
Abstract
Hypoxic ischemia is a common cause of damage to the fetal and neonatal brain. Although systemic and cerebrovascular physiologic factors play an important role in the initial phases of hypoxic-ischemic injuries, the intrinsic vulnerability of specific cell types and systems in the developing brain may be more important in determining the final pattern of damage and functional disability. Excitotoxicity, a term applied to the death of neurons and certain other cells caused by overstimulation of excitatory, mainly glutamate, neurotransmitter receptors, plays a critical role in these processes. Selected neuronal circuits as well as certain populations of glia such as immature periventricular oligodendroglia may die from excitotoxicity triggered by hypoxic ischemia. These patterns of neuropathologic vulnerability are associated with clinical syndromes of neurologic disability such as the extrapyramidal and spastic diplegia forms of cerebral palsy. The cascade of biochemical and histopathologic events triggered by hypoxic ischemia can extend for days to weeks after the insult is triggered, creating the potential for therapeutic interventions.
Similar content being viewed by others

Main
Our understanding of the pathogenesis of hypoxic-ischemic brain injury in the fetus and neonate has increased considerably over the last two decades related to both clinical and laboratory observations (1, 2). This work has led to substantial conceptual agreement on a general outline of how this type of injury is triggered and evolves to produce neuropathologic lesions and neurodevelopmental disabilities (Table 1). Although cerebrovascular factors contribute to the pathophysiology of hypoxic-ischemic brain injury and have played a dominant role in thinking about pathogenesis in the past, more recent studies are uncovering important cellular and molecular aspects of injury.
A fundamental process believed to be responsible for hypoxic-ischemic damage to neurons is called excitotoxicity (3). Excitotoxicity, a term popularized in the 1970s by John Olney, refers to cell death mediated by excessive stimulation of extracellular excitatory amino acid receptors (3). Normally these receptors mediate physiologic excitatory effects of the dicarboxylic acid glutamate, one of the most ubiquitous and versatile neurotransmitters in the brain. When excessively stimulated by combinations of elevated synaptic levels of glutamate and membrane depolarization associated with ischemia, channels associated with these receptors allow a lethal flood of Ca2+ and sodium to enter neurons. In the developing brain, excitotoxicity is the Achilles heel of neurons that normally benefit from the trophic stimulation provided by well-modulated excitatory stimulation (4). Excitotoxicity appears to be even more intimately involved in the pathogenesis of cell destruction from hypoxic ischemia in the developing brain than in the adult, and, for that reason, this brief review focuses heavily on this process. Although we focus here on neuronal systems, recent evidence suggests that immature white matter can also be damaged by excitotoxicity triggered through glutamate receptors by hypoxia-ischemia (5, 6).
A Cascade of Neurotoxic Events Triggered by Hypoxia-Ischemia
A great deal of laboratory work on cerebral blood flow and perfusion suggests that most hypoxic injuries in fetuses and infants reflect combinations of hypoxia and ischemia rather than hypoxia alone (1). It is unlikely that acute hypoxemia will damage the fetal or neonatal brain unless there is superimposed ischemia (4). This reflects both the enhanced resistance of the immature brain to hypoxia compared with the adult and the robustness of its protective mechanisms (e.g. capacity to increase cerebral blood flow). There is also general agreement that the syndrome of HIE after a severe asphyxial insult is an integral component of the evolving injury, reflecting a cascade of biochemical events that evolves over hours to several days (7–9). It is widely accepted that the evolving injury is accompanied by enhanced neuronal excitement with frequent seizures and electroencephalographic abnormalities (7, 10). These clinical observations are complemented by evidence from the laboratory that drugs that block NMDA-type glutamate channels can protect the brain from severe hypoxic-ischemic insults if given before or shortly after the insult (11–13). Finally, there is a growing awareness among clinicians based on the use of MR brain imaging that HIE targets special brain structures or groups of neurons, the concept of selective vulnerability (14).
To highlight the recent advances in hypoxia-ischemia research in the developing brain, we focus on two rapidly evolving areas: the pathogenesis of selective neuronal vulnerability from near-total asphyxia and the apoptosis-necrosis continuum that could become a target for novel neuroprotective strategies.
Relatively Selective Neuronal Vulnerability from Near-Total Asphyxia
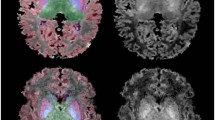
Most episodes of hypoxia-ischemia severe enough to damage the immature brain cause variable injury to selected groups of structures rather than uniform or global brain injury (15, 16). This phenomenon results in the clinical patterns of disability seen after these insults, such as spastic diplegia associated with periventricular white matter injury in premature infants (15). MRI over the last few years has also revealed a special pattern of symmetric injury to the thalamus, putamen, and peri-Rolandic cerebral cortex after severe or near-total asphyxia in term infants (Fig. 1) (17–22). Associated injury in the brain stem is also commonly present. The periventricular white matter is usually spared in these cases, but a transient alteration in the MR signal from the posterior internal capsule has been reported to be a sign of injury to adjacent neurons (21, 23). This neuroimaging pattern of selective injury in the basal ganglia and cortex is commonly associated with severe permanent motor impairment that includes rigidity, impairment of upper extremities more than lower extremities, and motor speech impairment (17, 19). This pattern of relatively selective vulnerability resembles the pattern of diencephalic and brain stem injury observed by Myers (24) in his studies of acute total asphyxia in subhuman primates in the early 1970s.

Brain MR images of a term infant after an episode of severe near-total asphyxia. Image on the left is a T1-weighted image at 6 d after the insult and shows increased signal in the putamen and thalamus bilaterally (arrows). Image on the right is a T2-weighted image 1 y later showing increased signal in the posterior putamen and ventrolateral thalamus bilaterally. The child had severe extrapyramidal cerebral palsy with rigidity.
