
Abstract
Although modern multimodal treatment of pediatric cancer has resulted in long-term cure of many patients, clinical success has come with significant acute and chronic morbidity. Targeted therapy using anticancer agents encapsulated in nanoparticles holds considerable promise in further improving efficacy and reducing toxic side effects. This review highlights the current strategies toward developing such therapeutic tools with an emphasis on using liposomes as flexible delivery vehicles. Potential strengths and technical difficulties encountered in advancing this platform are summarized. Critical functional determinants of nanoparticle delivery systems and future strategies to improve efficacy and specificity are described.
Similar content being viewed by others

Main
Pediatric Anticancer Therapies—Success at a Cost
Before 1950, the outcome for children and young adults diagnosed with cancer was dismal with cure rates as low as 5%. Today, the overall survival for children with malignancies approaches 80% and is truly one of the success stories in modern day medicine (1). The progress in the treatment of pediatric malignancy is partly because of the use of combination chemotherapy, multimodality treatments, a better understanding of the molecular pathogenesis of disease, improvements in supportive care, and interventions validated through large cooperative clinical trial groups, such as the Children's Oncology Group (COG). However, success has come with limitations. During the past few decades, the incremental improvement in overall survival has slowed. This is, in part, because of the poor prognosis of refractory, metastatic, and recurrent solid tumors where survival has not improved despite increasingly more aggressive therapies. For those patients who do survive into adulthood, treatment-related late onset toxicities are being recognized with greater frequency. These trends reinforce the need for development of therapeutic strategies that would be both more efficacious and more specific than current treatments.
Our current anticancer armamentarium does not lack for bombs, but the majority of our guidance systems are remarkably primitive. Systemic cytotoxic chemotherapy is administered with the implicit hope that enough of the active agent will percolate into tumor cells to kill them without wreaking irreversible havoc on the rest of the body. Strategies that improve the delivery of a particular anticancer agent to pediatric malignancies will hopefully not only increase the dose effectiveness of chemotherapy but also reduce the systemic toxicity to normal cells.
Nanotechnology provides a viable platform for the development of targeted therapeutic approaches to pediatric malignancies. Nanoparticles can be defined as synthetic structures, organic or inorganic, with loosely defined dimensions ranging from 1 to 1000 nm whose unique properties at least in part depend on size and component chemistry (2). There is now a wide variety of different nanoparticles that have been formulated as potential therapeutic delivery vehicles but none more intensively studied than liposomes. Liposomes are primarily organic nanoparticles composed of combinations of lipid molecules that self-assemble into hollow spherical structures into which a wide range of cargo molecules can be packaged. During the last several decades, the development of supporting chemistries and components has resulted in liposomes evolving into extremely flexible delivery vehicles. As a result, the liposome platform holds tremendous promise in the future of targeted cancer therapeutics to improve drug delivery while minimizing unwanted systemic bystander effects. This review will highlight our current knowledge, applications, challenges, and unanswered questions in the developing field of targeted anticancer liposomal nanoparticles.
Optimizing Liposomal Formulations—To PEGylate or Not to PEGylate
Since the serendipitous discovery of highly purified phospholipid dispersions, now called liposomes, in 1965 by Alec Bangham (3), various liposomal formulations encapsulating cytotoxic chemotherapy drugs have been studied and are becoming widely available to adult cancer patients. Of these liposomal formulations, conventional liposomes (“naked” liposomes) and polyethylene glycol (PEG)ylated liposomes (coated with PEG) have been clinically used most extensively. More recently, there is a growing interest in immunoliposomes and cationic liposomes as improvements on the conventional formulation to improve tumor targeting. Liposomal formulations of the anthracycline cytotoxic drugs, doxorubicin and daunorubicin, have been tested extensively in clinical trials and have shown both safety and efficacy, particularly in metastatic breast cancer (4). Both non-PEGylated and PEGylated versions of liposomal doxorubicin are in use although there are at least theoretically and, perhaps, clinically significant advantages to PEGylation.
