
Abstract
Reactive oxygen species (ROS) have been implicated in the pathogenesis of pulmonary hypertension. ROS might mediate vascular responses, at least in part, by stimulating prostanoid production. Our goals were to determine whether the effect of ROS on vascular tone is altered in resistance pulmonary arteries (PRAs) of newborn piglets with chronic hypoxia-induced pulmonary hypertension and the role, if any, of prostanoids in ROS-mediated responses. In cannulated, pressurized PRA, ROS generated by xanthine (X) plus xanthine oxidase (XO) had minimal effect on vascular tone in control piglets but caused significant vasoconstriction in hypoxic piglets. Both cyclooxygenase inhibition with indomethacin and thromboxane synthase inhibition with dazoxiben significantly blunted constriction to X+XO in hypoxic PRA. X+XO increased prostacyclin production (70 ± 8%) by a greater degree than thromboxane production (50 ± 6%) in control PRA; this was not the case in hypoxic PRA where the increases in prostacyclin and thromboxane production were not statistically different (78 ± 13% versus 216 ± 93%, respectively). Thromboxane synthase expression was increased in PRA from hypoxic piglets, whereas prostacyclin synthase expression was similar in PRA from hypoxic and control piglets. Under conditions of chronic hypoxia, altered vascular responses to ROS may contribute to pulmonary hypertension by a mechanism that involves the prostanoid vasoconstrictor, thromboxane.
Similar content being viewed by others


Main
Reactive oxygen species (ROS) have been implicated in the pathogenesis of a number of vascular diseases (1–6). We and others have provided evidence that ROS are involved in the development of pulmonary hypertension in both neonates and adults (7–14). However, whether ROS mediate different responses in the hypertensive versus normotensive pulmonary circulation is unknown. Aberrant responses to ROS could underlie abnormal pulmonary vascular tone and reactivity and thereby contribute to the progressive development of chronic pulmonary hypertension. Furthermore, the mechanisms by which ROS alter pulmonary vascular tone are not yet clear. This knowledge could have important clinical implications. Sudden elevations in ROS occur with sepsis (15,16) and abrupt increases in oxygen administration (17) in critically ill patients.
The purpose of this study was to evaluate whether responses to ROS were altered in resistance level pulmonary arteries (PRAs) of piglets with chronic hypoxia-induced pulmonary hypertension. ROS can stimulate release of arachidonic acid (18,19), the substrate for production of the prostanoid metabolites, including the potent dilator, prostacyclin (PGI2), and the potent constrictor, thromboxane (TXA2). Hence, an additional goal was to determine whether prostanoids are involved in the responses of normoxic, normotensive and hypoxic, hypertensive PRA to ROS.
METHODS
Animals: Chronic hypoxia model of pulmonary hypertension.
Newborn pigs (2 d old) were placed in a hypoxic normobaric environment for 10 d. Oxygen content was regulated at 10–12% O2 (inspired PO2 64–78 Torr). CO2 was absorbed with soda lime and inspired PCO2 was maintained at 3–6 Torr. Healthy, normoxic piglets were used as controls on the day of arrival from the vendor, at postnatal age of 12 d, i.e. comparable postnatal age to the hypoxic piglets on the day of study. We have previously found no differences in vascular responses between piglets raised in a room-air environment and piglets raised by the vendor (20,21). At the time of study, all piglets were preanesthetized with ketamine (30 mg/kg i.m.) and acepromazine (2 mg/kg i.m.) and then anesthetized with pentobarbital (10 mg/kg i.v.). All animals were given heparin (1000 IU/kg i.v.) and then exsanguinated. The thorax was opened, and the lungs were removed and placed in cold (4°C) Krebs until use. All experimental protocols were performed in adherence to the National Institutes of Health guidelines for the use of experimental animals and approved by the Animal Care and Use Committee of Vanderbilt University Medical Center. This animal resource facility is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Use.
Cannulated artery preparation.
Using our previously published methods (9,22), piglet resistance level PRAs (80–200 μm diameter) were isolated, cannulated, and pressurized for continuous measurement of diameter.
Cannulated artery protocols.