Although the pathophysiology of this injury is incompletely understood in human infants, there is good evidence from the laboratory as well as human experimental studies that this special pattern of damage reflects the dysfunction of a selected set of excitatory neuronal circuits triggering selective neuronal death (25). The major neurotransmitter-specific connections between the vulnerable regions are shown schematically in Figure 2 (26). It is noteworthy that each of the vulnerable regions is the target of a major excitatory glutamatergic input as depicted by the sharp arrows (26). If these circuits are hyperactive during the period of HIE, it will predispose each of these areas to glutamate-mediated excitotoxicity (7, 27). On the other hand, hyperactivity in these circuits would inhibit the internal and external segments of the globus pallidus, which tend not to be injured during near-total asphyxia, at least as visualized by MRI (19). Inhibition of the globus pallidus interna would also potentially reduce its inhibition of the thalamus, allowing it to more actively stimulate the cerebral cortex. This has been proposed as a working model for selective neuronal vulnerability from near-total asphyxia, and it provides a rationale for differences between asphyxia and disorders that target the globus pallidi such as kernicterus (28).

Schematic of circuitry of the basal ganglia and regions especially vulnerable to near-total asphyxia. Pointed arrows indicate excitatory synapses using glutamate as their neurotransmitter, and blunted lines indicate inhibitory synapses that use gamma-aminobutyric acid (GABA) as their neurotransmitter. The peri-Rolandic cortex, putamen, and thalamus are especially vulnerable to near-total asphyxia. Gpi indicates globus pallidus interna;Gpe, globus pallidus externa;STN, subthalamic nucleus.
Functional Imaging Evidence that Vulnerable Regions Are Hyperactive during HIE
Hyperactivity of cerebral cortex manifested by seizures and abnormal electrical activity is a well-established component of the syndrome of HIE after asphyxia severe enough to damage the brain (4, 7, 10). There is also evidence from functional imaging that selectively vulnerable regions in the basal ganglia and cerebral cortex are also hypermetabolic (7, 29). In a study of glucose metabolism in infants with HIE after severe asphyxia, Blennow et al.(29) examined [18F]FD-glucose PET in six full-term infants at a median age of 2.5 (2–5) d after the insult. They found focal elevations in the regional cerebral metabolism of glucose (rCMRgl) in the basal ganglia and cerebral cortex in five infants who later had severe neurologic deficits but no increase in one infant who was normal at follow up. The areas of increased rCMRgl generally corresponded to the areas in the basal ganglia and peri-Rolandic cerebral cortex that are commonly abnormal on MR images, although only computerized tomographic images were performed in this study. Similar regions of cerebral glucose hypermetabolism have been reported in an infant rat model of hypoxia-ischemia (30). Although these results originally suggested a general increase in neuronal metabolism after HIE, recent experimental work using MRS suggests that they actually point more specifically to an increase in glutamate release and reuptake at excitatory synapses (31).
The proposal that glucose metabolic rate is coupled directly to the activity of glutamate-using excitatory synapses rather than neuronal cell bodies is supported by several lines of evidence (32–34). Sokoloff (32) has pointed out that glucose utilization as measured by rCMRgl increases linearly with spike frequency in neuropil-containing synapses but not neuronal cell bodies in functionally activated neural tissues. Using MRS to follow the flow of carbon derived from [1–13C]glucose in brain glutamate and glutamine pools in rodents, Sibson et al.(33) found a direct 1:1 stoichiometric coupling between brain glucose metabolism and glutamate neurotransmitter cycling during neuronal activation. It has been proposed that this coupling takes place in perisynaptic astrocytes where a molecule of glucose anaerobically generates two ATPs that power glutamate reuptake from the synapse into glia and conversion of glutamate to glutamine (31). Pfund et al.(34) also recently reported that 2-deoxy-2[F-18]fluoro-d-glucose PET (FDG-PET) measurement of glucose metabolism in children with epilepsy and controls correlated with glutamate/glutamine concentrations using proton MRS. These observations suggest that focal glucose hypermetabolism in infants with HIE after asphyxia reflects enhanced release of glutamate at hyperactive excitatory synapses in the selectively vulnerable regions.
These observations are consistent with several human and animal studies demonstrating high extracellular and cerebrospinal fluid levels of excitatory neurotransmitters during HIE (35–39). Pu et al.(40) also recently reported that the proton MRS peak for glutamate/glutamine was elevated in the basal ganglia and thalami of four infants with moderate or severe HIE after asphyxia but not in eight with mild HIE or in normal infants. The most widely accepted explanation for this elevation is that hypoxia-ischemia inhibits the activity of energy-dependent glutamate reuptake pumps. Silverstein et al.(41) originally reported that perinatal hypoxia-ischemia disrupts high-affinity reuptake of 3H-glutamate into synaptosomes prepared from the basal ganglia of infant rats exposed to unilateral hypoxia-ischemia. They also reproduced the same effect of inhibiting the maximal velocity of the glutamate reuptake system by injecting the neurotoxic glutamate agonist NMDA into the neonatal basal ganglia (42). Recently, Jabaudon et al.(43) used an elegant organotypic hippocampal culture system to demonstrate that energy deprivation produces an early severe reduction in glutamate reuptake and promotes reversal of the transporter. Similarly, Rossi et al.(44) reported that glutamate release in severe brain ischemia occurs mainly by reversal of the transporter. In a newborn piglet model of asphyxia that replicates the selective basal ganglia and peri-Rolandic lesions seen in human infants, Martin et al.(45) reported evidence of early astroglial degeneration and loss of the astroglial (GLT 1) transporter in striatum that could have contributed to neurodegeneration. Therefore, these data suggest that power failure from hypoxic ischemia causes excessive release of glutamate from nerve terminals combined with reduced activity of the glial pumps that normally keep synaptic glutamate levels low. Through these mechanisms, glutamate can reach high levels, triggering excessive activity at glutamate receptors and eventually excitotoxicity.
