
Abstract
Duchenne muscular dystrophy (DMD) is a progressive, lethal, muscle wasting disease that affects 1 of 3500 boys born worldwide. The disease results from mutation of the dystrophin gene that encodes a cytoskeletal protein associated with the muscle cell membrane. Although gene therapy will likely provide the cure for DMD, it remains on the distant horizon, emphasizing the need for more rapid development of palliative treatments that build on improved understanding of the complex pathology of dystrophin deficiency. In this review, we have focused on therapeutic strategies that target downstream events in the pathologic progression of DMD. Much of this work has been developed initially using the dystrophin-deficient mdx mouse to explore basic features of the pathophysiology of dystrophin deficiency and to test potential therapeutic interventions to slow, reverse, or compensate for functional losses that occur in muscular dystrophy. In some cases, the initial findings in the mdx model have led to clinical treatments for DMD boys that have produced improvements in muscle function and quality of life. Many of these investigations have concerned interventions that can affect protein balance in muscle, by inhibiting specific proteases implicated in the DMD pathology, or by providing anabolic factors or depleting catabolic factors that can contribute to muscle wasting. Other investigations have exploited the use of anti-inflammatory agents that can reduce the contribution of leukocytes to promoting secondary damage to dystrophic muscle. A third general strategy is designed to increase the regenerative capacity of dystrophic muscle and thereby help retain functional muscle mass. Each of these general approaches to slowing the pathology of dystrophin deficiency has yielded encouragement and suggests that targeting downstream events in dystrophinopathy can yield worthwhile, functional improvements in DMD.
Similar content being viewed by others


Main
Duchenne muscular dystrophy (DMD) is the most common, lethal, inherited disease of childhood, affecting 1 of 3500 boys born worldwide. This too-common disease results from mutations in the dystrophin gene that lead to either a complete loss or a loss of critical functional domains of the membrane-associated protein, dystrophin (1). Loss of dystrophin from the cell membrane results in a mechanically weaker membrane that is more easily damaged during muscle contraction. Muscle inflammation, necrosis, and fibrosis occur as direct or indirect consequences of dystrophin deficiency so that DMD patients experience severe and progressive loss of muscle mass and function. Typically, DMD patients are limited to wheelchair mobility by 9–12 y of age, and die by the late teens or 20s, often because of complications that are secondary to respiratory muscle wasting (2).
Although the ultimate cure for DMD will lie in the stable and systemic introduction of a functional dystrophin gene into the muscles of DMD boys, it is unpredictable when therapeutic strategies based on gene therapy or on the transplantation of stem cells or muscle precursor cells will be clinically available. In the interim, improved therapeutics to protect muscle mass and function could improve the quality and length of life for DMD boys, by reducing secondary features of the pathologic progression of dystrophin deficiency. Several observations emphasize that secondary features of dystrophin deficiency may be of great importance in determining the severity of the disease. For example, null mutation of dystrophin produces an early-onset, progressive disease in humans and dogs that greatly reduces life expectancy (3,4) but causes a late-onset progressive pathology in mice, so that dystrophin-deficient mice live a nearly normal lifespan (4). Remarkably, dystrophin deficiency produces muscle hypertrophy rather than atrophy in cats (5). Severity of pathology can also differ between dystrophin-deficient muscles in the same individual, with extraocular muscles spared pathology and limb muscles severely affected in humans (6), and diaphragm muscles in mice experiencing an early-onset, progressive pathology that differs from the late-onset of progressive pathology seen in limb muscles (7) (Fig. 1). Together, these muscle-type and species-specific differences in the pathology of dystrophinopathy show that the loss of dystrophin per se is not sufficient to explain the course, severity, or lethality of muscular dystrophy, which emphasizes the potential importance of epigenetic factors. Thus, control of secondary pathologic features in DMD has the potential to reduce the severity of muscular dystrophy, and possibly add years of mobility and longevity.

Sections of skeletal muscle from healthy, control mouse (A) and dystrophin-deficient, mdx mouse (B). The control mouse muscle shows close-packing of muscle fibers that are nearly uniform in diameter. The dystrophic mouse muscle shows that the muscle fibers have been replaced by connective tissue. Dystrophic fibers are scattered, highly variable in size, and show disrupted cytoplasmic structure, which are signs of previous and ongoing pathology. Bar = 60 μm.
PROTECTING MUSCLE MASS IN DYSTROPHINOPATHIES
Increasing Protein Synthesis
Anabolic steroids.
Loss of functional muscle mass is a primary defect that reduces quality and length of life in DMD. Defects in gait and posture that are attributable to muscle weakness appear between 3 and 6 y of age, after which muscle strength and functional muscle mass decline progressively and continuously until about 11 y of age (8,9). This loss of muscle is accompanied first by a loss of the ability to climb stairs, then lost ability to arise from supine to standing, and then inability to walk even short distances. Conserving muscle mass in DMD patients would slow this functional decline, and would also preserve potential for future gene therapies or stem cell therapies, which require availability of target muscle cells.
Treatment of DMD with anabolic steroids has provided an obvious, and perennially explored approach for protecting muscle mass. Synthetic steroids have been tested in DMD boys, in hopes of identifying agents that can provide desirable anabolic effects with minimal androgenic side-effects. However, the early attempts to identify an anabolic steroid with therapeutic promise for DMD were discouraging. Norethandrolone (10) and methandrostenolone (11) were found to cause initial, modest improvements in muscle strength, but this was accompanied by androgenic side effects, but even worse, cessation of treatment led to a rapid and severe deterioration in muscle mass and function. Subsequent examination of 1-methyl-δ-androstenolone treatments of DMD patients (12) showed that daily administration of 0.25 mg/kg for 1 y yielded no apparent improvement in function; three of five DMD boys who were ambulatory at the beginning of the treatment period lost their ability to walk within the first 2 mo of the study, and the other two patients lost ambulation by mo 7. Although no serious toxic effects were observed, androgenic effects such as priapism, acne, and growth of pubic hair resulted from the treatment.
More recent studies have shown greater promise for the anabolic steroid oxandrolone for use in the treatment of DMD. Oxandrolone may help protect muscle mass through two interacting pathways. Not only can it bind to androgen receptors, to activate transcription of genes involved in anabolic pathways, but it also antagonizes cortisol binding to glucocorticoid receptors to decrease catabolic pathways (13). Oxandrolone has been used with success in increasing muscle mass and strength in HIV patients (14) and burn victims (15), and it produces relatively minor androgenic side-effects in children (16). Importantly, the increase in lean body mass that is achieved by burn victims receiving oxandrolone is retained for at least 6 mo after cessation of treatment (15), which suggests that DMD patients may not experience the rapid loss of muscle mass that occurs at the end of treatment with other anabolic steroids.
Oxandrolone can produce significant improvements in muscle function in DMD patients. Quantitative muscle testing of elbow flexion and knee extension showed that DMD patients receiving daily oxandrolone over a 6-mo trial were stronger than control subjects who received placebo (17). However, function testing (e.g. time to climb stairs) showed no differences between the treated and control groups. No significant, adverse effects were reported, and the height and weight of the treated boys increased linearly during the study, as occurred in placebo-control subjects. Although the beneficial effects of oxandrolone treatment were not large, the ability to achieve these benefits without the negative effects on growth provides some advantage over corticosteroid use, which can significantly slow normal growth.
Growth factors.
An alternative approach for shifting dystrophin-deficient muscle toward a positive protein balance has tested whether systemic administration of growth factors can maintain muscle mass and function. Based on early success that showed exogenous GH can cause a positive nitrogen balance in myotonic dystrophy patients (18), the possibility that GH could have similar effects on DMD boys was tested (19). However, 12 d of GH treatment to seven DMD patients unexpectedly produced undesired, catabolic effects. Those findings coupled with a subsequent observation that a DMD patient with GH deficiency had a relatively benign phenotype (20) suggested that inhibiting GH could reduce the pathology of DMD. However, subsequent clinical studies have produced mixed results. Nine DMD patients receiving a GH inhibitor, mazindol, daily for 6 mo showed no improvement in muscle function (21), although another study of two identical twins with DMD showed much higher levels of motor function in the twin receiving mazindol for 12 mo than the sibling receiving placebo (22). Conversely, daily administration of mazindol to five DMD patients for 3 mo yielded no improvement in function but did produce adverse effects such as irritability, sleep disturbances, and elevated heart rate (23). In addition, a subsequent investigation showed that mazindol treatment of 83 boys with DMD for a 12-mo trial did not slow the progression of muscle weakness, but did slow gain in height of the boys (24). More recently, the possibility that exogenous GH could reduce DMD pathology has been revisited, with a particular focus on a potential ameliorative effect on DMD cardiomyopathy (25). However, only slight improvement in cardiac systolic function and no improvement in skeletal muscle function were observed after 3 mo of treatment. Collectively, these mixed findings indicate that perturbation of GH concentration in DMD patients does not yield a predictable or clearly beneficial effect.
