SNO-IRE1 inhibits ribonuclease activity and attenuates XBP1 splicing; however, it does not affect the phosphorylation or oligomerization of IRE1α, while the SNO of PERK activates its kinase activity, leading to the phosphorylation of its downstream target eIF2α. Researchers have demonstrated that the site-directed mutagenesis of IRE1α at cysteine 931 (Cys931), a predominant nitrosylation site in IRE1α, averts SNO and increases IRE1α ribonuclease activity107,108. SNO of IRE1 has also been observed in metabolic diseases, such as genetic and dietary models of obesity, where downstream XBP1 splicing activity is inhibited108. Under this condition, other canonical ER stress sensors, such as PERK and IRE1, are phosphorylated, and other ER stress-related events, such as JNK phosphorylation and UPR gene expression, are also increased. These data suggest that SNO of IRE1 inhibits the adaptive pathway of XBP1 but promotes the JNK pathway (upon its activation), which further damages hepatic cells by inducing apoptosis. Moreover, SNO of IRE1 alpha attenuated the downstream target ER chaperone genes of XBP1s, such as GRP78, PDI, and EDEM, and we suspect that the preventive effect of the ER chaperones may be abrogated during the NO-mediated SNO of IRE1α (ref. 108). Another report suggests that the inhibition of XBP1 increases oxidative stress, inflammation, and apoptosis in ob/ob mice, where JNK regulates the transition from adaptive to apoptotic UPR109.
A recent paper also demonstrated that Sdf2l1, an ER-resident molecule with a chaperoning function, was decreased in obese and diabetic mice91. These data were correlated with a decreased level of nuclear XBP1s, possibly because of the disruption of the ERAD pathway. An insufficient level of Sdf2l1 was also correlated with insulin resistance and steatohepatitis. The ER stress response pathway was also observed to be impaired in obesity and obesity-related diabetes, where the downstream target proteins of the main ER stress sensors were insufficiently activated. This impairment leads to ER stress response failure, which exacerbates ER stress. Furthermore, a decreased level of nuclear XBP1s is observed in diabetic patients and nephropathy murine models110. In this study, the authors observed a maladaptive ER stress response in the disease model, which indicated that simultaneously none of the UPR elements were activated; for instance, nuclear XBP1s was reduced and nuclear ATF6 and CHOP were increased. However, the mRNA expression of the UPR target genes that enhance the protein folding function of molecular chaperones (DNAJB9, DNAJC3, PDIA4, and Ero1b) and ERAD (Edem1) was increased in patients with diabetic nephropathy. These data suggest that the decreased nuclear translocation of XBP1s and the activation of its target genes are impaired during ER stress response failure or that the ER response does not depend on downstream XBP1 target genes. Nuclear XBP1 is impaired in diabetic mice and in cells treated with high levels of glucose, a finding confirmed by podocyte-specific deletion of XBP1, with its deficiency promoting ER stress in diabetic nephropathy. However, ATF6 overexpression in podocytes increased diabetic nephropathy through ATF6-dependent CHOP activation. These researchers explained that XBP1s lies downstream of insulin signaling and that the disturbance to insulin signaling or its sensitivity is caused by the impairment of the insulin receptor or the regulatory subunits of PI3K p85α and p85β. The interaction of XBP1s with p85α and p85β is critical to prevent diabetic nephropathy, and this disruption enhances pathogenesis. The pathogenesis of diabetic nephropathy and the disruption of XBP1s and P13K subunit interactions are also linked with obesity-associated insulin resistance. In an ob/ob mouse model, the interaction between p85 and XBP1s was lost in the liver, thereby reducing the nuclear translocation of XBP1s and inducing severe ER stress111. These data collectively suggest that nuclear XBP1 is critical for maintaining ER homeostasis, and its disruption can cause severe ER stress. The maladaptive ER stress response can limit the adaptive response and may follow ER stress-mediated cell death signaling. Interestingly, the same group of researchers found that bromodomain-containing protein 7 (BRD7), a tumor suppressor gene, is a component of UPR signaling and can regulate the nuclear translocation of XBP1. In addition, BRD7 interacts with the regulatory subunits of PI3K and enhances the nuclear translocation of p85α, p85β, and XBP1s. Furthermore, the in vivo data show that BRD7 expression was low in obese livers, and that its overexpression in the