
Abstract
Although tyrosine is considered indispensable during the neonatal period, its poor solubility has limited its inclusion in parenteral amino acid solutions to less than 1% of total amino acids. Dipeptides of tyrosine are highly soluble, have been shown to be well used and safe in animal models and humans, and, therefore, may be used as an effective means of providing tyrosine in the parenterally fed neonate. The goal of the present study was to determine the tyrosine requirement of the parenterally fed neonate receiving graded intakes of glycyl-l-tyrosine as a source of tyrosine. Thirteen infants receiving adequate energy (340 ± 38 kJ·kg-1·d-1) and protein (2.4 ± 0.4 g·kg-1·d-1) were randomized to receive parenteral nutrition with one of five graded levels of glycyl-l-tyrosine. The mean requirement and safe level of intake were estimated using a 1-13C-phenylalanine tracer and linear regression cross-over analysis that identified a break point in the response of label appearance in breath CO2 (F13CO2) and phenylalanine oxidation to graded tyrosine intake. Based on the mean estimates of whole-body phenylalanine oxidation, the tyrosine mean requirement and safe level of intake were found to be 74 mg·kg-1·d-1 and 94 mg·kg-1·d-1, respectively. This represents 3.1 and 3.9% of total amino acids, respectively, considerably higher than levels found in present commercially available pediatric amino acid solutions. These data raise concern regarding the adequacy of aromatic amino acid intake in the parenterally fed neonate.
Similar content being viewed by others
Main
Nearly three decades ago, Snyderman (1) suggested that the neonate requires a preformed source of tyrosine. Presently, the main impediment to meeting total aromatic amino acid requirements in the parenterally fed neonate is the poor solubility of tyrosine (0.453 g/L in H2O at 25°C) (2), limiting its inclusion in parenteral amino acid solutions to <1% of total amino acids. When compared with neonatal reference proteins such as human milk or human fetal tissue, which contain approximately equal quantities of phenylalanine and tyrosine (3.7–4.1 and 2.9–4.6 g/100 g protein, respectively) (3–5), the levels provided by these synthetic nutrient solutions appear grossly inadequate. To circumvent this problem, increased phenylalanine has been included in some solutions in hope that the neonate will endogenously hydroxylate sufficient phenylalanine to tyrosine to meet total aromatic amino acid needs. However, elevated plasma phenylalanine levels have been observed with this approach (6–11), even though it has been shown that increased phenylalanine intake is associated with an absolute greater rate of in vivo phenylalanine hydroxylation (12).
Derivatives of tyrosine have also been investigated as soluble precursors of tyrosine. N-acetyl-tyrosine, for example, has been included in one of the commercially available pediatric amino acid solutions, TrophAmine (McGaw Inc., Irvine, CA, U.S.A.) (13). Unfortunately, N-acetyl-tyrosine has been shown to be poorly used in the parenterally fed piglet, which excreted 65% of intake (14), and in the human neonate who also has demonstrated poor utilization through the excretion of large quantities of the derivative (13, 15–17). In contrast, there is evidence that dipeptides of tyrosine can serve as a soluble source of tyrosine in pigs (14, 18, 19), primates (20), and adult humans (21–25). To improve the utilization of the presently available pediatric amino acid solutions, a soluble source of tyrosine must be included. At present, dipeptides seem to be the most likely candidate to meet this objective.
Using graded intakes of tyrosine in the form of GT, House et al.(18) estimated the mean requirement and safe tyrosine intake (95% upper confidence limit) in the piglet receiving a fixed phenylalanine intake of 4.1% of total amino acids to be 2.7 and 3.2% of total amino acids, respectively. This is significantly higher than the level of tyrosine in currently available pediatric amino acid solutions (0.9% of total amino acids). The lower level of total aromatic amino acids is responsible for the reduced growth and nitrogen retention in piglets receiving the pediatric amino acid solution Vaminolact (Pharmacia and Upjohn, Stockholm) versus the general purpose solution Vamin (Pharmacia and Upjohn, Stockholm) (26).
Due to the immaturities in the neonatal tyrosine catabolic enzyme pathway, tolerance of tyrosine intakes at levels greatly over requirement is limited (27). In addition, due to the known neurologic impairment caused by hypertyrosinemia to the developing brain as assessed by lower IQ and psychologic tests, excess intakes must be avoided (28, 29).
The tyrosine requirement in the neonate receiving parenteral nutrition has not been determined. In the present study, this was determined by partitioning the oxidation of phenylalanine to graded intake of tyrosine at a fixed phenylalanine intake.
METHODS
Patients and nutrient intake.
Thirteen parenterally fed neonates (Table 1) who had been referred to the Neonatal Intensive Care Unit at The Hospital for Sick Children were enrolled into the study. All infants were clinically stable during the study period and were maintained in standard servo-controlled incubators or infant cribs. Intestinal atresia, necrotizing enterocolitis, gastroschisis, and malrotation were the diagnoses of infants enrolled in the study (Table 1). The study protocol was reviewed and approved by the Human Subject Review Committee of The Hospital for Sick Children, and written informed consent was obtained from one or both parents.