Although manipulations of GH levels in DMD patients do not appear to have great promise as a therapeutic treatment for DMD, IGF-1 may have potential for protecting muscle mass and function in DMD. Observations that IGF-1 functions as a potent anabolic agent for skeletal muscle in vivo (26,27) by stimulating protein synthesis (28,29) and by promoting regeneration (30) suggested that IGF-1 could also have beneficial effects on dystrophic muscle. Thus far, experimentation on the mdx mouse has provided encouragement for a potential therapeutic use for IGF-1 in DMD. IGF-1 delivery to mdx muscle through the expression of a muscle-specific transgene (31) or by delivery with a miniosmotic pump (32) significantly increased muscle mass (31,32), increased specific force in diaphragm muscle (32), and induced muscle hyperplasia (31). These findings indicate that increased delivery of IGF-1 to dystrophic muscle can reduce pathology by shifting muscle toward a positive protein balance and by stimulating regeneration. In addition, subcutaneous injections of IGF-1 into mdx mice reduced exercise-induced weakness (33), which suggests that the systemic delivery of IGF-1 could have beneficial effects in the treatment of muscular dystrophy.
Although no detrimental effects of IGF-1 treatments of mdx mice were noted, IGF-1 receptors are expressed on numerous cell types that could potentially experience disruptions of normal homeostasis as a consequence of elevated IGF-1. Because production of collagen by fibroblasts is increased by IGF-1, there was a particular concern that the pathologic fibrosis of dystrophin-deficient muscle would be exacerbated by IGF-1 treatments. On the contrary, overexpression of IGF-1 reduced fibrosis of mdx muscle (31), suggesting this could provide a further, unanticipated benefit of IGF-1 treatments. IGF-1 has already been tested clinically in the treatment of amyotrophic lateral sclerosis, and found to produce no clinically significant, negative side-effects during 7- and 9-mo clinical trials (34,35), which supports the possibility that it could be used safely for treating DMD. However, other experimental and epidemiologic findings emphasize that long-term IGF-1 treatments should be used with caution, especially in children. IGF-1 can increase the proliferation and metastasis of cancer cells, at least in model systems (36), and there may be a significant relationship between levels of circulating IGF-1 and occurrence of cancer in humans (37,38).
Decreasing Proteolysis
Inhibiting calcium-dependent proteases.
Additional strategies for protecting muscle mass in muscular dystrophy have targeted proteolytic systems that are important in dystrophinopathies. In addition to slowing catabolic processes, antiproteolytic strategies may provide unpredicted benefits by reducing the degradation of specific substrates, the loss of which may contribute significantly to defects in muscle function. A potential role for calcium-dependent proteases (calpains) in the progression of dystrophinopathies has been anticipated for years, even before the discovery of dystrophin. Early studies noted the influx of extracellular calcium into dystrophin-deficient muscle (39–41), leading to speculations that the elevated calcium could promote muscle death by activating proteases. Further investigations showed that calpains in necrotic fibers of mdx mice have higher levels of activation (42), but only recently have experimental data addressed whether the activation of calpains in dystrophic muscle were important in muscular dystrophy. Generation of mdx mouse lines in which there was a muscle-specific overexpression of calpastatin, the endogenous inhibitor of calpains, showed that transgene expression reduced necrosis in mdx muscle and reduced the size of muscle lesions (43). In addition, the beneficial effects occurred without reducing the efflux of extracellular marker dyes (and presumably calcium) into the muscle fibers, which indicated that calpain-mediated pathology in mdx muscle is an event that occurs downstream of membrane damage (43).
A recent pilot study has suggested that therapies that may reduce calpain activity in vivo can improve muscle function in DMD patients. Because β2-adrenergic agonists can increase calpastatin expression in muscle, reduce the proteolysis of muscle contractile proteins, and increase muscle mass in mammals (44), Spencer and colleagues (45) tested whether oral administration of β2-adrenergic agonists to DMD boys could improve muscle function. After 12 wk of daily, oral administration of albuterol, patients showed increased muscle strength in knee extension and during manual muscle testing, with no adverse side-effects reported (46). However, no significant difference in muscle function tests (e.g. time to climb stairs or rise to standing from a supine position) was observed. Although this investigation suggests that calpain-targeted therapeutics can yield benefits for DMD patients, it is possible that the albuterol-mediated effects may not occur exclusively through influences on the calpain system. For example, T-lymphocytes and macrophages promote muscle pathology in mdx mice (47,48), and β2-adrenergic agonists can inhibit the activation of T cells and macrophages (49–51).
Inhibiting the ubiquitin/proteasome system.
The ubiquitin/proteasome system is the primary proteolytic system in all eukaryotic cells, which can lead to the breakdown of polypeptides to small peptides and amino acids. A recent report (52) presented evidence that the systemic inhibition of the ubiquitin/proteasome system using the MG-132 protease inhibitor can diminish pathology in mdx mice. Systemic administration of the inhibitor was also reported to increase localization at the muscle cell membrane of the N-terminal portion of dystrophin (52), which is upstream of the mutation that introduces a premature stop codon into exon 23 of the mdx dystrophin gene (53). This suggests that dystrophin has an unidentified domain that associates with the cell membrane because the domain that binds β-dystroglycan at the membrane lies within the cysteine-rich domain encoded by exons 64–67 (54,55). The ability of dystrophin that lacks its C-terminus and its β-dystroglycan binding domain to localize at the cell membrane has been observed previously in DMD patients. For example, one DMD patient was identified in whom a truncated dystrophin that lacked everything downstream of exon 50 was expressed and correctly localized at the cell membrane (56). However, loss of the C-terminus of dystrophin, and especially the cysteine-rich domain that binds β-dystroglycan, is associated with severe pathology in DMD (57,58). These findings in DMD patients indicate that preventing dystrophin proteolysis may not provide rescue from pathology unless critical functional domains of the molecule are spared the effects of the mutation.
DECREASING THE INFLAMMATORY RESPONSE
The aggressive inflammatory response associated with dystrophin deficiency, although secondary to the etiology, is a significant component of the pathophysiology and an attractive target for pharmacologic therapeutics. Although dystrophin-deficient muscular dystrophy is not categorized as an inflammatory myopathy, immune cells that infiltrate dystrophin-deficient skeletal muscle promote the pathology in mdx mice. In vivo, depletions of CD4+ or CD8+ T cells or macrophages significantly reduced the pathology in mdx mice, illustrating the role of those cell types in aggravating the disease (47,48). Furthermore, mdx mice show increased susceptibility to mast cell granule-induced damage suggesting a role for mast cells in the pathology (59). To investigate the feasibility of immunosuppressive/anti-inflammatory therapies in DMD, numerous studies have focused on the role of immune cells and cytokines in dystrophin deficiency as well as directly testing the efficacy of anti-inflammatory and immunosuppressive therapies in patients.
Immunosuppressants
Strong support for the possibility that immune-based interventions can be beneficial in DMD as well as mdx dystrophy come from findings on DMD patients receiving the immunosuppressant glucocorticoid, prednisone. Prednisone-treated DMD patients experience a significant delay in the disease progression that includes prolongation of ambulation, maintenance of strength and function, and a recently documented delay in or prevention of the development of scoliosis (60). Prednisone may also be beneficial in preserving ventilatory function in dystrophin deficiency by attenuating fibrosis of the diaphragm (61). However, daily prednisone treatment is often associated with adverse side-effects including weight gain, suppression of growth, bone demineralization, cushingoid appearance, hirsutism, and behavioral changes that often prompt cessation of treatment. Nondaily dosing can reduce these adverse side-effects while still yielding functional benefits (60,62–65). The benefits of prednisone treatments can also be optimized by initiating treatment before the onset of functional decline in DMD patients. Long-term evaluations of DMD patients who started therapy between 2 and 5 y of age showed prednisone was well tolerated, beneficial in maintaining and prolonging function, and elicited more robust responses than when treatment was initiated in later years (63–65). The superior treatment effects caused by early treatment have been attributed to the relatively greater efficacy of prednisone in maintaining function than recovering function (63–65).
