
Abstract
The pathogenesis of bilirubin encephalopathy appears to result from accumulation of unconjugated bilirubin (UCB), which, in turn, may cause mitochondrial perturbation, release of intermembrane proteins, and, ultimately, cell death. Aging imparts to cells a different susceptibility to this toxic stimulus, as neonates are particularly vulnerable to the accumulation of UCB in the CNS. In this paper, we further characterize UCB-induced toxicity in isolated neuronal and glial cells according to age in culture. In addition, we investigate sensitivity of mitochondria derived from young and old rats to UCB-induced membrane permeabilization and, finally, evaluate whether age-dependent changes in UCB toxicity are accompanied by alterations in the mitochondrial content of cytochrome c. The results showed that UCB is more toxic to immature neural cells after 4–5 d in culture (p < 0.001), whereas neurons were more sensitive than astrocytes (p < 0.05). In fact, approximately 40% of cells were apoptotic in immature cultures compared with 20% in mature cultures. Unexpectedly, mitochondrial swelling and subsequent efflux of cytochrome c induced by UCB were 2-fold greater in organelles derived from older rats (p < 0.01). In conclusion, UCB toxicity of isolated rat neuronal and glial cells is modulated by age in culture in that immature cells are more susceptible. Mitochondria derived from younger rats are nevertheless more resistant to membrane permeabilization and cytochrome c release induced by UCB. The data indicate that the cells of young animals are relatively resistant to UCB toxicity, through a protective mechanism at the mitochondrial level; however, this is not sufficient to prevent apoptosis of cells in the young animal. Thus, although playing a role, direct mitochondrial injury may not be the sole mechanism of UCB cytotoxicity.
Similar content being viewed by others


Main
Toxicity of UCB to neurons is typically characterized by disruption of plasma membrane integrity and release of intracellular constituents, and therefore, cell death has been defined as loss of neuronal function associated with necrosis. In fact, bilirubin has been found to modulate several crucial aspects of cell function, including inhibition of DNA and protein synthesis (1–3) and impairment of neurotransmitter synthesis, release, and uptake (4–6), as well as inhibition of protein phosphorylation (5, 7, 8), which may all culminate in cell death. Only recently, UCB cytotoxicity to isolated rat neural cells has been attributed to apoptotic processes (9, 10).
Despite the striking heterogeneity of cell death induction pathways, the execution of the death program is often associated with mitochondrial perturbation. This appears to be the consequence of an increase in the mitochondrial membrane permeability (11, 12), involving nonspecific injury to the membrane lipids or opening of a cyclosporin A-sensitive multiprotein pore complex. As a result of membrane permeabilization, the mitochondrial transmembrane potential collapses, thereby uncoupling the respiratory chain, and reactive oxygen species production increases, causing subsequent release of intermembranous space proteins, such as cytochrome c and the apoptosis-inducing factor (13–16). On entering the cytosol, cytochrome c was suggested to promote the formation of a multiprotein complex that induces proteolytic activation of cell death proteases known as caspases (17, 18). Several consequences of increased mitochondrial permeability, such as changes in redox potentials, primary activation of caspases, modification of the Bcl-2 complex, and sustained increase in cytosolic calcium levels, also favor permeabilization, thereby creating positive feedback loops (19). We have previously shown that UCB could activate apoptosis in neural cells through the induction of mitochondrial permeability transition (9).
Although jaundiced neonates show some evidence of cell injury because of the accumulation of UCB, extensive damage such as that found in cell culture models of the disease does not occur either in animals or in patients. This suggests that adaptive responses influenced by factors such as age may take place, leading to resistance to cell death by the toxic stimulus. Therapeutic insights to treat bilirubin encephalopathy and kernicterus may result from a better understanding of potential mechanisms of adaptive response. Much as during cholestasis, in which mitochondria isolated from bile duct-ligated rats have been shown to be resistant to bile acid-induced permeability (20), inhibition of the mitochondrial permeabilization depending on cell age may be an adaptive mechanism to resist to UCB-induced cell death.
The purpose of this study was to 1) further characterize UCB-induced toxicity in isolated neurons and astrocytes according to cell age in culture;2) investigate sensitivity of mitochondria derived from young and old rats to UCB-induced membrane permeabilization; and 3) evaluate whether age-dependent changes in UCB toxicity are accompanied by alterations in the mitochondrial content of cytochrome c. The results demonstrate that UCB is more toxic to immature neural cells after 4–5 d in primary culture. Interestingly, the extent of UCB-induced membrane permeabilization and subsequent cytochrome c release was significantly higher in mitochondria isolated from older rats. These results suggest that the cells of young animals are relatively resistant to UCB toxicity, exhibiting a protective mechanism at the mitochondrial level.