liver enhanced glucose homeostasis in diabetic and obese mice by restoring the nuclear translocation of XBP1s (ref. 112). Obesity needs to be controlled to maintain the health and lifespan because obesity accelerates the aging process113. In obesity, ER protein folding is impaired, and the UPR is induced, leading to hepatic steatosis, while overexpressing the ER chaperone (GRP78) prevents hepatic steatosis. Increased free fatty acids increase ROS and ER stress, and diminish SERCA activity. Several inflammatory modulators are also involved in obesity and its associated conditions. However, increased inducible nitric oxide synthase (iNOS) activity plays a major role in causing SNO of IRE1α, which further impairs ER function and prolongs ER stress108. Furthermore, iNOS induces ER stress-associated insulin resistance, where suppression or deletion of iNOS significantly enhances insulin sensitivity under HFD-fed conditions108,114. Similarly, an increase in misfolded proteins and loss of chaperones, or diminished proteasomal degradation are observed in aging and related pathologies, such as Parkinson’s or Alzheimer’s disease93,115,116. Subsequently, increased protein misfolding leads to cell death by enhancing apoptosis-inducing proteins107,108. For example, in a recent study, the loss of the ER chaperone GRP78 induced pulmonary fibrosis through increased ER stress, apoptosis, and senescence. Decreased GRP78 expression and increased ER stress-mediated apoptosis were revealed, as shown by increased CHOP and cleaved caspase-3 levels, suggesting that impaired ER stress or impaired UPR response reduces the function of old alveolar type II cells117.
The mechanisms described are promising for the study of ER stress response failure in metabolic diseases, where the downstream ER stress sensors fail to be fully activated. Further investigation of how ER stress response failure contributes to cell death during metabolic disturbances is needed. Considering the evidence, we can hypothesize that the expression of apoptotic proteins involved in ER stress is predominantly increased, whereas the expression of proteins involved in the adaptive response is diminished. In addition, ER stress response failure and its mechanism have been connected and applied to treating certain malignancies. Regarding potential therapies, protein disulfide isomerase inhibitors increased PERK dimerization and IRE1α oligomerization, decreasing the effect of inactive XBP1s on the accumulation of misfolded ER proteins118. The downstream signaling molecule of PERK-CHOP remained mostly intact. In other arms of the UPR, the upstream IRE1α was activated but not the downstream transcription factor XBP1s, which has a role in the adaptive ER stress response. The reduction in nuclear XBP1s was correlated with elevated poly (ADP-ribose) polymerase cleavage, suggesting the involvement of ER stress-mediated apoptosis118. These data provide strong evidence implicating ER stress response failure-mediated apoptosis signaling as a means to treat cancer phenotypes.
While ER stress has been linked with various diseases, such as diabetes, obesity, or aging, the specific regulatory mechanism of the UPR pathways that lead to pathogenesis still needs to be determined in further research. Whether the involvement of all UPR pathways is necessary to induce ER stress, or whether one specific pathway is enough to induce ER stress, the disease progression needs to be elucidated in future studies.
References
Rutkowski, D. T. & Kaufman, R. J. A trip to the ER: coping with stress. Trends Cell Biol. 14, 20–28 (2004).
Martinez, G., Khatiwada, S., Costa-Mattioli, M. & Hetz, C. ER proteostasis control of neuronal physiology and synaptic function. Trends Neurosci. 41, 610–624 (2018).
Lai, E., Teodoro, T. & Volchuk, A. Endoplasmic reticulum stress: signaling the unfolded protein response. Physiology 22, 193–201 (2007).
Dubnikov, T., Ben-Gedalya, T. & Cohen, E. Protein quality control in health and disease. Cold Spring Harb. Perspect. Biol. 9, a023523 (2017).
Hwang, J. & Qi, L. Quality control in the endoplasmic reticulum: crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 43, 593–605 (2018).
Wang, M. & Kaufman, R. J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529, 326–335 (2016).
Baiceanu, A., Mesdom, P., Lagouge, M. & Foufelle, F. Endoplasmic reticulum proteostasis in hepatic steatosis. Nat. Rev. Endocrinol. 12, 710–722 (2016).
Walter, P. & Ron, D. The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 (2011).
Glembotski, C. C. Roles for ATF6 and the sarco/endoplasmic reticulum protein quality control system in the heart. J. Mol. Cell Cardiol. 71, 11–15 (2014).