How do these experimental data relate to the pattern of brain injury seen clinically in infants with near-total asphyxia as shown in Figure 1 ? We hypothesize that the primary factor that makes the putamen, thalamus, and peri-Rolandic cortex more vulnerable than surrounding regions to hypoxic-ischemic injury is their positions within maturing excitatory neuronal circuits (28). Other areas may be less vulnerable because their excitatory neurotransmitter circuitry is less well established in the neonatal period. This is consistent with PET studies of normal newborn infants who have relatively high glucose metabolic rates in the basal ganglia, brain stem, and sensorimotor cortex with much lower rates elsewhere (46). It seems far more likely that this pattern of vulnerability is related to the intrinsic properties of vulnerable neurons (e.g. excitatory receptors, neurotransmitter reuptake pumps, ability to fire repetitively) than it is to a pattern of vascular supply or redistribution of cerebral blood flow.
The Neurotoxic Cascade
When excessively excited by high levels of glutamate, neurons and other cells with appropriate receptors can be sent into a death spiral that results in their demise. Excessive levels of synaptic glutamate and possibly other excitatory neurotransmitters such as glycine together with membrane depolarization can contribute to the opening of NMDA-type glutamate receptors, flooding cells with Ca2+(25, 47). NMDA receptors open passively when membrane potential is reduced by hypoxia even if glutamate levels are not high (48, 49). Developmentally determined expression of NMDA receptor subunits that favor channels with longer open times and larger Ca2+ fluxes may be responsible for greatly enhanced NMDA-mediated neurodegeneration in neonatal animals (25, 50, 51). Other glutamate neurotransmitter-specific receptors and ion channels [e.g. alpha-amino-3-hydroxy-5-methyl-isoazole-4-propionic acid (AMPA) receptors] may also contribute to trigger a neurotoxic cascade of events that can result in cell death as depicted in Figure 3 (25, 52). Direct effects of Ca2+ flooding and Ca2+-mediated generation of nitric oxide and peroxynitrite contribute to damage depending on the severity of the insult and other factors such as tissue redox state (52–58). Activation of lipases and proteases, including those involved in proinflammatory cytokine cascades, also contribute to excitotoxic injury triggered by hypoxia-ischemia (59–63). The experimental observation that NMDA-mediated neuroprotection becomes ineffective if postponed more than 1–2 h after a hypoxic-ischemic insult suggests that these downstream events quickly become self-sustaining (11–13). The evolution of these events is probably responsible for the delayed expression of neurodegeneration that is seen during HIE.

Schematic of events that may contribute to the evolution of HIE as described in the text. Hypoxic ischemia triggers opening of NMDA-type glutamate receptor-operated channels in the cytoplasmic membrane, allowing Ca2+ to flood into the cytoplasm (upper left of diagram). The cascade unfolds over time from left to right in the diagram over a period that may extend for days to weeks depending on the species, the nature and severity of the initial insult, the brain region, and other factors such as the temperature and supply of trophic factors. Ca2+ fluxed through NMDA channels can activate Ca2+-sensitive enzymes such as nitric oxide synthase (NOS), producing the free radical gas nitric oxide (NO), which is toxic alone or when combined with superoxide ions to form even more toxic peroxynitrite. One target of NO and peroxynitrite is mitochondria, which generate their own supply of oxygen free radicals under hypoxic conditions. Very severe hypoxic-ischemic insults can cause total mitochondrial failure, leading promptly to destruction of cellular membranes and histologic necrosis. Less severe degrees of hypoxic ischemia can trigger delayed programmed cell death or apoptosis. One potential scenario is depicted here in which cytochrome c protein released from distressed mitochondria triggers the activation of cysteine-dependent aspartate-directed proteases (caspases) such as caspase 3 that lead to fragmentation of DNA and many other actions. DNA fragmentation in turn can trigger activation of poly(ADP-ribose)polymerase (PARP), a nuclear enzyme that facilitates DNA repair. However, this process consumes NAD, possibly limiting its concentration within mitochondria. Oxidative failure from hypoxic ischemia combined with a reduction in NAD can further impair mitochondrial function and reduce energy needed to maintain membrane potentials. A fall in membrane potential leads to passive opening of NMDA channels, worsening and extending the excitotoxic cascade.
Mitochondria May Play a Pivotal Role in Neurodegeneration
Mitochondria appear to play a central role to determine the fate of cells subjected to hypoxia-ischemia (64–66). Mitochondria handle multiple oxidation reactions that can yield highly toxic oxygen free radicals under conditions of oxidative stress. They are major buffers of intracellular Ca2+ and can become overloaded by cytoplasmic Ca2+ flooding secondary to opening of NMDA and voltage-sensitive Ca2+ channels. Diminished mitochondrial function can lead to decreased energy to maintain membrane ion gradients, potentially perpetuating a vicious cycle of membrane depolarization and NMDA receptor channel opening (Fig. 3) (25, 67). MRS studies in animals and human infants indicate that delayed energy failure and persistently elevated brain lactate levels are associated with neurodegeneration after asphyxia (68). Therefore, disrupted mitochondrial function during HIE could contribute to persistent seizures and electroencephalographic abnormalities such as the burst-suppression pattern (69). In experimental animals of asphyxia, NMDA antagonist drugs can improve disrupted mitochondrial function (66).