METHODS
Isolation of rat astrocytes and neurons and cell culture.
Astrocytes were isolated from 2-d-old Wistar rats as described previously (21) with minor modifications. Briefly, the rat brain was collected after decapitation in Dulbecco's modified Eagle's medium (Life Technologies Inc., Grand Island, NY, U.S.A.) containing 11 mM sodium bicarbonate, 71 mM glucose, and 1% antibiotic and antimycotic solution (Sigma Chemical Co., St. Louis, MO, U.S.A.). Meninges, blood vessels, and white matter were then removed. The cortex was homogenized by mechanical fragmentation, and the cell suspension was passed sequentially through steel screens of 230-, 104-, and 73.3-μm pore size. Cells were collected by centrifugation at 700 ×g for 10 min and resuspended in Dulbecco's modified Eagle's medium supplemented with 10% FCS (Life Technologies Inc.). Finally, 2.0 × 105 cells/cm2 were plated on 12-well tissue culture plates (Corning Costar Corp., Cambridge, MA, U.S.A.) and maintained at 37°C in a humidified atmosphere of 5% CO2. Culture medium was replaced every 3 d, and cells were used after 5, 10, or 20 d in culture. Cells were morphologically characterized by phase-contrast microscopy, and by indirect immunocytochemistry for glial fibrillary acidic protein.
Rat neurons were isolated from fetuses of 17–18 d pregnant Wistar rats as previously described (22) with minor modifications. In short, pregnant rats were anesthetized with ether and decapitated. The fetuses were collected in Hanks' balanced salt solution (HBSS-1; Life Technologies Inc.) and rapidly decapitated. After removal of meninges and white matter, the brain cortex was collected in Hanks' balanced salt solution without Ca2+ and Mg2+ (HBSS-2). Brain cortexes were mechanically fragmented, transferred to a 0.025% trypsin in HBSS-2 solution and incubated for 15 min at 37°C. After trypsinization, cells were washed twice in HBSS-2 containing 10% FCS, and resuspended in Neurobasal medium (Life Technologies Inc.) supplemented with 0.5 mM l-glutamine, 25 μM l-glutamic acid, 2% B-27 Supplement (Life Technologies Inc.), and 12 mg/mL gentamicin. Aliquots of 1 × 105 cells/cm2 were plated on 12-well tissue culture plates (Corning Costar Corp.) and maintained at 37°C in a humidified atmosphere of 5% CO2. Every 3 d, 0.5 mL of old medium was removed by aspiration and replaced by the same volume of fresh medium without glutamic acid. Cells were used after 4, 8, or 18 d in culture. Neurons were morphologically characterized by phase-contrast microscopy, and by indirect immunocytochemistry for neurofilaments.
Incubation of neural cells with UCB.
UCB (Sigma Chemical Co.) was purified (23) and dissolved in 0.1 N NaOH, immediately before use, to prepare a stock solution at a concentration of 8.6 mM. The stock solution was added to culture medium (without FCS), containing human serum albumin (Sigma Chemical Co.) to obtain a final UCB concentration of 86 μM, at a bilirubin-to-albumin molar ratio of 3:1. Although rarely found in jaundiced newborns, this molar ratio was selected to achieve, in short incubation periods, significant interaction of UCB with cells, mimicking the toxic conditions of a prolonged severe hyperbilirubinemia. Isolated rat astrocytes and neurons were cultured as described above and then incubated with 86 μM UCB for 4 h. The medium was gently removed at the indicated time point and scored for nonviable cells by trypan blue dye exclusion. Attached cells were fixed with 4% formaldehyde in PBS, pH 7.4, for 10 min at room temperature for morphologic assessment of apoptosis.
Morphologic evaluation of apoptosis.