Although prednisone's clinical efficacy in dystrophin deficiency is unequivocal, the degree to which prednisone's immunosuppressive function contributes to its functional benefits in vivo is unknown. Quantitative, histologic examination of dystrophin-deficient tissue shows that prednisone reduces the infiltration of inflammatory cells into muscle (66,67) and recent micro-array data from prednisone-treated mdx muscle demonstrates that prednisone down-regulates the expression of genes involved in the immune response (68). These data suggest that prednisone's immunosuppressive function significantly alters the pathogenic profile of the disease. However, DMD patients treated with the immunosuppressant azathioprine failed to demonstrate clinical improvements similar to prednisone-treated patients (69), although the concentration of leukocytes in muscle biopsies from DMD patients treated with prednisone or azathioprine did not differ significantly (70). These findings led the investigators to conclude that immunosuppression is probably not the primary mechanism of prednisone-induced clinical improvement in DMD. However, azathioprine is not an immunosuppressant per se; it is a purine analog that inhibits DNA synthesis. Thus, azathioprine can reduce leukocyte populations by preventing their proliferation, but it will also inhibit the proliferation of muscle satellite cells and thereby inhibit muscle repair and regeneration. This latter effect may explain the lack of clinical improvement in azathioprine-treated DMD patients. In addition, prednisone functions as an immunosuppressant by reducing the expression of genes involved in leukocyte activation (68), as well as inhibiting their invasion into tissue. Thus, the level of activation of inflammatory cells in muscles of prednisone-treated DMD patients could be lower than in azathioprine-treated patients, which may further account for the functional improvement in prednisone-treated but not azathioprine-treated patients.
In addition to its immunosuppressive functions, prednisone may have direct effects on muscle cells that could contribute to its efficacy in DMD. For example, prednisone can modulate proteolysis and calcium handling, enhance myogenesis, and inhibit apoptosis (71–76). The ability of prednisone to have direct effects on muscle cells is further supported by findings using Caenorhabditis elegans, which lack an immune system. A mutant C. elegans line that experienced muscle degeneration and impaired locomotion was treated with prednisone, which reduced muscle degeneration (77), although improvements in locomotion were not observed. Prednisone may also directly effect muscle by increasing expression of utrophin, a dystrophin homologue, by increasing the activity of a utrophin promoter (78). Up-regulation of utrophin could have tremendous importance in the treatment of DMD because utrophin can compensate for the lack of dystrophin (79). However, a biopsy from a prednisone-treated patient did not show elevated utrophin (64), indicating that, at least in that patient, prednisone did not affect utrophin expression.
Deflazacort, an oxazoline derivative of prednisone, is another glucocorticoid immunosuppressant with significant benefits in DMD patients. Not only is deflazacort effective in maintaining muscle strength and function, but it also improves pulmonary and cardiac function and attenuates the development of scoliosis, which makes it valuable for the treatment of DMD even after ambulation is lost (80–85). Compared with prednisone, deflazacort is equally effective in maintaining strength and function (86) and is generally more tolerable because adverse side-effects, such as weight gain, are less severe (82,84–86). Whether deflazacort induces less bone demineralization than prednisone is not clear. Hand x-rays showed no difference between prednisone- and deflazacort-treated patients (86), although dual photon absorptiometry showed less bone loss in deflazacort patients (87). Nondaily dosing was tested in an attempt to alleviate the adverse side-effects from deflazacort treatment and, although the DMD patients on a lower and intermittent deflazacort-dosing regimen experienced less growth suppression, functional and strength benefits were reduced suggesting that daily deflazacort treatment is more favorable (85).
The beneficial effects of prednisone and deflazacort in the treatment of DMD led to the search for other immunosuppressants that could prove therapeutically useful. The history of success of cyclosporine as an immunosuppressant following tissue and organ transplantation suggested that it could also be valuable in decreasing the immune cell contribution to dystrophinopathy. However, current findings concerning the effect of cyclosporine treatments on dystrophin deficiency are in conflict. An initial, open-label, 2-mo trial with cyclosporine-treated DMD patients was encouraging in demonstrating strength increases in the single muscle tested (88). However, the unblinded design of this study could have been influenced by a placebo effect, though the authors argue against this because strength increases were observed during electrically stimulated as well as voluntary contractions. Subsequent clinical trials with cyclosporine produced variable results. However, cyclosporine treatments in these latter studies were performed in conjunction with myoblast transfer therapy and, in some cases, intermittent prednisone treatment. The compounded variables could obscure any specific cyclosporine treatment effects and limit the extent to which conclusions regarding cyclosporine's efficacy in DMD can be made (89–92). Animal studies testing cyclosporine are equally difficult to interpret. Histologic analyses and strength testing of cyclosporine-treated mdx mice produced no effect (93,94) or a detrimental effect (95), which may reflect variability in dosing regimens. Studies in which no significant effects were observed used only 20–50% of the human dosage, which may not have been sufficient to induce a response (93,94), whereas the study in which detrimental effects were observed (95) used 600% of the human cyclosporine dosage.
The conflicting findings concerning the effect of cyclosporine on dystrophinopathy may result, in part, from the broad, dose-dependent effects of cyclosporine that are independent of its function as an immunosuppressant. Because cyclosporine functions by inhibiting calcineurin, and calcineurin mediates multiple signaling pathways through its phosphatase activity in multiple cell types, systemic delivery of cyclosporine may have multiple unpredictable effects, especially in the context of a progressive, muscle-wasting disease. Contention over whether calcineurin plays a significant role in the regulation of the growth of healthy muscle has centered, in part, on questions concerning the magnitude and frequency of dosing, and the vehicle used for drug delivery (96,97). Because cyclosporine has the potential to inhibit muscle growth and regeneration, under at least some treatment regimens, other immunosuppressants without potential, detrimental effects on muscle could be better choices for use in DMD patients.
Chinese Herbal Medicines
In the search for novel DMD therapeutics, Chinese herbal medicines used to treat nonspecific muscle weakness have been examined. Functional improvements observed in some DMD patients treated with herbal medicines (63,98) were accompanied by signs of hypercorticism (e.g. weight gain, cushingoid appearance, hirsutism, and shortness of stature) (98), which suggested the presence of glucocorticoids that was later confirmed by chemical analysis (99). Although some DMD patients showed promising results with Chinese herbal medicine treatment, there is a lack of quantitative, clinical data to confirm those findings. Additionally, because Chinese herbal medicines are not regulated and their glucocorticoid content is unknown, there is potential for harmful, toxic side-effects due to improper dosing.
Cytokine Modulation
TNF-α.
Tumor necrosis factor-α (TNF-α) is a cytokine that is up-regulated in DMD patients (100) and may function as a mediator of the dystrophic pathology by enhancing the inflammatory response and through direct cytotoxicity. Modulating the bioavailability of TNF-α has proved successful in the treating of autoimmune diseases such as rheumatoid arthritis (101), which has suggested that this strategy may also be useful in controlling inflammatory cell-mediated damage to dystrophic muscle. However, modulating TNF-α levels in mdx mice via genetic removal or by treatment with anti-TNF-α antibodies has produced unpredictable and conflicting results. For example, null mutation of TNF-α in dystrophin-deficient (TNF-/mdx) mice worsened diaphragm pathology at 4 wk of age, although older TNF-/mdx mice showed decreased quadriceps pathology and improved ventilatory function (102,103). Furthermore, mdx mice receiving weekly doses of infliximab, an anti-TNF-α antibody, showed delayed appearance of muscle pathology (104), at the age that TNF-/mdx mice exhibited severe pathology. Infliximab treatment also yielded a cyclic pattern of inflammation and necrosis that may have been an effect of the weekly dosing regimen. Other recent findings show that TNF-α can also play a role in promoting muscle repair. Recovery of muscle strength after injury was impaired in mice that were null mutants for the TNF receptor and in mice treated with neutralizing anti-TNF-α, compared with injured wild-type mice (105). Together, these findings show that TNF-α is likely to serve more than one role in the mdx pathophysiology and suggest that it is not yet possible to predict whether modulating TNF-α levels in DMD patients will have a net beneficial or detrimental effect.
Normalizing nitric oxide production.
The discovery that most nNOS is lost from dystrophin-deficient muscle (106) led to recent studies of whether NOS deficiency contributes significantly to dystrophinopathy and whether NO-based therapeutics could have value in treating DMD. Loss of NOS from dystrophin-deficient muscle has now been shown to contribute significantly to muscle pathology both through loss of NO-mediated vasodilation (107,108), which could contribute to muscle ischemia, and through loss of normal NO regulation of the inflammatory response (48). NO derived from healthy muscle can regulate muscle inflammation by inhibiting inflammatory cell invasion, inhibiting enzymes that generate cytolytic molecules and by scavenging cytotoxic free radicals (109–111). Thus, some of the increased susceptibility of dystrophin-deficient muscle to damage by free radicals (112) may result from loss of normal levels of nNOS. Loss of NO production by dystrophin-deficient muscle may also negatively affect the regenerative capacity of muscle. NO can increase the release of hepatocyte growth factor from muscle, which can then increase activation of muscle satellite cells (113). Thus, NO-based therapeutics have the potential to promote the repair of dystrophin-deficient muscle, as well as decreasing damage.