Shore, G. C., Papa, F. R. & Oakes, S. A. Signaling cell death from the endoplasmic reticulum stress response. Curr. Opin. Cell Biol. 23, 143–149 (2011).
Calfon, M. et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96 (2002).
Karagoz, G. E., Acosta-Alvear, D. & Walter, P. The Unfolded protein response: detecting and responding to fluctuations in the protein-folding capacity of the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 11, a033886 (2019).
Acosta-Alvear, D. et al. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 27, 53–66 (2007).
Maurel, M., Chevet, E., Tavernier, J. & Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 39, 245–254 (2014).
Kim, I., Xu, W. & Reed, J. C. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 7, 1013–1030 (2008).
Kaneko, M., Niinuma, Y. & Nomura, Y. Activation signal of nuclear factor-kappa B in response to endoplasmic reticulum stress is transduced via IRE1 and tumor necrosis factor receptor-associated factor 2. Biol. Pharm. Bull. 26, 931–935 (2003).
Algire, M. A. & Lorsch, J. R. Where to begin? The mechanism of translation initiation codon selection in eukaryotes. Curr. Opin. Chem. Biol. 10, 480–486 (2006).
Krishnamoorthy, T., Pavitt, G. D., Zhang, F., Dever, T. E. & Hinnebusch, A. G. Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol. Cell Biol. 21, 5018–5030 (2001).
Bogorad, A. M., Lin, K. Y. & Marintchev, A. eIF2B mechanisms of action and regulation: a thermodynamic view. Biochemistry 57, 1426–1435 (2018).
Glembotski, C. C. The role of the unfolded protein response in the heart. J. Mol. Cell Cardiol. 44, 453–459 (2008).
Xu, C., Bailly-Maitre, B. & Reed, J. C. Endoplasmic reticulum stress: cell life and death decisions. J. Clin. Investig. 115, 2656–2664 (2005).
Sano, R. & Reed, J. C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 1833, 3460–3470 (2013).
Basseri, S. & Austin, R. C. Endoplasmic reticulum stress and lipid metabolism: mechanisms and therapeutic potential. Biochem. Res. Int. 2012, 841362 (2012).
Bhardwaj, M., Leli, N. M., Koumenis, C. & Amaravadi, R. K. Regulation of autophagy by canonical and non-canonical ER stress responses. Semin Cancer Biol. 66, 116–128. https://doi.org/10.1016/j.semcancer.2019.11.007 (2019).
Pakos-Zebrucka, K. et al. The integrated stress response. EMBO Rep. 17, 1374–1395 (2016).
Hetz, C., Martinon, F., Rodriguez, D. & Glimcher, L. H. The unfolded protein response: integrating stress signals through the stress sensor IRE1alpha. Physiol. Rev. 91, 1219–1243 (2011).
Chang, T. K. et al. Coordination between two branches of the unfolded protein response determines apoptotic cell fate. Mol. Cell 71, 629–636 e625 (2018).
Halliwell, B. Free radicals and antioxidants - quo vadis? Trends Pharm. Sci. 32, 125–130 (2011).
Winterbourn, C. C. Are free radicals involved in thiol-based redox signaling? Free Radic. Biol. Med. 80, 164–170 (2015).
Kaminskyy, V. O. & Zhivotovsky, B. Free radicals in cross talk between autophagy and apoptosis. Antioxid. Redox Signal. 21, 86–102 (2014).
Zhang, J. et al. ROS and ROS-mediated cellular signaling. Oxid. Med. Cell Longev. 2016, 4350965 (2016).
Kosuri, P. et al. Protein folding drives disulfide formation. Cell 151, 794–806 (2012).
Qin, M., Wang, W. & Thirumalai, D. Protein folding guides disulfide bond formation. Proc. Natl Acad. Sci. USA 112, 11241–11246 (2015).
Berridge, M. J. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32, 235–249 (2002).
Lakkaraju, A. K., Mary, C., Scherrer, A., Johnson, A. E. & Strub, K. SRP keeps polypeptides translocation-competent by slowing translation to match limiting ER-targeting sites. Cell 133, 440–451 (2008).
Araki, K. et al. Ero1-alpha and PDIs constitute a hierarchical electron transfer network of endoplasmic reticulum oxidoreductases. J. Cell Biol. 202, 861–874 (2013).