Morphology was performed as described previously (24). Briefly, medium was gently removed at the indicated time point to prevent detachment of cells. Fixed cells were incubated with Hoechst dye 33258 (Sigma Chemical Co.) at 5 μg/mL in PBS for 5 min, washed with PBS, and mounted with PBS-glycerol (3:1, vol/vol). Fluorescence was visualized using an Axioskop fluorescence microscope (Carl Zeiss GmbH, Jena, Germany). Photographs were taken with Kodak GOLD ultra400 films (Eastman Kodak Co., Rochester, NY, U.S.A.). Fluorescent nuclei were scored by three different people, blinded as to the treatment received by cells, and categorized according to condensation and staining characteristics of chromatin. Normal nuclei were identified by uncondensed chromatin dispersed over the entire nucleus. Apoptotic nuclei were identified by condensed chromatin, contiguous to the nuclear membrane, as well as nuclear fragmentation of condensed chromatin. Three random microscopic fields per sample of approximately 500 nuclei were counted, and mean values were expressed as the percentage of apoptotic nuclei.
Mitochondria isolation.
Low-calcium brain and liver mitochondria were isolated from young (1-mo-old) and older adult (18-mo-old) Wistar rats as previously described (25, 26). In short, animals were killed by exsanguination under ether anesthesia, and the brains and livers were removed and rinsed in normal saline. The tissue was homogenized using six complete up and down strokes with a speed-controlled mechanical skill drill and a Teflon pestle in an ice-cold solution of 70 mM sucrose, 220 mM mannitol, 1 mM EGTA, and 10 mM HEPES, pH 7.4, as a 10% (wt/vol) homogenate. The homogenate was centrifuged at 600 ×g for 10 min at 4°C in a Sigma 3K 30 (B. Braun-Biotech Inc., Allentown, PA, U.S.A.), and the postnuclear supernatant was centrifuged at 7,000 ×g for 10 min at 4°C. The crude mitochondrial pellet was further purified by sucrose-Percoll gradient centrifugation (27). The pellet was resuspended in 2 mL of homogenate buffer, and 1 mL of the resuspended pellet was carefully layered onto a 20-mL self-generating gradient containing 0.25 M sucrose, 1 mM EGTA, and Percoll (Sigma Chemical Co.; 75:25, vol/vol). The mitochondria were purified by centrifugation at 43,000 ×g for 30 min at 4°C. The clear supernatant solution was removed, and the lower turbid layer was resuspended in 30 mL of wash buffer containing 0.1 M KCl, 5 mM 3-(N-morpholino)-propane sulfonic acid (MOPS), and 1 mM EGTA, at pH 7.4, and centrifuged at 7,000 ×g for 10 min at 4°C. The resulting mitochondria pellet was washed in wash buffer two times. A final wash was carried out in chelex-100-treated buffer (Bio-Rad Laboratories, Richmond, CA, U.S.A.; 200–400 mesh, potassium form) without EGTA. The pellet was suspended in 4 mL of chelex-100-treated resuspension buffer containing 125 mM sucrose, 50 mM KCl, 5 mM HEPES, and 2 mM KH2PO4. After isolation, mitochondria were maintained on ice and used for experiments within 3 h of isolation. Aliquots were removed for examining the purity of the mitochondria preparation as previously described (28).
Measurement of mitochondrial membrane permeability.
The membrane permeability was assessed using a spectrophotometric assay measuring high-amplitude rapid swelling of mitochondria as previously described (26, 28) with minor modifications. An increase in mitochondrial swelling results in a decrease in OD. In brief, mitochondria (0.1 mg of protein) were incubated in 1 mL of chelex-100-treated respiration buffer (10 mM HEPES, 10 mM succinate, 215 mM mannitol, 71 mM sucrose, pH 7.4) for 10 min at 25°C, and swelling was monitored at 540 nm in a Unicam Spectrometer UV2 (Unicam, Portsmouth, NH, U.S.A.). Basal values of mitochondria absorbance were measured for 5 min, and the OD was monitored for an additional 5 min after addition of 4.3 μM UCB. We have chosen to use lower concentrations of UCB because, unlike in the cell culture studies, albumin was not included in the respiration buffer.
Determination of cytochrome c content in supernatants and mitochondrial pellets after the permeability assay.