Each artery was equilibrated for 30 min to establish basal tone. Control and hypoxic arteries were equilibrated at transmural pressures that represent in vivo pressures (20,21): the control arteries at a transmural pressure of 15 cm H2O and the hypoxic arteries at a transmural pressure of 25 cm H2O. We have previously shown no effect between these transmural pressures on pulmonary arterial responses to acetylcholine (ACh) (23). After establishment of basal tone, all arteries were tested for viability by contraction to the thromboxane mimetic, U46619 (10−8 M). To check for a functional endothelium in control arteries, responses to ACh (10−6 M) were evaluated. Responses to A23187 were used to check for a functional endothelium in hypoxic arteries as we previously found that hypoxic arteries constricted to ACh but dilated to the calcium ionophore, A23187 (23).
To evaluate responses to ROS, xanthine (X, 10−4 M) was added to the reservoir, after which changes in vessel diameter to cumulative doses of xanthine oxidase (XO, 0.0001–0.005 IU/mL) were measured. Changes in vessel diameter to cumulative doses of H2O2 (10−6–10−3 M) were measured in other vessels. To evaluate the influence of prostanoid production on ROS responses, the cyclooxygenases inhibitor, indomethacin (10−5 M), was added to the reservoir 20 min before measuring changes in vessel diameter to H2O2 or X+XO. To evaluate the influence of thromboxane production, similar studies were performed after adding the thromboxane synthase inhibitor, dazoxiben (10−5 M).
For all of the above studies, vessel responses to the vehicle used for solubilization of the ROS, indomethacin, or dazoxiben were evaluated.
RIA of the stable metabolite of TXA2, TXB2, and enzyme immunoassay of the stable metabolite of PGI2, 6-keto-PGF1α.
These studies were performed to assess whether ROS stimulated production of either PGI2 and/or TXA2 differs between PRAs of hypoxic piglets and PRAs of comparable age control piglets. Dissected PRAs (≤300 μm diameter) were first incubated for 15 min at 37°C in HEPES buffer. The supernatant was discarded and replaced with new HEPES buffer containing vehicle, H2O2 (10−3 M), or xanthine (10−4 M) plus xanthine oxidase (0.005 IU/mL). The vessels were incubated for a second 15-min time period at 37°C. After the second incubation period, the supernatant was collected and stored at −20°C until the time of assay. The vessels were dried for at least 72 h. TXB2 synthesis was measured by the method of Campbell and Ojeda (24). The antibody for TXB2 was from Dr. Pfister's laboratory. Sensitivity of the assay is 1 pg/0.3 mL for TXB2. 6-keto-PGF1α synthesis was measured by enzyme immunoassay using kits (Cayman Chemical Co, Ann Arbor, MI). Sensitivity of the assay is 11 pg/mL. RIA determinations of TXB2 and enzyme immunoassay determinations of 6-keto-PGF1α were normalized to vessel dry weight.
Immunoblot analysis of PGI2 synthase (PGIS) and TXA2 synthase (TXAS).
These studies were performed to determine whether differences in PGI2 and TXA2 production involve altered expression of their synthases, PGIS and TXAS, respectively. Pulmonary arteries (≤300 μm diameter) were dissected from lungs of control and hypoxic piglets, frozen in liquid nitrogen and stored at −80°C until use for immunoblot analysis.
We performed preliminary studies to determine the dynamic range of the immunoblot analysis. Accordingly, 10 μg protein for PGIS and 30 μg protein for TXAS were used to compare protein abundances between pulmonary artery homogenates from control and hypoxic piglets.
Frozen samples of small pulmonary arteries from both groups of piglets were used for immunoblot analysis of PGIS (antibody from Caymen Chemical Co.) and TXAS (antibody from Dr. Pfister's laboratory) using our previously published methods (9,22). The membranes were developed using enhanced chemiluminescence reagents and captured on X-Ray film. Similar procedures were followed to reprobe the membranes for β-actin (Sigma Chemical Co.-Aldrich, St. Louis, MO). The bands for each protein were quantified using densitometry.
Drugs.
H2O2, xanthine, xanthine oxidase, and indomethacin were from Sigma Chemical Co.-Aldrich. Dazoxiben was a generous gift from Pfizer (Groton, CT). Concentrations for each drug listed in cannulated artery protocols and vessel incubations for TXB2 and 6-keto-PGF1α determinations are final concentrations in the vessel bath or incubation solutions.