The addition of PEG, a large synthetic hydrophilic polymer, to the liposomal surface reduces nonspecific interactions between liposomes and cells. PEGylation also decreases the binding of hydrophobic serum proteins that can act as opsonins, which can potentiate consumption of liposomes by phagocytes of the reticuloendothelial system (RE). In effect, PEGylation creates a “stealth” nanoparticle, which in comparison with its unPEGylated counterpart, has a prolonged circulation time, an increased bioavailability, and a greater potential for tumor targeting (5). For example, the PEGylated liposomal doxorubicin has a half life in children of 36 h (6). This is in contrast to the 2- to 3-h plasma half life of the non-PEGylated liposomal formulation (7). The optimal amount of surface PEG-lipid complex necessary for creating a stealth nanoparticle varies with different liposome formulations. In general, most PEGylated liposomes contain 5 to 10 mol% of PEG molecules with polymer lengths up to a molecular weight of 2000 D (8). Although PEG clearly extends the in vivo circulation time of liposomes, they are eventually removed and metabolized by the RE in the liver and spleen (9). Other synthetic polymers have been used in place of PEG to provide a liposomal cloak, such as poly(acryloyl) morpholine and poly(acrylamide) (10). However, there is such a bulk of safety information and clinical use of PEG in children and adults that this is the liposomal formulation with the greatest clinical applicability in the development of pediatric nanocancer therapeutics.
Passive Tumor Targeting and the EPR Effect—Close Enough for Government Work?
In 1991, Maeda et al. (11) discovered that in animal model systems, high molecular weight drug conjugates preferentially accumulated in tumors to greater levels than free drug did. To describe this effect, these investigators coined the term the “enhanced permeability and retention (EPR)” effect. Although primarily studied in animal model systems, there is preliminary evidence that the EPR might also exist in human solid tumors (12). Although precise mechanisms responsible for the EPR effects are still be elucidated, it is currently thought that inherent differences in tumor vascular organization from normal tissues play a dominant role.
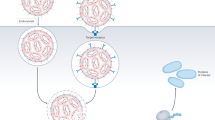
Similar to the tumors that they feed, tumor vasculature is frequently immature, disorganized, and chaotic (Fig. 1). In many solid tumors, the capillary bed is haphazardly constructed with an assortment of malformed branching structures juxtaposing vessels of random caliber and dimension. The vessels themselves are immature, consisting of loosely fitted endothelial cells lacking pericyte support. Tumor vessels have large endothelial fenestrations ranging in size from 100 to 600 nm (13). As a result, there is an increased capillary permeability leading to extravasation of plasma proteins and a proportionate increase in extravascular pressure within many solid tumors. It is still unclear that which of these features is important to establish an EPR effect.

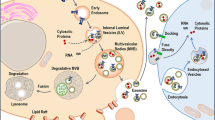
Schematic of targeted liposomal nanoparticle delivery to cancer cells. A, The nanoparticles circulate from the intact circulatory system into the disordered tumor vasculature. Nanoparticles are able to accumulate and extravasate through these large fenestrations, illustrating the EPR effect. B, Nanoparticles enter tumor cells predominantly through receptor-mediated endocytosis. Nanoparticles are internalized through receptor-ligand interactions in a primary endosome, then forming an acidified endosome initiating payload release. Fusion with lysosomes causes further enzymatic digestion of nanoparticles.
Nanoparticles, like other high–molecular-weight bioconjugates, can be designed to exploit the EPR effect and preferentially enter the tumors (14). At least two critical parameters need to be optimized for this to happen: (i) nanoparticles must be able to exist in the circulation for a sufficient period of time; (ii) nanoparticles need to be able to cross the vascular endothelial barrier into the tumor interstitium. From this perspective, nanoparticle size matters. Liposomes that are too large (>250 nm) will not be able to pass through the fenestrations between the tumor endothelial cells. Liposomes that are too small (10 nm) are rapidly filtered out of the circulation by the kidney (15).