After membrane permeabilization, mitochondria were spun down at 12,000 ×g for 3 min at 4°C. Aliquots of the supernatant and pellet were subjected to SDS-PAGE for detection of cytochrome c release. Proteins were separated on a 15% gel and transferred onto nitrocellulose membranes, and the immunoblots were treated with 15% H2O2 for 15 min at room temperature. Blots were then sequentially incubated with 5% milk blocking solution, primary MAb to cytochrome c (PharMingen, San Diego, CA, U.S.A.) at a dilution of 1:5,000 overnight at 4°C, and finally with secondary goat anti-mouse IgG antibody conjugated with horseradish peroxidase (Bio-Rad Laboratories, Hercules, CA, U.S.A.) for 2 h at room temperature. Membranes were processed for cytochrome c detection using the enhanced chemiluminescence light reagent (Amersham Life Science, Inc., Arlington Heights, IL, U.S.A.).
Densitometry and statistical analysis.
Densitometry was accomplished using a personal computer coupled to a PRIMAX 9600 Profi VM6575 scanner (Primax International B.V., Utrecht, The Netherlands). Quantitation of the autoradiograms was performed using the ImageMater 1D Elite densitometric analysis program (Amersham Pharmacia Biotech, Uppsala, Sweden). The fold change in protein levels was calculated based on the corresponding controls. All data are expressed as mean ± SEM from at least three separate experiments. Differences between groups were compared using the unpaired two-tailed t test performed on the basis of equal or unequal variance as appropriate. We considered p < 0.05 to be statistically significant.
RESULTS
Apoptosis induced by UCB in primary rat astrocytes and neurons depends on cell age in culture.
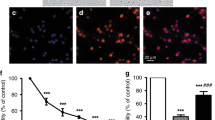
Apoptosis in rat astrocytes and neurons induced by UCB was characterized by condensation of chromatin and nuclear fragmentation with formation of apoptotic bodies as revealed by Hoechst staining. In addition, DNA fragmentation was assessed by the terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labeling assay, and the results were comparable to those obtained by the evaluation of nuclear morphology (data not shown). As shown in Figure 1, astrocytes incubated with UCB exhibited profound changes in nuclear morphology consistent with apoptosis. When cultures exposed to UCB were scored for nonviable cells by trypan blue dye exclusion, we found <20% loss in cell viability, compared with approximately 2% in controls (p < 0.01). Moreover, toxicity was very marked in cells after 5 d in culture, but less pronounced in astrocytes cultured for 10 or 20 d before the incubation studies. When the number of condensed and fragmented nuclei was expressed as percentage of the total number, control astrocytes showed <5% of apoptosis irrespective of cell age in culture (Fig. 2A). In contrast, after incubation with UCB, astrocytes cultured for 5 d exhibited approximately 40% of apoptotic cell death (p < 0.001) whereas glial cells cultured for 10 or 20 d exhibited <20% (p < 0.01). This indicates that astrocytes of younger age in culture are twice as susceptible to the cytotoxic effects of UCB. Finally, similar results were obtained when neuronal cultures were prepared and incubated with UCB, albeit there was increased sensitivity of neurons compared with astrocytes (Fig. 2B). The different susceptibility of neurons and astrocytes was even better highlighted using the terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labeling assay, in which sensitivity to DNA fragmentation was approximately 20% greater in neuronal cells (p < 0.05). Nevertheless, UCB is more harmful to immature cultured neuronal cells.

Apoptosis in primary rat astrocytes incubated with UCB. Isolated astrocytes were cultured for 5, 10, and 20 d before incubation with either no addition (control) or 86 μM UCB for 4 h, and fixed for morphologic analysis as described in “Methods.” Apoptotic cells were identified by fluorescence microscopy of Hoechst-stained nuclei according to the presence of nuclear fragmentation and condensed chromatin.

Apoptosis induced by UCB in primary rat astrocytes and neurons depends on cell age in culture. Astrocytes (A) were cultured for 5, 10, and 20 d and neurons (B) were cultured for 4, 8, and 18 d before incubation with either no addition (control) or 86 μM UCB for 4 h, and fixed for morphologic analysis as described in “Methods.” The percentage of apoptotic cells was determined by fluorescence microscopy of Hoechst-stained nuclei. The results are means ± SEM from at least three separate experiments. *p < 0.001 indicates difference from astrocytes cultured for 5 d; †p < 0.01 indicates difference from neurons cultured for 4 d. Significant differences were observed between UCB and same-age controls (p < 0.001).
Mitochondrial membrane permeability transition induced by UCB in isolated mitochondria depends on the age of the animal from which organelles were derived.
