
Abstract
There is a high prevalence of growth retardation in children with type 1 Gaucher disease. The cause of this poor growth is not yet known; however, studies have shown acceleration of growth with enzyme replacement therapy (ERT). IGF are recognized as important determinants of somatic growth. It has been proven that chronic diseases with liver involvement might cause IGF deficiency. The aim of this study was to assess the IGF system in patients with childhood-onset Gaucher disease, before and after ERT, and its association with other clinical and analytical parameters. Twenty-two patients with type I Gaucher disease were included. The diagnosis was established before 14 y of age in all patients. Baseline determinations of total IGF-I, free IGF-I, and IGF binding protein 3 (IGFBP-3) were obtained in 19 patients before starting ERT at a mean age of 13.8 ± 11.2 y. A Spearman test was performed to establish the association with other clinical and analytical parameters. In a group of 13 patients receiving IGF, changes were evaluated during the initial 2 y of treatment. A Wilcoxon test was performed for the statistical analysis. Total IGF-I, free IGF-I, and IGFBP-3 were expressed as SD scores (SDS). We found low levels of IGF and its binding proteins before ERT. A significant association was found between the total IGF-I SDS before treatment and the age-adjusted severity score index:r = −0.62, p < 0.05. Total IGF-I and IGFBP-3 SDS correlated negatively with the presence of the L444P mutation (r = −0.53 and −0.5, respectively, p < 0.05). Height SDS correlated with total IGF-I and IGFBP-3 SDS in eight children (r = 0.84 and 0.78, respectively, p < 0.05). Total IGF-I SDS increased from −1.8 ± 0.8 to −0.8 ± 1.4 (p = 0.005) and free IGF-I increased from −1.2 ± 1 to 1.1 ± 2.1 after 12 ± 6.8 mo (p = 0.011) of ERT. IGFBP-3 SDS increased from −1.3 ± 0.6 to −0.2 ± 1.2 (p = 0.012) after 12 ± 4.5 mo of ERT. Type 1 Gaucher disease is associated with low levels of IGF and its binding proteins, which could be a consequence of liver involvement. Total IGF-I deficiency is associated with the severity of the illness. Growth retardation in pediatric patients with Gaucher disease is related to the alterations in IGF axis. Total IGF-I and IGFBP-3 are the two parameters that better correlate with height before treatment. ERT results in significant increase of total IGF-I, free IGF-I, and IGFBP-3 during the first year of treatment.
Similar content being viewed by others
Main
Gaucher disease, the most common lysosomal storage disease, is an autosomal recessive disorder. The primary biochemical defect is the deficiency of glucocerebrosidase, a lysosomal acid β-glucosidase, which results in the accumulation of glucosylceramide in lysosomes of reticuloendothelial cells (1).
The gene locus for glucocerebrosidase has been mapped to chromosome 1 in the q21 region (2). Several point mutations have been described, and the presence of two gene defects has been associated with the clinical expression of the disease (3). However, differences in the phenotype expression cannot be always explained by the amino acid changes or truncated proteins resulting from those gene defects (4).
Gaucher disease has several clinical subtypes. Type 1 Gaucher disease represents non-neuronopathic disease and it is characterized by splenomegaly, hepatomegaly, pancytopenia, and osteolytic and osteopenic degeneration of the skeleton. The clinical expression of the disease is variable and may manifest during the first years of life or remain undetected until the last decades of life (5). Type 2 is less common and is characterized by a rapidly progressive neurovisceral storage disorder and early death. Type 3 is less rapidly progressive and also involves a neurovisceral storage disorder and early death (6, 7).
Childhood-onset Gaucher disease has similar features, but there are a number of indirect manifestations of Gaucher disease in the affected child that also provide indicators of disease activity (8). One of these manifestations is growth retardation, which seems to be especially valuable in assessing severity as well as rate of progression of Gaucher disease in children. Nearly half of pediatric type 1 patients have anthropometric evidence of growth retardation, which correlates with disease severity as measured by other parameters (8, 9). In some children, growth retardation may be the sole overt manifestation of type 1 Gaucher disease (8). The cause of this poor growth is not yet elucidated. It has been reported that ERT accelerates growth in these patients (9). In a cohort of 15 Spanish patients with type 1 Gaucher disease who were receiving ERT, we have observed a significant increase in height z score. The estimated z score before treatment was −1.65, and after 13 ± 5.6 mo of ERT it increased to −1.25 (p < 0.05) (unpublished results).