Enyedi, B., Varnai, P. & Geiszt, M. Redox state of the endoplasmic reticulum is controlled by Ero1L-alpha and intraluminal calcium. Antioxid. Redox Signal 13, 721–729 (2010).
Borges, C. R. & Lake, D. F. Oxidative protein folding: nature’s knotty challenge. Antioxid. Redox Signal 21, 392–395 (2014).
Kozlov, G., Maattanen, P., Thomas, D. Y. & Gehring, K. A structural overview of the PDI family of proteins. FEBS J. 277, 3924–3936 (2010).
Ellgaard, L. & Ruddock, L. W. The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 6, 28–32 (2005).
Lee, E. & Lee, D. H. Emerging roles of protein disulfide isomerase in cancer. BMB Rep. 50, 401–410 (2017).
Darby, N. J. & Creighton, T. E. Functional properties of the individual thioredoxin-like domains of protein disulfide isomerase. Biochemistry 34, 11725–11735 (1995).
Klappa, P., Ruddock, L. W., Darby, N. J. & Freedman, R. B. The b’ domain provides the principal peptide-binding site of protein disulfide isomerase but all domains contribute to binding of misfolded proteins. EMBO J. 17, 927–935 (1998).
Riemer, J., Bulleid, N. & Herrmann, J. M. Disulfide formation in the ER and mitochondria: two solutions to a common process. Science 324, 1284–1287 (2009).
Frand, A. R. & Kaiser, C. A. The ERO1 gene of yeast is required for oxidation of protein dithiols in the endoplasmic reticulum. Mol. Cell 1, 161–170 (1998).
Zito, E., Chin, K. T., Blais, J., Harding, H. P. & Ron, D. ERO1-beta, a pancreas-specific disulfide oxidase, promotes insulin biogenesis and glucose homeostasis. J. Cell Biol. 188, 821–832 (2010).
Appenzeller-Herzog, C. et al. Disulphide production by Ero1alpha-PDI relay is rapid and effectively regulated. EMBO J. 29, 3318–3329 (2010).
Kodali, V. K. & Thorpe, C. Oxidative protein folding and the Quiescin-sulfhydryl oxidase family of flavoproteins. Antioxid. Redox Signal 13, 1217–1230 (2010).
Sevier, C. S. Erv2 and quiescin sulfhydryl oxidases: Erv-domain enzymes associated with the secretory pathway. Antioxid. Redox Signal 16, 800–808 (2012).
Rancy, P. C. & Thorpe, C. Oxidative protein folding in vitro: a study of the cooperation between quiescin-sulfhydryl oxidase and protein disulfide isomerase. Biochemistry 47, 12047–12056 (2008).
Bedard, K. & Krause, K. H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87, 245–313 (2007).
Sciarretta, S. et al. Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2alpha/activating transcription factor 4 pathway. Circ. Res. 113, 1253–1264 (2013).
Cadenas, E., Boveris, A., Ragan, C. I. & Stoppani, A. O. Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch. Biochem. Biophys. 180, 248–257 (1977).
Han, D., Williams, E. & Cadenas, E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem J. 353, 411–416 (2001).
Piskernik, C. et al. Antimycin A and lipopolysaccharide cause the leakage of superoxide radicals from rat liver mitochondria. Biochim. Biophys. Acta 1782, 280–285 (2008).
Brand, M. D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 45, 466–472 (2010).
Jezek, P. & Hlavata, L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int. J. Biochem. Cell Biol. 37, 2478–2503 (2005).
Li, J. M. & Shah, A. M. ROS generation by nonphagocytic NADPH oxidase: potential relevance in diabetic nephropathy. J. Am. Soc. Nephrol. 14, S221–S226 (2003).
Velaithan, R. et al. The small GTPase Rac1 is a novel binding partner of Bcl-2 and stabilizes its antiapoptotic activity. Blood 117, 6214–6226 (2011).
Block, K., Gorin, Y. & Abboud, H. E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl Acad. Sci. USA 106, 14385–14390 (2009).
Doughan, A. K., Harrison, D. G. & Dikalov, S. I. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 102, 488–496 (2008).
Starkov, A. A. et al. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J. Neurosci. 24, 7779–7788 (2004).
Rakhit, R. D., Mojet, M. H., Marber, M. S. & Duchen, M. R. Mitochondria as targets for nitric oxide-induced protection during simulated ischemia and reoxygenation in isolated neonatal cardiomyocytes. Circulation 103, 2617–2623 (2001).