Charge is also important in the design of these tumor-targeting liposomes because it can effect the ultimate circulation life of the nanoparticles and the potential for EPR. Although anionic liposomes have the benefit of decreased self-aggregation in suspension, they seem to have increased nonspecific cellular uptake. Cationic liposomes, on the other hand, can be cleared by the kidney having the ability to filter positively charged particles. Furthermore, large amounts of cationic liposomes may cause a tissue inflammatory response (16). Highly charged liposomes, whether positively or negatively charged, can trigger opsonization by fixing complement proteins and, hence, increasing RE clearance (17). Neutrally charged liposomes have the longest circulation times and least amount of RE uptake but greatest aggregation, which may limit tumor penetration. Thus, it seems that the optimal configuration of liposomal nanoparticles is maintaining a size somewhere between 50 and 100 nm with either a neutral or slightly anionic charge (18).
Active Tumor Targeting—Pursuing Anticancer's Holy Grail
When it comes to targeting tumors with therapeutic nanoparticles, the EPR effect is probably necessary but may not be sufficient to accomplish the task. First, there is variability to the EPR. It is anticipated that across a wide range of solid malignancies there will be a differential susceptibility to the EPR effect, and even the permeability of the vasculature may be heterogeneous within the tumor itself (19,20). Certainly, the mere feat of achieving passive targeting of a liposome to a tumor does not necessarily mean that the particle will enter the cancer cell, nor that it will deliver its cytotoxic payload. Simply put, the EPR may get a particular nanoparticle into tumor tissue but not necessarily into tumor cells. To achieve the latter, a strategy of active targeting needs to be used.
The most widely studied strategy of active liposomal targeting has been attaching ligands or specific binding molecules, such as antibodies, to the surface lipids of the nanoparticle. These liposomal-ligand complexes are then internalized within the tumor cell by receptor-mediated endocytosis. Whether these “immunoliposomes” improve tumor localization in vivo is debated, it seems that, at least, the receptor-ligand interactions facilitates internalization and payload delivery into tumor cells (21). A summary of some of the targeting ligands that have been used in liposomal nanoparticles to achieve active targeting in malignancies is listed in Table 1.
Once affixed to the tumor cell surface through multivalent ligand-receptor interactions, liposomes can enter the cell through a variety of internalization pathways, such as clathrin-mediated endocytosis, caveolae-mediated endocytosis, macropinocytosis, and other clathrin- and caveolae-independent pathways. Receptor-dependent, clathrin-mediated endocytosis is a major mechanism for internalization of ligand-receptor complexes (32). Ligand-conjugated liposomes bind to their cognate receptors and endocytosis takes place in clathrin-rich areas of the cell's cytoplasmic membrane forming an endocytic vesicle. Sequential fusion with cytoplasmic vesicles produces a harsher acidic environment that initiates liposomal degradation and payload release. Acidified endosomes can go on to fuse with lysosomes resulting in enzymatic dissolution of the liposomes and potentially destruction of payload molecules as well (33). For this reason, strategies that facilitate payload release before lysosomal attack have been investigated for particular nanoparticles. Clathrin-mediated endocytosis of liposomal nanoparticles can also occur in a receptor-independent manner although in a nonspecific fashion with a much slower internalization rate (33,34).
Although endocytosis through clathrin-coated pits seems to be the most common mechanism of nanoparticle entry into cells, other mechanisms exist (Fig. 1). Caveolae-mediated endocytosis is an alternative pathway that uses regions of the cytoplasmic membrane rich in the calveolin-1 protein. Finally, there are less well-characterized endocytic mechanisms that are independent of either clathrin or caveolin. The potential benefit of nonclathrin endocytic pathways is that, under certain circumstances, they can bypass harsh lysosomal degradation. This may be a preferable route for liposomal delivery of pH sensitive cytotoxic agents, nucleic acids, and peptides.