Permeabilization of the mitochondrial membrane appears to be a key event in apoptosis induced by several stimuli and may mark the commitment to the apoptotic death process (12). Thus, to characterize the direct effect of UCB on mitochondrial membranes as well as the influence of aging on organelle resistance to UCB-induced toxicity, we assayed membrane permeability in mitochondria isolated from young and old rats. Isolation of mitochondria from rat liver was performed with better yield and purity than from rat brain (data not shown). Inasmuch as results of mitochondrial swelling from preliminary studies using liver and brain mitochondria showed similar trends, we decided to use liver organelles for the remainder of the experiments.
Although we have previously shown that volumes of control mitochondria are not significantly altered during the permeability assay (9, 28), we had not compared mitochondria isolated from rats with different ages. In the present study, having verified no age difference on membrane permeabilization in controls, we next assayed mitochondrial volume after incubation with UCB in organelles derived from young versus old rats (Fig. 3). UCB induced significant mitochondrial swelling, resulting in markedly decreased absorbance of mitochondrial suspensions compared with same-age controls. These results suggest a direct effect of UCB on mitochondrial membranes. Interestingly, swelling was greater in mitochondria derived from older rats versus younger animals. Indeed, after 5 min of incubation, the permeabilization induced by UCB was almost 2-fold greater in mitochondria from older rats compared with younger animals (p < 0.01). The increase in organelle volume was inhibited by cyclosporin A in both age groups by at least 50% (p < 0.05), indicating that mitochondrial swelling was mediated, at least in part, by the opening of the permeability transition megapore. In addition, these results suggest that mitochondria from younger animals adapt during early life by increasing their threshold for membrane permeabilization to prevent cell death induced by UCB. This adaptive phenomenon does not abolish apoptosis induced by UCB, as cell death is still greater in younger cells (Fig. 1), but it may increase resistance to toxicity.

Mitochondrial membrane permeability transition induced by UCB in isolated mitochondria depends on the age of the animal from which organelles were derived. Mitochondria were isolated from 1- and 18-mo-old rats and incubated with either no addition (control) or 4.3 μM UCB in respiration buffer as described in “Methods.” At 5 min, the apoptotic agent was added and the decrease in absorbance at 540 nm was monitored for an additional 5 min. A, effect of UCB on OD of mitochondrial suspensions isolated from young and old rats. B, percent change in swelling from 5 to 10 min in mitochondria isolated from young (black bars) and old (gray bars) rats. The results are means ± SEM from at least three separate experiments.†p < 0.01 indicates difference from mitochondria isolated from young rats. Significant differences were observed between UCB and same-age controls (p < 0.001).
Release of cytochrome c induced by UCB in isolated mitochondria depends on the mitochondrial resistance to membrane permeabilization.
Having shown a relative resistance of younger rat mitochondria to membrane permeabilization induced by UCB, we next sought to determine the potential implications of these findings to the mitochondrial release of inducers of apoptosis such as cytochrome c. Although precise biochemical pathways mediating UCB-induced apoptosis remain a matter of debate, both the mitochondrial permeability transition and the subsequent release of cytochrome c may be key events. As expected, control mitochondria exhibited no detectable release of cytochrome c, irrespective of the animal age (Fig. 4, top). Conversely, mitochondrial-associated levels of cytochrome c were decreased after incubation with UCB, whereas accumulation in the supernatant was similarly increased. Consistent with the data on mitochondrial permeabilization, cytochrome c efflux was more evident in mitochondria isolated from older rats. Indeed, densitometry analysis of blots showed almost a 2-fold decrease in cytochrome c in mitochondria from old rats in the presence of UCB but only a 1.1-fold decrease in organelles from young animals (p < 0.01; Fig. 4, bottom). Thus, these results show an association between decreased mitochondrial cytochrome c content and relative resistance of mitochondria to the mitochondrial membrane permeability transition.

Release of cytochrome c induced by UCB in isolated mitochondria depends on the mitochondrial resistance to membrane permeabilization. Mitochondria were isolated from 1- and 18-mo-old rats and incubated with either no addition (control) or 4.3 μM UCB in respiration buffer as described in “Methods.” The mitochondrial pellets and supernatants were examined for cytochrome c levels by Western blot analysis. After SDS-PAGE and transfer, the nitrocellulose membranes were incubated with the MAb to cytochrome c, and the 15-kD protein was detected using enhanced chemiluminescence light reagent. Representative Western blots (top) and densitometric means ± SEM relative to controls (bottom) from at least three separate experiments. †p < 0.01 indicates difference from mitochondria isolated from young rats. Significant differences were observed between UCB and same-age controls (at least p < 0.05).