Brown, G. C. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim. Biophys. 1504, 46–57 (2001).
Nisoli, E. et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 299, 896–899 (2003).
Burwell, L. S., Nadtochiy, S. M. & Brookes, P. S. Cardioprotection by metabolic shut-down and gradual wake-up. J. Mol. Cell Cardiol. 46, 804–810 (2009).
Radi, R., Cassina, A., Hodara, R., Quijano, C. & Castro, L. Peroxynitrite reactions and formation in mitochondria. Free Radic. Biol. Med. 33, 1451–1464 (2002).
Kuzkaya, N., Weissmann, N., Harrison, D. G. & Dikalov, S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. J. Biol. Chem. 278, 22546–22554 (2003).
Eletto, D., Chevet, E., Argon, Y. & Appenzeller-Herzog, C. Redox controls UPR to control redox. J. Cell Sci. 127, 3649–3658 (2014).
Booth, D. M., Enyedi, B., Geiszt, M., Varnai, P. & Hajnoczky, G. Redox nanodomains are induced by and control calcium signaling at the ER-mitochondrial interface. Mol. Cell 63, 240–248 (2016).
Leadsham, J. E. et al. Loss of cytochrome c oxidase promotes RAS-dependent ROS production from the ER resident NADPH oxidase, Yno1p, in yeast. Cell Metab. 18, 279–286 (2013).
Shimizu, Y. & Hendershot, L. M. Oxidative folding: cellular strategies for dealing with the resultant equimolar production of reactive oxygen species. Antioxid. Redox Signal 11, 2317–2331 (2009).
Malhotra, J. D. & Kaufman, R. J. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid. Redox Signal. 9, 2277–2293 (2007).
Tavender, T. J., Springate, J. J. & Bulleid, N. J. Recycling of peroxiredoxin IV provides a novel pathway for disulphide formation in the endoplasmic reticulum. EMBO J. 29, 4185–4197 (2010).
Toppo, S., Vanin, S., Bosello, V. & Tosatto, S. C. Evolutionary and structural insights into the multifaceted glutathione peroxidase (Gpx) superfamily. Antioxid. Redox Signal. 10, 1501–1514 (2008).
Chakravarthi, S., Jessop, C. E. & Bulleid, N. J. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep. 7, 271–275 (2006).
Cubillos-Ruiz, J. R., Bettigole, S. E. & Glimcher, L. H. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell 168, 692–706 (2017).
Higa, A. et al. Endoplasmic reticulum stress-activated transcription factor ATF6alpha requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol. Cell Biol. 34, 1839–1849 (2014).
Gogala, M. et al. Structures of the Sec61 complex engaged in nascent peptide translocation or membrane insertion. Nature 506, 107–110 (2014).
Haynes, C. M., Titus, E. A. & Cooper, A. A. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol. Cell 15, 767–776 (2004).
Hetz, C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102 (2012).
Cao, S. S. & Kaufman, R. J. Unfolded protein response. Curr. Biol. 22, R622–R626 (2012).
Tabas, I. & Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 13, 184–190 (2011).
Li, G., Scull, C., Ozcan, L. & Tabas, I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J. Cell Biol. 191, 1113–1125 (2010).
Chong, W. C., Shastri, M. D. & Eri, R. Endoplasmic reticulum stress and oxidative stress: a vicious nexus implicated in bowel disease pathophysiology. Int. J. Mol. Sci. 18, 771 (2017).
Zeeshan, H. M., Lee, G. H., Kim, H. R. & Chae, H. J. Endoplasmic reticulum stress and associated ROS. Int. J. Mol. Sci. 17, 327 (2016).
Bhandary, B., Marahatta, A., Kim, H. R. & Chae, H. J. An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int. J. Mol. Sci. 14, 434–456 (2012).
Bhattarai, K. R., Chaudhary, M., Kim, H. R. & Chae, H. J. Endoplasmic reticulum (ER) stress response failure in diseases. Trends Cell Biol. 30, 672–675. https://doi.org/10.1016/j.tcb.2020.05.004 (2020).
Ozcan, U. et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461 (2004).