Study subjects were enrolled into the study when receiving adequate amino acid and energy intake (30); targeted minimal intakes were 2 g·kg-1·d-1 of amino acid and 270 kJ·kg-1·d-1. Infants were randomized to receive TPN containing a pediatric amino acid solution, Primene (Baxter, Deerfield, IL, U.S.A.), with one of five levels of GT supplement (ICN Biomedicals, Mississauga, Ontario) as the source of tyrosine for 2 d. The base amino acid profile of Primene is shown in Table 2. The five levels of GT were 0.18, 0.35, 0.52, 0.68, and 1.01 mg/mL of parenteral amino acid solution. Nutrient intake was as prescribed by the attending physician. Actual individual intakes of amino acid, lipid, carbohydrate, phenylalanine, and tyrosine are described in Table 3. There were no enteral feeds consumed by the subjects.
Study protocol.
Isotopes used for the tracer study were l-[1-13C]phenylalanine (99%, 13C, Tracer Technologies, Sommerville, MA, U.S.A.) and l-[3,3-2H2]tyrosine (98%, 2H2, Cambridge Isotope Laboratories, Andover, MA, U.S.A.). Quality control tests were performed by the manufacturers. Chemical purity, specified isotopic enrichment, and position were confirmed by nuclear magnetic resonance, whereas isomeric purity and a second confirmation of isotopic enrichment were performed by gas chromatography mass spectrometry. Solutions of each tracer amino acid in 3.3% dextrose and 0.3% NaCl were prepared by the Manufacturing Pharmacy at The Hospital for Sick Children. Isotope solutions were sterilized by passage through a 0.22-μm filter before being dispensed into single-use vials for storage at 4°C until use. All solutions were demonstrated to be sterile and free of bacterial growth over 7 d in culture and to be pyrogen-free by the Limulus amebocyte lysate test (31).
The phenylalanine dose required to achieve a measurable expired 13CO2 was determined in a previous study (12). At least 24 h after the start of the infusion of TPN, phenylalanine flux, hydroxylation, oxidation, percent dose oxidized, and tyrosine flux were determined using a primed i.v. 24-h constant infusion of l-[1-13C]phenylalanine (15.6 μmol/kg and 13 μmol·kg-1·h-1, respectively) and l-[3,3-2H2]tyrosine (3.6 μmol/kg and 3.0 μmol·kg-1·h-1, respectively). The priming dose was infused over the first 15 min of isotope infusion. Urine samples were collected for the measurement of background and steady state tracer amino acid enrichment and creatinine. Urine was used for measuring amino acid enrichment, as it is considered to be a valid representation of plasma enrichment (32, 33), provided the isotopically labeled amino acids are d-isomer-free (34). One to three baseline urine samples were collected before parenteral isotope infusion, followed by one sample every 3–4 h throughout the infusion. Urine was stored at −20°C until analyses were performed. Expired CO2 was collected, and the production rate measured before and after 17–20 h of constant isotope infusion.
Analytical procedures and calculations.
Urine was analyzed for phenylalanine and tyrosine enrichment as described previously (12).
Urinary creatinine was measured by the Jaffe reaction by using a Kodak Ektachem 700 Analyzer (Johnson and Johnson, Rochester, NY, U.S.A.). Urinary phenylalanine, tyrosine, glycine, and GT concentrations were analyzed by ion-exchange chromatography (Beckman 7300 Amino Acid Analyzer), expressed in micromoles per millimole of creatinine. Urinary excretion was determined using standards of known amounts of amino acid or dipeptide. The concentration, standardized to creatinine excretion, was determined using an external standard and was calculated with the following equation: EQUATION

The concentration of the external standard was 5 μg/μL. S2-aminoethyl-cysteine (400 μmol/L) was used as internal standard.
Expired CO2 production collected from a ventilated hood system was measured directly using a portable CO2 analyzer (1400 series, Servomex, Westech Industrial Ltd., Mississauga, Ontario) and mass flowmeter (5860 series, Brooks, Trillium Measurement and Control, Stouffville, Ontario). Expired 13CO2 enrichment was determined using isotope-ratio mass spectrometry as previously described (12).
The model of amino acid metabolism used in this study is a modification of the Waterlow et al.(35) model: EQUATION where Q is the rate of phenylalanine flux (μmol·kg-1·h-1);S is the rate of phenylalanine nonoxidative disposal, a measure of the rate of phenylalanine incorporation into body protein;PAH is the rate of phenylalanine hydroxylation to tyrosine;B is the rate of phenylalanine release from body protein; and I is the rate of exogenous phenylalanine intake.

Whole-body phenylalanine and tyrosine turnover was calculated from dilution of the isotope in the body amino acid pool at isotopic steady state (32, 33, 36) : EQUATION

where Qphe is the rate of phenylalanine or tyrosine (Qtyr) flux, i is the isotope infusion rate (μmol·kg-1·h-1), Ei is the enrichment of the infused isotope, and Ep is the enrichment of the amino acid in the urine at the isotopic steady state.
To measure the rate of phenylalanine hydroxylation, the model of Clarke and Bier (37) with the modification of Thompson et al.(38) was used.
EQUATION

where Etyr and Ephe are the urinary enrichments of l-[1-13C]tyrosine and l-[1-13C]phenylalanine, respectively; and iphe is the rate of infusion of the labeled phenylalanine.