Statistics.
Data are presented as mean ± SEM. For cannulated artery studies, the % change from baseline diameter in response to each dose of H2O2 or X+XO was compared between control and hypoxic groups or between untreated and treated vessels using linear mixed effects models. Concentration of H2O2 and X+XO were included as unordered categorical variables. We included a random intercept in the regression model to account for taking repeated observations on the same vessel. All models were fit using the R statistics program. For the RIA and ELISA assay, a Wilcoxon signed-rank test was used to compare basal- and ROS-stimulated values for both groups. In addition, the % change between basal- and ROS-stimulated values of prostacyclin and thromboxane were calculated and compared using a Wilcoxon signed-rank test. For Western blot analysis, a Mann-Whitney U test was used to compare densitometry values for PGIS and TXAS between control and hypoxic piglets.
RESULTS
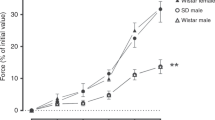
As shown in Figure 1, responses to X+XO differed between PRAs from hypoxic and comparable age normoxic piglets. Arteries from normoxic piglets had minimal responses to X+XO, whereas arteries from hypoxic piglets constricted to X+XO. Both indomethacin treatment (Fig. 2B) and dazoxiben treatment (Fig. 2D) markedly reduced the degree of PRA constriction to X+XO in PRAs from hypoxic piglets. In normoxic piglets, treatment with indomethacin (Fig. 2A) or dazoxiben (Fig. 2C) had minimal to no impact on PRA responses to X+XO.

Responses to X+XO in PRA from piglets raised in normoxia (n = 26 arteries, •) and hypoxia (n = 21 arteries, ▪). All values are mean ± SEM *p < 0.05 by the linear mixed effects model.

X+XO-induced responses in untreated and indomethacin-treated PRA from piglets raised in (A) normoxia (n = 26 untreated arteries, •; n = 10 treated arteries, ▪) and (B) hypoxia (n = 21 untreated arteries, •; n = 8 treated arteries, ▪), and X+XO-induced responses in untreated and dazoxiben-treated PRA from piglets raised in (C) normoxia (n = 26 untreated arteries, •, solid line; n = 5 treated arteries, ▪) and (D) hypoxia (n = 26 untreated arteries, •; n = 6 treated arteries, ▪). All values are mean ± SEM *p < 0.05 by the linear mixed effects model.
X+XO increased both 6-keto-PGF1α and TXB2 in PRA from normoxic and hypoxic piglets (Table 1). In PRA from normoxic piglets stimulated with X+XO, the relative increase in 6-keto-PGF1α was greater than that of TXB2 (Fig. 3A). This was not the case in PRA from hypoxic piglets (Fig. 3B). This differential impact of X+XO on PGI2 and TXA2 production in normoxic and hypoxic PRA could contribute to the altered vascular responses observed (Fig. 1).

% change from basal values of 6-keto-PGF1α and TXB2 with X+XO stimulation in arteries from piglets raised in (A) normoxia (n = 14 arteries) and (B) hypoxia (n = 12 arteries). All values are mean ± SEM *p < 0.05; Wilcoxon signed-rank test.
H2O2 is known to be one of the ROS generated by X+XO (25,26). We therefore evaluated whether responses to H2O2 differed between PRAs from hypoxic and normoxic piglets. In response to all concentrations of H2O2, PRAs from both groups exhibited a brief, initial constriction (data not shown) with return to near baseline within 30 s. After return to near baseline, the higher concentrations of H2O2 elicited a dilator response in PRAs from both groups (Fig. 4). Dilation to H2O2 was greater in PRAs from normoxic than hypoxic piglets (Fig. 4). Neither indomethacin treatment (Fig. 5A and B) nor dazoxiben treatment (Fig. 5C and D) had a significant effect on responses to H2O2 in PRAs from normoxic or hypoxic piglets.

Responses to H2O2 in PRA from piglets raised in normoxia (n = 31 arteries, •) and hypoxia (n = 21 arteries, ▪). All values are mean ± SEM *p < 0.05 by the linear mixed effects model.