Neurodegeneration from hypoxia-ischemia can take the form of necrosis or apoptosis, and the choice may depend on the intensity of mitochondrial dysfunction (64). In neuronal cell culture, Ankarcrona et al. (64) used an excitatory amino acid agonist to produce a very intense or a milder excitotoxic insult and found that they produced either rapid necrosis or delayed apoptosis. The more intense insult produced rapid loss of mitochondrial membrane potential, loss of ATP production, and explosion of nuclear and cytoplasmic membranes consistent with necrosis. On the other hand, neurons subjected to the less intense excitotoxic insult initially lost and recovered their mitochondrial membrane potentials but ultimately developed nuclear fragmentation of chromatin and shrinkage of nuclear and cytoplasmic contents typical of apoptosis. It has been suggested that damaged mitochondria may signal the apoptotic process by release of cytochrome c or other intramitochondrial proteins, in turn activating cysteine protease enzymes or caspases that fragment DNA and execute apoptotic programs (64, 65, 70, 71). In adult animal models, DNA fragmentation produced by this process as well as through free radical oxidative stress can also trigger the repair enzyme poly(ADP-ribose)polymerase, which may further impair mitochondrial function by depleting NAD needed to maintain mitochondrial energetics (72). This can create another vicious cycle of death in the neurotoxic cascade and is an area of active investigation in the immature brain.
Apoptosis May Be More Prominent in the Immature Brain
Apoptosis, which involves activation of genetically determined cell-suicide programs, has been observed in postmortem brain tissue from infants after hypoxic-ischemic insults as well as in immature animal models of hypoxia-ischemia (73–76). Comparison of adult and immature animal models of hypoxia-ischemia suggests that apoptosis may be more prevalent in the immature brain (77, 78). Our recent studies in which we quantitated the relative numbers of apoptotic versus necrotic cells in a rodent model of hypoxic ischemia indicate that many regions such as the cerebral cortex and basal ganglia contain high numbers of apoptotic cells for over 7 d after hypoxia-ischemia (79).
Using multiple markers for apoptosis including electron microscopy, we found apoptotic cells in all regions of the hypoxic-ischemic 7-d-old rat brain, with apoptosis exceeding 50% of degenerating cells in several regions (79). In contrast, one detailed study of an adult middle cerebral ischemia model showed ratios of apoptosis to necrosis of less than 1:6 (78). The classic eosinophilic ischemic neurons that are a consistent feature of ischemia in adult models are relatively rare in the newborn rat, but necrotic cells with aggregates of irregularly shaped chromatin are common (Fig. 4). Most brain regions of the hypoxic-ischemic 7-d-old rat brain contain a continuum of apoptotic-to-necrotic cells with hybrid cells that contain morphologic features of both types of degeneration. A similar continuum has been observed after injections of excitotoxins or trauma in the immature rodent brain, suggesting that it is triggered by the excitotoxic cascade (80, 81). This continuum would be consistent with a potential continuum of injury to mitochondria, as suggested by the experiments of Ankarcrona et al. (64) mentioned above. Given the relatively short time period over which cells retain their apoptotic morphology before being phagocytosed, this may indicate that many cells delay their commitment to apoptosis for several days after the insult.

Electron microscopic images of injured or dying neurons in neocortex from an infant rat 48 h after hypoxic ischemia ipsilateral to unilateral carotid artery ligation plus 8% oxygen for 2 h at 7 d of age. (A) Apoptotic neuron with one large black apoptotic body including condensed chromatin; (B) necrotic neuron with chromatin dispersed into numerous small irregularly shaped structures and disrupted nuclear and cytoplasmic membranes; (C) hybrid neuron: chromatin bodies are smaller than in apoptotic neurons but rounder and larger than in necrotic ones. Cytoplasmic organelles are condensed and more intact than in the necrotic neuron. Scale bar = 1 μm.
The prominence of apoptosis and the apoptotic-necrotic continuum in neurodegeneration after hypoxia-ischemia in the immature brain suggests that it will be important to understand these processes to develop effective neuroprotective strategies. The cysteine protease (caspase) enzyme caspase 3 plays an important role to execute apoptotic programs in many cells (70). Cheng et al.(70) demonstrated the efficacy of inhibiting caspase-3 enzyme activity for protecting the 7-d-old rat brain from hypoxic-ischemic injury. These data and others suggest that activation of apoptosis-executing caspases is much greater in the immature brain than in the adult (82). A combination of NMDA antagonists and caspase inhibitors may provide a longer therapeutic window than either administered alone, suggesting that the two mechanisms interact with each other (83, 84). Activation of proapoptotic caspases may also be linked to activation of calpains, a related family of Ca2+-sensitive cysteine proteases that regulate cytoskeletal function (85, 86).
Control of apoptosis and the apoptosis/necrosis continuum involves a balance between expression of numerous apoptotic and antiapoptotic proteins after injury, providing many potential approaches to modifying outcome (70, 87, 88). Several protein growth factors that have been reported to protect against hypoxic-ischemic injury in immature animal models may act by inhibiting apoptosis (89–93). The protective effects of some of these growth factors may be mediated by the Ras-MAP (mitogen-activated protein kinase) kinase signaling pathway to the nucleus (94). Glucocorticoids have also been reported to have a potent neuroprotective effect when given at certain times before hypoxic ischemia in rodents, possibly mediated through a nuclear receptor mechanism (95). It is also plausible that hypothermia, which is currently receiving considerable clinical attention as a neuroprotective strategy, may slow or reduce the excitotoxic cascade by altering processes favoring apoptosis (96–100) or through other mechanisms such as reducing glutamate release. The discovery that preconditioning with exposure to hypoxia before an experimental hypoxic-ischemic insult can reduce the extent of damage also suggests that endogenous neuroprotective mechanisms are being activated (101).