The rate of phenylalanine oxidation was calculated as described by Matthews et al.(36) : EQUATION

where Ophe represents phenylalanine oxidation, and F13CO2 represents the rate of 13CO2 release by phenylalanine tracer oxidation (μmol·kg-1·h-1) calculated from the following equation: EQUATION

where FCO2 is the CO2 production rate (cm3/min), and ECO2 is the 13CO2 enrichment in expired breath at the isotopic steady state. The constants 44.6 μmol/cm3 and 60 min/h convert FCO2 to micromoles per hour, and the factor of 100 changes the enrichment in atoms percent into a fraction. W is the weight (kg) of the infant. 13CO2 retained by the body as bicarbonate is corrected for by using a retention factor calculated from a regression equation, using energy intake as the predictor variable (39).
Statistical analyses.
The study followed a completely randomized design with tyrosine intake as the independent variable. Regression models (linear, quadratic, linear regression cross-over model) were fitted to the data to determine the relationship that best described the response of the dependent variables phenylalanine hydroxylation, phenylalanine F13CO2 and oxidation, and tyrosine flux to increasing tyrosine intake. The regression analyses were performed by the method of least squares by use of the regression procedure of SAS (40). Selection of the best model was determined by factors relating to fit (significance of the model and r2) and estimates of variation about the model (coefficient of variation and SE of the estimate).
A two-phase regression cross-over model (41) was found to be the most appropriate analysis for the data (phenylalanine F13CO2 and oxidation), and a break point was objectively identified. As described previously (42, 43), the analysis involves partitioning the response of the dependent variables to tyrosine intake into data points fitting either a flat line (zero slope) representing obligatory phenylalanine oxidation when tyrosine intake is inadequate or a sloped line representing phenylalanine oxidation when tyrosine intake is above requirement. The break point of the two linear regression lines is considered representative of the group mean tyrosine requirement. The equation for the statistical model is: EQUATION

where Y is the individual observation of the dependent variable phenylalanine F13CO2 or oxidation;A1 and A2 are the intercepts of the first and second line, respectively;B1 and B2 are the slopes of the first and second lines, respectively;D equals 0 if the observation is from the first line and 1 if it is from the second line;x is tyrosine intake; and E is the residual error associated with the model. The safe population intake was estimated by determining the upper 95% confidence limits of the break point estimate (41).
RESULTS
Clinical characteristics and nutrient intake.
Subject information including clinical characteristics, nutrient intake, and diagnoses are presented in Table 1. Parenteral nutrition was as prescribed by the attending physician, and, therefore, the exact nutrient intakes were dependent on the total volume of parenteral nutrition infused. The average energy and protein intakes were 340 ± 38 kJ·kg-1·d-1 (81 ± 9 kcal·kg-1·d-1) and 2.4 ± 0.4 g·kg-1·d-1, respectively (Table 3). Mean phenylalanine intake was 97 ± 13 mg·kg-1·d-1. The tyrosine intake reflected the level of GT to which the neonate was assigned and ranged from 29 to 108 mg·kg-1·d-1, whereas the phenylalanine intake was that present in the base parenteral amino acid solution (Table 3).
Urinary amino acid and expired CO2 enrichment.
Plateau in amino acid enrichment was achieved for all neonates by 9 h and was defined by the absence of a significant slope. The variation in phenylalanine and tyrosine amino acid enrichment within the plateau was <12%. Individual enrichments of l-[1-13C]phenylalanine, l-[1-13C]tyrosine, l-[3,3-2H2]tyrosine, and expired 13CO2 are shown in Table 4.
Phenylalanine and tyrosine kinetics.
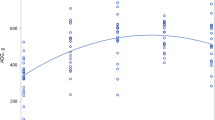
There was no significant relationship between tyrosine intake and phenylalanine flux (Table 5). There was, however, a significant quadratic response between phenylalanine hydroxylation and tyrosine intake (p < 0.004, r2 = 0.59; Table 5) with hydroxylation being lowest and equal at the lowest and highest tyrosine intake levels. There was also a significant linear relationship between tyrosine intake and tyrosine flux (p < 0.006, r2 = 0.51). The two-phase linear regression cross-over model proved to be the best fit for the F13CO2 data and the phenylalanine oxidation data. A break point in phenylalanine F13CO2 at 66 mg·kg-1·d-1 (p < 0.008, r2 = 0.7; Fig. 1) was identified, and in phenylalanine oxidation at 82 mg·kg-1·d-1 (Fig. 2). Increasing tyrosine intake above the break point level resulted in increased F13CO2 and oxidation. These break points, objectively identified by the two-phase linear regression cross-over model, are representative of the mean tyrosine requirement for the neonate. The 95% upper confidence limits were found to be 90 and 97 mg·kg-1·d-1 for the F13CO2 and oxidation estimate, respectively (Figs. 1 and 2).

The effect of increasing tyrosine intake via GT on F13CO2 production from phenylalanine oxidation. The dotted vertical lines represent the 95% confidence limits of the break point estimate.

The effect of increasing tyrosine intake via GT on phenylalanine oxidation. The dotted vertical lines represent the 95% confidence limits of the break point estimate.
Urinary amino acid concentrations.