H2O2-induced responses in untreated and indomethacin-treated PRA from piglets raised in (A) normoxia (n = 31 untreated arteries, •; n = 18 treated arteries, ▪) and (B) hypoxia (n = 21 untreated arteries, •; n = 9 treated arteries, ▪) and H2O2-induced responses in untreated and dazoxiben-treated PRA from piglets raised in (C) normoxia (n = 30 untreated arteries, •; n = 6 treated arteries, ▪) and (D) hypoxia (n = 21 untreated arteries, •; n = 5 treated arteries, ▪). All values are mean ± SEM. No statistical differences between groups by the linear mixed effects model.
H2O2 increased 6-keto-PGF1α in PRAs from normoxic piglets but had no significant effect on 6-keto-PGF1α in PRAs from hypoxic piglets (Table 2). In contrast, H2O2 significantly increased TXB2 from PRAs only in the hypoxic group (Table 2). Therefore, the relative magnitude of change in PGI2 was greater than that of TXB2 in normoxic PRA (Fig. 6A) but not in hypoxic PRA (Fig. 6B). The absence of an H2O2 effect on PGI2 in hypoxic piglets (Table 2, Fig. 6B) might unmask a greater functional effect from the increase in TXA2 (Fig. 6B) and thereby contribute to the difference in responses to H2O2 noted between normoxic and hypoxic piglets (Fig. 4).

% change from basal values of 6-keto-PGF1α and TXB2 with H2O2 stimulation in arteries from piglets raised in (A) normoxia (n = 14 arteries) and (B) hypoxia (n = 14 arteries). All values are mean ± SEM *p < 0.05; Wilcoxon signed-rank test.
As shown in Figure 7, immunoblot analyses demonstrate that TXAS expression was increased (Fig. 7B) while PGIS expression was unchanged (Fig. 7A) in PRA homogenates from piglets exposed to 10 d of hypoxia.

Immunoblot results and corresponding densitometry for (A) PGIS and (B) TXAS in PRA homogenates from piglets raised in normoxia (control, n = 5) and hypoxia (n = 5). All values are means ± SEM *p < 0.05, Mann-Whitney U test.
DISCUSSION
One of the major findings in this study is that responses to ROS differ between PRAs from normoxic, normotensive piglets and piglets with chronic hypoxia-induced pulmonary hypertension. We also provide evidence that the prostanoid constrictor, thromboxane, is involved in the disparate functional responses to ROS found between normoxic and hypoxic vessels. These data provide an important, new mechanistic link between ROS and the prostanoid pathway in mediating aberrant pulmonary vascular responses in piglets with chronic hypoxia-induced pulmonary hypertension.
The ability of ROS to modify vascular tone has been a topic of great interest for the past several decades (10,27–39). Yet, the effect of ROS on vascular tone from animals with either systemic (40–44) or pulmonary hypertension has not been well studied (10). Others have found that responses to ROS differ between normotensive and hypertensive beds of the systemic circulation (40–45). Ours is the first report of a differential impact of ROS on pulmonary vascular responses in normoxic, normotensive animals and those with chronic hypoxia-induced pulmonary hypertension.
Like us, other investigators have been interested in evaluating a role for prostanoids in mediating vascular responses to ROS (30–32,37,39–50). Supportive of this possibility, prostanoid inhibitors have been shown to alter responses to ROS in both the systemic (31,32,39–49) and pulmonary circulations (30,37,50). Our findings with the cyclooxygenases inhibitor, indomethacin, clearly implicate a role for prostanoids in mediating PRA responses to X+XO in the neonate, particularly in the hypertensive pulmonary circulation (Fig. 2). Moreover, findings with the thromboxane synthase inhibitor, dazoxiben, point to a contribution from thromboxane in mediating the constrictor response to X+XO in hypoxic arteries (Fig. 2). However, the failure of indomethacin and dazoxiben treatments to completely abolish constriction to X+XO in hypoxic vessels implicates additional constrictor(s). Furthermore, our data suggest that pulmonary arterial responses to H2O2 largely depend on other vasoactive metabolites as neither indomethacin nor dazoxiben significantly altered vascular responses. Other investigators have also provided evidence that nonprostanoid metabolites are involved with H2O2 responses in the pulmonary circulation (28,51). These vasoactive metabolites could include other arachidonic acid metabolites, including those of nonenzymatic, i.e. isoprostanes (52), and enzymatic, i.e. leukotriene (28), pathways.