Several experimental strategies have been examined to test whether increasing NO levels in dystrophin-deficient muscle can reduce pathology. In one approach, skeletal muscle NO production in mdx mice was normalized with the expression of a muscle-specific, nNOS transgene. NOS transgene expression significantly reduced the concentration of macrophages and ameliorated muscle damage, which indicated the possible benefits of an NO-based anti-inflammatory therapy for DMD (48). Another treatment strategy has used systemic administration of l-arginine, the substrate of nNOS, which was reported to reduce damage of mdx muscle by increasing expression of utrophin (114,115). However, this finding differs from another report that expression of a nNOS transgene in mdx muscle reduced utrophin concentration in muscle fibers (116). Systemic administration of NO donors could provide a third, alternative strategy for NO-based treatments, although this approach may present the most risk for adverse side-effects. Exceeding normal NO levels can diminish any beneficial effect (117) and may be cytotoxic to the muscle. Moreover, the possibility of using NO-releasing compounds to treat DMD patients will depend upon the development of a systemic delivery system that does not perturb other NO-dependent physiologic systems.
INCREASING MUSCLE CELL PROLIFERATION AND REGENERATION
Blocking Myostatin Function
A door to new, potential, therapeutic strategies for treating downstream events in DMD was opened in 1997 by the discovery of myostatin, a member of the transforming growth factor-β family of proteins that regulate the proliferation and differentiation of numerous cell types (118). Myostatin, also called growth and differentiation factor 8 (GDF-8), is a potent, negative regulator of muscle mass. Unlike other factors that regulate the proliferation and differentiation of muscle cells, and typically promote one process at the expense of the other, myostatin inhibits both proliferation and differentiation. The net result of its duel inhibitory roles is that satellite cells are maintained in a quiescent state in which they increase neither the number nor size of muscle cells. Myostatin's ability to inhibit the proliferation of satellite cells is the best characterized of its negative regulatory roles. Satellite cells produce myostatin, which can then inhibit their progression from the G0 of G1 phase of the cell cycle to the S phase by increasing the expression of cyclin-dependent kinase inhibitors, leading to reduced expression of cyclin-dependent kinases and withdrawal from the cell cycle (119–122). These in vitro findings are consistent with in vivo observations that show that null mutation of myostatin causes muscle cell hyperplasia (118). Although the withdrawal of satellite cells from the cell cycle typically leads to their differentiation, myostatin can also inhibit differentiation. Application of myostatin to muscle cells in vitro decreases expression of MyoD (123) and myogenin (121), both of which are transcription factors that promote the expression of muscle specific genes necessary for muscle differentiation. This effect on inhibiting differentiation in vitro is consistent with in vivo observations that show loss of myostatin causes muscle hypertrophy (118,124). Thus, myostatin could have the capacity to inhibit muscle regeneration after injury or disease by reducing both the proliferation and differentiation of satellite cells required for muscle repair.
The powerful, negative regulatory effects of myostatin on muscle mass and strength and the apparent specificity of myostatin action on muscle have fueled recent and ongoing studies that test whether preventing the expression or activation of myostatin could have therapeutic value in muscle-wasting diseases, including DMD. Strong support for this hope was provided independently by two investigative teams who showed that mdx mice that were null mutants for myostatin (125) and mdx mice that received intraperitoneal injections of anti-myostatin (126) showed increases in muscle mass and strength, with no reported, adverse side effects. In addition, the recent exciting case report of a 41/2-y-old boy with mutated myostatin gene and no detectable myostatin in his sera (127) suggests that treatments that reduce myostatin may not have important detrimental effects on children. The subject, who showed normal levels of circulating testosterone and IGF-1, had unusually large muscles at birth. No health problems have been observed, and cardiac function tests showed no cardiomyopathy or cardiac conduction defects. Although the child was originally admitted to the neonatal ward several hours after birth because of stimulus-induced myoclonus, the myoclonus subsided after 2 mo of age. Although these findings support the hope that myostatin-targeted therapeutics can ameliorate the pathology of DMD without other significant, negative side-effects, important questions remain to be addressed. For example, although no negative effects were observed in the 41/2-y-old boy who lacked myostatin, unanticipated consequences may yet appear. In addition, even though myostatin-targeted therapeutics can increase muscle mass and force production, the muscles will still be dystrophin deficient. It is feasible that increases in force production by dystrophin-deficient muscle could increase mechanically induced damage. Nevertheless, myostatin presents an important and ripe target for therapeutic interventions for DMD.
Myostatin's appeal as a therapeutic target in DMD is enhanced by the variety of strategies that could be used to regulate its function. Anti-myostatin injections have already been shown to produce desirable effects in mdx muscles (126), and a similar strategy could be attempted using a humanized antibody provided to DMD boys. Alternative approaches could exploit any of several negative regulators of myostatin function. For example, binding of myostatin to its receptor can be inhibited by follistatin binding to the latent, myostatin propeptide dimer (128). Similarly, follistatin-related gene product (129) and growth and differentiation factor-associated serum protein 1 (130) bind the myostatin propeptide dimer and inhibit its activity. Therapies based on increasing follistatin, follistatin-related gene product, growth and differentiation factor-associated serum protein, or the myostatin-binding domains of any of these proteins could have potential benefits for DMD patients.
Delivery of Mitogens to Muscle
Exogenous mitogens may also provide a means to slow the progressive wasting of DMD muscle. Part of the beneficial effects of potential therapeutic agents such as IGF-1 may reflect their mitogenic effects on satellite cells, in addition to increasing muscle protein synthesis. Much of the experimental work examining the utility of mitogens on reducing dystrophinopathy has centered on the effects of LIF on mdx muscle pathology. LIF is a glycosylated cytokine that is a potent mitogen, especially for undifferentiated cells, including embryonic stem cells (131), hematopoietic stem cells (132,133) and primordial germ cells (134). The finding that LIF also stimulated the proliferation of muscle cells introduced the possibility that it could have therapeutic potential for treating muscle-wasting diseases, including DMD.
Initial studies of LIF's potential to promote muscle growth and repair indicated that LIF could have therapeutic potential for the treatment of muscle injury and disease. Delivery of exogenous LIF to acutely injured muscle increased muscle regeneration (135), and mice that were null mutants for LIF showed slower muscle repair following injury than wild-type mice (136). In addition, exogenous LIF prevented muscle atrophy that occurred after muscle denervation (137). Based on these studies, the palliative effect of LIF on mdx muscle pathology was tested and yielded encouraging findings. In an initial investigation (138), LIF was delivered to mdx mouse diaphragms by diffusion from an alginate rod sutured to the diaphragm, which permitted approximately 20 ng of LIF release per day. Three months of treatment yielded diaphragm muscle hyperplasia and hypertrophy, as well as significant reduction of fibrosis. Subsequently, delivery of only 5 ng of LIF per day by the same mechanism was tested for 3 mo, which yielded an increase in myoblast proliferation, but no increase in fiber size was reported (139). Although these findings indicate that LIF has therapeutic potential for the treatment of dystrophinopathies, developing a mechanism for its systemic delivery to muscle without causing undesirable effects on nonmuscle tissues may present a tremendous impediment for its use. For example, systemic administration of LIF by the intravenous delivery of immortalized, murine hematopoietic cells that expressed high levels of LIF was lethal for the recipient mice (140). Mice receiving the cells with the retroviral construct containing LIF cDNA died within 12 to 70 d, showing cachexia, pancreatitis, and calcification in the heart and skeletal muscle. In addition, LIF has been tested clinically in patients with advanced cancer in a phase 1 trial in which some negative, autonomic effects (dizziness, impotence) Table 1 were produced by relatively high dosages of LIF (up to 16 μg/kg/d delivered s.c.) (141). However, no serious negative side-effects were reported for mice receiving LIF at lower doses for longer periods when released from a osmotic pump (136,142) or injected intraperitoneally (143), indicating that it may be possible for LIF to be administered safely, and perhaps with benefit to DMD patients.
CONCLUSIONS
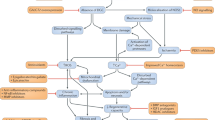
The complex epigenetic factors that contribute to the pathophysiology of muscular dystrophy create both obstacles and opportunities for developing useful palliative treatments. On one hand, the complicated matrix of interacting systems that modulate the severity of the disease, including endocrine, immune, and musculoskeletal systems, makes it difficult to identify single, best strategies for developing treatments (Fig. 2). On the other hand, the existence of multiple factors that contribute to affecting disease severity provides multiple, potential therapeutic targets.