Urinary phenylalanine, tyrosine, and glycine excretion were generally within the normal range for neonates (phenylalanine = 0–151 μmol/mmol creatinine, tyrosine = 0–200 μmol/mmol creatinine, glycine = 254–2341 μmol/mmol creatinine) (4). However, subject 4, receiving a tyrosine intake of 52.5 mg·kg-1·d-1, excreted an above average amount of phenylalanine and tyrosine in the urine, suggesting that the plasma concentration of these amino acids may also have been elevated (Table 6). This neonate did not demonstrate any evidence of abnormal liver function based on results of serum alanine aminotransferase or serum aspartate aminotransferase measurements performed near the time of the study. The individual receiving the highest tyrosine intake (108.1 mg·kg-1·d-1) demonstrated the greatest excretion of tyrosine in the urine, although levels remained within the expected ranges for the neonate (Table 6). The additional glycine intake from the dipeptide increased the glycine intake from 4.2 to 5.3% total amino acids. Urinary glycine excretion did not differ in relation to the level of GT intake (Table 6). Four (subjects 3, 6, 9, 11) of the thirteen subjects demonstrated a small amount of dipeptide in the urine, in each case representing <10 μmol/mmol creatinine (Table 6).
DISCUSSION
Using nitrogen balance and growth measures, Snyderman (1) identified that the enterally fed neonate may require a preformed source of tyrosine at levels ranging from 50 to 120 mg·kg-1·d-1. The estimate of the tyrosine requirement for the enterally fed neonate does not necessarily apply to neonates fed parenterally because recent evidence has demonstrated that the splanchnic organs play a major role in amino acid metabolism, and its bypass can significantly alter the needs of individual amino acids (44–50). Starting with a piglet model, we have studied the aromatic amino acid requirements of the parenterally fed neonate. First, we showed in the piglet that the current pediatric TPN amino acid mixtures are limiting in total aromatic amino acids (14), a finding which we have recently confirmed in human neonates (12). Further, we showed that both the piglet and human neonates have active hydroxylation of phenylalanine to tyrosine (12, 14). Further, in the piglet, we showed that phenylalanine was an effective source of tyrosine for protein synthesis and growth measured by nitrogen balance, but at the cost of high plasma phenylalanine levels as well as elevated urinary excretion of alternate catabolites of phenylalanine and tyrosine (14). We have recently confirmed this observation in human neonates (12). In the piglet, we have determined both tyrosine (18) and phenylalanine (19) requirements by using amino acid oxidation techniques. The present work was designed to determine whether the human neonate used GT and to obtain an estimate of tyrosine requirements at the moderate phenylalanine intake level in a commercial pediatric TPN mixture. The isotope model we used took advantage of the fact (based on our piglet work) that the phenylalanine concentration in Primene was modestly higher than requirement. After sufficient tyrosine was given, the excess phenylalanine would be metabolically channeled within the liver to oxidation (51). Therefore, the break point in either the F13CO2 or phenylalanine oxidation versus tyrosine intake (Figs. 1 and 2) would be an estimate of the population average tyrosine needs, and the 95% confidence interval would cover 95% of the population and, hence, would be a safe level for the population as a whole.
The present data are, to our knowledge, the first estimate of tyrosine requirement in the parenterally fed human neonate. Using a linear regression cross-over analysis, the estimated average tyrosine requirement at a fixed and adequate but not excessive phenylalanine intake of 4.0% of total amino acids was 66 and 82 mg·kg-1·d-1 for the estimates based on 13CO2 production (F13CO2) and phenylalanine oxidation, respectively. These intakes represent 2.8 and 3.4% of total amino acid intake, respectively. The upper 95% confidence limit, representative of the safe population intake, was estimated as 90 and 97 mg·kg-1·d-1 for phenylalanine F13CO2 and oxidation data, respectively. These intakes represent between 3.8 and 4.0% of total amino acids. Another way of examining these results is to compare the net protein deposition occurring in the three infants receiving tyrosine intake above the 95% confidence interval with the three infants receiving the lowest intakes of tyrosine. Net protein deposition was calculated from phenylalanine balance (intake − hydroxylation) and assuming there are 272 μmol of phenylalanine per gram of mixed body protein (52). Net protein deposition in the three infants receiving the highest tyrosine intakes was 50% higher (1.02 g·kg-1·d-1) than the group receiving the lowest tyrosine intake (0.68 g·kg-1·d-1). By using the value of 272 μmol of phenylalanine per gram of mixed body protein, it is also possible to convert the mean phenylalanine flux rate of 125 μmol·kg-1·h-1 to a whole-body protein flux (turnover) rate of 11.0 g·kg-1·d-1. This value is fairly similar to an earlier estimate of 9.5 g·kg-1·d-1 that we obtained for parenterally fed neonates by using 15N-glycine as a tracer and 15N-urea as an end-product (53).
The present results are supported by similar estimates of tyrosine in the parenterally fed piglet. The estimated mean requirement and safe level of intake in the piglet model was found to represent 2.1 and 2.4% total amino acids at a fixed phenylalanine intake of 4.0% of total amino acids. Further, the present results are within the estimated range of tyrosine requirement in the orally fed infant receiving adequate phenylalanine intake (50–120 mg·kg-1·d-1) (1). Nonetheless, the method used in the present study is novel and has not been previously used. It would, therefore, be desirable in future studies to confirm our estimate of tyrosine requirements by using other established methods such as indicator amino acid oxidation (42), which directly reflects the use of the test amino acid (in this case tyrosine) for whole-body protein synthesis.