There are limited reports of ROS-stimulated prostanoid production in the pulmonary (37,48,53) and systemic circulations (42,43,45,46,49). It has been reported that H2O2-stimulated TXB2 production is increased in mesenteric arteries from rats with systemic hypertension (42,45). All other reports to date have been limited to measurements of ROS-stimulated prostanoid production in the circulatory bed from either normotensive (37,46,48,49,53) or hypertensive (43) animals but not from both. To our knowledge, we are the first to show that the effect of ROS on prostanoid production differs between the normotensive and hypertensive pulmonary vascular bed.
Our finding that hypoxia increases TXAS expression provides a mechanism for the differential effects of ROS on prostanoid production in pulmonary vessels from piglets exposed to normoxia versus hypoxia for 10 d. ROS stimulate the release of arachidonic acid that is metabolized to PGH2 by cyclooxygenases. PGH2 is subsequently metabolized to a number of prostanoids, including PGI2 and TXA2, in accordance with the presence and abundance of their synthases. In the case of 10 d exposure to hypoxia, the increase in TXAS expression would lead to preferential metabolism of PGH2 to TXA2. Thus, the net effect of hypoxia on expression of PGIS and TXAS would result in a change in ROS-stimulated production of these prostanoids favoring the constrictor, TXA2.
Other mechanisms by which ROS mediate vascular tone could also contribute to the different functional effects of ROS on hypoxic and normoxic vessels. For example, it has been shown that the same concentration of H2O2 can act as a relaxing factor or as a vasoconstrictor depending on whether hyperpolarization is compromised or not (34,35). We previously found that PRA smooth muscle cells from hypoxic piglets are depolarized (54). Therefore, hypoxia-related differences in the resting membrane potential of smooth muscle cells could contribute to the differential responses to ROS observed in this study. H2O2 has also been found to directly relax smooth muscle by hyperpolarization through KCa channel activation (49). Hence, differences between KCa channel expression between normoxic and hypoxic PRAs could also contribute to differences in responses to H2O2. This possibility remains to be explored.
There are limitations of our study. One is that our findings were generated with exogenous ROS. Precise levels of endogenous ROS in normoxic and hypoxic vessels are not known. Indeed, there continues to be great controversy as to whether and which ROS levels are altered by hypoxia (34,55). In addition, we are not certain which of the ROS generated by X+XO, which include superoxide, H2O2, and hydroxyl radicals, underlie the different responses to X+XO in PRAs from normoxic and hypoxic piglets. Nonetheless, we have previously provided evidence suggesting that endogenous ROS mediate vasodilation in the PRAs from normoxic piglets, whereas endogenous ROS seem to mediate vasoconstriction in PRAs from hypoxic piglets (8,9). Findings in this study add to these previous findings and provide further support to the notion that ROS elicit aberrant responses in hypoxic vessels.
In conclusion, our study demonstrates differential functional effects of ROS on the normoxic and hypoxic pulmonary circulation. Although the mechanisms mediating ROS responses differ with the specific ROS studied, we provide evidence of a role for thromboxane in the altered vascular responses to ROS generated by X+XO in PRA from hypoxic piglets. These findings may have potential therapeutic implications in the NICU. Although precise ROS levels are not known, local concentrations of ROS are certain to be increased in the presence of activated inflammatory cells, such as neutrophils, reaching millimolar concentrations (4,15,56,57). Thus, our findings may be particularly applicable to situations of increased inflammatory stress where the aberrant pulmonary vascular response to ROS which are mediated, at least in part, by prostanoid production, could help explain the poor ability of infants with chronic pulmonary hypertension to tolerate acute episodes of infection. Taken together, our findings continue to support the pursuit of both prostanoid and ROS signaling as important therapeutic targets for the treatment of chronic hypoxia-induced pulmonary hypertension in newborns.