Deldicque, L., Van Proeyen, K., Francaux, M. & Hespel, P. The unfolded protein response in human skeletal muscle is not involved in the onset of glucose tolerance impairment induced by a fat-rich diet. Eur. J. Appl. Physiol. 111, 1553–1558 (2011).
Sasako, T. et al. Hepatic Sdf2l1 controls feeding-induced ER stress and regulates metabolism. Nat. Commun. 10, 947 (2019).
Mishra, D., Patel, V. & Banerjee, D. Nitric oxide and S-nitrosylation in cancers: emphasis on breast cancer. Breast Cancer 14, 1178223419882688 (2020).
Uehara, T. et al. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441, 513–517 (2006).
Matus, S., Valenzuela, V., Medinas, D. B. & Hetz, C. ER dysfunction and protein folding stress in ALS. Int. J. Cell Biol. 2013, 674751 (2013).
Yoboue, E. D., Sitia, R. & Simmen, T. Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 9, 1–14 (2018).
Zhang, Z. et al. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 25, 101047 (2019).
Ly, L. D. et al. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 49, e291 (2017).
Liu, X. et al. Excessive oxidative stress contributes to increased acute ER stress kidney injury in aged mice. Oxid. Med. Cell Longev. 2019, 2746521 (2019).
Poplawski, T., Pytel, D., Dziadek, J. & Majsterek, I. Interplay between redox signaling, oxidative stress, and unfolded protein response (UPR) in pathogenesis of human diseases. Oxid. Med. Cell Longev. 2019, 6949347 (2019).
Lee, S., Kim, S., Hwang, S., Cherrington, N. J. & Ryu, D. Y. Dysregulated expression of proteins associated with ER stress, autophagy and apoptosis in tissues from nonalcoholic fatty liver disease. Oncotarget 8, 63370–63381 (2017).
Wang, S. & Kaufman, R. J. The impact of the unfolded protein response on human disease. J. Cell Biol. 197, 857–867 (2012).
Chu, Q. et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat. Commun. 10, 4883 (2019).
Rao, R. V. & Bredesen, D. E. Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr. Opin. Cell Biol. 16, 653–662 (2004).
Qi, L., Tsai, B. & Arvan, P. New insights into the physiological role of endoplasmic reticulum-associated degradation. Trends Cell Biol. 27, 430–440 (2017).
Walker, A. K. et al. Protein disulphide isomerase protects against protein aggregation and is S-nitrosylated in amyotrophic lateral sclerosis. Brain 133, 105–116 (2010).
Forrester, M. T., Benhar, M. & Stamler, J. S. Nitrosative Stress in the ER: A New Role for S-nitrosylation in neurondegenerative diseases. ACS Chem Biol. 1, 6, 355–358 (2006).
Nakato, R. et al. Regulation of the unfolded protein response via S-nitrosylation of sensors of endoplasmic reticulum stress. Sci. Rep. 5, 14812 (2015).
Yang, L. et al. METABOLISM. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science 349, 500–506 (2015).
Chan, J. Y. et al. The balance between adaptive and apoptotic unfolded protein responses regulates beta-cell death under ER stress conditions through XBP1, CHOP and JNK. Mol. Cell. Endocrinol. 413, 189–201 (2015).
Madhusudhan, T. et al. Defective podocyte insulin signalling through p85-XBP1 promotes ATF6-dependent maladaptive ER-stress response in diabetic nephropathy. Nat. Commun. 6, 6496 (2015).
Park, S. W. et al. The regulatory subunits of PI3K, p85α and p85β, interact with XBP-1 and increase its nuclear translocation. Nat. Med. 16, 429 (2010).
Park, S. W. et al. BRD7 regulates XBP1s’ activity and glucose homeostasis through its interaction with the regulatory subunits of PI3K. Cell Metab. 20, 73–84 (2014).
Salvestrini, V., Sell, C. & Lorenzini, A. Obesity may accelerate the aging process. Front. Endocrinol. 10, 266 (2019).
Zanotto, T. M. et al. Blocking iNOS and endoplasmic reticulum stress synergistically improves insulin resistance in mice. Mol. Metab. 6, 206–218 (2017).
Brown, M. K. & Naidoo, N. The endoplasmic reticulum stress response in aging and age-related diseases. Front. Physiol. 3, 263 (2012).
Naidoo, N., Ferber, M., Master, M., Zhu, Y. & Pack, A. I. Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J. Neurosci. Off. J. Soc. Neurosci. 28, 6539–6548 (2008).