Schematic illustrating some of the possible routes through which downstream events in dystrophinopathies have been targeted experimentally or therapeutically. Red lines indicate processes that increase pathology. Green lines indicate processes that decrease pathology. Lines ending in arrowheads promote the subsequent event. Lines ending in bars inhibit the subsequent event.
Current evidence suggests that several routes for developing useful palliative treatments deserve further exploration. Experimental findings in mdx mice and limited observations in DMD patients indicate that dystrophinopathy can be reduced by increasing the regenerative capacity of dystrophic muscle, slowing the catabolic processes that are important in the dystrophic progression and preventing the exacerbation of dystrophic muscle cell death by the immune system. However, in each case, experimental treatments need further examination to determine efficacy, safety and reliability. Furthermore, there has been little investigation of the benefit of combining multiple treatment strategies in parallel, which may magnify treatment effects, or in series, which may minimize adverse side-effects that may arise from the prolonged use of a single treatment. In the end, genetic interventions will provide cures for muscular dystrophy, but manipulating down-stream events in the dystrophic pathology currently provides the best opportunities for slowing the disease progression and improving quality of life for DMD patients.
Abbreviations
- DMD:
-
Duchenne muscular dystrophy
- LIF:
-
leukemia inhibitory factor
- TNF-α:
-
tumor necrosis factor-α
- nNOS:
-
neuronal nitric oxide synthase
References
Hoffman EP, Brown RH Jr, Kunkel LM 1987 Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51: 919–928
Emery AE 1987 Duchenne Muscular Dystrophy. Oxford University Press New York
Cooper BJ, Winand NJ, Stedman H, Valentine BA, Hoffman EP, Kunkel LM, Scott MO, Fischbeck KH, Kornegay JN, Avery RJ, Williams JR, Schmickel RD, Sylvester JE 1988 The homologue of the Duchenne locus is defective in X-linked muscular dystrophy of dogs. Nature 334: 154–156
Cooper BJ 1989 Animal models of Duchenne and Becker muscular dystrophy. Br Med Bull 45: 703–718
Gaschen FP, Hoffman EP, Gorospe JR, Uhl EW, Senior DF, Cardinet GH III, Pearce LK 1992 Dystrophin deficiency causes lethal muscle hypertrophy in cats. J Neurol Sci 110: 149–159
Khurana TS, Prendergast RA, Alameddine HS, Tome FM, Fardeau M, Arahata K, Sugita H, Kunkel LM 1995 Absence of extraocular muscle pathology in Duchenne's muscular dystrophy: role for calcium homeostasis in extraocular muscle sparing. J Exp Med 182: 467–475
Stedman HH, Sweeney HL, Shrager JB, Maguire HC, Panettieri RA, Petrof B, Narusawa M, Leferovich JM, Sladky JT, Kelly AM 1991 The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 352: 536–539
Allsop KG, Ziter FA 1981 Loss of strength and functional decline in Duchenne's dystrophy. Arch Neurol 38: 406–411
Cohen L, Morgan J, Babbs R, Gilula Z, Karrison T, Meier P 1982 A statistical analysis of the loss of muscle strength in Duchenne's muscular dystrophy. Res Commun Chem Pathol Pharmacol 37: 123–138
Dowben RM, Perlstein MA 1961 Muscular dystrophy treated with norethandrolone. Arch Intern Med 107: 245–251
Gamstorp I 1964 Clinical evaluation of an oral anabolic steroid (methandrostenolone, dianabol CIBA) in children with muscular weakness and wasting. Acta Paediatr 53: 570–577
Charash L 1965 Anabolic steroids in the management of muscular dystrophy. Pediatrics 36: 402–405
Zhao J, Bauman WA, Huang R, Caplan AJ, Cardozo C 2004 Oxandrolone blocks glucocorticoid signaling in an androgen receptor-dependent manner. Steroids 69: 357–366
Mulligan K, Schambelan M 2002 Anabolic treatment with GH, IGF-I, or anabolic steroids in patients with HIV-associated wasting. Int J Cardiol 85: 151–159
Demling RH, DeSanti L 2003 Oxandrolone induced lean mass gain during recovery from severe burns is maintained after discontinuation of the anabolic steroid. Burns 29: 793–797
Church JA 2004 Oxandrolone treatment of childhood hereditary angioedema. Ann Allergy Asthma Immunol 92: 377–378
Fenichel GM, Griggs RC, Kissel J, Kramer TI, Mendell JR, Moxley RT, Pestronk A, Sheng K, Florence J, King WM, Pandya S, Robison VD, Wang H 2001 A randomized efficacy and safety trial of oxandrolone in the treatment of Duchenne dystrophy. Neurology 56: 1075–1079
Rudman D, Chyatte SB, Patterson JH, Gerron GG, O'Beirne I, Barlow J, Ahmann P, Jordan A, Mosteller RC 1971 Observations on the responsiveness of human subjects to human growth hormone. Effects of endogenous growth hormone deficiency and myotinic dystrophy. J Clin Invest 50: 1941–1949
Rudman D, Chyatte SB, Patterson JH, Gerron GG, O'Beirne I, Barlow J, Jordan A, Shavin JS 1972 Metabolic effects of human growth hormone and of estrogens in boys with Duchenne muscular dystrophy. J Clin Invest 51: 1118–1124
Zatz M, Betti RT, Levy JA 1981 Benign Duchenne muscular dystrophy in a patient with growth hormone deficiency. Am J Med Genet 10: 301–304
Collipp PJ, Kelemen J, Chen SY, Castro-Magana M, Angulo M, Derenoncourt A 1984 Growth hormone inhibition causes increased selenium levels in Duchenne muscular dystrophy: a possible new approach to therapy. J Med Genet 21: 254–256
Zatz M, Betti RT, Frota-Pessoa O 1986 Treatment of Duchenne muscular dystrophy with growth hormone inhibitors. Am J Med Genet 24: 549–566
Coakley JH, Moorcraft J, Hipkin LJ, Smith CS, Griffiths RD, Edwards RH 1988 The effect of mazindol on growth hormone secretion in boys with Duchenne muscular dystrophy. J Neurol Neurosurg Psychiatry 51: 1551–1557
Griggs RC, Moxley RT III, Mendell JR, Fenichel GM, Brooke MH, Miller PJ, Mandel S, Florence J, Schierbecker J, Kaiser KK, King W, Pandya S, Robinson J, Signore L 1990 Randomized, double-blind trial of mazindol in Duchenne dystrophy. Muscle Nerve 13: 1169–1173
Cittadini A, Comi L, Longobardi S, Rocco Petretta V, Casaburi C, Passamano L, Merola B, Durante-Mangoni E, Sacca L, Politano L 2003 A preliminary randomized study of growth hormone administration in Becker and Duchenne muscular dystrophies. Eur Heart J 24: 664–672
Bark TH, McNurlan MA, Lang CH, Garlick PJ 1998 Increased protein synthesis after acute IGF-I or insulin infusion is localized to muscle in mice. Am J Physiol 275: E118–E123
Musaro A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, Barton ER, Sweeney HL, Rosenthal N 2001 Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet 27: 195–200
Russell-Jones DL, Umpleby AM, Hennessy TR, Bowes SB, Shojaee-Moradie F, Hopkins KD, Jackson NC, Kelly JM, Jones RH, Sonksen PH 1994 Use of a leucine clamp to demonstrate that IGF-I actively stimulates protein synthesis in normal humans. Am J Physiol 267: E591–E598
Adams GR, McCue SA 1998 Localized infusion of IGF-I results in skeletal muscle hypertrophy in rats. J Appl Physiol 84: 1716–1722
Barton-Davis ER, Shoturma DI, Musaro A, Rosenthal N, Sweeney HL 1998 Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proc Natl Acad Sci U S A 95: 15603–15607
Barton ER, Morris L, Musaro A, Rosenthal N, Sweeney HL 2002 Muscle-specific expression of insulin-like growth factor I counters muscle decline in mdx mice. J Cell Biol 157: 137–148
Gregorevic P, Plant DR, Leeding KS, Bach LA, Lynch GS 2002 Improved contractile function of the mdx dystrophic mouse diaphragm muscle after insulin-like growth factor-I administration. Am J Pathol 161: 2263–2272