IGF are recognized as important determinants of somatic growth (10). Recent research has proven that IGF have an anabolic effect even in adults (11). These peptides stimulate whole-body and muscle protein synthesis. IGF exist in the circulation complexed to IGFBP, which have been shown to increase the half-lives of IGF, and to modulate their bioactivities and bioavailabilities (12). Six high-affinity IGFBP have been classified; the predominant serum IGFBP is IGFBP-3, a glycosylated protein of 40–45 kD. IGFBP-3 is the only IGFBP that can circulate as part of a large 130–150 kD ternary complex in association with IGF and an ALS. The role of ALS has not been completely determined, but ALS may have a central role in regulating the bioavailability of IGF by stabilizing the IGF–IGFBP-3 complex (12, 13). These proteins, produced by liver, are GH-, age-, and nutritionally dependent (14). The major correlates of growth are changes in the concentrations of circulating IGFBP-3 and IGF-I. It has been proven that chronic diseases with liver involvement can cause IGF deficiency (15, 16).
The aim of this study was to asses the IGF axis in patients with childhood-onset Gaucher disease, before and after ERT, and its association with other clinical and analytical parameters.
METHODS
Twenty-two Spanish patients with Gaucher disease were studied. The diagnosis was established before 14 y of age in all patients. Biochemical and genetic studies were performed with the informed consent of all participants. Diagnosis of Gaucher disease was based on clinical features, appearance of Gaucher cells in samples of bones marrow, and low activity of acid β -glucosidase in leukocytes. Our experimental protocol was approved by the Committee on Human Experimentation at the University of Zaragoza, Zaragoza, Spain, and was in accordance with the Helsinki Declaration of 1975, as revised in 1983.
Clinical and analytical data were obtained from a questionnaire sent to pediatricians and hematologists who managed these patients with Gaucher disease in Spanish hospitals. The age-adjusted severity score index of Zimran et al.(17) was used to evaluate the clinical status. Enzymatic activity, genetic studies, and IGF determinations were done in a reference laboratory (the same for all the samples). Blood samples were obtained from patients after an overnight fast and stored in test tubes containing EDTA. Plasma was separated by low-speed centrifugation at 2000 g for 30 min at 4°C. The activity of glucocerebrosidase was measured on leukocytes with 4-methylumbelliferyl-β -d-glycopyranoside as substrate (18). Genomic DNA of all participants was isolated from white blood cells by standard methods (19, 20). The mismatched PCR method followed by digestion with Xho I was used to detect the N370S mutation (21); restriction analysis with Pvu II was used to detect the G377S mutation (22) and with Nci I and Msp I to analyze the L444P mutation (23). Major glucocerebrosidase gene rearrangements were determined with the procedure described by Beutler and Gelbart (24).
Baseline determinations in plasma samples of total IGF-I, free IGF-I, and IGFBP-3 were performed in 19 patients before starting ERT at 13.8 ± 11.2 y of age by immunoradiometric assay (IRMA) using the DSL-2800 ACTIVE Non-Extraction IGF-I Coated-Tube IRMA Kit, the DSL-9400 ACTIVE Free IGF-I Coated-Tube IRMA Kit, and the DSL-6600 ACTIVE IGF Binding Protein-3 Coated-Tube IRMA Kit, respectively (all from Diagnostic Systems Laboratories, Inc., Webster, TX, U.S.A.). The Spearman test was performed to establish the association with other clinical and analytical parameters. In a group of 13 patients receiving ERT, total IGF-I, free IGF-I, and IGFBP-3 were also measured during the first 2 y of treatment. The Wilcoxon test was performed for the statistical analysis. Total IGF-I and IGFBP-3 were expressed as SD score (SDS) using total IGF-I and IGFBP-3 standardized normal values by age and sex provided by Diagnostic Systems Laboratories and free IGF-I normal values provided by J. Argente and V. Barrios (“Niño Jesús” Hospital, Madrid, Spain). We have expressed the results in SDS because these proteins are age dependent.