Although basic pathways by which nanoparticles bind and enter cells are well described, the critical determinants dictating the effectiveness of a particular liposomal formulation are less well understood. For example, the characteristics of specific cell-surface targets may play a role in how efficiently a liposome enters a cell. Doxorubicin-loaded anti-CD19 immunoliposomes proved more effective in antagonizing the growth of a mouse lymphoma model than similarly loaded immunoliposomes directed against the noninternalizing CD20 epitope (35). On this basis, actively targeting liposomes using ligands or receptors that get internalized as part of normal physiologic processes [e.g. folate, transferrin (TF), or receptor tyrosine kinases] have been favored by many. Finally, devising hard and fast rules that dictate how a nanoparticle is processed once it enters a cell have proved elusive. It seems that particles with a size of <200 nm are endocytosed primarily through clathrin-mediated processes, whereas larger particles are increasingly endocytosed through caveolae vesicles (36). However, size is likely to be only one of many factors influencing liposomal fate in cells.
Penetrating the Blood-Brain Barrier—Breaking on Through to the Other Side
As a group, few tumors conjure more frustration, even despair, among pediatric oncologists than brain tumors. The dramatic responses to chemotherapy, which can be seen with many pediatric cancers, rarely occur with the majority of primary CNS tumors. In part, this may be due to the relative resistance of brain tumors to current cytotoxic agents. However, a major problem is surmounting the blood-brain barrier (BBB), a physiologic system that normally protects the brain from potentially noxious exposures. The inherent problem of transducing cytotoxic chemotherapeutic agents across the BBB and maintenance of therapeutic drug concentrations in the CNS has been a major concern in pediatric primary CNS malignancies. This has required the use of direct intrathecal administration of chemotherapy in some cases and suboptimal CNS concentrations of chemotherapeutic agents in others. A non-PEGylated liposomal formulation of the chemotherapeutic agent, cytarabine, has been available and tested in children and adults. This agent does not effectively cross the BBB but, when given intrathecally, results in sustained drug levels in the cerebrospinal fluid for ∼8 d (37,38). However, traversing the BBB is another issue.
There are several ways through which molecules can cross the BBB: carrier-mediated transport, active efflux transport (from brain to blood), and receptor-mediated transport (39). Liposomes, whether PEGylated or not, generally have a limited ability to cross the BBB in most situations. However, by targeting the large number of TF receptors on the endothelial capillary surfaces of the BBB with a number of TF-receptor ligands has resulted in efficient transport of liposomes across this barrier into the CNS (40,41). Other strategies using immunoliposomes to cross the BBB have used coating the liposome surface with ligand to the insulin receptor and E-selectin also highly expressed on the endothelial surface participating in receptor-mediated transport (42,43). Targeted immunoliposomes with TF ligands or other ligands to BBB endothelial receptors is a developing field and promising strategy for delivering traditionally impenetrable molecules, such as nucleic acids, peptides, proteins, and small and large molecule inhibitors, into the pediatric CNS.
Liposomal Therapeutic Payloads—Getting the Most Bang for the Buck
Liposomes have a vast potential for delivering therapeutic payloads to malignancies. Much of the early development of liposomal formulations to cancers have centered on the delivery of well-known and widely applicable cytotoxic agents, such as doxorubicin, daunorubicin, vinorelbine, paclitaxel, cytarabine, and vincristine. Some of the available liposomal chemotherapeutic agents approved by the Food and Drug Administration in adult cancers, currently in pediatric clinical trials, and others that have received orphan drug designation are listed in Table 2.
Liposomes can be loaded with drug either passively or actively. In passive loading or entrapment, the liposomal formulation is prepared with the chemotherapeutic agent of choice in the incubation mixture. Drug is then encapsulated into the liposome as the nanoparticle is formed. The free, nonentrapped drug is then washed away by gel-filtration or other dialysis method. Lipophilic and ampiphilic cytotoxic drugs, such as paclitaxel (highly lipophilic), and doxorubicin (ampiphilic), are loaded somewhat more efficiently because they partition stably in both the lipid membrane and internal compartment of the liposomes. However, drug loading through passive entrapment is less efficient for water soluble drugs, such as methotrexate and cytarabine, in which the concentration in the liposome is directly proportional to the external concentration of the solution (44,45). As expected, chemotherapies with limited biphasic solubility (e.g. 6-Mercaptopurine) are poorly incorporated into the liposome aqueous and lipid compartments (45).