De Luca A, Pierno S, Liantonio A, Cetrone M, Camerino C, Fraysse B, Mirabella M, Servidei S, Ruegg UT, Conte Camerino D 2003 Enhanced dystrophic progression in mdx mice by exercise and beneficial effects of taurine and insulin-like growth factor-1. J Pharmacol Exp Ther 304: 453–463
Lai EC, Felice KJ, Festoff BW, Gawel MJ, Gelinas DF, Kratz R, Murphy MF, Natter HM, Norris FH, Rudnicki SA 1997 Effect of recombinant human insulin-like growth factor-I on progression of ALS. A placebo-controlled study. The North America ALS/IGF-I Study Group. Neurology 49: 1621–1630
Borasio GD, Robberecht W, Leigh PN, Emile J, Guiloff RJ, Jerusalem F, Silani V, Vos PE, Wokke JH, Dobbins T 1998 A placebo-controlled trial of insulin-like growth factor-I in amyotrophic lateral sclerosis. European ALS/IGF-I Study Group. Neurology 51: 583–586
Khandwala HM, McCutcheon IE, Flyvbjerg A, Friend KE 2000 The effects of insulin-like growth factors on tumorigenesis and neoplastic growth. Endocr Rev 21: 215–244
Chan JM, Stampfer MJ, Giovannucci E, Gann PH, Ma J, Wilkinson P, Hennekens CH, Pollak M 1998 Plasma insulin-like growth factor-I and prostate cancer risk: a prospective study. Science 279: 563–566
Pollak MN, Schernhammer ES, Hankinson SE 2004 Insulin-like growth factors and neoplasia. Nat Rev Cancer 4: 505–518
Bodensteiner JB, Engel AG 1978 Intracellular calcium accumulation in Duchenne dystrophy and other myopathies: a study of 567,000 muscle fibers in 114 biopsies. Neurology 28: 439–446
Turner PR, Fong PY, Denetclaw WF, Steinhardt RA 1991 Increased calcium influx in dystrophic muscle. J Cell Biol 115: 1701–1712
McArdle A, Edwards RH, Jackson MJ 1992 Accumulation of calcium by normal and dystrophin-deficient mouse muscle during contractile activity in vitro. Clin Sci (Lond) 82: 455–459
Spencer MJ, Croall DE, Tidball JG 1995 Calpains are activated in necrotic fibers from mdx dystrophic mice. J Biol Chem 270: 10909–10914
Spencer MJ, Mellgren RL 2002 Overexpression of a calpastatin transgene in mdx muscle reduces dystrophic pathology. Hum Mol Genet 11: 2645–2655
Bardsley RG, Allcock SM, Dawson JM, Dumelow NW, Higgins JA, Lasslett YV, Lockley AK, Parr T, Buttery PJ 1992 Effect of beta-agonists on expression of calpain and calpastatin activity in skeletal muscle. Biochimie 74: 267–273
Fowler EG, Graves MC, Wetzel GT, Spencer MJ 2004 Pilot trial of albuterol in Duchenne and Becker muscular dystrophy. Neurology 62: 1006–1008
Spencer MJ, Walsh CM, Dorshkind KA, Rodriguez EM, Tidball JG 1997 Myonuclear apoptosis in dystrophic mdx muscle occurs by perforin-mediated cytotoxicity. J Clin Invest 99: 2745–2751
Spencer MJ, Montecino-Rodriguez E, Dorshkind K, Tidball JG 2001 Helper (CD4(+)) and cytotoxic (CD8(+)) T cells promote the pathology of dystrophin-deficient muscle. Clin Immunol 98: 235–243
Wehling M, Spencer MJ, Tidball JG 2001 A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J Cell Biol 155: 123–131
Sekut L, Champion BR, Page K, Menius JA Jr, Connolly KM 1995 Anti-inflammatory activity of salmeterol: down-regulation of cytokine production. Clin Exp Immunol 99: 461–466
Panina-Bordignon P, Mazzeo D, Lucia PD, D'Ambrosio D, Lang R, Fabbri L, Self C, Sinigaglia F 1997 Beta2-agonists prevent Th1 development by selective inhibition of interleukin 12. J Clin Invest 100: 1513–1519
Malfait AM, Malik AS, Marinova-Mutafchieva L, Butler DM, Maini RN, Feldmann M 1999 The beta2-adrenergic agonist salbutamol is a potent suppressor of established collagen-induced arthritis: mechanisms of action. J Immunol 162: 6278–6283
Bonuccelli G, Sotgia F, Schubert W, Park DS, Frank PG, Woodman SE, Insabatio L, Cammer M, Minetti C, Lisanti MP 2003 Proteasome inhibitor (MG-132) treatment of mdx mice rescues the expression and membrane localization of dystrophin and dystrophin-associated proteins. Am J Pathol 163: 1663–1675
Sicinski P, Geneg Y, Ryder-Cook A, Barnard EA, Darlison MG, Barnard PJ 1989 The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science 244: 1578–1580
Suzuki A, Yoshida M, Hayashi K, Mizuno Y, Hagiwara Y, Ozawa E 1994 Molecular organization at the glycoprotein-complex-binding site of dystrophin. Three dystrophin-associated proteins bind directly to the carboxy-terminal portion of dystrophin. Eur J Biochem 220: 283–292
Jung D, Yang B, Meyer J, Chamberlain JS, Campbell KP 1995 Identification and characterization of the dystrophin anchoring site on beta-dystroglycan. J Biol Chem 270: 27305–27310
Helliwell TR, Ellis JM, Mountford RC, Appleton RE, Morris GE 1992 A truncated dystrophin lacking the C-terminal domains is localized at the muscle membrane. Am J Hum Genet 50: 508–514
Hoffman EP, Garcia CA, Chamberlain JS, Angelini C, Lupski JR, Fenwick R 1991 Is the carboxyl-terminus of dystrophin required for membrane association? A novel, severe case of Duchenne muscular dystrophy. Ann Neurol 30: 605–610
Bies RD, Caskey CT, Fenwick R 1992 An intact cysteine-rich domain is required for dystrophin function. J Clin Invest 90: 666–672
Gorospe JR, Tharp M, Demitsu T, Hoffman EP 1994 Dystrophin-deficient myofibers are vulnerable to mast cell granule-induced necrosis. Neuromuscul Disord 4: 325–333
Yilmaz O, Karaduman A, Topaloglu H 2004 Prednisolone therapy in Duchenne muscular dystrophy prolongs ambulation and prevents scoliosis. Euro J Neurol 11: 541–544
Gosselin LE, McCormick KM 2004 Targeting the immune system to improve ventilatory function in muscular dystrophy. Med Sci Sports Exerc 36: 44–51
Connolly AM, Schierbecker J, Renna R, Florence J 2002 High dose weekly oral prednisone improves strength in boys with Duchenne muscular dystrophy. Neuromuscul Disord 12: 917–925
Dubowitz V, Kinali M, Main M, Mercuri E, Muntoni F 2002 Remission of clinical signs in early Duchenne muscular dystrophy on intermittent low-dosage prednisolone therapy. Eur J Paediatr Neurol 6: 153–159
Kinali M, Mercuri E, Main M, Muntoni F, Dubowitz V 2002 An effective, low-dosage, intermittent schedule of prednisolone in the long-term treatment of early cases of Duchenne dystrophy. Neuromuscul Disord 12 Suppl 1: S169–S174
Merlini L, Cicognani A, Malaspina E, Gennari M, Gnudi S, Talim B, Franzoni E 2003 Early prednisone treatment in Duchenne muscular dystrophy. Muscle Nerve 27: 222–227
Kissel JT, Burrow KL, Rammohan KW, Mendell JR, CIDD Study Group 1991 Mononuclear cell analysis of muscle biopsies in prednisone-treated and untreated Duchenne muscular dystrophy. CIDD Study Group. Neurology 41: 667–672
Wehling-Henricks M, Lee JJ, Tidball JG 2004 Prednisolone decreases cellular adhesion molecules required for inflammatory cell infiltration in dystrophin-deficient skeletal muscle. Neuromuscul Disord 14: 483–490
Muntoni F, Fisher I, Morgan JE, Abraham D 2002 Steroids in Duchenne muscular dystrophy: from clinical trials to genomic research. Neuromuscul Disord 12( Suppl 1): S162–S165
Griggs RC, Moxley RT III Mendell JR, Fenichel GM, Brooke MH, Pestronk A, Miller JP, Cwik VA, Pandya S, Robison J, King W, Signore L, Schierbecker J, Florence J, Matheson-Burden N, Wilson B 1993 Duchenne dystrophy: randomized, controlled trial of prednisone (18 months) and azathioprine (12 months). Neurology 43: 520–527
Kissel JT, Lynn DJ, Rammohan KW, Klein JP, Griggs RC, Moxley RT III Cwik VA, Brooke MH, Mendell JR 1993 Mononuclear cell analysis of muscle biopsies in prednisone- and azathioprine-treated Duchenne muscular dystrophy. Neurology 43: 532–536