RESULTS
The mean age at diagnosis of patients was 7.25 y (range, 0.3–14 y), with a male/female ratio of 12/10. The clinical and analytical data are summarized in Tables 1 and 2. Splenomegaly was present in all patients at diagnosis. In 18.1% of patients, the spleen was removed. Fifty percent of patients had Hb levels <11 g/dL, 45.4% had low platelet count (<100 × 109/ L) and 18.1% had low leukocyte count (<4.0 × 109/L). High acid phosphatase levels were observed in 100% of cases, and, in 63.6%, mild biochemical hepatic dysfunction was observed as a result of the alterations of liver transaminases. All of the studied patients on ERT experienced normalization of liver transaminases levels. Albumin, alkaline phosphatase, and bilirubin levels did not alter in the studied patients. The test for acid glucosidase showed a marked decrease in enzymatic activity in all patients. The two most common glucocerebrosidase mutations were N370S and the L444P, which accounted for 43.18% and 20.45% of all mutations, respectively (Table 3).
Total IGF-I, free IGF-I, and IGFBP-3 were determined in 19 patients before ERT. We found a significant association between total IGF-I SDS and the age-adjusted severity score index in this group before ERT (r = −0.62, p < 0.05). In the same group, total IGF-I and IGFBP-3 SDS correlated negatively with the presence of L444P mutation (r = −0.53 and −0.5, respectively, p < 0.05). Height SDS correlated with total IGF-I and IGFBP-3 SDS (r = 0.84 and 0.78, respectively, p < 0.05).
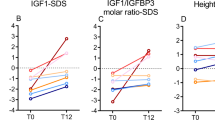
Changes in IGF axis during ERT during the first years of treatment were also studied. Total IGF-I SDS increased from −1.8 ± 0.8 to −0.8 ± 1.4 (p = 0.005) and free IGF-I SDS increased from −1.2 ± 1 to 1.1 ± 2.1 (p = 0.011) at 12 ± 6.8 mo of ERT (n = 10). IGFBP-3 showed the same response as it increased from −1.3 ± 0.6 to −0.2 ± 1.2 SDS (p = 0.012) at 12 ± 4.5 mo of ERT (n = 8) (Table 4). During the second year, total IGF-I SDS increased from −1.3 ± 0.7 to −0.5 ± 0.8 (p = 0.012) (n = 8). No significant changes were found during the second year or ERT for free IGF-I and IGFBP-3.
DISCUSSION
Type 1 Gaucher disease is characterized by splenomegaly, hepatomegaly, pancytopenia, and osteolytic and osteopenic degeneration of the skeleton (1). These are the classical features of Gaucher disease, but in some children growth retardation may be the sole manifestation of the disease. In these children, growth retardation has a valuable role in assessing severity and progression of Gaucher disease (8). Kaplan et al.(9) found a significant incidence of severe retardation of linear growth in untreated patients during the two first decades of life. They observed the following three patterns of growth: (1) in 28%, there was normal growth compared with both the general population and the children's parents; (2) in 22%, the growth pattern was normal for the general population but was low in comparison with the family; and (3) in 50%, the growth pattern was abnormal compared with both the general population and the child's family. Recently, Kauli et al.(25) has proven that growth retardation in childhood and delay of puberty are more frequent with severe disease. In 52.6% of a group of patients followed since early childhood, a deceleration of growth was observed at 3–5 y of age. Height SDS decreased from 0.34 at age 0–3 y to −1.93 (p < 0.01) at age 7–10 y, and was more pronounced with severe disease. With ERT, growth velocity increased dramatically at all ages in most children whose growth velocity was below normal (9).