CONCLUSION
Although the immature brain is relatively protected from hypoxia-ischemia by powerful adaptive mechanisms and relatively low energetic demands, severe insults can trigger a self-sustaining cascade of neurotoxic events lasting several days or longer. In the term infant, neurons connected in established neuronal circuits appear to be especially vulnerable to excitotoxic damage. Compared with the adult nervous system, apoptosis and an apoptosis/necrosis continuum may be more prominent in the developing brain. Combinations of antiexcitotoxic and antiapoptotic therapies my hold promise for salvaging brain tissue after hypoxic-ischemic insults.
Abbreviations
- Ca2+:
-
calcium
- FD-glucose:
-
fluorodeoxyglucose
- HIE:
-
hypoxic-ischemic encephalopathy
- MR:
-
magnetic resonance
- MRI:
-
magnetic resonance imaging
- MRS:
-
magnetic resonance spectroscopy
- NMDA:
-
N-methyl-d-aspartate a subtype of glutamate receptor
- PET:
-
positron emission tomography
References
Vannucci RC 1990 Experimental biology of cerebral hypoxia-ischemia: relation to perinatal brain damage. Pediatr Res 27: 317–326
Johnston MV, Trescher WH, Ishida A, Nakajima W 2000 Novel treatments after experimental brain injury. Semin Neonatol 5: 75–86
Choi DW, Rothman SM 1990 The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Ann Rev Neurosci 13: 171–182
Johnston MV, Trescher WH, Taylor GA 1995 Hypoxic and ischemic central nervous system disorders in infants and children. Adv Ped 42: 1–45
Follett PL, Rosenberg PA, Volpe JJ, Jensen FE 2000 NBQX alternates excitotoxic injury in developing white matter. J Neuroscience 20: 9235–9241
Gressens P 1999 VIP neuroprotection against excitotoxic lesions in developing mouse brain. Ann NY Acad Sci 897: 109–124
Sarnat HB, Sarnat MS 1976 Neonatal encephalopathy following fetal distress: a clinical and electroencephalographic study. Arch Neurol 33: 696–705
Towfighi J, Zec N, Yager J, Housman C, Vannucci RC 1995 Temporal evolution of neuropathologic changes in an immature animal model of cerebral hypoxia: a light microscopic study. Acta Neuropathol Berl 90: 375–386
Albensi BC, Schweizer MP, Rarick TM, Filloux F 1999 Unilateral hypoxic-ischemic injury in the neonatal rat brain evaluated by in vivo MRI. Invest Radiol 34: 249–261
Williams CE, Gunn AJ, Mallard C, Gluckman PD 1992 Outcome after ischemia in the developing sheep brain: an electroencephalographic and histological study. Ann Neurol 31: 14–21
McDonald JW, Silverstein FS, Johnston MV 1987 MK-801 protects the neonatal brain from hypoxic-ischemic damage. Eur J Pharmacol 140: 359–361
Hattori H, Morin AM, Schwartz PH, Fujikawa DG, Wasterlain CG 1989 Posthypoxic treatment with MK-801 reduces hypoxic-ischemic damage in the neonatal rat. Neurology 39: 713–718
Hagberg H, Gilland E, Diemer NH, Andine P 1994 Hypoxia-ischemia in the neonatal rat brain: histopathology after post-treatment with NMDA and non-NMDA receptor antagonists. Biol Neonate 66: 206–213
Johnston MV 1998 Selective vulnerability in the neonatal brain. Ann Neurol 44: 155–156
Volpe JJ 1997 Brain injury in the premature infant: neuropathology, clinical aspects, and prevention. Clin Perinatol 24: 567–588
Rivkin MJ 1997 Hypoxic-ischemic brain injury in the term newborn: neuropathology, clinical aspects, and neuroimaging. Clin Perinatol 24: 607–626
Menkes JH, Curran J 1994 Clinical and MR correlates in children with extrapyramidal cerebral palsy. AJNR Am J Neuroradiol 15: 451–457
Barkovich AJ, Westmark K, Partridge C, Sola A, Ferriero DM 1995 Perinatal asphyxia: MR findings in the first 10 days. AJNR Am J Neuroradiol 16: 427–438
Hoon AH, Reinhardt EM, Kelley RI, Breiter SN, Morton DH, Naidu S, Johnston MV 1997 Brain MRI in suspected extrapyramidal cerebral palsy: observations in distinguishing genetic-metabolic from acquired causes. J Pediatr 131: 240–245
Roland EH, Poskitt K, Rodriguez E, Lupton BA, Hill A 1998 Perinatal hypoxic-ischemic thalamic injury: clinical features and neuroimaging. Ann Neurol 44: 161–166
Pasternak JF, Gorey MT 1998 The syndrome of near-total intrauterine asphyxia in the term infant. Pediatr Neurol 18: 391–398
Maller AI, Hankins LL, Yeakley JW, Butler IJ 1998 Rolandic-type cerebral palsy in children as a pattern of hypoxic-ischemic injury in the full-term neonate. J Child Neurol 13: 313–321
Rutherford MA, Pennock JM, Counsell SJ, Mercuir E, Cowan FM, Dubowitz LMS, Edwards AD 1998 Abnormal magnetic resonance signal in the internal capsule predicts poor neurodevelopmental outcome in infants with hypoxic-ischemic encephalopathy. Pediatrics 102: 323–328
Myers RE 1972 Two patterns of brain damage and their conditions of occurrence. Am J Obstet Gynecol 112: 245–276
McDonald JW, Johnston MV 1990 Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Res Rev 15: 41–70
Alexander GE, Crutcher MD 1990 Functional architectural of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci 13: 266–271
