
Abstract
Although germline alterations and somatic mutations in disease cells have been extensively analyzed, molecular changes in immune cells associated with disease conditions have not been characterized in depth. It is clear that our immune system has a critical role in various biological and pathological conditions, such as infectious diseases, autoimmune diseases, drug-induced skin and liver toxicity, food allergy, and rejection of transplanted organs. The recent development of cancer immunotherapies, particularly drugs modulating the immune checkpoint molecules, has clearly demonstrated the importance of host immune cells in cancer treatments. However, the molecular mechanisms by which these new therapies kill tumor cells are still not fully understood. In this regard, we have begun to explore the role of newly developed tools such as next-generation sequencing in the genetic characterization of both cancer cells and host immune cells, a field that is called immunogenomics/ immunopharmacogenomics. This new field has enormous potential to help us better understand changes in our immune system during the course of various disease conditions. Here we report the potential of deep sequencing of T-cell and B-cell receptors in capturing the molecular contribution of the immune system, which we believe plays critical roles in the pathogenesis of various human diseases.
Similar content being viewed by others
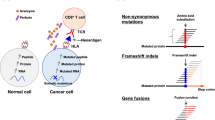


Introduction
The recent development of high-throughput DNA sequencing technologies has driven rapid progress in genetics and genomics research, enabling us to address a multitude of biological questions. In the field of population and medical genetics, including cancer genomics, sequencing of the whole-genome or whole-exome as well as genome-wide association studies using single-nucleotide polymorphisms (SNPs) has allowed us to characterize disease-associated genetic loci and variants [1]. In the past decade, the International Cancer Genome Consortium (ICGC) as well as The Cancer Genome Atlas (TCGA) have characterized the genetic drivers of disease by performing comprehensive genomic characterization of various types of human cancer [2, 3].
Although germline and somatic mutations have been extensively analyzed, changes in the molecular makeup of immune cells associated with certain disease conditions have not been characterized in depth. The immune system plays a critical role in various biological and pathological conditions, such as infectious disease, autoimmune disease, drug-induced skin and liver toxicity, food allergy, and rejection after organ transplantation. In addition, recent developments in cancer immunotherapy have highlighted the importance of host immune cells in the fight against cancer. For example, through a process known as immune surveillance, the immune system eliminates nascent tumor cells from our body [4], and serves as a primary defense against cancer. The recent success of antibodies targeting immune checkpoint molecules such as cytotoxic T lymphocyte antigen 4 (CTLA-4), programmed cell death protein 1 (PD-1) and its ligand PD-L1, have clearly demonstrated that our immune system has the ability to eradicate cancer cells [5]. However, the molecular mechanisms by which those immune checkpoint-blocking antibodies kill tumor cells are still not fully characterized.
The adaptive immune system cells, the T and B lymphocytes, are selectively activated by recognition of an antigen through their trans-membrane receptors. These receptors are known as the T-cell receptors (TCRs) and B-cell receptors (BCRs), respectively, and are important for inducing various immunological reactions as described above. TCRs and BCRs are also known as the ‘signatures’ of T and B lymphocytes. Considering the extremely high complexity of immune responses in our body, a comprehensive approach to fully characterize changes in TCR and BCR repertoires is urgently needed.
During lymphocyte differentiation, genes encoding TCRs and BCRs, which include variable (V), diversity (D), and joining (J) exon segments, undergo a complex rearrangement process to generate functional receptors. Extremely diverse TCR and BCR repertories are created by combinatorial diversity generated by the presence of a large number of distinct V, D, and J gene segments, and also by junctional diversity created by the template-independent insertion and deletion of nucleotides at the V–D, D–J, or V–J rearranged junctions during V–D–J recombination. This rearrangement process generates the highly variable complementary determining region 3 (CDR3), which determines specificity and affinity for antigen recognition [6]. Theoretically, a repertoire of ~1018 different TCRs, heterodimers of alpha and beta-chains, can be generated in a human [7]. In addition to TCR diversity, an even more diverse BCR repertoire is expected due to further somatic hypermutation [8].
To characterize the complex structure of our immune system and investigate the underlying mechanisms related to various disease conditions, two strategies have previously been applied. The first strategy aimed to detect the presence of different TCR families by comparing the usage of different TCR variable (V) sequences [9]. A second strategy, called CDR3 size spectratyping, was used to determine the clonality of the repertoire using fluorescent primers to measure length variation of the CDR3 region within each TCR V family [10, 11]. Although these approaches can provide a certain level of information, such as the proportion of each V(D)J combination, they do not provide detailed information about the CDR3 region sequences. In addition, these methods are not applicable if unidentified exons for V and/or J segments are present in certain individuals or populations.
To overcome these technical limitations, a systematic, accurate and unbiased analysis of TCR and BCR transcripts is needed. Although the earlier generation of high-throughput DNA sequencers had limitations due to their short read-lengths (50–100 bp long) or low sequencing output, the development of longer read-lengths has enabled us to analyze millions of TCRs and BCRs in a single experiment [12,13,14,15].
In this review article, we describe the current status and applications of TCR and BCR repertoire deep sequencing, and discuss the future potential of immunogenomics/immunopharmacogenomics studies (Fig. 1).

Scientific areas covered by immunogenomics/immunopharmacogenomics. Immunogenomics (IG)/immunopharmacogenomics (IPG) approaches can be applied to better understand the pathogenesis of autoimmune diseases, immune rejection after organ transplantation, graft-versus-host disease (GVHD) and graft-versus-leukemia (GVL) effects after bone marrow transplantation (BMT) or hematopoietic stem cell transplantation (HSCT), food allergy, and immune responses following vaccine treatments and cancer treatments including immunotherapy. These approaches are also important for the establishment of methods to predict efficacy or adverse events after treatment by various immune-modulating agents
TCR and BCR sequencing with next-generation sequencers (NGS)
For TCR and BCR cDNA sequencing by NGS, we have applied a method known as 5′ rapid amplification of cDNA end (5′RACE) PCR [13, 16], in which one common forward primer is designed as the adapter sequence at the 5′ end, and another primer corresponding to the C region of the TCR, or of each BCR isotype, is used as a reverse primer (Fig. 2). This method allows efficient and less-biased PCR amplification of TCR and BCR cDNA.