The cause of this poor growth is not yet understood. It has been proven that chronic diseases with liver involvement are associated with growth retardation because of IGF deficiency (16, 17). Kaplan et al.(9) found a significant association between the z scores for height before treatment and liver enlargement (r = 0.57).
Mandel et al.(12), when examining the changes in IGF-I, IGFBP-3, GHBP, erythrocyte IGF-I receptors, and the growth rate in prepubertal GHD children during GH therapy, showed that the major correlates of growth are changes in the concentrations of circulating IGFBP-3 and IGF-I. IGFBP-3 acts synergistically with IGF-I in promotion of growth in GH deficiency, as well as normal children. In patients with type 1 Gaucher disease, we demonstrated that these two parameters, total IGF-I and IGFBP-3, show the best correlation with children's height before treatment (r = 0.84 and 0.78, respectively). IGF-I and IGFBP-3 are significantly reduced as the child's height is more affected. Total IGF-I has a significant negative correlation with the severity of the disease using Zimran's age-adjusted severity score index (r = −0.62). Both total IGF-I and IGFBP-3 correlate negatively with the presence of L444P mutation (r = −0.53 and −0.5, respectively). The presence of this mutation in type 1 Gaucher disease has been related with poor outcome (26). In this study, we found that the IGF axis is affected before treatment. This could be a consequence of liver involvement that could affect the synthesis of proteins, including IGF-I and IGFBP-3. The reduced levels of IGF-I and IGFBP-3 may reflect a GH insensitivity state, as has been shown in other chronic diseases.
The main consequence of IGF-I deficiency in childhood is growth retardation. However, recent research has proven that IGF have an anabolic effect even in adults. These peptides stimulate whole-body and muscle protein synthesis. The reduced IGF-I concentrations are associated with reduced muscle mass and strength (12). In GHD adults, it has been proven that the increase in IGF-I levels correlates with the changes in body composition (27) and serum lipid profile. Bengtsson et al.(27) demonstrated that quality of life, determined by a specific questionnaire, improved after 6 mo of GH therapy in those patients. These results could also be expected in Gaucher patients receiving ERT, because IGF-I is normalized.
When we analyzed the IGF axis response during ERT, we found that ERT results in significant increase of free IGF-I and IGFBP-3 during the first year of treatment and total IGF-I during the first 2 y of treatment. This could be explained by the normalization of liver function that would normalize the GH insensitivity state in the first months of ERT. There is a significant increase in IGF-I and IGFBP-3 levels that results in adequate IGF-I bioavailability.
In conclusion, we consider growth retardation in pediatric patients with Gaucher disease to be related to the alterations in the IGF axis. ERT results in significant increase of IGF during the first years of treatment, and further studies are necessary to clarify long-term response.
Abbreviations
- ERT:
-
enzyme replacement therapy
- IGFBP-3:
-
IGF binding protein 3
- ALS:
-
acid-labile subunit
- GHBP:
-
GH binding protein
- GHD:
-
GH deficiency
References
Beutler E, Grabowski GA 1983 Gaucher disease. In: Scriver CR, Beudet AL, Sly WS, Valle D (eds) The Metabolic Basis of Inherited Disease, 7th Ed. McGraw-Hill, New York, pp 2641–2670