Based on the data of the present study, the proportional balance of the mean aromatic amino acid requirements is 56% phenylalanine and 44% tyrosine. This ratio is similar to that found in the piglet model of the neonate (59:41) (18, 19) and to the proportional phenylalanine to tyrosine content of fetal tissue (59:41) (52). Provision of aromatic amino acids in this recommended balance should lower the incidence of hyperphenylalaninemia and hypertyrosinemia observed in neonates receiving high intakes of phenylalanine (6–8, 11, 26, 54, 55) or tyrosine (14). Because the long-term neurologic effects of low or elevated tyrosine intake are unknown, efforts should be made to provide intakes that meet needs without great excess.
The present estimate of tyrosine requirement was determined at a phenylalanine intake of 97 ± 13 mg·kg-1·d-1, representing a phenylalanine intake of 4% of total amino acids. This intake is above the level determined by House et al.(18) in the piglet model to be the safe level of phenylalanine requirement (3.2% of total amino acids). The impact of such a small excess of phenylalanine on the tyrosine requirement is unknown but may be that the tyrosine requirement was underestimated, because phenylalanine can be hydroxylated through hepatic phenylalanine hydroxylase to form tyrosine. Whether this is so remains to be determined; however, because the phenylalanine excess was small, any underestimation of tyrosine needs should also be small. In addition, a small excess of phenylalanine would not change the estimate of total aromatic amino acid requirements, only the proportion of phenylalanine to tyrosine. Also, because the estimated balance of phenylalanine to tyrosine is so similar to the piglet data and the composition of human fetal tissue, the phenylalanine excess must have been very small.
Although a significant quadratic response of phenylalanine hydroxylation to increasing tyrosine intake was observed, the estimate of phenylalanine hydroxylation did not provide meaningful results toward estimating the tyrosine requirement. The response of phenylalanine hydroxylation to graded tyrosine intake was unexpected. The rate of phenylalanine hydroxylation was similar at both the lowest and highest intake of tyrosine. The predicted response was that hydroxylation would be higher at the lowest intake of tyrosine and decrease as tyrosine intake approached requirement. These data reveal that the estimate of phenylalanine hydroxylation by use of plasma amino acid enrichments and the Clarke and Bier (37) model as modified by Thompson et al.(38) cannot be used as an index of the adequacy of tyrosine intake in the neonate. Phenylalanine hydroxylation estimates have been previously shown to poorly reflect net tissue phenylalanine and tyrosine accumulation in the parenterally fed piglet receiving a high level of phenylalanine intake (56). Such discrepancies may be attributed to limitations of the model, possibly due to the fact that plasma (or urine as a surrogate) enrichment does not accurately reflect the precursor pool enrichment. As new approaches to quantify intrahepatocyte precursor pool enrichment become available, such as using the rapid turnover hepatic protein apolipoprotein B100 (57), the hydroxylation estimate will need to be reassessed as a measure of the adequacy of tyrosine intake.
Urinary excretion of phenylalanine, tyrosine, glycine, and GT were monitored to provide some insight into the metabolic handling of the doses of aromatic amino acids and GT. Plasma levels were not measured to minimize the invasiveness of the study protocol. In general, the excretion of the three amino acids was within normal ranges, and the excretion of GT was either nondetectable or appeared in trace amounts. These data suggest that the intakes of these amino acids were not at levels that cause overload to the neonate's metabolic pathways. The lack of significant excretion of the dipeptide, even at the highest intake of GT supplementation, suggests that it was rapidly hydrolyzed into its respective amino acids.
One subject (subject 4) who received a tyrosine intake below the estimated requirement level (GT = 52.5 mg·kg-1·d-1) experienced urinary phenylalanine and tyrosine levels that were above expected norms for the neonate. This suggests that the plasma concentrations of phenylalanine and tyrosine were elevated (4). The greater than normal excretion of phenylalanine and tyrosine with a modest aromatic amino acid intake in conjunction with the lack of evidence of impaired liver function from liver enzyme tests suggests that this neonate may have had an immaturity in the tyrosine catabolic pathway. In fact, tyrosine aminotransferase (E.C. 2.6.1.5) (27, 58) and 4-hydroxyphenylpyruvate dioxygenase (E.C. 1.13.11.27) (58, 59) are considered to be immature during the neonatal period.
Most of the subjects had undetectable excretion of the dipeptide GT in the urine. Four individuals excreted trace amounts of GT, and, within the subjects with dipeptide excretion, the presence of the dipeptide was not related to the intake. In fact, the two individuals with the greatest intake of the dipeptide did not present with detectable amounts of GT in the urine. This efficient handling of the dipeptide provides further evidence that GT is well used in the human neonate.
Additional glycine associated with the dipeptide was infused. This amounted to 4 to 29 mg·kg-1·d-1 and reflected an increase of glycine intake from 4.2 to 5.3% of total amino acids. Although the glycine intake was increased, the excretion of glycine did not correlate with increasing GT infused, suggesting that the additional glycine load did not greatly impact the plasma glycine pool.