Abbreviations
- ACh:
-
acetylcholine
- PGI2:
-
prostacyclin
- PGIS:
-
prostacyclin synthase
- PRAs:
-
resistance pulmonary arteries
- ROS:
-
reactive oxygen species
- TXA2:
-
thromboxane
- TXB2:
-
stable metabolite of thromboxane
- TXAS:
-
thromboxane synthase
- X+XO:
-
xanthine plus xanthine oxidase
References
Buetler TM, Krauskopf A, Ruegg UT 2004 Role of superoxide as a signaling molecule. News Physiol Sci 19: 120–123
Lambeth JD 2007 Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic Biol Med 43: 332–347
Lyle AN, Griendling KK 2006 Modulation of vascular smooth muscle signaling by reactive oxygen species. Physiology (Bethesda) 21: 269–280
Rhee SG 2006 H2O2, a necessary evil for cell signalling. Science 312: 1882–1883
Thannickal VJ, Fanburg BL 2000 Reactive oxygen species in cell signalling. Am J Physiol Lung Cell Mol Physiol 279: L1005–L1028
Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J 2007 Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39: 44–84
Brennan LA, Steinhorn RH, Wedgwood S, Mata-Greenwood E, Roark EA, Russell JA, Black SM 2003 Increased superoxide generation is associated with pulmonary hypertension in fetal lambs: a role for NADPH oxidase. Circ Res 92: 683–691
Dennis KE, Aschner JL, Milatovic D, Schmidt JW, Aschner M, Kaplowitz MR, Zhang Y, Fike CD 2009 NADPH oxidases and reactive oxygen species at different stages of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Lung Cell Mol Physiol 297: L596–L607
Fike CD, Slaughter JC, Kaplowitz MR, Zhang Y, Aschner JL 2008 Reactive oxygen species from NADPH oxidase contribute to altered pulmonary vascular responses in piglets with chronic hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 295: L881–L888
Fresquet F, Pourageaud F, Leblais V, Brandes RP, Savineau J-P, Marthan R, Muller B 2006 Role of reactive oxygen species and gp91 phox in endothelial dysfunction of pulmonary arteries induced by chronic hypoxia. Br J Pharmacol 148: 714–723
Jankov RP, Kantores C, Pan J, Belik J 2008 Contribution of xanthine oxidase-derived superoxide to chronic hypoxic pulmonary hypertension in neonatal rats. Am J Physiol Lung Cell Mol Physiol 294: L233–L245
Liu JQ, Zelko IN, Erbynn EM, Sham JS, Folz RJ 2006 Hypoxic pulmonary hypertension: role of superoxide and NADPH oxidase (gp91 phox). Am J Physiol Lung Cell Mol Physiol 290: L2–L10
Mittal M, Roth M, Konig P, Hofmann S, Dony E, Goyal P, Selbitz A-C, Schermuly RT, Ghofrani HA, Kwapiszewska G, Kummer W, Klepetko W, Hoda MA, Fink L, Hanze J, Seeger W, Grimminger F, Schmidt HH, Weissmann N 2007 Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ Res 101: 258–267
Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, Karwande SV, Stringham JC, Bull DA, Gleich M, Kennedy TP, Hoidal JR 2006 Transforming growth factor β-1 induces Nox4 NADPH oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 290: L661–L673
El-Benna J, Dang PM, Gougerot-Pocidalo M 2008 Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin Immunopathol 30: 279–289
Suzuki YJ, Forman HJ, Sevanian A 1997 Oxidants as stimulators of signal transduction. Free Radic Biol Med 22: 269–285
Lakshminrusimha S, Russell JA, Wedgwood S, Gugino SF, Kazzaz JA, Davis JM, Steinhorn RH 2006 Superoxide dismutase improves oxygenation and reduces oxidation in neonatal pulmonary hypertension. Am J Respir Crit Care Med 174: 1370–1377
Boyer CS, Bannenberg GL, Neve EP, Ryrfeldt A, Moldeus P 1995 Evidence for the activation of the signal-responsive phospholipase A2 by exogenous hydrogen peroxide. Biochem Pharmacol 50: 753–761
Rao GN, Runge MS, Alexander RW 1995 Hydrogen peroxide activation of cytosolic phospholipase A2 in vascular smooth muscle cells. Biochim Biophys Acta 1265: 67–72