Kawai H, Adachi K, Nishida Y, Inui T, Kimura C, Saito S 1993 Decrease in urinary excretion of 3-methylhistidine by patients with Duchenne muscular dystrophy during glucocorticoid treatment. J Neurol 240: 181–186
Rafai Z, Welle S, Moxley RT III, Lorenson M, Griggs RC 1995 Effect of prednisone on protein metabolism in Duchenne dystrophy. Am J Physiol 268: E67–E74
Banik NL, Matzelle D, Terry E, Hogan EL 1997 A new mechanism of methylprednisolone and other corticosteroids action demonstrated in vitro: inhibition of a proteinase (calpain) prevents myelin and cytoskeletal protein degradation. Brain Res 748: 205–210
Metzinger L, Passaquin AC, Leijendekker WJ, Poindron P, Ruegg UT 1995 Modulation by prednisolone of calcium handling in skeletal muscle cells. Br J Pharmacol 116: 2811–2816
Passaquin AC, Metzinger L, Leger JJ, Warter JM, Poindron P 1993 Prednisolone enhances myogenesis and dystrophin-related protein in skeletal muscle cell cultures from mdx mouse. J Neurosci Res 35: 363–372
Sklar RM, Brown RH Jr 1991 Methylprednisolone increases dystrophy levels by inhibiting myotube death during myogenesis of normal human muscle in vitro. J Neurol Sci 101: 73–81
Gaud A, Simon JM, Witzel T, Carre-Pierrat M, Wermuth CG, Segalat L 2004 Prednisone reduces muscle degeneration in dystrophin-deficient Caenorhabditis elegans. Neuromuscul Disord 14: 365–370
Courdier-Fruh I, Barman L, Briguet A, Meier T 2002 Glucocorticoid-mediated regulation of utrophin levels in human muscle fibers. Neuromuscul Disord 12( Suppl 1): S95–S104
Tinsley JM, Potter AC, Phelps SR, Fisher R, Trickett JI, Davies KE 1996 Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature 384: 349–353
Mesa LE, Dubrovsky AL, Corderi J, Marco P, Flores D 1991 Steroids in Duchenne muscular dystrophy—deflazacort trial. Neuromuscul Disord 4: 261–266
Angelini C, Pegoraro E, Turella E, Intino MT, Pini A, Costa C 1994 Deflazacort in Duchenne dystrophy: study of long-term effect. Muscle Nerve 17: 386–391
Biggar WD, Gingras M, Fehlings DL, Harris VA, Steele CA 2001 Deflazacort treatment of Duchenne muscular dystrophy. J Pediatr 138: 45–50
Silversides CK, Webb GD, Harris VA, Biggar WD 2003 Effects of deflazacort on left ventricular function in patients with Duchenne muscular dystrophy. Am J Cardiol 91: 769–772
Alman BA, Raza SN, Biggar WD 2004 Steroid treatment and the development of scoliosis in males with Duchenne muscular dystrophy. J Bone Joint Surg Am 86A: 519–524
Biggar WD, Politano L, Harris VA, Passamano L, Vajsar J, Alman B, Palladino A, Comi LI, Nigro G 2004 Deflazacort in Duchenne muscular dystrophy: a comparison of two different protocols. Neuromuscul Disord 14: 476–482
Bonifati MD, Ruzza G, Bonometto P, Berardinelli A, Gorni K, Orcesi S, Lanzi G, Angelini C 2000 A multicenter, double-blind, randomized trial of deflazacort versus prednisone in Duchenne muscular dystrophy. Muscle Nerve 23: 1344–1347
Gennari C, Imbimbo B 1985 Effects of prednisone and deflazacort on vertebral bone mass. Calcif Tissue Int 37: 592–593
Sharma KR, Mynhier MA, Miller RG 1993 Cyclosporine increases muscular force generation in Duchenne muscular dystrophy. Neurology 43: 527–532
Law PK, Goodwin TG, Fang Q, Duggirala V, Larkin C, Florendo JA, Kirby DS, Deering MB, Li HJ, Chen M, Yoo TJ, Cornett J, Li LM, Shirzad A, Quinley T, Holcomb RL 1992 Feasibility, safety, and efficacy of myoblast transfer therapy on Duchenne muscular dystrophy boys. Cell Transplant 1: 235–244
Law PK, Goodwin TG, Fang Q, Deering MB, Duggirala V, Larkin C, Florendo JA, Kirby DS, Li HJ, Chen M, Cornett J, Li LM, Shirzad A, Quinley T, Yoo TJ, Holcomb R 1993 Cell transplantation as an experimental treatment for Duchenne muscular dystrophy. Cell Transplant 2: 485–505
Mendell JR, Kissel JT, Amato AA, King W, Signore L, Prior TW, Sahenk Z, Benson S, McAndrew PE, Rice R, Nagaraja H, Stephens R, Lantry L, Morris GE, Burghes AHM 1995 Myoblast transfer in the treatment of Duchenne's muscular dystrophy. N Engl J Med 333: 832–838
Miller RG, Sharma KR, Pavlath GK, Gussoni E, Mynhier M, Lanctot AM, Greco CM, Steinman L, Blau HM Myoblast implantation in Duchenne muscular dystrophy: the San Francisco study. 220: 469–478 1997
Weller B, Massa R, Karpati G, Carpenter S 1991 Glucocorticoids and immunosuppressants do not change the prevalence of necrosis and regeneration in mdx skeletal muscles. Muscle Nerve 14: 771–774
Granchelli JA, Pollina C, Hudecki MS 2000 Pre-clinical screening of drugs using the mdx mouse. Neuromuscul Disord 10: 235–239
Stupka N, Gregorevic P, Plant DR, Lynch GS 2004 The calcineurin signal transduction pathway is essential for successful muscle regeneration in mdx dystrophic mice. Acta Neuropathol (Berl) 107: 299–310
Dunn SE, Simard AR, Prud'homme RA, Michel RN 2002 Calcineurin and skeletal muscle growth. Nat Cell Biol 4: E46
Yancopoulos GD, Glass DJ 2002 Calcineurin and skeletal muscle growth. Nat Cell Biol 4: E46–E47
Urtizberea JA, Fan QS, Vroom E, Recan D, Kaplan JC 2003 Looking under every rock: Duchenne muscular dystrophy and traditional Chinese medicine. Neuromuscul Disord 13: 705–707
Courdier-Fruh I, Barman L, Wettstein P, Meier T 2003 Detection of glucocorticoid-like activity in traditional Chinese medicine used for the treatment of Duchenne muscular dystrophy. Neuromuscul Disord 13: 699–704
Porreca E, Guglielmi MD, Uncini A, Di Gregorio P, Angelini A, Di Febbo C, Pierdomenico SD, Baccante G, Cuccurullo F 1999 Haemostatic abnormalities, cardiac involvement and serum tumor necrosis factor levels in X-linked dystrophic patients. Thrombosis Haemostasis 81: 543–546
Toussirot E, Wendling D 2004 The use of TNF-alpha blocking agents in rheumatoid arthritis: an overview. Expert Opin Pharmacother 5: 581–594
Spencer MJ, Marino MW, Winkler WM 2000 Altered pathological progression of diaphragm and quadriceps muscle in TNF-deficient, dystrophin-deficient mice. Neuromuscul Disord 10: 612–619
Gosselin LE, Barkley JE, Spencer MJ, McCormick KM, Farkas GA 2003 Ventilatory dysfunction in mdx mice: impact of tumor necrosis factor-alpha deletion. Muscle Nerve 28: 336–343
Grounds MD, Torrisi J 2004 Anti-TNFα (Remicade) therapy protects dystrophic skeletal muscle from necrosis. FASEB J 18: 676–682
Warren GL, Hulderman T, Jensen N, McKinstry M, Mishra M, Luster MI, Simeonova PP 2002 Physiological role of tumor necrosis factor α in traumatic muscle injury. FASEB J 16: 1630–1632
Brenman JE, Chao DS, Xia H, Aldape K, Bredt DS 1995 Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 82: 743–752
Thomas GD, Sander M, Lau KS, Huang PL, Stull JT, Victor RG 1998 Impaired metabolic modulation of alpha-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc Natl Acad Sci U S A 95: 15090–15095
Sander M, Chavoshan B, Harris SA, Iannaccone ST, Stull JT, Thomas GD, Victor RG 2000 Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc Natl Acad Sci U S A 97: 13818–13823
Kubes P, Granger DN 1992 Nitric oxide modulates microvascular permeability. Am J Physiol 262: H611–H615
Clancy RM, Leszczynska-Piziak J, Abramson SB 1992 Nitric oxide, an endothelial cell relaxation factor, inhibits neutrophil superoxide anion production via a direct action on the NADPH oxidase. J Clin Invest 90: 1116–1121
Wink DA, Cook JA, Pacelli R, Liebmann J, Krishna MC, Mitchell JB 1995 Nitric oxide (NO) protects against cellular damage by reactive oxygen species. Toxicol Lett 82/83: 221–226