DISCUSSION
The neurotoxicity of UCB has been well established using embryonic cell culture models [for review, see Cashore (29)]. However, if high levels of UCB are frequently more cytotoxic in neonates than later in life, it is not surprising that embryonic and older neural cells do not respond to this toxic stimulus in a similar manner. Because influences of an aging environment can be separated from changes inherent to aging cells, culture of cells during different periods provides a simple procedure, frequently used, to investigate age-related cellular responses to various toxic stimuli. The present study demonstrates that incubation of neuronal and glial cells with UCB results in greater cytotoxicity in 4- to 5-d cultures compared with older cultures, suggesting increased sensitivity of immature cells to death by apoptosis. In addition, our results show that cytochrome c is released from mitochondria in response to direct incubation with UCB and that, unexpectedly, mitochondria derived from older rats liberate more cytochrome c, suggesting that membrane permeabilization occurs by a mechanism sensitive to the age of the animal. Finally, the decreased neurotoxic effects of UCB with cell age in culture along with increased responsiveness of mitochondria may also indicate that bilirubin-induced cell injury is not solely mediated through mitochondrial disturbances. In this regard, it has recently been shown that bilirubin induces apoptosis via activation of N-methyl-d-aspartate receptors and inhibition of protein kinase C activity (30). Mitochondria may, nevertheless, be a common target for different apoptotic pathways.
Cytochrome c redistribution from mitochondria to cytosol has been shown to be a critical event in the cell death effector pathway initiated by several apoptotic stimuli (31–33). However, the molecular mechanism(s) for release of caspase-activating proteins remains controversial. Opening of the mitochondrial permeability transition pore is thought to cause cytochrome c efflux and cell death by leading to a collapse of the mitochondrial inner transmembrane potential, expansion of the matrix space, and subsequent rupture of the outer membrane (11–14). Inhibitors of the permeability transition pore prevent apoptosis in some systems, supporting a central role for mitochondrial permeabilization in apoptotic processes (15). Furthermore, Bax interacts directly with pores in isolated mitochondria to induce permeability transition and cytochrome c release (34). However, other studies reported that cytochrome c release and caspase activation occur independently of the mitochondrial permeability and without any detectable mitochondrial depolarization, implying that opening of the permeability transition pore is an event downstream to caspase activation (31, 32, 35). Thus, the efflux of cytochrome c would also occur through a mechanism independent of the mitochondrial permeability transition pore. Supporting this idea, it has been demonstrated that Bax overexpression triggers the release of cytochrome c in different cell types independently of the permeability transition (36, 37). Our results, using isolated mitochondria, indicate that the onset of membrane permeabilization by UCB is required for cytochrome c release. Moreover, we found that mitochondria isolated from young rats were relatively resistant to the membrane permeability transition induced by UCB cytotoxicity, which explains the increased mitochondrial-associated levels of cytochrome c. This apparently adaptive phenomenon does not abolish apoptosis induced by UCB, as death in younger cells was still greater than in older cultures (Fig. 1), but it may increase resistance to toxicity.
Several lines of evidence suggest that aging imparts to cells a different susceptibility to toxic agents relevant to several diseases. It is generally accepted that neonates are particularly vulnerable to the accumulation of UCB in the CNS, where it may lead to the yellow staining and neurologic dysfunction characterizing UCB encephalopathy. In fact, in vitro studies have shown that premature neural cells are more sensitive to bilirubin toxicity than mature cells (38–40). Our hypothesis is that during neonatal hyperbilirubinemia, toxic UCB accumulates within neural cells. Thus, increased concentration of UCB in young-age cells represents a potent cytotoxic stimulus to which the cell will have to adapt. The greater mitochondrial threshold for the membrane permeability transition renders the cell less vulnerable to death by the toxic substance. Indeed, the ability of mitochondria isolated from younger rats to resist to the permeability induced by UCB may represent a potential adjustment of cells to function despite the accumulation of the cytotoxic agent. The increased threshold for the mitochondrial membrane permeability suggested by the present study is not without precedent. In this regard, it has been demonstrated that cholestasis increases the mitochondrial cardiolipin content as an adaptive mechanism that confers resistance to the mitochondrial permeability transition (20).