Strategy of TCR and BCR sequencing. The PCR adapter is tailed to cDNA that is reversely transcribed from mRNA isolated from B or T cells, whole lymphocytes, white blood cells, or tissues of interest. For BCR sequencing, PCR is carried out using a forward primer for the PCR adapter (red) and a reverse primer for the constant region of each BCR isotype (green). Quantitative PCR (qPCR) is used to estimate the expression level of each BCR isotype. For TCR sequencing, PCR is carried out using a forward primer for the PCR adapter and a reverse primer for the TCR constant region. The PCR products are subject to BCR or TCR sequencing with next-generation sequencers
The combination of NGS technology with this cDNA library construction method enables us to obtain an unprecedented amount of information about TCRs and BCRs. For a single sample, we can generate at most 10 million sequence reads of the expressed receptor genes, which should provide a comprehensive characterization of the TCR or BCR repertoires. We have applied this approach to clinical samples to determine specific T-cell populations that have infiltrated into tumor tissues or malignant ascites [17, 18], or to identify specific T-cell populations that have expanded or decreased during the course of treatment [19,20,21,22,23,24,25,26,27]. Furthermore, we have utilized it to characterize the BCR repertoire to diagnose and monitor B-cell-related disease conditions such as food allergy as well as autoimmune and infectious diseases [28].
Identifying neoantigen-specific TCRs for cancer immunotherapy
Recently in the field of oncology, monoclonal antibodies that block immune checkpoint molecules such as CTLA-4, PD-1, and PD-L1, have shown significant clinical benefit, and have revolutionized cancer immunotherapy [29].
Tumor tissues often evolve multiple mechanisms to escape from immune-mediated destruction of tumor cells [30]. One of these mechanisms involves cell-surface expression of immune checkpoint molecules, such as CTLA-4, PD-1, and PD-L1 [31]. Durable responses have been frequently observed with antibodies against these molecules [32]. A higher number of somatic mutations has been shown to be correlated with better clinical responses to these anti-immune checkpoint antibodies [33,34,35]. It is suspected that higher numbers of somatic mutations generate higher numbers of good immunogenic neoantigens, which are somatic-mutation-derived cancer-specific antigens expressed on HLA molecules of cancer cells. These antigens are capable of activating T cells, which are considered to drive the clinical effects of immune checkpoint inhibitors. There has been significant interest in harnessing the neoantigen-specific immune response in clinical settings. Leisegang et al. [36] have reported the efficacy of TCR-engineered T cells targeting the cancer-specific p68 mutation (mp68) in eradicating very large solid tumors in mice. Adoptive transfer of patient-derived tumor-infiltrating lymphocytes (TILs) has also yielded positive clinical results in a small subset of patients with solid tumors [37, 38]. To facilitate the translation of these promising findings into clinical use, we have reported a time-efficient approach to identify neoantigen-specific TCRs using blood [39]. Notably, our protocol requires a total of two weeks, beginning from T-cell priming with candidate neoantigen peptides to identifying neoantigen-specific TCRs [39]. This rapid process is critical for the application of neoantigen-specific TCR-engineered T cells in clinical settings.
Characterizing T-cell changes during cancer immunotherapy
Given the complex and dynamic nature of the tumor immune microenvironment, it is critically important to analyze the molecular nature of immune responses in patients who are treated with cancer immunotherapy. In particular, the pre-existing balance between immune-active and immune-suppressive molecules in the tumor microenvironment mediates clinical response (Fig. 3). In the context of melanoma, several immunotherapies, including ipilimumab-targeting CTLA-4, pembrolizumab and nivolumab-targeting PD-1, have been approved by the U.S. Food and Drug Administration. However, only a fraction of patients respond to these immunotherapies. Previous studies have shown that PD-L1 expression as well as the density and location of T cells in metastatic melanomas were predictive of response to PD-1 blockade [40]. Our analysis of the tumor TCRβ repertoire of melanoma patients undergoing nivolumab treatment revealed that oligoclonal expansion of TILs as well as increased expression of PD-L1, granzyme A (GZMA), and HLA-A were associated with treatment response [22].

Characterization of the tumor immune microenvironment and immune responses induced by cancer immunotherapy. The pre-existing balance between immune-active and immune-suppressive cells/molecules in the tumor microenvironment affects clinical responses to immune checkpoint blockades in cancer immunotherapy. In the state in which cancer progresses, the immune-suppressive side is more dominant compared to the immune-active side in the tumor microenvironment. After treatment with immune checkpoint blockades, if the immune-suppressive side is still dominant compared to the immune-active side, a poor clinical response is expected. However, if the immune-active side becomes more dominant compared to the immune-suppressive side, a favorable clinical response is expected
Since it is technically possible to obtain comprehensive information about T lymphocytes in cancer tissues, blood, and cancer-related effusions by sequencing of TCR, these approaches should enable us to further characterize the detailed mechanisms of immune responses in these treatments.
Characterizing T-cell changes during non-immune-targeted cancer therapy
Using NGS to evaluate the immune components in cancer patients who are treated with non-immune-targeted therapies has yielded extremely useful information for disease monitoring and clinical outcome prediction. The relationship between TILs and patient prognosis was first reported in a study of epithelial ovarian cancer [41]. Patients exhibiting an increased fraction of CD3+ TILs had better survival, indicating that the immune response may have an important role in determining clinical outcomes. Subsequent characterization of the infiltrated lymphocytic fraction in these cases revealed the importance of CD8+ T-cell populations in determining better outcomes [42]. It was hypothesized that the lack of effector CD8+ T cells in the malignant pleural effusion resulted from defects in CD8+ T-cell recruitment due to the immunosuppressive effects of the disease [43], allowing the malignant cancer cells to proliferate. The use of TCR sequencing may help to further differentiate the T-cell populations that are present in a particular environment. As compared to conventional flow cytometry techniques or immunological assays, comprehensive characterization of the T-cell repertoire by NGS provides information about specific subpopulations that are present in tumor tissues or ascitic/pleural effusions, enabling us to decipher which populations have key roles in treatment response. In our study of malignant effusions from ovarian cancer patients, we observed enriched T-cell clones that were not common to those found in tumors [17]. Furthermore, the abundant TCR clonotypes in CD4+, CD8+, and CD4+CD25+ T-cell populations in these samples were mutually exclusive, indicating that the immune microenvironments in tumors and ascites are entirely distinct [17].