Active entrapment, also termed “remote loading,” is a more efficient and preferable strategy for liposomal drug loading. In this method, a pH or ion gradient is created, which efficiently drives a molecule of choice across the lipid membrane leading to up to 100% loading efficiency of chemotherapeutic agents, such as doxorubicin and vincristine, with stable retention (46,47). In the end, efficiency and ability to actively load a liposomal nanoparticle depends on the individual chemotherapeutic drug characteristics and reaction conditions.
The discovery of RNAi a decade ago by Fire et al. (48) opened up an exciting field of cancer therapeutics with vast clinical application theoretically being able to silence any cancer-related gene pathway. In pediatric bone and soft tissue sarcomas associated with distinct fusion genes, such as Ewing sarcoma, rhabdomyosarcoma, and synovial sarcoma, the potential therapeutic use of siRNA holds promise for tumor-specific therapy. However, the safe and effective delivery of siRNA to malignancies has been the major hurdle in the translation of this technology to clinical application. “Naked” siRNAs are subjected to immune system activation and recognition, endogenous enzymatic degradation, and generally are too negatively charged to cross the cell membrane (49).
Liposomes are attractive delivery vehicles for siRNA in their inherent ability to protect the nucleic acid payload, camouflage the siRNA from the RE system, and inhibit potentially harmful nonspecific delivery to normal tissues. There is substantial literature and experience during the past several decades using liposomes as nanocarriers for nucleic acids both in vitro and in vivo (50). In models of adult-type malignancies, melanoma, lung cancer, breast cancer, and ovarian cancer, targeted liposomes have been regularly used to deliver siRNA constructs (51–53). Currently, exciting applications to pediatric cancer therapeutics using liposomes as nanocarriers for siRNA involve neuroblastoma, chronic myeloid leukemia, and hepatoblastoma. In neuroblastoma, an aggressive small round blue cell cancer of childhood, antisense oligonucleotides to c-myb have been encapsulated in anti-GD2 coated liposomes. These liposomes target neuroblastoma cells expressing the disialoganglioside GD2 and result in inhibition of growth and increased apoptosis (54). With in vitro chronic myeloid leukemia cell lines, TF receptor-targeted liposomes containing anti-BCR-ABL siRNA have been used to efficiently knockdown BCR-ABL mRNA resulting in increased cytotoxicity. However, there were reported off target effects related to nonspecific gene silencing (55). In an in vitro model of hepatoblastoma, a liver cancer diagnosed almost exclusively in young children and liposomal transfer of anti-BCL2 siRNA resulted in improved sensitivity of hepatoblastoma to cisplatin (56).
Concluding Remarks—“Candygram for Mongo … ”
In Mel Brooks' ground breaking film “Blazing Saddles,” the indestructible villain is brought to heel not by brute force but by using an irresistible package to penetrate his defenses. Targeted anticancer therapy is aiming for the same outcome. In constructing targeted nanoparticles, it is hoped that anticancer agents can be delivered directly to tumor cells with greater efficiency and precision. To achieve this goal, successful nanoparticle-based therapies will rely on optimizing two effects: (i) passive accumulation of nanoparticles in tumor tissues by the EPR effect and (ii) active targeting of nanoparticles to tumor surface markers to promote intracellular delivery of therapeutics. It seems doubtful that a single nanoparticle preparation will be optimal for all tumors. However, with greater understanding, the rules for successfully tuning specific formulations to particular cancers will be forthcoming.