Rando TA, Disatnik MH, Yu Y, Franco A 1998 Muscle cells from mdx mice have an increased susceptibility to oxidative stress. Neuromuscul Disord 8: 14–21
Tatsumi R, Hattori A, Ikeuchi Y, Anderson JE, Allen RE 2002 Release of hepatocyte growth factor from mechanically stretched skeletal muscle satellite cells and role of pH and nitric oxide. Mol Biol Cell 13: 2909–2918
Chaubourt E, Fossier P, Baux G, Leprince C, Israel M, De La Porte S 1999 Nitric oxide and L-arginine cause an accumulation of utrophin at the sarcolemma: a possible compensation for dystrophin loss in Duchenne muscular dystrophy. Neurobiol Dis 6: 499–507
Chaubourt E, Voisin V, Fossier P, Baux G, Israel M, de La Porte S 2000 The NO way to increase muscular utrophin expression?. CR Acad Sci III 323: 735–740
Tidball JG, Wehling-Henricks M 2004 Expression of a NOS transgene in dystrophin-deficient muscle reduces muscle membrane damage without increasing the expression of membrane-associated cytoskeletal proteins. Mol Genet Metab 82: 312–320
Zhuang W, Eby JC, Cheong M, Mohapatra PK, Bredt DS, Disatnik MH, Rando TA 2001 The susceptibility of muscle cells to oxidative stress is independent of nitric oxide synthase expression. Muscle Nerve 24: 502–511
McPherron AC, Lawler AM, Lee SJ 1997 Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 387: 83–90
Thomas M, Langley B, Berry C, Sharma M, Kirk S, Bass J, Kambadur R 2000 Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J Biol Chem 275: 40235–40243
Taylor WE, Bhasin S, Artaza J, Byhower F, Azam M, Willard DH Jr ., Kull FC Jr ., Gonzalez-Cadavid N 2001 Myostatin inhibits cell proliferation and protein synthesis in C2C12 muscle cells. Am J Physiol Endocrinol Metab 280: E221–E228
Joulia D, Bernardi H, Garandel V, Rabenoelina F, Vernus B, Cabello G 2003 Mechanisms involved in the inhibition of myoblast proliferation and differentiation by myostatin. Exp Cell Res 286: 263–275
McCroskery S, Thomas M, Maxwell L, Sharma M, Kambadur R 2003 Myostatin negatively regulates satellite cell activation and self-renewal. J Cell Biol 162: 1135–1147
Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R 2002 Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem 277: 49831–49840
Zhu X, Hadhazy M, Wehling M, Tidball JG, McNally EM 2000 Dominant negative myostatin produces hypertrophy without hyperplasia in muscle. FEBS Lett 474: 71–75
Wagner KR, McPherron AC, Winik N, Lee SJ 2002 Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann Neurol 52: 832–836
Bogdanovich S, Krag TO, Barton ER, Morris LD, Whittemore LA, Ahima RS, Khurana TS 2002 Functional improvement of dystrophic muscle by myostatin blockade. Nature 420: 418–421
Schuelke M, Wagner KR, Stolz LE, Hubner C, Riebel T, Komen W, Braun T, Tobin JF, Lee SJ 2004 Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med 350: 2682–2688
Lee SJ, McPherron AC 2001 Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci U S A 98: 9306–9311
Hill JJ, Davies MV, Pearson AA, Wang JH, Hewick RM, Wolfman NM, Qiu Y 2002 The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J Biol Chem 277: 40735–40741
Hill JJ, Qiu Y, Hewick RM, Wolfman NM 2003 Regulation of myostatin in vivo by growth and differentiation factor-associated serum protein-1: a novel protein with protease inhibitor and follistatin domains. Mol Endocrinol 17: 1144–1154
Williams RL, Hilton DJ, Pease S, Willson TA, Stewart CL, Gearing DP, Wagner EF, Metcalf D, Nicola NA, Gough NM 1988 Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature 336: 684–687
Leary AG, Wong GG, Clark SC, Smith AG, Ogawa M 1990 Leukemia inhibitory factor differentiation-inhibiting activity/human interleukin for DA cells augments proliferation of human hematopoietic stem cells. Blood 75: 1960–1964
Verfaillie C, McGlave P 1991 Leukemia inhibitory factor/human interleukin for DA cells: a growth factor that stimulates the in vitro development of multipotential human hematopoietic progenitors. Blood 77: 263–270
Resnick JL, Bixler LS, Cheng L, Donovan PJ 1992 Long-term proliferation of mouse primordial germ cells in culture. Nature 359: 550–551
Barnard W, Bower J, Brown MA, Murphy M, Austin L 1994 Leukemia inhibitory factor (LIF) infusion stimulates skeletal muscle regeneration after injury: injured muscle expresses lif mRNA. J Neurol Sci 123: 108–113
Kurek JB, Bower JJ, Romanella M, Koentgen F, Murphy M, Austin L 1997 The role of leukemia inhibitory factor in skeletal muscle regeneration. Muscle Nerve 20: 815–822
Finkelstein DI, Bartlett PF, Horne MK, Cheema SS 1996 Leukemia inhibitory factor is a myotrophic and neurotrophic agent that enhances the reinnervation of muscle in the rat. J Neurosci Res 46: 122–128
Austin L, Bower JJ, Bennett TM, Lynch GS, Kapsa R, White JD, Barnard W, Gregorevic P, Byrne E 2000 Leukemia inhibitory factor ameliorates muscle fiber degeneration in the mdx mouse. Muscle Nerve 23: 1700–1705
White JD, Davies M, McGeachie J, Grounds MD 2002 An evaluation of leukaemia inhibitory factor as a potential therapeutic agent in the treatment of muscle disease. Neuromuscul Disord 12: 909–916
Metcalf D, Gearing DP 1989 Fatal syndrome in mice engrafted with cells producing high levels of the leukemia inhibitory factor. Proc Natl Acad Sci U S A 86: 5948–5952
Gunawardana DH, Basser RL, Davis ID, Cebon J, Mitchell P, Underhill C, Kilpatrick TJ, Reardon K, Green MD, Bardy P, Amor P, Crump D, Ng S, Nation RL, Begley CG 2003 A phase I study of recombinant human leukemia inhibitory factor in patients with advanced cancer. Clin Cancer Res 9: 2056–2065
Leong J, Hayes A, Austin L, Morrison W 1999 Muscle protection following motor nerve repair in combination with leukemia inhibitory factor. J Hand Surg [Am] 24: 37–45
Gregorevic P, Hayes A, Lynch GS, Williams DA 2000 Functional properties of regenerating skeletal muscle following LIF administration. Muscle Nerve 23: 1586–1588
Acknowledgements
The authors thank Dr. Melissa Spencer, who provided valuable comments during the preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by the National Institutes of Health (AR40343 and AR47721), the Muscular Dystrophy Association, and the American Heart Association (0325146Y).
Rights and permissions
About this article
Cite this article
Tidball, J., Wehling-Henricks, M. Evolving Therapeutic Strategies for Duchenne Muscular Dystrophy: Targeting Downstream Events. Pediatr Res 56, 831–841 (2004). https://doi.org/10.1203/01.PDR.0000145578.01985.D0
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000145578.01985.D0
This article is cited by
-
sPIF promotes myoblast differentiation and utrophin expression while inhibiting fibrosis in Duchenne muscular dystrophy via the H19/miR-675/let-7 and miR-21 pathways
Cell Death & Disease (2019)
-
The role of oxidative stress in skeletal muscle injury and regeneration: focus on antioxidant enzymes
Journal of Muscle Research and Cell Motility (2015)
-
In vitro myoblast motility models: investigating migration dynamics for the study of skeletal muscle repair
Journal of Muscle Research and Cell Motility (2013)
-
Extrinsic regulation of satellite cell specification
Stem Cell Research & Therapy (2010)
-
Protein–DNA array-based identification of transcription factor activities differentially regulated in skeletal muscle of normal and dystrophin-deficient mdx mice
Molecular and Cellular Biochemistry (2008)