The presence of TILs is associated with favorable clinical outcomes in several tumor types. In the context of bladder cancer, a small cohort study has documented an increase in both infiltrative B cell and T cells into the bladder tissues after BCG intravesical treatment, and those cases revealed a lower incidence of disease recurrence [44]. In our study investigating immune regulation in muscle-invasive bladder cancer, we found that oligoclonal expansion of TILs was significantly associated with longer recurrence-free survival (RFS) of patients who underwent definitive surgery [20]. Higher neoantigen load was also associated with longer RFS. These molecular patterns may thus provide useful prognostic markers and serve as a tool for prediction of disease recurrence following each respective treatment.
Regulatory T cells (Tregs) in tumors were shown to be associated with poor clinical outcomes in various cancer types [45]. However, one study found that high numbers of Tregs in follicular lymphoma (FL) tissues were associated with better clinical outcomes [46]. A subsequent report concluded that a specific intrafollicular Treg pattern, rather than number of Tregs, was correlated with poor survival [47]. In addition, it remains unclear whether Tregs suppress the immune system in an antigen-specific manner. Interestingly, we found that Tregs characterized from pretreatment FL biopsy specimens were highly clonal [18]. In line with previous research, perifollicular CD8+ T cells in tumors showed stronger clonal expansion compared to the intrafollicular CD8+ T cells, suggesting that antigen-specific CD8+ T cells capable of recognizing FL cells may be excluded from malignant follicles by some undetermined mechanism [18].
While nivolumab and pembrolizumab have been approved for the second-line treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (SCCHN), SCCHN in the locoregionally advanced stage is currently treated with chemoradiation therapy and surgery. We investigated tumor tissues from patients with locoregionally advanced SCCHN prior to chemoradiotherapy to examine the possible roles of the host immune system in SCCHN [26]. Interestingly, clonal expansion of T cells was significantly stronger in human papilloma virus (HPV)-negative tumors compared with HPV-positive tumors. HLA-A expression levels were also significantly higher in HPV-negative tumors. Additionally, higher GMZB levels in tumor tissues were significantly correlated with longer RFS independent of other clinicopathologic parameters. These findings imply differences in immune microenvironment between HPV-negative and HPV-positive tumors. Additionally, pretreatment levels of immune markers, such as GZMB, might serve as one of the predictors of recurrence risk for patients with locoregionally advanced disease.
Cryoablation is used for the treatment of renal-cell carcinoma (RCC) as well as other cancer types. This type of treatment is expected to attract immune cells through inflammatory signals. Studies of cryoablation in RCC and prostate cancer have indeed demonstrated its ability to induce a tumor-specific cytotoxic T-cell response [48, 49]. Moreover, a study in a mouse melanoma model showed that cryoablation resulted in an increase of infiltration of macrophages and dendritic cells (DCs) in the tumor microenvironment [50]. We analyzed the TCRβ repertoire in tumor tissues and blood samples from kidney cancer patients before and 3 months after cryoablation to better characterize the tumor and peripheral immune response during the treatment. Clonal expansion of certain T-cell clones was observed in the tissues after the treatment, and some of these clonotypes also expanded in peripheral blood samples. This would suggest that cryoablation can induce not only local but also systemic T-cell responses. In line with previous findings, we observed an increase in CD11c+ cells (macrophages and DCs) in the post-cryoablation tissues. Interestingly, CD8+ expression levels (CD8+ T-cell infiltration) were significantly associated with antigen presentation, which was measured by HLA-A expression. Altogether, these molecular changes have enabled us to better characterize the immune-mediated response to cryoablation (Fig. 4). In the clinical context, expanded TCR sequences that emerge after cryoablation might be applied for cancer-antigen-specific TCR-engineered T-cell therapy.

Possible mechanism of cryoablation-induced immune-mediated elimination of cancer cells in distant lesions. Cryoablation damages tumor cells while proteins are likely to be kept intact. Damaged cells release inflammatory signals like cytokines and chemokines. This stimulates infiltration of antigen-presenting cells (APCs) and lymphocytes into the tumor microenvironment. Dead cancer cells are phagocytosed by APCs, and proteins in cancer cells are processed and presented on HLA molecules of APCs as tumor-antigen-specific peptides. These antigens are recognized by tumor-antigen-specific CD8+ lymphocytes, resulting in activation and proliferation of tumor-antigen-specific T cells. Expanded T cells may go into circulation in blood, and eliminate cancer cells in distant tumor lesions
Applications of TCR/BCR sequencing in other diseases
Pathogenesis of autoimmune diseases
Tissue infiltrating lymphocytes in an affected lesion have an important role in the pathogenesis of autoimmune diseases. For example, CD20+ B cells and CD8+ T cells have been identified as the major infiltrating lymphocytes in the thyroid of patients with autoimmune thyroid disease (AITD) [51]. Infiltration of CD4+ and CD8+ T cells is related to progression of renal dysfunction and enrichment of CD3+CD4−CD8− (double negative) T cells in salivary gland is suggested to contribute to the damage of the tissues in patients with systemic lupus erythematosus [52,53,54]. Furthermore, persistent activation of certain clonal T and B cells has been observed in various autoimmune disease conditions. Hence, characterizing the human immune cells including Type 1T helper cells (Th1), Type 2T helper cells (Th2), and B cells in blood and/or affected lesions of patients with autoimmune diseases should provide valuable information to aid in understanding the molecular pathogenesis of autoimmune diseases. In this regard, quantification of unique T-cell and B-cell subpopulations through the use of high-throughput sequencing of TCRs and BCRs in a temporal and spatial manner should contribute to a better understanding of these diseases. In addition, this type of knowledge can be applied to improvements in diagnosis, prognosis and selection of patients for appropriate therapies [55].