Abbreviations
- EPR:
-
enhanced permeability and retention
- BBB:
-
blood-brain barrier
- PEG:
-
polyethylene glycol
- RE:
-
reticuloendothelial system
- TF:
-
transferrin
References
Linabery AM, Ross JA 2008 Childhood and adolescent cancer survival in the US by race and ethnicity for the diagnostic period 1975–1999. Cancer 113: 2575–2596
Whitesides GM 2003 The ‘right' size in nanobiotechnology. Nat Biotechnol 21: 1161–1165
Bangham AD 1995 Surrogate cells or Trojan horses. The discovery of liposomes. Bioessays 17: 1081–1088
Rivera E 2003 Liposomal anthracyclines in metastatic breast cancer: clinical update. Oncologist 8: 3–9
Hamidi M, Azadi A, Rafiei P 2006 Pharmacokinetic consequences of pegylation. Drug Deliv 13: 399–409
Marina NM, Cochrane D, Harney E, Zomorodi K, Blaney S, Winick N, Bernstein M, Link MP 2002 Dose escalation and pharmacokinetics of pegylated liposomal doxorubicin (Doxil) in children with solid tumors: a pediatric oncology group study. Clin Cancer Res 8: 413–418
Cowens JW, Creaven PJ, Greco WR, Brenner DE, Tung Y, Ostro M, Pilkiewicz F, Ginsberg R, Petrelli N 1993 Initial clinical (Phase I) trial of Tlc D-99 (doxorubicin encapsulated in liposomes). Cancer Res 53: 2796–2802
Lian T, Ho RJ 2001 Trends and developments in liposome drug delivery systems. J Pharm Sci 90: 667–680
Litzinger DC, Buiting AM, van Rooijen N, Huang L 1994 effect of liposome size on the circulation time and intraorgan distribution of amphipathic poly(ethylene glycol)-containing liposomes. Biochim Biophys Acta 1190: 99–107
Torchilin VP, Shtilman MI, Trubetskoy VS, Whiteman K, Milstein AM 1994 Amphiphilic vinyl-polymers effectively prolong liposome circulation time in-vivo. Biochim Biophys Acta 1195: 181–184
Maeda H, Bharate GY, Daruwalla J 2009 Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm 71: 409–419
Harrington KJ, Mohammadtaghi S, Uster PS, Glass D, Peters AM, Vile RG, Stewart JS 2001 Effective targeting of solid tumors in patients with locally advanced cancers by radiolabeled pegylated liposomes. Clin Cancer Res 7: 243–254
Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, Jain RK 1995 Vascular-permeability in a human tumor xenograft—molecular-size dependence and cutoff size. Cancer Res 55: 3752–3756
Maeda H, Wu J, Sawa T, Matsumura Y, Hori K 2000 Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release 65: 271–284
Alexis F, Pridgen E, Molnar LK, Farokhzad OC 2008 Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm 5: 505–515
Scheule RK, StGeorge JA, Bagley RG, Marshall J, Kaplan JM, Akita GY, Wang KX, Lee ER, Harris DJ, Jiang CW, Yew NS, Smith AE, Cheng SH 1997 Basis of pulmonary toxicity associated with cationic lipid-mediated gene transfer to the mammalian lung. Hum Gene Ther 8: 689–707
Chonn A, Cullis PR, Devine DV 1991 The role of surface charge in the activation of the classical and alternative pathways of complement by liposomes. J Immunol 146: 4234–4241
Gullotti E, Yeo Y 2009 Extracellularly activated nanocarriers: a new paradigm of tumor targeted drug delivery. Mol Pharm 6: 1041–1051
Jain RK 1994 Barriers to drug-delivery in solid tumors. Sci Am 271: 58–65
Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R 2007 Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol 2: 751–760
Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong KL, Nielsen UB, Marks JD, Benz CC, Park JW 2006 Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res 66: 6732–6740