From these studies, UCB induces apoptosis in both neuronal and glial cells through a mechanism that may involve mitochondrial membrane perturbation. In fact, these toxic stimuli directly interfere with the membrane permeability transition in isolated mitochondria, thereby increasing cytochrome c efflux. The data support the concept that the cells of young animals are relatively resistant to UCB toxicity, through a protective mechanism at the mitochondrial level; however, this is not sufficient to prevent apoptosis of cells in the young animal. Thus, although playing a role, direct mitochondrial injury may not be the sole mechanism of UCB cytotoxicity.
Abbreviations
- UCB:
-
unconjugated bilirubin
References
Majumdar APN 1974 Bilirubin encephalopathy: effect on RNA polymerase activity and chromatin template activity in the brain of Gunn rat. Neurobiology 4: 425–431
Yamada N, Sawasaki Y, Nakajima H 1977 Impairment of DNA synthesis in Gunn rat cerebellum. Brain Res 126: 295–307
Schiff D, Chan G, Poznansky MJ 1985 Bilirubin toxicity in neuronal cell lines N-115 and NBR-10A. Pediatr Res 19: 908–911
Ohno T 1980 Kernicterus: effect on choline acetyltransferase, glutamic acid decarboxylase and tyrosine hydroxylase activities in the brain of Gunn rat. Brain Res 196: 282–285
Hansen TWR, Bratlid D, Walaas SI 1988 Bilirubin decreases phosphorylation of synapsin I, a synaptic vesicle-associated neuronal phosphoprotein, in intact synaptosomes from rat cerebral cortex. Pediatr Res 23: 219–223
Silva R, Mata LR, Gulbenkian S, Brito MA, Tiribelli C, Brites D 1999 Inhibition of glutamate uptake by unconjugated bilirubin in cultured cortical rat astrocytes: role of concentration and pH. Biochem Biophys Res Comm 265: 67–72
Morphis L, Constantopoulos A, Matsaniotis N 1982 Bilirubin-induced modulation of cerebral protein phosphorylation in neonate rabbits in vivo. Science 218: 156–158
Sano K, Nakamura H, Matsuo T 1985 Mode of inhibitory action of bilirubin on protein kinase C. Pediatr Res 19: 587–590
Rodrigues CMP, Solá S, Silva R, Brites D 2000 Bilirubin and amyloid-β peptide induce cytochrome c release through mitochondrial membrane permeabilization. Mol Med 6: 936–946
Silva RFM, Rodrigues CMP, Brites D 2001 Bilirubin-induced apoptosis in glial and nerve cells is aggravated by chenodeoxycholic acid but prevented by ursodeoxycholic acid. J Hepatol 34: 402–408
Kroemer G, Zamzami N, Susin SA 1997 Mitochondrial control of apoptosis. Immunol Today 18: 44–51
Green DR, Reed JC 1998 Mitochondria and apoptosis. Science 281: 1309–1312
Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B, Kroemer G 1995 Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med 182: 367–377
Rodrigues CMP, Fan G, Ma X, Kren BT, Steer CJ 1998 A novel role of ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane perturbation. J Clin Invest 101: 2790–2799
Zamzami N, Marchetti P, Castedo M, Hirsch T, Susin SA, Masse B, Kroemer G 1996 Inhibitors of permeability transition interfere with the disruption of the mitochondrial transmembrane potential during apoptosis. FEBS Lett 384: 53–57
Susin AS, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G 1999 Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397: 441–446
Liu X, Kim CN, Yang J, Jemmerson R, Wang X 1996 Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86: 147–157
Zou H, Henzel WJ, Liu X, Lutschg A, Wang X 1997 Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 90: 405–413
Kroemer G 1997 The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med 3: 614–620
Lieser MJ, Park J, Natori S, Jones BA, Bronk SF, Gores GJ 1998 Cholestasis confers resistance to the rat liver mitochondrial permeability transition. Gastroenterology 115: 693–701
Blondeau JP, Beslin A, Chantoux F, Francon J 1993 Triiodothyronine is a high-affinity inhibitor of amino acid transport system L1 in cultured astrocytes. J Neurochem 60: 1407–1413
Brewer GJ, Torricelli JR, Evege EK, Price PJ 1993 Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res 35: 567–576
McDonagh AF, Assisi F 1972 The ready isomerization of bilirubin IX− in aqueous solution. Biochem J 129: 797–800