Olney JW, Collins RC, Sloviter RS 1986 Excitotoxic mechanisms of epileptic brain damage. Adv Neurol 44: 857–877
Johnston MV, Hoon A 2000 Possible mechanisms for selective basal ganglia damage in infants from asphyxia, kernicterus, or mitochondrial encephalopathies. J Child Neurol 15: 588–591
Blennow M, Ingvar M, Lagercrantz H, Stone-Elander S, Eriksson L, Forsberg H, Ericson K, Flodmark O 1995 Early [18F]FDG positron emission tomography in infants with hypoxic-ischemic encephalopathy shows hypermetabolism during the postasphyctic period. Acta Paediatr 84: 1289–1295
Gilland E, Hagberg H 1996 NMDA-dependent increase of cerebral glucose utilization after hypoxia-ischemia in the immature rat. J Cereb Blood Flow Metab 16: 1005–1013
Magistretti PJ, Pellerin L, Rothman DL, Shulman RG 1999 Energy on demand. Science 283: 496–497
Sokoloff L 1999 Energetics of functional activation in neural tissues. Neurochem Res 24: 321–329
Sibson NR, Dhankhar A, Mason GF, Rothman DL, Behar KL, Shulman RG 1998 Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc Natl Acad Sci USA 95: 316–321
Pfund Z, Chugani DC, Juhasz C, Musik O, Chugani HT, Wilds IB, Se Bozorgzad N, Moore GJ 2000 Evidence for coupling between glucose metabolism and glutamate cycling using FDG-PET and 1H magnetic resonance spectroscopy in patients with epilepsy. J Cereb Blood Flow Metab 20: 871–878
Andine P, Sandberg M, Bagenholm R, Lehmann A, Hagberg H 1991 Intra- and extracellular changes of amino acids in the cerebral cortex of the neonatal rat during hypoxia-ischemia. Dev Brain Res 64: 115–120
Hagberg H, Andersson P, Kjellmer I, Thuringer K, Thordstein M 1987 Extracellular overflow of glutamate, aspartate, GABA, and taurine in the cortex and basal ganglia of fetal lambs during hypoxia-ischemia. Neurosci Lett 78: 311–317
Riikonen RS, Kero PO, Simell OG 1992 Excitatory amino acids in cerebrospinal fluid in neonatal asphyxia. Pediatr Neurol 8: 37–40
Silverstein FS, Naik B, Simpson J 1991 Hypoxia-ischemia stimulates hippocampal glutamate efflux in perinatal rat brain: an in vivo microdialysis study. Pediatr Res 30: 587–590
Hagberg H, Thornberg E, Blennow M, Kjellmer I, Lagercrantz H, Thiringer K, Hamberger A, Sandberg M 1993 Excitatory amino acids in the cerebral spinal fluid of asphyxiated infants: relationship to hypoxic-ischemic encephalopathy. Acta Paediatr 82: 925–929
Pu Y, Li QF, Zeng CM, Gao J, Qi J, Luo DX, Mahankali S, Fox PT, Gao JH 2000 Increased detectability of alpha brain glutamate/glutamine in neonatal hypoxic-ischemic encephalopathy. AJNR Am J Neuroradiol 21: 203–212
Silverstein FS, Buchanan K, Johnston MV 1986 Perinatal hypoxia-ischemia disrupts striatal high affinity 3H-glutamate uptake into synaptosomes. J Neurochem 47: 1614–1619
Hu B, McDonald JW, Johnston MV, Silverstein FS 1991 Excitotoxic brain injury suppresses striatal high affinity glutamate uptake in perinatal rats. J Neurochem 56: 933–937
Jabaudon D, Scanziani M, Gahwiler BH, Gerber U 2000 Acute decrease in net glutamate uptake during energy deprivation. Proc Natl Acad Sci USA 97: 5610–5615
Rossi DJ, Oshima T, Attwell D 2000 Glutamate release in severe brain ischemia is mainly by reversed uptake. Nature 403: 316–321
Martin LJ, Brambrink AM, Lehmann C, Portera-Cailliau C, Koehler Rothstein J, Traystman RJ 1997 Hypoxia-ischemia causes abnormalities in glutamate transport and death of astroglia and neurons in newborn striatum. Ann Neurol 43: 335–348
Chugani HT 1999 Metabolic imaging: a window on brain development and plasticity. The Neuroscientist 5: 29–40
Delivoria-Papadopoulos M, Mishra OP 1998 Mechanisms of cerebral injury in perinatal asphyxia and strategies for prevention. J Pediatr 132: S30–S34
Novelli A, Reilly JA, Lysko PG, Henneberry RC 1988 Glutamate becomes neurotoxic via the NMDA receptor when intracellular energy levels are reduced. Brain Res 451: 205–212
Hammond C, Crepel V, Gozlan H, Ben-Ari Y 1994 Anoxic LTP sheds light on the multiple facets of NMDA receptors. Trends Neurosci 17: 497–503
McDonald JW, Silverstein FS, Johnston MV 1988 Neurotoxicity of N-methyl-d-aspartate is markedly enhanced in developing rat central nervous system. Brain Res 459: 200–203
Monyer H, Brunashev N, Laurie DJ 1993 Development and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12: 529–540
Strijbos PJ, Leach MJ, Garthwaite J 1996 Vicious cycle involving Na+ channels, glutamate release, and NMDA receptors mediates delayed neurodegeneration through nitric oxide formation. J Neurosci 16: 5004–5013
Strijbos PJLM 1998 Nitric oxide in cerebral ischemic neurodegeneration and excitotoxicity. Crit Rev Neurobiol 12: 223–243
Almeida A, Heales SJR, Bolanos JP, Medina JM 1999 Nitric oxide mediates glutamate-induced mitochondrial depolarization in rat cortical neurons. Brain Res 239: 183–201
Ferriero DM, Holtman DM, Black SM, Sheldon RA 1996 Neonatal mice lacking neuronal nitric oxide synthase are less vulnerable to hypoxic-ischemic injury. Neurobiol Dis 3: 652–656