In conclusion, this is the first estimate of tyrosine requirement in the parenterally fed human neonate. These data specifically demonstrate that the mean tyrosine requirement of the parenterally fed neonate lies between 66 and 82 mg·kg-1·d-1, representing 2.8 to 3.4% of total amino acids. The corresponding safe estimate of intake was 90 to 97 mg·kg-1·d-1, corresponding to 3.8 to 4.0% of total amino acids. The ideal balance of phenylalanine to tyrosine was found to be 56:44 based on the mean requirement estimate. This balance of aromatic amino acid intake is similar to that found in the piglet model of the neonate (59:41) (18, 19) as well as that of fetal tissue composition (59:41) (52). Therefore, the ratio may be similar even for lower-birth-weight infants. However, because the total amino acid (protein) needs of the very-low-birth-weight infant are higher (52, 60), the total safe aromatic amino acid intake may also be higher. On the basis of the present research, the total safe aromatic amino acid intake that meets the needs of the neonate is approximately 7.9% of total amino acids. This total aromatic amino acid intake is below the intake of the phenylalanine-supplemented solutions such as Vamin, which contain 8.6% of the total amino acids as phenylalanine and tyrosine. In contrast, the requirement estimate in the present experiment is well above that found in current pediatric amino acid solutions (5.1% of total amino acids). These data provide valuable information toward the development of a parenteral amino acid solution that better meets the needs of the neonate.
Abbreviations
- TPN:
-
total parenteral nutrition
- FCO2:
-
breath carbon dioxide production
- F13CO2:
-
breath 13CO2 production
- GT:
-
glycyl-l-tyrosine
References
Snyderman SE 1971 The protein and amino acid requirements of the premature infant. In: Jonxis JHP, Visser HKA, Troelstra JA(eds) Metabolic Processes in the Foetus and Newborn. Stenfert Kroese, Leiden, The Netherlands, 128–141.
Merck Index 1983 10th Ed. Merck and Co., Inc., Rahway, NJ
Pencharz PB, House JD, Wykes LJ, Ball RO 1996 What are the essential amino acids for the preterm and term infant? In: Bindels JG, Goedhart AC, Visser HKA(eds) Recent Developments in Infant Nutrition (10th Nutricia Symposium). Kluwer Academic Publishers, Dordrech, 278–296.
Bremer HJ, Duran M, Kamerling JP, Przyrembel H, Wadman SK 1981 Disturbances of Amino Acid Metabolism: Clinical Chemistry and Diagnosis. Urban and Schwarzenberg, Baltimore, MD.
Rassin DK 1989 Amino acid metabolism in total parenteral nutrition during development. In: Friedman M(ed) Absorption and Utilization of Amino Acids. CRC Press, Boca Raton, FL, 71–85.
Chessex P, Zebiche H, Pineault M, Lepage D, Dallaire L 1989 Effect of amino acid composition of parenteral solutions on nitrogen retention and metabolic response in very-low-birth-weight infants. J Pediatr 106: 111–117
Puntis JW, Edwards MA, Green A, Morgan I, Booth IW, Ball PA 1986 Hyperphenylalaninaemia in parenterally fed newborn babies. Lancet 2: 1105–1106
Rigo J, Senterre J 1987 Significance of plasma amino acid pattern in preterm infants. Biol Neonate 52: 41–49
Mitton SG, Burston D, Brueton MJ 1988 Hyperphenylalaninaemia in parenterally fed newborn infants. Lancet 2: 1497–1498
Puntis JW, Ball PA, Preece MA, Green A, Brown GA, Booth IW 1989 Egg and breast milk-based nitrogen sources compared. Arch Dis Child 64: 1472–1477
Walker V, Mills GA 1990 Metabolism of intravenous phenylalanine by babies born before 33 weeks of gestation. Biol Neonate 57: 155–166
Roberts SA, Ball RO, Filler RM, Moore AM, Pencharz PB 1998 Phenylalanine and tyrosine metabolism in neonates receiving parenteral nutrition differing in pattern of amino acids. Pediatr Res 44: 907–914
Heird WC, Dell RB, Helms RA, Greene HL, Ament ME, Karna P, Storm MC 1987 Amino acid mixture designed to maintain normal plasma amino acid patterns in infants and children requiring parenteral nutrition. Pediatrics 80: 401–408
Wykes LJ, House JD, Ball RO, Pencharz PB 1994 Aromatic amino acid metabolism of neonatal piglets receiving TPN: effect of tyrosine precursors. Am J Physiol 267: E672–E679
Adamkin DH, McClead RE, Desai NS, McCulloch KM, Marchildon MB 1991 Comparison of two neonatal intravenous amino acid formulations in preterm infants: a multicenter study. J Perinatol 11: 375–382
Hanning RM 1993 Total parenteral nutrition for the low-birth-weight infant: retention of N-acetyl tyrosine. J Can Diet Assoc 54: 25–28
van Goudoever JB, Sulkers EJ, Timmerman M, Huijmans JGM, Langer K, Carnielli VP, Sauer PJJ 1994 Amino acid solutions for premature neonates during the first week of life: the role of N-acetyl-l-cysteine and N-acetyl-l-tyrosine. JPEN 18: 404–408
House JD, Pencharz PB, Ball RO 1997 Tyrosine kinetics and requirement during total parenteral nutrition in the neonatal piglet: the effect of glycyl-l-tyrosine supplementation. Pediatr Res 41: 575–583