Gabizon A, Horowitz AT, Goren D, Tzemach D, Shmeeda H, Zalipsky S 2003 In vivo fate of folate-targeted polyethylene-glycol liposomes in tumor-bearing mice. Clin Cancer Res 9: 6551–6559
Chen H, Ahn R, Van den Bossche J, Thompson DH, O'Halloran TV 2009 Folate-mediated intracellular drug delivery increases the anticancer efficacy of nanoparticulate formulation of arsenic trioxide. Mol Cancer Ther 8: 1955–1963
Shmeeda H, Tzernach D, Mak L, Gabizon A 2009 Her2-targeted pegylated liposomal doxorubicin: Retention of target-specific binding and cytotoxicity after in vivo passage. J Control Release 136: 155–160
Goren D, Horowitz AT, Zalipsky S, Woodle MC, Yarden Y, Gabizon A 1996 Targeting of stealth liposomes to erbB-2 (Her/2) receptor: in vitro and in vivo studies. Br J Cancer 74: 1749–1756
Chang DK, Lin CT, Wu CH, Wu HC 2009 A novel peptide enhances therapeutic efficacy of liposomal anti-cancer drugs in mice models of human lung cancer. PLoS One 4: e4171
Eavarone DA, Yu X, Bellamkonda RV 2000 Targeted drug delivery to C6 glioma by transferrin-coupled liposomes. J Biomed Mater Res 51: 10–14
Uyama I, Kumai K, Yasuda T, Tagawa T, Ishibiki K, Kitajima M, Tadakuma T 1994 Improvement of therapeutic effect by using Fab' fragment in the treatment of carcinoembryonic antigen-positive human solid tumors with adriamycin-entrapped immunoliposomes. Jpn J Cancer Res 85: 434–440
Mamot C, Drummond DC, Greiser U, Hong K, Kirpotin DB, Marks JD, Park JW 2003 Epidermal growth factor receptor (EGFR)-targeted immunoliposomes mediate specific and efficient drug delivery to EGFR- and EGFRvIII-overexpressing tumor cells. Cancer Res 63: 3154–3161
Xiong XB, Huang Y, Lu WL, Zhang X, Zhang H, Nagai T, Zhang Q 2005 Intracellular delivery of doxorubicin with RGD-modified sterically stabilized liposomes for an improved antitumor efficacy: in vitro and in vivo. J Pharm Sci 94: 1782–1793
Garg A, Tisdale AW, Haidari E, Kokkoli E 2009 Targeting colon cancer cells using PEGylated liposomes modified with a fibronectin-mimetic peptide. Int J Pharm 366: 201–210
Bareford LM, Swaan PW 2007 Endocytic mechanisms for targeted drug delivery. Adv Drug Deliv Rev 59: 748–758
Hillaireau H, Couvreur P 2009 Nanocarriers' entry into the cell: relevance to drug delivery. Cell Mol Life Sci 66: 2873–2896
Stromhaug PE, Berg TO, Gjoen T, Seglen PO 1997 Differences between fluid-phase endocytosis (pinocytosis) and receptor-mediated endocytosis in isolated rat hepatocytes. Eur J Cell Biol 73: 28–39
Sapra P, Allen TM 2002 Internalizing antibodies are necessary for improved therapeutic efficacy of antibody-targeted liposomal drugs. Cancer Res 62: 7190–7194
Rejman J, Oberle V, Zuhorn IS, Hoekstra D 2004 Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J 377: 159–169
Bomgaars L, Geyer JR, Franklin J, Dahl G, Park J, Winick NJ, Klenke R, Berg SL, Blaney SM 2004 Phase I trial of intrathecal liposomal cytarabine in children with neoplastic meningitis. J Clin Oncol 22: 3916–3921
Lassaletta A, Lopez-Ibor B, Mateos E, Gonzalez-Vicent M, Perez-Martinez A, Sevilla J, Diaz MA, Madero L 2009 Intrathecal liposomal cytarabine in children under 4 years with malignant brain tumors. J Neurooncol 95: 65–69
Pardridge WM 2003 Blood-brain barrier drug targeting: the future of brain drug development. Mol Interv 3: 90–105, 151
Soni V, Kohli DV, Jain SK 2005 Transferrin coupled liposomes as drug delivery carriers for brain targeting of 5-florouracil. J Drug Target 13: 245–250