For example, inflammatory bowel disease (IBD), such as Crohn’s disease (CD) and ulcerative colitis, is a chronic inflammatory condition of intestines likely caused by dysfunction of innate and adaptive immune responses that are likely related to commensal microbiota [56]. In these disease conditions, oligoclonally expanded CD4+ T cells were persistently present in preoperative and postoperative disease lesions [56]. Our study of inflamed tissue specimens from CD patients demonstrated expansion of oligoclonal T cells whose TCR sequences were not observed in corresponding normal tissues [57], suggesting that certain disease-causative T cells could be culprits for intestinal inflammation in CD. Further analysis of sorted T-cell subpopulations will provide a better understanding of immune dysfunctions that contribute to CD/IBD pathogenesis.
Pathogenesis of food allergy
In the United States, ~15 million people suffer from food allergies, including 1 in 13 children. Interestingly, there are unique geographic patterns of possible antigens causing food allergy [58]. For instance, peanut allergies kill 100–150 people in the US each year [59], but are uncommon in Asian countries. For example, in Japan, the estimated number of deaths in children from peanut allergies was 14 in the seven years between 1995 and 2001 [60]. On the other hand, allergy to shellfish is highly prevalent in Asian countries such as Japan, Philippines and Thailand [61]; population surveys show the prevalence rate in teenagers in Singapore to be 5.23% [58]. Allergy can be characterized into two major phases; the sensitization phase and the effector phase. Dendritic cells with food-specific antigens on their HLA molecules stimulate CD4+ Th2 cells that have TCRs recognizing these antigens. These activated CD4+ cells promote B cells to produce allergen-specific IgE in the sensitization stage [62]. These T cells also help maintain the allergen-specific IgE level in the late phase. A number of studies have indicated various potential roles of Th1, Th9, Th17, and Th22 effector T cells in food allergy [63,64,65]. Hence, the quantitative analysis of TCR and BCR repertoires in patients with food allergy will help to determine the inter-individual differences and intra-individual time-course changes. We conducted a preliminary analysis of the BCR repertoire in patients with peanut allergy prior to and post oral immunotherapy (OIT) to determine changes in the BCR repertoire during the course of treatment [28]. Through OIT, certain immunoglobulin heavy chain alpha (IGHA) and IGH gamma (IGHG) clones were oligoclonally enriched, and overall diversity of the BCR repertoire was significantly reduced. The identification of specific BCR sequences that are involved in the development of peanut allergy is a critical next step, as this information can help shed light on the detailed mechanism of the molecular pathology of food allergy.
Pathogenesis of GVHD after HCT
Hematopoietic cell transplantation (HCT) is now one of the essential treatment options for patients with hematologic diseases. The obstacles in this treatment are graft rejection or graft-versus-host disease (GVHD), which are an impediment to the success of HCT. T cells are known to play a key role in both graft rejection and GVHD [66, 67].
The immune response of host cells against the donor cells is initiated by recognition of the antigen(s) presented on HLA molecules of the donor cells. This is referred to as the sensitization stage. The host T cells recognize the donor cells and start to proliferate. The cells then destroy and eliminate the donor cells from the host, which is termed the effector stage [68]. There are three general forms of graft rejection. They are hyperacute, acute, or chronic [68]. Hyperacute rejection normally occurs within minutes to hours after transplantation due to pre-existing antibodies and effector T cells against the graft cells. Acute rejection usually occurs within six months following transplantation, while chronic rejection manifests months to years after transplantation. Several studies have shown that recipient-derived cytotoxic T cells might cause acute graft rejection through recognition of HLA-A, HLA-B, or HLA-C antigens of donor cells [69,70,71]. The disparity of these HLA alleles increases the risk of acute graft rejection.
GVHD occurs when the donor T cells attack the host tissue and the host cells are unable to mount an immunological response against the graft cells [72]. GVHD can be classified into two phases, acute or chronic. These two phases differ in the symptoms, time of onset and target tissues. Acute GVHD usually occurs within 3 months after the transplantation and often causes severe damage to the skin, liver, and gastrointestinal tract. Chronic GVHD is observed after 100 days of the transplantation and displays a more diverse set of clinical manifestations similar to the systemic autoimmune syndromes [72, 73]. The development of GVHD and its severity is likely to depend on various factors including age and source of the graft. Generally, the incidence of GVHD is higher in patients whose donor is unrelated or whose HLA types are not well matched. Older patients are likely to have a higher risk of developing GVHD compared to younger patients. Having a donor with a different sex also increases risk. The pathophysiology of GVHD is very complex. Prior to transplantation, patients receive a conditioning regimen which consists of chemotherapy and/or radiotherapy and is used to suppress the host immune function to prevent graft rejection. However, this treatment may in fact cause damage to tissues and induce inflammation. Tissue damage and proinflammatory cytokines will then trigger the activation of donor-derived T cells, which will mediate cytotoxicity against the target host cells [73]. Several studies have shown that the activated donor-derived cytotoxic T cells play an important role in the development of acute GVHD [74, 75]. Moreover, a lower level of regulatory T cells [76] is suggested to be associated with acute and chronic GVHD [77].
We have performed a comprehensive high-throughput analysis of the TCR repertoire in acute GVHD patients as well as in non-GVHD patients using the next-generation sequencing platform. We found that certain T-cell clones were activated in the course of GVHD development. Moreover, stronger oligoclonal enrichment was observed in GVHD patients around the time when they were diagnosed with GVHD, compared to the controls [27] (Fig. 5), suggesting that clonal expansion immediately following HSCT transplantation might serve as a biomarker of risk for GVHD. Characterization of the TCR repertoire by deep sequencing is therefore informative and may contribute to a better understanding of the molecular mechanisms of GVHD pathophysiology.