House JD, Pencharz PB, Ball RO 1997 Phenylalanine requirements determined by using l-[1- 14C]phenylalanine in neonatal piglets receiving total parenteral nutrition supplemented with tyrosine. Am J Clin Nutr 65: 984–993
Steinhardt HJ, Paleos GA, Brandl M, Fekl WL, Adibi SA 1984 Efficacy of a synthetic dipeptide mixture as the source of amino acids for total parenteral nutrition in a subhuman primate (baboon). Plasma concentration, metabolic clearance, and urinary excretion of a series of dipeptides. Gastroenterology 86: 1562–1569
Albers S, Wernerman J, Stehle P, Vinnars E, Furst P 1989 Availability of amino acids supplied by constant intravenous infusion of synthetic dipeptides in healthy man. Clin Sci 76: 643–648
Steininger R, Karner J, Roth E, Langer K 1989 Infusion of dipeptides as nutritional substrates for glutamine, tyrosine, and branched-chain amino acids in patients with acute pancreatitis. Metabolism 38: 78–81
Druml W, Lochs H, Roth E, Hubl W, Balcke P, Lenz K 1991 Utilization of tyrosine dipeptides and acetyltyrosine in normal and uremic humans. Am J Physiol 260: E280–E285
Lochs H, Hubl W, Gasic S, Roth E, Morse EL, Adibi SA 1992 Glycylglutamine: metabolism and effects on organ balances of amino acids in postabsorptive and starved subjects. Am J Physiol 262: E155–E160
Morlion BJ, Stehle P, Wachtler P, Siedhoff HP, Koller M, Konig W, Furst P, Puchstein C 1998 Total parenteral nutrition with glutamine dipeptide after major abdominal surgery: a randomized, double-blind, controlled study. Ann Surg 227: 302–308
Wykes LJ, House JD, Ball RO, Pencharz PB 1994 Amino acid profile and aromatic amino acid concentration in total parenteral nutrition: effect on growth, protein metabolism, and aromatic amino acid metabolism in the neonatal piglet. Clin Sci 87: 75–84
Ohisalo JJ, Laskowska-Klita T, Andersson SM 1982 Development of tyrosine aminotransferase and para-hydroxyphenylpyruvate dioxygenase activities in fetal and neonatal human liver. J Clin Invest 70: 198–200
Menkes JH, Welcher DW, Levi HS, Dallas J, Gretsky NE 1972 Relationship of elevated blood tyrosine to the intellectual performance of premature infants. Pediatrics 49: 218–224
Mamunes P, Prince PE, Thorton NH, Hunt PA, Hitchcock ES 1976 Intellectual deficits after transient tyrosinemia in the term neonate. Pediatrics 57: 675–680
Zlotkin SH, Stallings VA, Pencharz PB 1985 Total parenteral nutrition in children. Pediatr Clin North Am 32: 381–400
Pearson FC 1979 The litmus amebocyte lysate endotoxin assay: current status. Am J Med Technol 45: 704–709
De Benoist B, Abdulrazzak Y, Brooke OG, Halliday D, Millward DJ 1984 The measurement of whole body protein turnover in the preterm infant with intragastric infusion of l-[1–13C]leucine and sampling of the urinary leucine pool. Clin Sci 66: 155–164
Wykes LJ, Ball RO, Menendez CE, Pencharz PB 1990 Urine collection as an alternative to blood sampling: a noninvasive means of determining isotopic enrichment to study amino acid flux in neonates. Eur J Clin Nutr 44: 605–608
Darling PB, Bross R, Wykes LJ, Ball RO, Pencharz PB 1999 Isotopic enrichment of amino acids in urine following oral infusions of l-[1-13C]phenylalanine and l-[1-13C] lysine in humans: confounding effect of d-[1-13C]amino acids. Metabolism 48: 732–737
Waterlow JC, Garlick PJ, Millward DJ 1978 Protein Turnover in Mammalian Tissues and in the Whole Body. Elsevier/North Holland, Amsterdam, The Netherlands, 179–223.
Matthews DE, Motil KJ, Rohrbaugh DK, Burke JF, Young VR, Bier DM 1980 Measurement of leucine metabolism in man from a primed continuous infusion of l-[1-13C]leucine. Am J Physiol 238: E473–E479
Clarke JT, Bier DM 1982 The conversion of phenylalanine to tyrosine in man. Direct measurement by continuous intravenous tracer infusions of l-[ring-2H5]phenylalanine and l-[1–13C]tyrosine in the postabsorptive state. Metabolism 31: 999–1005
Thompson GN, Pacy PJ, Merritt H, Ford GC, Read MA, Cheng KN, Halliday D 1989 Rapid measurement of whole body and forearm protein turnover using a [2H5]phenylalanine model. Am J Physiol 256: E631–E639
Van Aerde JE, Sauer PJ, Pencharz PB, Canagarayar U, Beesley J, Smith JM, Swyer PR 1985 Pediatr Res 19: 806–810
SAS Institute Inc. 1991 SAS/STAT Guide for Personal Computers, 9th Ed. SAS Institute, Inc., Cary, NC
Seber GAF 1977 Linear Regression Analysis. John Wiley, New York, NY
Kim KI, Elliott JI, Bayley HS 1983 Oxidation of an indicator amino acid by young pigs receiving diets with varying levels of lysine or threonine, and an assessment of amino acid requirements. Br J Nutr 50: 391–399
Zello GA, Pencharz PB, Ball RO 1990 Phenylalanine flux, oxidation, and conversion to tyrosine in humans studied with l-[1-13C]phenylalanine. Am J Physiol 259: E835–E843
Elwyn DH 1970 The role of the liver in regulation of amino acid and protein metabolism. In: Munro HN(ed) Mammalian Protein Metabolism. Academic Press, New York, NY, 523–557.