Cormand B, Monfort M, Chabas A, Vilageliu L, Ginberg D 1997 Genetic fine localization of the beta-glucocerebrosidase (GBA) prosaposin (PSAP) genes: implications for Gaucher disease. Hum Genet 100: 75–77
Beutler E, Demina A, Gelbert T 1994 Glucocerebrosidase mutations in Gaucher disease. Mol Med 1: 82–92
Mistry PK 1995 Genotype/phenotype correlations in Gaucher's disease. Lancet 346: 982–983
Cox TM, Schofield JP 1997 Gaucher's disease: clinical features natural history. Baillieres Clin Haematol 10: 657–689
Kaminsky P, Belmatoug N, Billette de Villemeur T 1996 Maladie de Gaucher. Presse Med 25: 108–112
Rice EO, Mifflin TE, Sakallah S, Lee RE, Sansieri CA, Barranger JA 1996 Gaucher disease: studies of phenotype, molecular diagnosis treatment. Clin Genet 49: 111–118
Mistry PK, Abrahamov A 1997 A practical approach to diagnosis management of Gaucher's disease. Baillieres Clin Haematol 10: 817–837
Kaplan P, Mazur A, Manor O, Charrow J, Esplin J, Gribble TJ, Wappner RS, Wisch JS, Wienreb NJ 1996 Acceleration of retarded growth in children with Gaucher disease after treatment with alglucerase. J Pediatr 129: 149–153
Gluckman PD, Douglas RG, Ambler GR, Breier BH, Hodgkinson SC, Koena JB, Shaw JHF 1991 The endocrine role of insulin-like growth factor I. Acta Paediatr Scand Suppl 372: 97–105
Welle S 1998 Growth hormone insulin-like growth factor I as anabolic agents. Curr Opin Clin Nutr Metab Care 1: 257–262
Mandel SH, Moreland E, Rosenfeld RG, Gargosky SE 1997 The effect of GH therapy on the immunoreactive forms distribution of IGFBP-3, IGF-I, the acid-labile subunit, growth rate in GH-deficient children. Endocrine 7: 351–360
Labarta JI, Gargosky SE, Simpson DM, Lee PDK, Argente J, Guevera J, Rosenfeld RG 1997 Immunoblot studies of the acid labile subunit (ALS) in biological fluids, normal human serum in children with GH deficiency GH receptor deficiency before after long term therapy with GH or IGF-1 respectively. Clin Endocrinol 47: 657–666
Thissen JP, Ketelsegers JM, Underwood LE 1994 Nutritional regulation of the insulin-like growth factors. Endocr Rev 15: 80–101
Smith WJ, Underwood LE, Clemmons DR 1995 Effects of caloric or protein restriction on insulin-like growth factor 1 (IGF-1) IGF binding proteins in children adults. J Clin Endocrinol Metab 80: 443–449
Tonshoff B, Blum WF, Wingen AM, Mehls O 1995 Serum insulin-like growth factors (IGFs) IGF binding proteins 1,2, 3 in children with chronic renal failure: relationship to height glomerular filtration rate. J Clin Endocrinol Metab 80: 2684–2691
Zimran A, Sorge J, Gross E, Kubitz M, West C, Beutler E 1989 Prediction of severity of Gaucher's disease by identification of mutations at DNA level. Lancet 2: 349–352
Raghavan SS, Topol J, Kolodny EH 1980 Leukocyte beta-glucosidase in homozygotes heterozygotes for Gaucher disease. Am J Hum Genet 32: 158–173
Miller S, Dykes D, Polensky H 1988 A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16: 1215
Pocovi M, Cenarro A, Civeira F, Torralba MA, Pérez-Calvo JI, Mozas P, Giraldo P, Giralt M, Myers RH, Cupples LA, Ordovas JM 1998 β-glucocerebrosidase gene locus as a link for Gaucher's disease familial hypo-α-lipoproteinaemia. Lancet 351: 1919–1923
Beutler E, Gelbart T, West C 1990 The facile detection of the nt 1226 mutation of glucocerebrosidase by mismatched PCR. Clin Chim Acta 194: 161–166
Laubscher KH, Glew RH, Lee RE, Okinaka RT 1994 Use of denaturing gradient gel electrophoresis to identify mutant sequences in the beta-glucocerebrosidase gene. Hum Mutat 3: 411–415
Uchiyama A, Tomasu S, Kondo N 1994 New Gaucher disease mutations in exon 10; a novel L444R mutation produces a new NciI site the same as L444P. Hum Mol Genet 3: 411–415