Trifiletti R 1992 Neuroprotective effects of N-nitro-l-arginine in focal stroke in the 7-day-old rat. Eur J Pharmacol 218: 197–198
Ashwal S, Cole DJ, Osborne S, Pearce WJ 1995 l-NAME reduces infarct volume in a filament model of transient middle cerebral artery occlusion in the rat pup. Pediatr Res 38: 652–656
Hamada Y, Hayakawa T, Hattori H, Mikawa H 1994 Inhibitor of nitric oxide synthesis reduces hypoxic-ischemic brain damage in the neonatal rat. Pediatr Res 35: 10–14
Hara H, Friedlander RM, Gagliardini V, Ayata C, Fink K, Huang Z, Shimizu-Sasamata M, Yuan J, Moskowitz MA 1997 Inhibition of interleukin-1-beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc Natl Acad Sci USA 94: 2007–2012
Hagan P, Barks JD, Yabut BL, Davidson BL, Roessler B, Silverstein F 1996 Adenovirus-mediated over-expression of interleukin-1-beta receptor antagonist reduces susceptibility to excitotoxic brain injury in perinatal rats. Neuroscience 75: 1033–1045
Silverstein FS, Barks JD, Hagan P, Liu XH, Szaflarski J 1997 Cytokines and perinatal brain injury. Neurochem Int 30: 375–383
Martin-Ancel A, Garcia-Alix A, Pascual-Salcedo D, Cabansa F, Valcarce M, Quero J 1997 Interleukin-6 in the cerebrospinal fluid after perinatal asphyxia is related to early and late neurological manifestations. Pediatrics 100: 789–794
Hagberg H, Gilland E, Bona E, Hanson LA, Hahin-Zoric M, Blennow HM, McRae A, Soder O 1996 Enhanced expression of interleukin-1 (IL-1) and IL-6 messenger RNA and bioactive protein after hypoxia-ischemia in neonatal rats. Pediatr Res 40: 603–609
Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P 1995 Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron 15: 961–973
Abe K, Aoki M, Kawagoe J, Yosida T, Hattori A, Kogure K, Itoyan Y 1995 Ischemic delayed neuronal death, a mitochondrial hypothesis. Stroke 26: 1478–1489
Gilland E, Puka-Sundvall M, Hillered L, Hagberg H 1998 Mitochondrial function and energy metabolism after hypoxia-ischemia in the immature brain: involvement of NMDA receptors. J Cereb Blood Flow Metab 18: 297–304
Novelli A, Reilly JA, Lysko PG, Henneberry RC 1988 Glutamate becomes neurotoxic via NMDA receptors when intracellular energy levels are reduced. Brain Res 451: 205–212
Hanrahan J, Cox IJ, Edwards AD, Cowan IFM, Sargentoni J, Bell J, Bryant DJ, Rutherford MA, Azzopardi D 1998 Persistent increases in cerebral lactate concentration after birth asphyxia. Pediatr Res 44: 304–311
Thoresen M, Haaland K, Loberg EM, Whitelaw A, Apricena F, Hanko E, Steen PA 1996 A piglet survival model of posthypoxic encephalopathy. Pediatr Res 40: 738–748
Cheng Y, Deshmukh M, D'Costa A, Demaro JA, Gidday J, Shah A, Sun Y, Jacquin MF, Johnson EM, Holtzman DM 1998 Caspase inhibitor affords neuroprotection after delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. J Clin Invest 101: 1992–1999
Banasiak KJ, Xia Y, Haddad GG 2000 Mechanisms underlying hypoxia-induced neuronal apoptosis. Prog Neurobiol 62: 215–249
Eliasson MJL, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL 1997 Poly(ADP-ribose)polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med 3: 1089–1095
Edwards AD, Yue X, Cox P, Hope PL, Azzopardi DV, Squier MV, Mehmet H 1997 Apoptosis in the brains of infants suffering intrauterine cerebral injury. Pediatr Res 42: 684–689
Beilharz EJ, Williams CE, Dragunow M, Sirimanne ES, Gluckman PD 1995 Mechanisms of delayed cell death following hypoxic-ischemic injury in the immature rat: evidence for apoptosis during selective neuronal loss. Brain Res Mol Brain Res 29: 1–14
Pulera MR, Adams LM, Liu H, Santos DG, Nishimura RN, Yang F, Cole GM, Wasterlain CG 1998 Apoptosis in a neonatal rat model of cerebral hypoxia-ischemia. Stroke 29: 2622–2630
Yue X, Mehmet H, Penrice J, Cooper C, Cady E, Wyatt JS, Reynolds EO, Edwards AD, Squier MV 1997 Apoptosis and necrosis in the newborn piglet brain following transient cerebral hypoxia-ischemia. Neuropathol Appl Neurobiol 23: 16–25
McDonald HW, Behrens MI, Chung C, Bhattacharyya T, Choi DW 1997 Susceptibility to apoptosis is enhanced in immature cortical neurons. Brain Res 759: 228–232
Li Y, Sharov VG, Jiang N, Zaloga C, Sabbah HN, Chopp M 1998 Intact, injured, necrotic, and apoptotic cells after focal cerebral ischemia in the rat. J Neurol Sci 156: 119–132
Nakajima W, Ishida A, Lange MS, Gabrielson KL, Wilson MA, Martin LJ, Blue ME, Johnston MV 2000 Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. J Neurosci 20: 7994–8004
Martin LJ, Al-Abdulla NA, Brambrink AM, Kirsch JR, Sieber FE, Portera-Cailliau C 1998 Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: a perspective on the contributions of apoptosis and necrosis. Brain Res Bull 46: 281–309
Bittigau P, Sifringer M, Pohl D, Stadthus D, Ishimaru M, Shimizu H, Ikeda M, Lang D, Speer A, Olney JW, Ikonomidou C 1999 Apoptotic neurodegeneration following trauma is markedly enhanced in the immature brain. Ann Neurol 45: 724–735