Immune characterization of graft-versus-host disease (GVHD). GVHD occurs when donor T cells recognize host-cell-specific antigens. When host-antigen-specific T cells recognize antigens in certain tissues, T cells are activated and proliferate in host tissues. Such expanded T cells can be detected in blood samples after hematopoietic stem cell transplantation (HSCT) or bone marrow transplantation (BMT) before acute GVHD symptoms become overt
Challenges and future directions
Sample quality and sorting of different populations of immune cells
Sequencing data from sorted immune cells can enhance our understanding of the role of specific subsets of cells. However, obtaining a sufficient amount of tissue to quantify the functional subtypes of T cells is a challenge in certain disease conditions. In this regard, since the TCR repertoire of each subset of T cells is likely to be distinct, we may analyze the TCR information of each subset of T cells in blood and determine which subset of T cells has infiltrated into the cancer tissue. Indeed, in our analysis of serial tumor and peripheral blood mononuclear cell (PBMC) samples from non-small cell lung cancer (NSCLC) patients receiving anti-PD-1 therapy, we found in one case that dominantly expanded T-cell clones in the peripheral blood were concordant with those found within the tumor [78]. Thus, TCR sequencing of sorted blood samples may provide information about TIL subpopulations when the two repertoires are comparatively analyzed.
One major advantage of our cDNA sequencing approach is that we do not have to isolate T cells from the cancer tissue because TCRs are expressed only in T cells. This makes the characterization of TCR repertoires of TILs much easier. However, to obtain a more complete picture of the immune response mechanism in cancer, we will undoubtedly need a holistic approach that integrates TCR repertoire analysis with somatic-mutation analysis, determination of HLA types, and analysis of proteasome processing systems in the antigen presentation machinery. This poses a big technical challenge but will be crucial to accurately assess the cellular mechanisms that determine effective immune function in the cancer microenvironment.
Data analysis
Owing to the complexity and diversity of the TCR repertoire, TCR NGS data analysis represents a very challenging bioinformatics task. There are several publicly available software packages for TCR NGS data analysis, including IMGT/HighV-QUEST [79], MiXCR [80], Decombinator [81], and IgBlast [82]. IMGT/HighV-QUEST, developed by the international ImMunoGeneTics information system (IMGT), is a widely used online system for the standardized analysis of collections of rearranged nucleotide sequences of TCR and BCR. However, the input data is limited to less than 500,000 reads per run. Decombinator [81] was developed based on the classic approach of Aho and Corasick of pattern matching and it includes a novel modification to correct for sequencing error. MiXCR also considers sequencing quality. This package further performs correction of PCR and sequencing errors, as well as rescues low-quality sequencing data. In addition, MiXCR’s VDJTools package is available for data presentation.
Our group has developed a novel algorithm of V(D)J decomposition, Tcrip [83], which we have used in combination with a ‘remapping’ step for unmapped read analysis to effectively analyze the cDNA sequence of both the TCR α and β repertoires [83]. Compared to the widely used online software IMGT/HighV-QUEST [79], our algorithm has a much greater input capacity (up to millions per job), which is extremely critical for analyzing high-volume NGS data. Moreover, compared to MiXCR, our algorithm can provide additional information such as unmapped sequence reads, which has enabled us to discover molecular changes of biological and genetic importance. For example, we have observed a relatively large proportion of TCRβ reads containing intronic sequences in NSCLC patient samples who were treated with multiple chemotherapy regimens but not in the healthy donor samples [83]. In these intron-containing cDNA sequences, introns between the J–C segments are correctly spliced out. Hence, the contamination of genomic DNA is very unlikely. Given that those patients have received intensive chemotherapy (2–4 different regimens), we hypothesize that their T lymphocytes have been severely damaged by chemotherapy, which has caused impairment in their splicing machinery. For the analysis of the BCR repertoire sequencing data, we have developed an analogous algorithm, Bcrip [28], which provides highly concordant clonotype assignments (V–J–C and CDR3) as well as the potential to detect novel exons.
Pairing of TCRαβ sequencing—functional analysis for reconstruction of TCR
Although TCRβ sequencing alone is sufficient to determine TCR clonality, it is important to gather information on both the alpha and beta transcripts of the TCR since antigen specificity is determined by the conformational pairing of the two molecules [84]. Previous efforts to elucidate pairing information have involved single-cell sorting and Sanger sequencing of the isolated T-cell clone [85,86,87]. However, single-cell methods are limited in throughput and by expensive costs, and the field will inevitably require methods to examine millions of T-cell or B-cell repertoires to monitor the dynamic changes of our immune responses. In recent reports, investigators have scaled up the throughput of sequencing pair transcripts in B cells [88], however, this method still falls short on the total number of reads derived from each sample compared to our new method. Currently, we can predict the combination of α- and β-chains that constitute the TCR if the proportion of T cells of particular interest is highly expanded. However, when α- and β-chains are present at lower frequencies, it is almost impossible to predict the combination of the two chains.
In the future, it will be essential to develop more efficient methods for elucidating paired transcript information to decipher antigen specificity. A number of studies have proposed methods to address TCR pairing without the use of single-cell technologies [89, 90]. One such method relies on a pairing algorithm: T cells from the same individual are split into a subset of wells and amplified with barcode sequences to identify which well each α- or β-sequence came from [90]. Putative pairs are then identified based on the fact that the true TCRα and β pairs should occupy the same wells. By comparing the well occupancy patterns of each α- and β-sequence, those sharing the same barcode more frequently than expected by chance are determined to be pairs. This method has been used to identify 200,000 TCRα and β pairs from PBMC samples seeded at 160,000 cells per well, with a false discovery rate of 1 percent.