Oberhammer FA, Pavelka M, Sharma S, Tiefenbacher R, Purchio AF, Bursch W, Schulte-Hermann R 1992 Induction of apoptosis in cultured hepatocytes and in regressing liver by transforming growth factor beta 1. Proc Natl Acad Sci USA 89: 5408–5412
Walajtys-Rhode E, Zapatero J, Moehren G, Hoek JB 1992 The role of the matrix calcium level in the enhancement of mitochondrial pyruvate carboxylation by glucagon pretreatment. J Biol Chem 267: 370–379
Botla R, Spivey JR, Aguilar H, Bronk SF, Gores GJ 1995 Ursodeoxycholate (UDCA) inhibits the mitochondrial membrane permeability transition induced by glycochenodeoxycholate: a mechanism of UDCA cytoprotection. J Pharmacol Exp Ther 272: 930–938
Sokol RJ, Devereaux M, Mierau GW, Hambidge KM, Shikes RH 1990 Oxidant injury to hepatic mitochondrial lipids in rats with dietary copper overload. Modification by vitamin E deficiency. Gastroenterology 99: 1061–1071
Rodrigues CMP, Fan G, Wong PY, Kren BT, Steer CJ 1998 Ursodeoxycholic acid may inhibit deoxycholic acid-induced apoptosis by modulating mitochondrial transmembrane potential and reactive oxygen species production. Mol Med 4: 165–178
Cashore WJ 1990 The neurotoxicity of bilirubin. Clin Perinatol 17: 437–447
Grojean S, Koziel V, Vert P, Daval JL 2000 Bilirubin induces apoptosis via activation of NMDA receptors in developing rat brain neurons. Exp Neurol 166: 334–341
Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD 1997 The release of Cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 275: 1132–1136
Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng T-I, Jones DP, Wang X 1997 Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275: 1129–1132
Rodrigues CMP, Ma X, Linehan-Stieers C, Fan G, Kren BT, Steer CJ 1999 Ursodeoxycholic acid prevents cytochrome c release in apoptosis by inhibiting mitochondrial membrane depolarization and channel formation. Cell Death Differ 6: 842–854
Narita M, Shimizu S, Ito T, Chittenden T, Lutz RJ, Matsuda H, Tsujimoto Y 1998 Bax interacts with the permeability transition pore to induce permeability transition and cytochromE c release in isolated mitochondria. Proc Natl Acad Sci USA 95: 14681–14686
Bossy-Wetzel E, Newmeyer DD, Green DR 1998 Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J 17: 37–49
Eskes R, Antonsson B, Osen-Sand A, Montessuit S, Richter C, Sadoul R, Mazzei G, Nichols A, Martinou JC 1998 Bax-induced cytochrome c release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+ ions. J Cell Biol 143: 217–224
Jürgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC 1998 Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci USA 95: 4997–5002
Amit Y, Brenner T 1993 Age-dependent sensitivity of cultured rat glial cells to bilirubin toxicity. Exp Neurol 121: 248–255
Hansen TW, Allen JW 1997 Oxidation of bilirubin by brain mitochondrial membranes—dependence on cell type and postnatal age. Biochem Mol Med 60: 155–160
Rhine WD, Schmitter SP, Yu AC, Eng LF, Stevenson DK 1999 Bilirubin toxicity and differentiation of cultured astrocytes. J Perinatol 19: 206–211
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by EASL Research Fellowship from the European Association for the Study of the Liver, and grant PRAXIS/C/SAU/14311/1998 from Fundação para a Ciência e a Tecnologia (FCT), Lisbon, Portugal (to C.M.P.R.).
Rights and permissions
About this article
Cite this article
Rodrigues, C., Solá, S., Silva, R. et al. Aging Confers Different Sensitivity to the Neurotoxic Properties of Unconjugated Bilirubin. Pediatr Res 51, 112–118 (2002). https://doi.org/10.1203/00006450-200201000-00020
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200201000-00020
This article is cited by
-
Bilirubin metabolism: delving into the cellular and molecular mechanisms to predict complications
The Egyptian Journal of Internal Medicine (2024)
-
Molecular events in brain bilirubin toxicity revisited
Pediatric Research (2024)
-
Models of bilirubin neurological damage: lessons learned and new challenges
Pediatric Research (2023)
-
Experimental models assessing bilirubin neurotoxicity
Pediatric Research (2020)
-
High unbound bilirubin for age: a neurotoxin with major effects on the developing brain
Pediatric Research (2019)