Borok, Z. et al. Grp78 loss in epithelial progenitors reveals an age-linked role for endoplasmic reticulum stress in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 201, 198–211 (2020).
Elsevier Style 1 Numbered with Article and Chapter TitlesVatolin, S. et al. Novel protein disulfide isomerase inhibitor with anticancer activity in multiple myeloma. Cancer Res. 76, 3340–3350 (2016).
Santos, L. E. & Ferreira, S. T. Crosstalk between endoplasmic reticulum stress and brain inflammation in Alzheimer’s disease. Neuropharmacology 136, 350–360 (2018).
Lee, J. H. et al. Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Exp. Mol. Med. 42, 386–394 (2010).
Imai, Y., Soda, M. & Takahashi, R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J. Biol. Chem. 275, 35661–35664 (2000).
Saxena, S., Cabuy, E. & Caroni, P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 12, 627–636 (2009).
Yoshida, H. ER stress and diseases. FEBS J. 274, 630–658 (2007).
Pfaffenseller, B. et al. Impaired endoplasmic reticulum stress response in bipolar disorder: cellular evidence of illness progression. Int. J. Neuropsychopharmacol. 17, 1453–1463 (2014).
Lorz, C. et al. Paracetamol-induced renal tubular injury: a role for ER stress. J. Am. Soc. Nephrol. 15, 380–389 (2004).
Cybulsky, A. V., Takano, T., Papillon, J. & Bijian, K. Role of the endoplasmic reticulum unfolded protein response in glomerular epithelial cell injury. J. Biol. Chem. 280, 24396–24403 (2005).
Wang, M. G. et al. Activation of PERK-eIF2alpha-ATF4-CHOP axis triggered by excessive ER stress contributes to lead-induced nephrotoxicity. Biochim. Biophys. Acta Mol. Cell Res. 1866, 713–726 (2019).
Eizirik, D. L., Colli, M. L. & Ortis, F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat. Rev. Endocrinol. 5, 219–226 (2009).
Allagnat, F. et al. C/EBP homologous protein contributes to cytokine-induced pro-inflammatory responses and apoptosis in beta-cells. Cell Death Differ. 19, 1836–1846 (2012).
Chen, J. et al. Thioredoxin-interacting protein deficiency induces Akt/Bcl-xL signaling and pancreatic beta-cell mass and protects against diabetes. FASEB J. Off. Publ. Federation Am. Societies Exp. Biol. 22, 3581–3594 (2008).
Riaz, T. A. et al. Role of endoplasmic reticulum stress sensor IRE1alpha in cellular physiology, calcium, ROS signaling, and metaflammation. Cells 9, 1160 (2020).
Wang, Y., Vera, L., Fischer, W. H. & Montminy, M. The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature 460, 534–537 (2009).
Song, B., Scheuner, D., Ron, D., Pennathur, S. & Kaufman, R. J. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J. Clin. Investig. 118, 3378–3389 (2008).
Xu, J., Zhou, Q., Xu, W. & Cai, L. Endoplasmic reticulum stress and diabetic cardiomyopathy. Exp. Diabetes Res. 2012, 827971 (2012).
Tsukano, H. et al. The endoplasmic reticulum stress-C/EBP homologous protein pathway-mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques. Arterioscler. Thromb. Vasc. Biol. 30, 1925–1932 (2010).
Thorp, E. et al. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe-/- and Ldlr-/- mice lacking CHOP. Cell Metab. 9, 474–481 (2009).
Sanson, M. et al. Oxidized low-density lipoproteins trigger endoplasmic reticulum stress in vascular cells: prevention by oxygen-regulated protein 150 expression. Circ. Res. 104, 328–336 (2009).
Kammoun, H. L. et al. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J. Clin. Investig. 119, 1201–1215 (2009).
Kabala, P. A. et al. Endoplasmic reticulum stress cooperates with Toll-like receptor ligation in driving activation of rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Res Ther. 19, 207 (2017).
Ospelt, C. et al. Overexpression of toll-like receptors 3 and 4 in synovial tissue from patients with early rheumatoid arthritis: toll-like receptor expression in early and longstanding arthritis. Arthritis Rheum. 58, 3684–3692 (2008).
Savic, S. et al. TLR dependent XBP-1 activation induces an autocrine loop in rheumatoid arthritis synoviocytes. J. Autoimmun. 50, 59–66 (2014).