Fike CD, Kaplowitz MR 1994 Effect of chronic hypoxia on pulmonary vascular pressures in isolated lungs of newborn pigs. J Appl Physiol 77: 2853–2862
Fike CD, Kaplowitz MR 1996 Chronic hypoxia alters nitric oxide-dependent pulmonary vascular responses in lungs of newborn pigs. J Appl Physiol 81: 2078–2087
Fike CD, Kaplowitz MR, Zhang Y, Pfister SL 2005 Cyclooxygenase-2 and an early stage of chronic hypoxia-induced pulmonary hypertension in newborn pigs. J Appl Physiol 98: 1111–1118
Fike CD, Pfister SL, Kaplowitz MR, Madden JA 2002 Cyclooxygenase contracting factors and altered pulmonary vascular responses in chronically hypoxic newborn pigs. J Appl Physiol 92: 67–74
Campbell WB, Ojeda SR 1987 Measurement of prostaglandin by radioimmunoassay. Methods Enzymol 141: 323–341
Britigan BE, Pou S, Rosen GM, Lilleg DM, Buettner GR 1990 Hydroxyl radical is not a product of the reaction of xanthine oxidase and xanthine. J Biol Chem 265: 17533–17538
McCord JM, Fridovich I 1968 The reduction of cytochrome c by milk xanthine oxidase. J Biol Chem 243: 5753–5760
Ardanaz N, Pagano PJ 2006 Hydrogen peroxide as a paracrine vascular mediator: regulation and signaling leading to dysfunction. Exp Biol Med (Maywood) 231: 237–251
Burghuber OC, Strife R, Zirolli J, Mathias MM, Murphy RC, Reeves JT, Voelkel NF 1986 Hydrogen peroxide induced pulmonary vasoconstriction in isolated rat lungs is attenuated by U60,257, a leucotriene synthesis blocker. Wien Klin Wochenschr 98: 117–119
Burke TM, Wolin MS 1987 Hydrogen peroxide elicits pulmonary artery relaxation and guanylate cyclase activation. Am J Physiol 252: H721–H732
Burke-Wolin T, Abate CJ, Wolin MS, Gurtner GH 1991 Hydrogen peroxide-induced pulmonary vasodilation: role of guanosine 3′,5′-cyclic monophosphate. Am J Physiol 261: L393–L398
Fujimoto S, Asano T, Sakai M, Sakurai K, Takagi D, Yoshimoto N, Itoh T 2001 Mechanisms of hydrogen peroxide-induced relaxation in rabbit mesenteric small artery. Eur J Pharmacol 412: 291–300
Gao YJ, Lee RM 2005 Hydrogen peroxide is an endothelium-dependent contracting factor in rat renal artery. Br J Pharmacol 146: 1061–1068
Jin N, Rhoades RA 1997 Activation of tyrosine kinases in H2O2-induced contraction in pulmonary artery. Am J Physiol 272: H2686–H2692
Jones RD, Morice AH 2000 Hydrogen peroxide—an intracellular signal in the pulmonary circulation: involvement in hypoxic pulmonary vasoconstriction. Pharmacol Ther 88: 153–161
Lucchesi PA, Belmadani S, Matrougui K 2005 Hydrogen peroxide acts as both vasodilator and vasoconstrictor in the control of perfused mouse mesenteric resistance arteries. J Hypertens 23: 571–579
Sato A, Sakuma I, Gutterman DD 2003 Mechanism of dilation to reactive oxygen species in human coronary arterioles. Am J Physiol Heart Circ Physiol 285: H2345–H2354
Seeger W, Suttorp N, Schmidt F, Neuhof H 1986 The glutathione redox cycle as a defense system against hydrogen-peroxide-induced prostanoid formation and vasoconstriction in rabbit lungs. Am Rev Respir Dis 133: 1029–1036
Sheehan DW, Giese EC, Gugino SF, Russell JA 1993 Characterization and mechanisms of H2O2-induced contractions of pulmonary arteries. Am J Physiol 264: H1542–H1547
Yang ZW, Zheng T, Zhang A, Altura BT, Altura BM 1998 Mechanisms of hydrogen peroxide-induced contraction of rat aorta. Eur J Pharmacol 344: 169–181
Auch-Schwelk W, Katusic ZS, Vanhoutte PM 1989 Contractions to oxygen-derived free radicals are augmented in aorta of the spontaneously hypertensive rat. Hypertension 13: 859–864