Summary
The use of next-generation sequencing for the genetic characterization of the immune system, known as immunogenomics/immunopharmacogenomics, will be important for a deeper understanding of the pathogenesis of various disease conditions. Abnormal immune responses in our body lead to the development of autoimmune diseases and food allergy. Rejection of recipient cells or donor cells is also caused by uncontrolled immune responses in the recipient. There have been many reports indicating that activated immune responses through the drug–HLA interaction are present in drug-induced skin hypersensitivity and liver toxicity. The importance of host immune responses has been recognized in cancer treatments, not only for immunotherapy but also for cytotoxic agents and molecular targeted drugs. Thus, characterization of the TCR and BCR repertoire by means of NGS will ultimately enable us to identify the molecular mechanisms underlying various diseases. In addition, this approach may contribute to the identification of antigens that are associated with disease onset or progression. Computational analyses to draw meaningful inferences of functional recognition receptors on immune cells, however, remain a major challenge. In this review, we have tried to summarize the importance of TCR and BCR deep sequencing, and propose immunogenomics/immunopharmacogenomics as the next frontier in scientific discovery, as it continues to uncover the complex nature of our immune system, which has critical roles in the pathogenesis of various diseases.
References
Rabbani B, Tekin M, Mahdieh N. The promise of whole-exome sequencing in medical genetics. J Human Genet. 2014;59:5–15.
Cancer Genome Atlas Research N, Weinstein JN, Collisson EA, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113–20.
International Cancer Genome C, Hudson TJ, Anderson W, et al. International network of cancer genome projects. Nature. 2010;464:993–8.
Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Investig. 2007;117:1137–46.
Mullard A. New checkpoint inhibitors ride the immunotherapy tsunami. Nat Rev Drug Discov. 2013;12:489–92.
Morris GP, Allen PM. How the TCR balances sensitivity and specificity for the recognition of self and pathogens. Nat Immunol. 2012;13:121–8.
Venturi V, Price DA, Douek DC, Davenport MP. The molecular basis for public T-cell responses? Nat Rev Immunol. 2008;8:231–8.
Kim S, Davis M, Sinn E, Patten P, Hood L. Antibody diversity: somatic hypermutation of rearranged VH genes. Cell. 1981;27:573–81.
Langerak AW, van Den Beemd R, Wolvers-Tettero IL, et al. Molecular and flow cytometric analysis of the Vbeta repertoire for clonality assessment in mature TCR alphabeta T-cell proliferations. Blood . 2001;98:165–73.
Gorski J, Yassai M, Zhu X, et al. Circulating T cell repertoire complexity in normal individuals and bone marrow recipients analyzed by CDR3 size spectratyping. Correlation with immune status. J Immunol. 1994;152:5109–19.
Memon SA, Sportes C, Flomerfelt FA, Gress RE, Hakim FT. Quantitative analysis of T cell receptor diversity in clinical samples of human peripheral blood. J Immunol Methods. 2012;375:84–92.
Bashford-Rogers RJ, Palser AL, Huntly BJ, et al. Network properties derived from deep sequencing of human B-cell receptor repertoires delineate B-cell populations. Genome Res. 2013;23:1874–84.
Freeman JD, Warren RL, Webb JR, Nelson BH, Holt RA. Profiling the T-cell receptor beta-chain repertoire by massively parallel sequencing. Genome Res. 2009;19:1817–24.
Boyd SD, Gaeta BA, Jackson KJ, et al. Individual variation in the germline Ig gene repertoire inferred from variable region gene rearrangements. J Immunol. 2010;184:6986–92.
Lange V, Bohme I, Hofmann J, et al. Cost-efficient high-throughput HLA typing by MiSeq amplicon sequencing. BMC Genom. 2014;15:63.
Warren RL, Freeman JD, Zeng T, et al. Exhaustive T-cell repertoire sequencing of human peripheral blood samples reveals signatures of antigen selection and a directly measured repertoire size of at least 1 million clonotypes. Genome Res. 2011;21:790–7.
Jang M, Yew PY, Hasegawa K, et al. Characterization of T cell repertoire of blood, tumor, and ascites in ovarian cancer patients using next generation sequencing. Oncoimmunology. 2015;4:e1030561.
Liu X, Venkataraman G, Lin J, et al. Highly clonal regulatory T-cell population in follicular lymphoma - inverse correlation with the diversity of CD8(+) T cells. Oncoimmunology. 2015;4:e1002728.
Kato T, Iwasaki T, Uemura M, et al. Characterization of the cryoablation-induced immune response in kidney cancer patients. Oncoimmunology . 2017;6:e1326441.
Choudhury NJ, Kiyotani K, Yap KL, et al. Low T-cell receptor diversity, high somatic-mutation burden, and high neoantigen load as predictors of clinical outcome in muscle-invasive bladder cancer. Eur Urol Focus. 2016;2:445–52.
Ikeda Y, Kiyotani K, Yew PY, et al. Clinical significance of T cell clonality and expression levels of immune-related genes in endometrial cancer. Oncol Rep. 2017;37:2603–10.
Inoue H, Park JH, Kiyotani K, et al. Intratumoral expression levels of PD-L1, GZMA, and HLA-A along with oligoclonal T cell expansion associate with response to nivolumab in metastatic melanoma. Oncoimmunology. 2016;5:e1204507.
Kiyotani K, Park JH, Inoue H, et al. Integrated analysis of somatic mutations and immune microenvironment in malignant pleural mesothelioma. Oncoimmunology. 2017;6:e1278330.
Mai T, Takano A, Suzuki H, et al. Quantitative analysis and clonal characterization of T-cell receptor beta repertoires in patients with advanced non-small cell lung cancer treated with cancer vaccine. Oncol Lett. 2017;14:283–92.
Park J-H, Jang M, Tarhan YE, et al. Clonal expansion of antitumor T cells in breast cancer correlates with response to neoadjuvant chemotherapy. Int J Oncol. 2016;49:471–8.
Saloura V, Fatima A, Zewde M, et al. Characterization of the T-cell receptor repertoire and immune microenvironment in patients with locoregionally advanced squamous cell carcinoma of the head and neck. Clin Cancer Res. 2017;23:4897–907.