Martinon, F., Chen, X., Lee, A. H. & Glimcher, L. H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 11, 411–418 (2010).
Wang, J. et al. Deficiency of IRE1 and PERK signal pathways in systemic lupus erythematosus. Am. J. Med. Sci. 348, 465–473 (2014).
Guo, G. et al. Induction of apoptosis coupled to endoplasmic reticulum stress through regulation of CHOP and JNK in bone marrow mesenchymal stem cells from patients with systemic lupus erythematosus. J. Immunol. Res. 2015, 183738 (2015).
Lerner, A. G. et al. IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 16, 250–264 (2012).
Toosi, S., Orlow, S. J. & Manga, P. Vitiligo-inducing phenols activate the unfolded protein response in melanocytes resulting in upregulation of IL6 and IL8. J. Investig. Dermatol 132, 2601–2609 (2012).
Adolph, T. E. et al. Paneth cells as a site of origin for intestinal inflammation. Nature 503, 272–276 (2013).
Kaser, A. et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 134, 743–756 (2008).
Lenna, S. et al. Increased expression of endoplasmic reticulum stress and unfolded protein response genes in peripheral blood mononuclear cells from patients with limited cutaneous systemic sclerosis and pulmonary arterial hypertension. Arthritis Rheum. 65, 1357–1366 (2013).
Heindryckx, F. et al. Endoplasmic reticulum stress enhances fibrosis through IRE1alpha-mediated degradation of miR-150 and XBP-1 splicing. EMBO Mol. Med. 8, 729–744 (2016).
Manalo, R. V. M. & Medina, P. M. B. The endoplasmic reticulum stress response in disease pathogenesis and pathophysiology. Egypt. J. Med. Hum. Genet. 19, 59–68 (2018).
Li, Y. et al. Hepatitis B surface antigen activates unfolded protein response in forming ground glass hepatocytes of chronic hepatitis B. Viruses 11, 386 (2019).
Tardif, K. D., Mori, K., Kaufman, R. J. & Siddiqui, A. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 279, 17158–17164 (2004).
Ji, C. & Kaplowitz, N. ER stress: can the liver cope? J. Hepatol. 45, 321–333 (2006).
Rissanen, A., Sivenius, J. & Jolkkonen, J. Prolonged bihemispheric alterations in unfolded protein response related gene expression after experimental stroke. Brain Res. 1087, 60–66 (2006).
Tajiri, S. et al. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 11, 403–415 (2004).
So, A. Y., de la Fuente, E., Walter, P., Shuman, M. & Bernales, S. The unfolded protein response during prostate cancer development. Cancer Metastasis Rev. 28, 219–223 (2009).
Lee, A. S. GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Res. 67, 3496–3499 (2007).
Bhattarai, K. R. et al. Potential application of ixeris dentata in the prevention and treatment of aging-induced dry mouth. Nutrients 10, 1989 (2018).
Lee, H.-Y. et al. The correlation of IRE1α oxidation with Nox4 activation in aging-associated vascular dysfunction. Redox Biol. 37, 101727 (2020).
Acknowledgements
This study was supported by the National Research Foundation (NRF-2017M3A9G7072719, NRF-2017M3A9E4047243, and 2020R1A2B5B03094896), Republic of Korea.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bhattarai, K.R., Riaz, T.A., Kim, HR. et al. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp Mol Med 53, 151–167 (2021). https://doi.org/10.1038/s12276-021-00560-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-021-00560-8
This article is cited by
-
Metabolism of primary high-grade serous ovarian carcinoma (HGSOC) cells under limited glutamine or glucose availability
Cancer & Metabolism (2024)
-
Endoplasmic reticulum stress and the unfolded protein response: emerging regulators in progression of traumatic brain injury
Cell Death & Disease (2024)
-
Arnicolide D induces endoplasmic reticulum stress-mediated oncosis via ATF4 and CHOP in hepatocellular carcinoma cells
Cell Death Discovery (2024)
-
Pregnenolone sulfate induces transcriptional and immunoregulatory effects on T cells
Scientific Reports (2024)
-
Empagliflozin reduces endoplasmic reticulum stress associated TXNIP/NLRP3 activation in tunicamycin-stimulated aortic endothelial cells
Naunyn-Schmiedeberg's Archives of Pharmacology (2024)