Hu BR, Liu CL, Ouyang Y, Blomgren K, Siesjo BK 2000 Involvement of caspase-3 in cell death after hypoxia-ischemia declines during brain maturation. J Cereb Blood Flow Metab 20: 1294–1300
Ma J, Endres M, Moskowitz MA 1998 Synergistic effects of caspase inhibitors and MK-801 in brain injury after transient focal cerebral ischemia in mice. Br J Pharmacol 124: 756–762
Wang KKW, Yuen PW 1994 Calpain inhibition: an overview of its therapeutic potential. Trends Pharmacol Sci 15: 412–419
Blomgren K, McRae A, Elmered A, Bona E, Kawashima S, Saido TC, Ono T, Hagberg H 1997 The calpain proteolytic system in neonatal hypoxic-ischemia. Ann NY Acad Sci 825: 104–119
Kinloch RA, Treherne JM, Furness LM, Hajimahamadreza I 1999 The pharmacology of apoptosis. Trends Pharmacol Sci 20: 35–42
Schulz JB, Weller M, Moskowitz MA 1999 Caspases as treatment targets in stroke and neurodegenerative diseases. Ann Neurol 45: 421–429
Cheng Y, Gidday JM, Yan Q, Shah AR, Holtzman DM 1997 Marked age-dependent neuroprotection by BDNF against neonatal hypoxic-ischemic brain injury. Ann Neurol 41: 521–529
Nozaki K, Findlestein SP, Beal MF 1993 Basic fibroblast growth factor protects against hypoxia-ischemia and NMDA neurotoxicity in neonatal rats. J Cereb Blood Flow Metab 13: 221–228
Hossain MA, Fielding KE, Trescher WH, Ho T, Wilson MA, Laterra J 1998 Human FGF-1 gene delivery protects against quinolinate-induced striatal and hippocampal injury in neonatal rats. Eur J Neurosci 10: 2490–2499
Johnston BM, Mallard EC, Williams CE, Gluckman PD 1996 Insulin-like growth factor-1 is a potent neuronal rescue agent after hypoxic-ischemic injury in fetal lamb. J Clin Invest 97: 300–308
Gustafson K, Hagberg H, Bengtsson BA, Brantsing C, Isgaard J 1999 Possible protective role of growth hormone in hypoxia-ischemia. Pediatr Res 45: 318–323
Holtzman DM, Sheldon RA, Jaffe W, Cheng Y, Ferriero DM 1996 Nerve growth factor protects the neonatal brain against hypoxic-ischemic injury. Ann Neurol 39: 114–122
Han BH, Holtzman DM 2000 BDNF protects the neonatal brain from hypoxic-ischemic injury via the ERK pathway. J Neurosci 20: 5775–5781
Tuor UI 1997 Glucocorticoids and the prevention of hypoxic-ischemic brain damage. Neurosci Biobehav Rev 21: 175–179
Trescher WH, Ishiwa S, Johnston MV 1997 Brief post-hypoxic-ischemic hypothermia markedly delays neonatal brain injury. Brain Dev 19: 326–338
Dietrich WD, Lin B, Globus MYT, Green EJ, Ginsberg MD, Busto R 1995 Effect of delayed MK-801 (dizocilpine) treatment with or without immediate postischemic hypothermia on chronic neuronal survival after global forebrain ischemia in rats. J Cereb Blood Flow Metab 15: 960–968
Bona E, Hagberg H, Loberg EM, Bagenhoolm R, Thoresen M 1998 Protective effects of moderate hypothermia after neonatal hypoxia-ischemia: short- and long-term outcome. Pediatr Res 43: 738–745
Gunn AJ, Gunn TR, Guming MI, Williams CE, Gluckman PD 1998 Neuroprotection with prolonged head cooling started before postischemic seizures in fetal sheep. Pediatrics 102: 1098–1106
Guan J, Gunn AJ, Sirimanne ES, Tuffin J, Gunning MI, Clark R, Gluckman PD 2000 The window of opportunity for neuronal rescue with insulin-like growth factor-1 after hypoxia-ischemia in rats is critically modulated by cerebral temperatures during recovery. J Cereb Blood Flow Metab 20: 513–519
Gidday JM, Shah AR, Maceran RG, Wang O, Pelliguino DA, Holtzman DM, Park TS 1999 Nitric oxide mediates cerebral ischemic tolerance in a neonatal model of hypoxic preconditioning. J Cereb Blood Flow Metab 19: 331–340
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by NIH Grants R01 NS28208 and HD240611 (MRDDRC).
Rights and permissions
About this article
Cite this article
Johnston, M., Trescher, W., Ishida, A. et al. The Developing Nervous System: A Series of Review Articles: Neurobiology of Hypoxic-Ischemic Injury in the Developing Brain. Pediatr Res 49, 735–741 (2001). https://doi.org/10.1203/00006450-200106000-00003
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200106000-00003
This article is cited by
-
Knowledge mapping of spastic cerebral palsy. A bibliometric analysis of global research (2000–2022)
Italian Journal of Pediatrics (2024)
-
N6-methyladenosine demethylase FTO regulates synaptic and cognitive impairment by destabilizing PTEN mRNA in hypoxic-ischemic neonatal rats
Cell Death & Disease (2023)
-
Comparing the efficacy in reducing brain injury of different neuroprotective agents following neonatal hypoxia–ischemia in newborn rats: a multi-drug randomized controlled screening trial
Scientific Reports (2023)
-
Organotypic whole hemisphere brain slice models to study the effects of donor age and oxygen-glucose-deprivation on the extracellular properties of cortical and striatal tissue
Journal of Biological Engineering (2022)
-
Prenatal Hypoxia Induces Premature Aging Accompanied by Impaired Function of the Glutamatergic System in Rat Hippocampus
Neurochemical Research (2021)