Erdei N, Bagi Z, Edes I, Kaley G, Koller A 2007 H2O2 increases production of constrictor prostaglandins in smooth muscle leading to enhanced arteriolar tone in Type 2 diabetic mice. Am J Physiol Heart Circ Physiol 292: H649–H656
García-Redondo AB, Briones AM, Beltrán AE, Alonso MJ, Simonsen U, Salaices M 2009 Hypertension increases contractile responses to hydrogen peroxide in resistance arteries through increased thromboxane A2, Ca2+, and superoxide anion levels. J Pharmacol Exp Ther 328: 19–27
Hibino M, Okumura K, Iwama Y, Mokuno S, Osanai H, Matsui H, Toki Y, Ito T 1999 Oxygen-derived free radical-induced vasoconstriction by thromboxane A2 in aorta of the spontaneously hypertensive rat. J Cardiovasc Pharmacol 33: 605–610
Rodríquez-Martínez MA, García-Cohen EC, Baena AB, González R, Salaíces M, Marín J 1998 Contractile responses elicited by hydrogen peroxide in aorta from normotensive and hypertensive rats. Endothelial modulation and mechanism involved. Br J Pharmacol 125: 1329–1335
Gao YJ, Lee RM 2001 Hydrogen peroxide induces a greater contraction in mesenteric arteries of spontaneously hypertensive rats through thromboxane A2 production. Br J Pharmacol 134: 1639–1646
Leffler CW, Busija DW, Armstead WM, Mirro R 1990 H2O2 effects on cerebral prostanoids and pial arteriolar diameter in piglets. Am J Physiol 258: H1382–H1387
Sanderud J, Norstein J, Saugstad OD 1991 Reactive oxygen metabolites produce pulmonary vasoconstriction in young pigs. Pediatr Res 29: 543–547
Tate RM, Morris HG, Schroeder WR, Repine JE 1984 Oxygen metabolites stimulate thromboxane production and vasoconstriction in isolated saline-perfused rabbit lungs. J Clin Invest 74: 608–613
Thengchaisri N, Kuo L 2003 Hydrogen peroxide induces endothelium-dependent and -independent coronary arteriolar dilation: role of cyclooxygenase and potassium channels. Am J Physiol Heart Circ Physiol 285: H2255–H2263
Yamaguchi K, Asano K, Mori M, Takasugi T, Fujita H, Suzuki Y, Kawashiro T 1994 Constriction and dilatation of pulmonary arterial ring by hydrogen peroxide-importance of prostanoids. Adv Exp Med Biol 361: 457–463
Rhoades RA, Packer CS, Roepke DA, Jin N, Meiss RA 1990 Reactive oxygen species alter contractile properties of pulmonary arterial smooth muscle. Can J Physiol Pharmacol 68: 1581–1589
Gong Y, Yi M, Fediuk J, Lizotte PP, Dakshinamurti S 2010 Hypoxic neonatal pulmonary arterial myocytes are sensitized to ROS-generated 8-isoprostane. Free Radic Biol Med 48: 882–894
Sanderud J, Bjoro K, Saugstad OD 1993 Oxygen radicals stimulate thromboxane and prostacyclin synthesis and induce vasoconstriction in pig lungs. Scand J Clin Lab Invest 53: 447–455
Fike CD, Kaplowitz MR, Zhang Y, Madden JA 2006 Voltage-gated K+ channels at an early stage of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am J Physiol Lung Cell Mol Physiol 291: L1169–L1176
Waypa GB, Schumacker PT 2005 Hypoxic pulmonary vasoconstriction: redox events in oxygen sensing. J Appl Physiol 98: 404–414
Forman HJ 2007 Use and abuse of exogenous H2O2 in studies of signal transduction. Free Radic Biol Med 42: 926–932
Reth M 2002 Hydrogen peroxide as second messenger in lymphocyte activation. Nat Immunol 3: 1129–1134
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by RO1 HL68572 [C.D.F.] and by RO1 HL075511 [J.L.A.].
Rights and permissions
About this article
Cite this article
Fike, C., Aschner, J., Slaughter, J. et al. Pulmonary Arterial Responses to Reactive Oxygen Species Are Altered in Newborn Piglets With Chronic Hypoxia-Induced Pulmonary Hypertension. Pediatr Res 70, 136–141 (2011). https://doi.org/10.1203/PDR.0b013e3182207ce7
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3182207ce7