Yew PY, Alachkar H, Yamaguchi R, et al. Quantitative characterization of T-cell repertoire in allogeneic hematopoietic stem cell transplant recipients. Bone Marrow Transplant. 2015;50:1227–34.
Kiyotani K, Mai TH, Yamaguchi R, et al. Characterization of the B-cell receptor repertoires in peanut allergic subjects undergoing oral immunotherapy. J Human Genet. 2018;63:239–48.
Drake CG, Lipson EJ, Brahmer JR. Breathing new life into immunotherapy: review of melanoma, lung and kidney cancer. Nat Rev Clin Oncol. 2014;11:24–37.
Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol. 2006;90:51–81.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64.
Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33:1889–94.
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74.
Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8.
Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13.
Leisegang M, Engels B, Schreiber K, et al. Eradication of large solid tumors by gene therapy with a T-cell receptor targeting a single cancer-specific point mutation. Clin Cancer Res. 2016;22:2734–43.
Maoz A, Rennert G, Gruber SB. T-cell transfer therapy targeting mutant KRAS. New Engl J Med. 2017;376:e11.
Tran E, Turcotte S, Gros A, et al. Cancer immunotherapy based on mutation-specific CD4+T cells in a patient with epithelial cancer. Science. 2014;344:641–5.
Kato T, Matsuda T, Ikeda Y, et al. Effective screening of T cells recognizing neoantigens and construction of T-cell receptor-engineered T cells. Oncotarget. 2018;9:11009–19.
Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–71.
Zhang L, Conejo-Garcia JR, Katsaros D, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–13.
Sato E, Olson SH, Ahn J, et al. Intraepithelial CD8+tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA. 2005;102:18538–43.
Scherpereel A, Grigoriu BD, Noppen M, et al. Defect in recruiting effector memory CD8+T-cells in malignant pleural effusions compared to normal pleural fluid. BMC Cancer. 2013;13:324.
Chang SG, Lee SJ, Huh JS, Lee JH. Changes in mucosal immune cells of bladder tumor patient after BCG intravesical immunotherapy. Oncol Rep. 2001;8:257–61.
Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic value of tumor-infiltrating FoxP3+regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep. 2015;5:15179.
Carreras J, Lopez-Guillermo A, Fox BC, et al. High numbers of tumor-infiltrating FOXP3-positive regulatory T cells are associated with improved overall survival in follicular lymphoma. Blood . 2006;108:2957–64.
Farinha P, Al-Tourah A, Gill K, Klasa R, Connors JM, Gascoyne RD. The architectural pattern of FOXP3-positive T cells in follicular lymphoma is an independent predictor of survival and histologic transformation. Blood . 2010;115:289–95.
Thakur A, Littrup P, Paul EN, Adam B, Heilbrun LK, Lum LG. Induction of specific cellular and humoral responses against renal cell carcinoma after combination therapy with cryoablation and granulocyte-macrophage colony stimulating factor: a pilot study. J Immunother. 2011;34:457–67.
Si T, Guo Z, Hao X. Immunologic response to primary cryoablation of high-risk prostate cancer. Cryobiology. 2008;57:66–71.
Gazzaniga S, Bravo A, Goldszmid SR, et al. Inflammatory changes after cryosurgery-induced necrosis in human melanoma xenografted in nude mice. J Invest Dermatol. 2001;116:664–71.
Zha B, Huang X, Lin J, Liu J, Hou Y, Wu G. Distribution of lymphocyte subpopulations in thyroid glands of human autoimmune thyroid disease. J Clin Lab Anal. 2014;28:249–54.
Winchester R, Wiesendanger M, Zhang HZ, et al. Immunologic characteristics of intrarenal T cells: trafficking of expanded CD8+T cell beta-chain clonotypes in progressive lupus nephritis. Arthritis Rheum. 2012;64:1589–600.
Murata H, Matsumura R, Koyama A, et al. T cell receptor repertoire of T cells in the kidneys of patients with lupus nephritis. Arthritis Rheum. 2002;46:2141–7.
Alunno A, Bistoni O, Bartoloni E, et al. IL-17-producing CD4-CD8- T cells are expanded in the peripheral blood, infiltrate salivary glands and are resistant to corticosteroids in patients with primary Sjogren’s syndrome. Ann Rheum Dis. 2013;72:286–92.
Maecker HT, Lindstrom TM, Robinson WH, et al. New tools for classification and monitoring of autoimmune diseases. Nat Rev Rheumatol. 2012;8:317–28.
Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet. 2012;380:1590–605.
Chapman CG, Yamaguchi R, Tamura K, et al. Characterization of T-cell receptor repertoire in inflamed tissues of patients with Crohn’s disease through deep sequencing. Inflamm bowel Dis. 2016;22:1275–85.
Lee AJ, Thalayasingam M, Lee BW. Food allergy in Asia: how does it compare? Asia Pac Allergy. 2013;3:3–14.
Boyce JA, Assa’ad A, Burks AW, et al. Guidelines for the diagnosis and management of food allergy in the United States: report of the NIAID-sponsored expert panel. J Allergy Clin Immunol. 2010;126:S1–58.
Ho MH, Lee SL, Wong WH, Ip P, Lau YL. Prevalence of self-reported food allergy in Hong Kong children and teens--a population survey. Asian Pac J Allergy Immunol. 2012;30:275–84.
Lee AJ, Gerez I, Shek LP, Lee BW. Shellfish allergy—an Asia-Pacific perspective. Asian Pac J Allergy Immunol. 2012;30:3–10.
Palomares O. The role of regulatory T cells in IgE-mediated food allergy. J Investig Allergol Clin Immunol. 2013;23:371–82.
Palomares O, Yaman G, Azkur AK, Akkoc T, Akdis M, Akdis CA. Role of Treg in immune regulation of allergic diseases. Eur J Immunol. 2010;40:1232–40.