Visser CC, Stevanovic S, Voorwinden LH, van Bloois L, Gaillard PJ, Danhof M, Crommelin DJ, de Boer AG 2005 Targeting liposomes with protein drugs to the blood-brain barrier in vitro. Eur J Pharm Sci 25: 299–305
Zhang Y, Jeong Lee H, Boado RJ, Pardridge WM 2002 Receptor-mediated delivery of an antisense gene to human brain cancer cells. J Gene Med 4: 183–194
Tan PH, Manunta M, Ardjomand N, Xue SA, Larkin DF, Haskard DO, Taylor KM, George AJ 2003 Antibody targeted gene transfer to endothelium. J Gene Med 5: 311–323
Gulati M, Grover M, Singh S, Singh M 1998 Lipophilic drug derivatives in liposomes. Int J Pharm 165: 129–168
Schnyder A, Huwyler J 2005 Drug transport to brain with targeted liposomes. NeuroRx 2: 99–107
Mayer LD, Tai LC, Bally MB, Mitilenes GN, Ginsberg RS, Cullis PR 1990 Characterization of liposomal systems containing doxorubicin entrapped in response to pH gradients. Biochim Biophys Acta 1025: 143–151
Mayer LD, Bally MB, Loughrey H, Masin D, Cullis PR 1990 Liposomal vincristine preparations which exhibit decreased drug toxicity and increased activity against murine L1210 and P388 tumors. Cancer Res 50: 575–579
Fire A, Xu SQ, Montgomery MK, Kostas SA, Driver SE, Mello CC 1998 Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391: 806–811
Whitehead KA, Langer R, Anderson DG 2009 Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov 8: 129–138; erratum in: Nat Rev Drug Discov 8:516
Juliano R, Alam MR, Dixit V, Kang H 2008 Mechanisms and strategies for effective delivery of antisense and siRNA oligonucleotides. Nucleic Acids Res 36: 4158–4171
Mangala LS, Han HD, Lopez-Berestein G, Sood AK 2009 Liposomal siRNA for ovarian cancer. Methods Mol Biol 555: 29–42
Tran MA, Watts RJ, Robertson GP 2009 Use of liposomes as drug delivery vehicles for treatment of melanoma. Pigment Cell Melanoma Res 22: 388–399
Li SD, Chono S, Huang L 2008 Efficient oncogene silencing and metastasis inhibition via systemic delivery of siRNA. Mol Ther 16: 942–946
Pagnan G, Stuart DD, Pastorino F, Raffaghello L, Montaldo PG, Allen TM, Calabretta B, Ponzoni M 2000 Delivery of c-myb antisense oligodeoxynucleotides to human neuroblastoma cells via disialoganglioside GD(2)-targeted immunoliposomes: Antitumor effects. J Natl Cancer Inst 92: 253–261
Mendonca LS, Firmino F, Moreira JN, Pedroso de Lima MC, Simoes S 2010 Transferrin receptor-targeted liposomes encapsulating anti-BCR-ABL siRNA or asODN for chronic myeloid leukemia treatment. Bioconjug Chem 21: 157–168
Warmann SW, Fuchs J, Bitzer M, Lauer UM 2009 Emerging gene-directed anti-tumor strategies against human hepatoblastoma. Expert Opin Biol Ther 9: 1155–1161
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by the St. Baldrick's Foundation Career Development Award and the UCLA's Today's and Tomorrow's Childrens Fund [to N.F.].
Rights and permissions
About this article
Cite this article
Federman, N., Denny, C. Targeting Liposomes Toward Novel Pediatric Anticancer Therapeutics. Pediatr Res 67, 514–519 (2010). https://doi.org/10.1203/PDR.0b013e3181d601c5
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3181d601c5
This article is cited by
-
Recent advances in liposome formulations for breast cancer therapeutics
Cellular and Molecular Life Sciences (2021)
-
Liposomal borrelidin for treatment of metastatic breast cancer
Drug Delivery and Translational Research (2018)
-
A delivery system targeting bone formation surfaces to facilitate RNAi-based anabolic therapy
Nature Medicine (2012)