Beutler E, Gelbart T 1994 Erroneous assignment of Gaucher disease genotype as consequence of a gene deletion. Hum Mutat 4: 212–226
Kauli R, Zaizov R, Lazar L, Pertzelan A, Laron Z, Galatzer A, Phillip M, Yaniv Y, Cohen IJ 2000 Delayed growth puberty in patients with Gaucher disease type 1: natural history effect of splenectomy /or enzyme replacement therapy. Isr Med Assoc J 2: 158–163
Mistry PK, Smith SJ, Ali M, Hatton CSR, McIntyre N, Cox TM 1992 Genetic diagnosis of Gaucher's disease. Lancet 339: 889–892
Bengtsson BA, Abs R, Bennmarker H, Monson JP, Feldt-Rasmussen U, Hernberg-Stahl E, Westberg B, Wilton P, Wuster C 1999 The effects of treatment the individual responsiveness to growth hormone (GH) replacement therapy in 665 GH-deficient adults. J Clin Endocrinol Metab 84: 3929–3935
Acknowledgements
Spanish Gaucher's Disease Registry participants: Dr. Calabuig, Dr. Dalmau (Hospital Universitario La Fé, Valencia); Dr. Gracia Antequera (Hospital Doctor Peset, Valencia); Dr. Ferriols, Dr. Tomás (Hospital General, Valencia); Dr. Pastor (Hospital de Xativa); Dr. Martínez Odriozola (Hospital de Basurto, Bilbao); Dr. Sanjurjo (Hospital Universitario Cruces, Bilbao); Dr. Albadalejo (Hospital Sta. María del Rosell, Murcia); Dr. Martínez Pardos (Hospital Ramón y Cajal, Madrid); Dr. Aguado (Hospital La Paz, Madrid); Dr. Loyola (Hospital Montecelo, Pontevedra); Dr.Vale (Hospital Juan Canalejo/Virxe da Junqueira, A Coruña); Dr. Martín Núñez, Dr. Fernández Galán (Hospital Virgen del Puerto, Plasencia); Dr. Santos (Hospital San Pedro de Alcántara, Cáceres); Dr. Toll (Hospital San Juan de Diós, Barcelona); Dr. Domínguez, Dr. Castelló (Hospital Vall d'Hebrón, Barcelona); Dr. Salinas (Hospital General, Manresa); Dr. Romero Gómez (Hospital del Río Hortega, Valladolid); Dr. López González (Complejo Hospitalario de León, León); Dr. Aguiar (Hospital Ntra. Sra. del Pino, Las Palmas); Dr. Baldellou, Dr. Giraldo (Hospital Universitario Miguel Servet, Zaragoza); Dr. Figueredo, Dr. Reyes Santos (Hospital Virgen Macarena, Sevilla); Dr. Alonso, Dr. Plaza (Hospital Virgen del Rocío, Sevilla); Dr. Mendoza (Hospital Universitario Puerto Real, Cádiz); Dr. Fornell (Hospital Puerta del Mar, Cádiz); Dr. Romero Gallardo (Hospital de Jerez de la Frontera); Dr. Zarate (Hospital Punta Europa, Algeciras); Dr. López Muñoz (Hospital de Torrecárdenas, Almerí a).
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Supported by FIS 00/0546 and Spanish Foundation for the Study and Treatment of Gaucher Disease (FEETEG).
Rights and permissions
About this article
Cite this article
Rite, S., Baldellou, A., Giraldo, P. et al. Insulin-Like Growth Factors in Childhood-Onset Gaucher Disease. Pediatr Res 52, 109–112 (2002). https://doi.org/10.1203/00006450-200207000-00020
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200207000-00020
This article is cited by
-
Assessing the diagnostic utility of the Gaucher Earlier Diagnosis Consensus (GED-C) scoring system using real-world data
Orphanet Journal of Rare Diseases (2024)
-
Endocrine and metabolic disorders in patients with Gaucher disease type 1: a review
Orphanet Journal of Rare Diseases (2019)
-
Long-term effect of imiglucerase in Latin American children with Gaucher disease type 1: lessons from the International Collaborative Gaucher Group Gaucher Registry
BMC Hematology (2014)
-
Effects of imiglucerase on the growth and metabolism of Gaucher disease type I patients: a systematic review
Nutrition & Metabolism (2013)
-
Pediatric Gaucher disease type I and mild growth hormone deficiency: a new feature?
Journal of Inherited Metabolic Disease (2010)