Krempf M, Hoerr RA, Marks L, Young VR 1990 Phenylalanine flux in adult men: estimates with different tracers and route of administration. Metabolism 39: 560–562
Reeds PJ, Burrin DG, Jahoor F, Wykes L, Henry J, Frazer EM 1996 Enteral glutamate is almost completely metabolized in first pass by the gastrointestinal tract of infant pigs. Am J Physiol 270: E413–E418
Stoll B, Burrin DG, Henry J, Jahoor F, Reeds PJ 1997 Phenylalanine utilization by the gut and liver measured with intravenous and intragastric tracers in pigs. Am J Physiol 273: G1208–G1217
Dudley MA, Wykes LJ, Dudley AW, Burrin DG, Nichols BC, Rosenberger J, Jahoor F, Heird WC, Reeds PJ 1998 Parenteral nutrition selectively decreases protein synthesis in the small intestine. Am J Physiol 274: G131–G137
Stoll B, Henry J, Reeds PJ, Yu H, Jahoor F, Burrin DG 1998 Catabolism dominates the first-pass intestinal metabolism of dietary essential amino acids in milk protein-fed piglets. J Nutr 128: 606–614
Bertolo RFP, Pencharz PB, Ball RO 1999 A comparison of parenteral and enteral feeding in neonatal piglets, including an assessment of the utilization of a glutamine-rich, pediatric elemental diet. JPEN 23: 47–55
Shinman R, Gray DW 1998 Formation and fate of tyrosine-intracellular partitioning of newly synthesized tyrosine in mammalian liver. J Biol Chem 273: 34760–34769
Widdowson EM 1981 Changes in body composition during growth. In: David J, Dobbing J(eds) Scientific Foundations of Paediatrics, 2nd Ed. Heinemann, London, 330–342.
Duffy B, Gunn T, Collinge J, Pencharz P 1981 The effect of varying protein quality and energy intake on the nitrogen metabolism of parenterally fed very-low-birth-weight (<1600 g) infants. Pediatr Res 15: 1040–1044
Walker V, Hall MA, Bulusu S, Allan A 1986 Hyperphenylalaninaemia in parenterally fed newborn babies. Lancet 2: 1284
McIntosh N, Mitchell V 1990 A clinical trial of two parenteral nutrition solutions in neonates. Arch Dis Child 65: 692–699
House JD, Thorpe JM, Wykes LJ, Pencharz PB, Ball RO 1998 Evidence that phenylalanine hydroxylation rates are overestimated in neonatal subjects receiving total parenteral nutrition with a high phenylalanine content. Pediatr Res 43: 461–466
Reeds PJ, Hachey DL, Patterson BW, Motil KJ, Klein PD 1992 VLDL apolipoprotein B-100, a potential indicator of the isotopic labeling of the hepatic protein synthetic precursor pool in humans: studies with multiple stable isotopically labeled amino acids. J Nutr 122: 457–466
Delvalle JA, Greengard O 1977 Phenylalanine hydroxylase and tyrosine aminotransferase in human fetal and adult liver. Pediatr Res 11: 2–5
Kretchmer N, Levine SZ, McNamara H, Barnett HL 1956 Certain aspects of tyrosine metabolism in the young. I. The development of the tyrosine oxidizing system in human liver. J Clin Invest 35: 236–244
Zlotkin SH, Atkinson SA, Brennan J, Dunn M, Hanning R, Heim T, Innis SM, Lockitch G, Merko S, Pencharz PB, Perlman M, Radde I, Sauve R, Van Aerde JEE 1995 Nutrient needs and feeding of premature infants. A statement of the Nutrition Committee, Canadian Paediatric Society. Can Med Assoc J 152: 1765–1785
Acknowledgements
The authors thank the staff in the Neonatal Intensive Care Unit for their support in carrying out the study. We also thank the staff in the Pharmacy at The Hospital for Sick Children for their preparation of the stable isotopes and glycyl-l-tyrosine solution. A special “thank you” is extended to the families of the neonates participating in the study.
Author information
Authors and Affiliations
Additional information
Supported by the Medical Research Council, Grant MT 12928. Primene was generously donated by Baxter, Deerfield, Illinois, U.S.A. S.R. received partial support from the Fonds pour la Formation de Chercheurs a l'Aide a la Recherche, the Ontario Graduate Scholarship program, and The University of Toronto.
Rights and permissions
About this article
Cite this article
Roberts, S., Ball, R., Moore, A. et al. The Effect of Graded Intake of Glycyl-l-Tyrosine on Phenylalanine and Tyrosine Metabolism in Parenterally Fed Neonates with an Estimation of Tyrosine Requirement. Pediatr Res 49, 111–119 (2001). https://doi.org/10.1203/00006450-200101000-00022
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200101000-00022