Akdis M, Burgler S, Crameri R, et al. Interleukins, from 1 to 37, and interferon-gamma: receptors, functions, and roles in diseases. J Allergy Clin Immunol. 2011;127:701–21.
Akdis M, Palomares O, van de Veen W, van Splunter M, Akdis CA. TH17 and TH22 cells: a confusion of antimicrobial response with tissue inflammation versus protection. J Allergy Clin Immunol. 2012;129:1438–49.
Mattsson J, Ringden O, Storb R. Graft failure after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2008;14:165–70.
Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet. 2009;373:1550–61.
Wood KJ, Goto R. Mechanisms of rejection: current perspectives. Transplantation . 2012;93:1–10.
Petersdorf EW, Longton GM, Anasetti C, et al. Association of HLA-C disparity with graft failure after marrow transplantation from unrelated donors. Blood . 1997;89:1818–23.
Fleischhauer K, Kernan NA, O’Reilly RJ, Dupont B, Yang SY. Bone marrow-allograft rejection by T lymphocytes recognizing a single amino acid difference in HLA-B44. N Engl J Med. 1990;323:1818–22.
Anasetti C, Amos D, Beatty PG, et al. Effect of HLA compatibility on engraftment of bone marrow transplants in patients with leukemia or lymphoma. N Engl J Med. 1989;320:197–204.
Shlomchik WD. Graft-versus-host disease. Nat Rev Immunol. 2007;7:340–52.
Flowers ME, Inamoto Y, Carpenter PA, et al. Comparative analysis of risk factors for acute graft-versus-host disease and for chronic graft-versus-host disease according to National Institutes of Health consensus criteria. Blood. 2011;117:3214–9.
Schmaltz C, Alpdogan O, Muriglan SJ, et al. Donor T cell-derived TNF is required for graft-versus-host disease and graft-versus-tumor activity after bone marrow transplantation. Blood. 2003;101:2440–5.
Kataoka Y, Iwasaki T, Kuroiwa T, et al. The role of donor T cells for target organ injuries in acute and chronic graft-versus-host disease. Immunology. 2001;103:310–8.
Hau P, Jachimczak P, Schlingensiepen R, et al. Inhibition of TGF-beta2 with AP 12009 in recurrent malignant gliomas: from preclinical to phase I/II studies. Oligonucleotides. 2007;17:201–12.
Dong S, Maiella S, Xhaard A, et al. Multiparameter single-cell profiling of human CD4+FOXP3+regulatory T-cell populations in homeostatic conditions and during graft-versus-host disease. Blood. 2013;122:1802–12.
Olugbile S, Kiyotani K, Park JH, et al. PS01.05: early and persistent oligoclonal T cell expansion correlates with durable response to anti-PD1 therapy in NSCLC: topic: medical oncology. J Thorac Oncol. 2016;11:S272.
Alamyar E, Duroux P, Lefranc MP, Giudicelli V. IMGT((R)) tools for the nucleotide analysis of immunoglobulin (IG) and T cell receptor (TR) V-(D)-J repertoires, polymorphisms, and IG mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol Biol. 2012;882:569–604.
Bolotin DA, Poslavsky S, Mitrophanov I, et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods. 2015;12:380–1.
Thomas N, Heather J, Ndifon W, Shawe-Taylor J, Chain B. Decombinator: a tool for fast, efficient gene assignment in T-cell receptor sequences using a finite state machine. Bioinformatics. 2013;29:542–50.
Ye J, Ma N, Madden TL, Ostell JM. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 2013;41:W34–40.
Fang H, Yamaguchi R, Liu X, et al. Quantitative T cell repertoire analysis by deep cDNA sequencing of T cell receptor alpha and beta chains using next-generation sequencing (NGS). Oncoimmunology. 2014;3:e968467.
Davis MM, Bjorkman PJ. T-cell antigen receptor genes and T-cell recognition. Nature. 1988;334:395–402.
Dash P, McClaren JL, Oguin TH 3rd, et al. Paired analysis of TCR alpha and TCRbeta chains at the single-cell level in mice. J Clin Investig. 2011;121:288–95.
Kim SM, Bhonsle L, Besgen P, et al. Analysis of the paired TCR alpha- and beta-chains of single human T cells. PLoS ONE. 2012;7:e37338.
Seitz S, Schneider CK, Malotka J, et al. Reconstitution of paired T cell receptor alpha- and beta-chains from microdissected single cells of human inflammatory tissues. Proc Natl Acad Sci USA. 2006;103:12057–62.
DeKosky BJ, Ippolito GC, Deschner RP, et al. High-throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire. Nat Biotechnol. 2013;31:166–9.
Lee ES, Thomas PG, Mold JE, Yates AJ. Identifying T cell receptors from high-throughput sequencing: dealing with promiscuity in TCR alpha and TCRbeta pairing. PLoS Comput Biol. 2017;13:e1005313.
Howie B, Sherwood AM, Berkebile AD, et al. High-throughput pairing of T cell receptor alpha and beta sequences. Sci Transl Med. 2015;7:301ra131.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Yusuke Nakamura is a stockholder and an adviser of OncoTherapy Science Inc and an adviser of Cancer Precision Medicine Inc. Jae-Hyun Park, Poh Yin Yew, and Makiko Harada are employees of Cancer Precision Medicine Inc.
Rights and permissions
About this article

Cite this article
Zewde, M., Kiyotani, K., Park, JH. et al. The era of immunogenomics/immunopharmacogenomics. J Hum Genet 63, 865–875 (2018). https://doi.org/10.1038/s10038-018-0468-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-018-0468-1
This article is cited by
-
Immunogenomics in personalized cancer treatments
Journal of Human Genetics (2021)
-
Quantitative characterization of T-cell repertoire alteration in Chinese patients with B-cell acute lymphocyte leukemia after CAR-T therapy
Bone Marrow Transplantation (2019)
-
Back to the Colorectal Cancer Consensus Molecular Subtype Future
Current Gastroenterology Reports (2019)
