
Abstract
We investigated whether group B streptococcal (STREP) infusion impairs the cerebral blood flow (CBF) response to acute hypercarbia in piglets, and whether STREP-induced prostanoids or hemodynamic alterations could account for this impairment. Piglets, 2-3 wk old, were anesthetized, paralyzed, and mechanically ventilated (50% O2; partial pressure of arterial CO2 (Paco2) ≈ 40 torr). CBF was assessed by internal carotid artery blood flow (ICBF). Group 1 (n = 5) received a continuous infusion of STREP for 4 h (2.0-8.0 × 107 org/kg-min). Group 2(n = 5) was pretreated with indomethacin (5 mg/kg), then received the identical STREP infusion. Group 3 (n = 6) did not receive STREP, but cardiac output (CO) and systemic blood pressure (BP) were reduced to levels equal to that of group 1 by incremental inflation of a left atrial balloon (LAB) catheter. Cerebral vascular reactivity to acute hypercarbia(Paco2 ≈ 70 torr for 7.5 min) was assessed at baseline and after each hour of STREP infusion or LAB inflation. We found that 4 h of STREP infusion caused CO to fall significantly (634 ± 121 to 324 ± 172 mL/min, group 1; 600 ± 68 to 291 ± 80 mL/min, group 2) and BP to fall significantly (104 ± 20 to 57 ± 4 mm Hg, group 1; 91± 11 to 53 ± 16 mm Hg, group 2) By design, in group 3 LAB inflation caused CO (573 ± 181 to 375 ± 159 mL/min) and BP (104± 14 to 60 ± 9 mm Hg) to fall to values not significantly different from septic groups 1 and 2. At 4 h, unilateral ICBF decreased significantly during STREP infusion in group 1 (32.0 ± 10.8 to 21.0± 7.3 mL/min) and group 2 (22.9 ± 9.9 to 13.1 ± 4.3 mL/min), but not in nonseptic group 3 (23.1 ± 7.4 to 19.6 ± 6.3 mL/min). At baseline, hypercarbia induced an increase in ICBF (%ΔICBF = 68.7 ± 13.0% in group 1, 62.2 ± 15.6% in group 2, and 87.7± 34.0% in group 3). After 4 h of STREP, this response was completely ablated as ICBF fell during hypercarbia by -7.8 ± 23.2% (group 1). Indomethacin did not protect cerebral vascular reactivity after 4 h of STREP infusion, as%ΔICBF fell during hypercarbia by -10.9 ± 17.7%(group 2). In contrast, despite equivalent reductions in CO and BP after 4 h of LAB inflation in nonseptic group 3, ICBF rose during hypercarbia by 61.8± 23.2%, not significantly different from baseline, but significantly different from the decrease in%ΔICBF in groups 1 and 2. We conclude that STREP infusion reduces ICBF and cerebral vascular reactivity to acute hypercarbia in piglets. This phenomenon is not accounted for by STREP-induced reduction in CO or BP, and is not mediated by prostanoids.
Similar content being viewed by others
Main
Recently, the regulation of blood flow during bacterial sepsis has received intensified interest. Abnormal reactivity of organ bed vessels has been proposed, for example, to play a major role in tissue hypoxia and the pathogenesis of subsequent organ dysfunction(1–3). Specifically, concern has been focused on CBF regulation and cerebral vascular reactivity during sepsis, as CNS changes during sepsis are well described in humans(4). Presently, compared with septic adult humans(5), there exists a void in knowledge concerning the effect of bacterial sepsis on CBF regulation and cerebral vascular reactivity in neonates. This lack of information in newborn infants is particularly. disconcerting in light of the catastrophic CNS consequences which may result from systemic bacterial infection.
We have developed in piglets a model of neonatal sepsis by infusing STREP(6), and a measure of cerebral vascular responsiveness using the stimulus of acute hypercarbia. In the series of experiments reported here we first hypothesized that 4 h of STREP infusion markedly impaired cerebral vascular responsiveness to acute hypercarbia. In addition, we attempted to investigate what might mediate such a pathologic response. We hypothesized that altering systemic hemodynamic variables (i.e. reductions in systemic blood flow or BP) could account for the reduction in cerebral vascular responsiveness observed in septic piglets. For example, if the cerebral circulation had already dilated maximally in an attempt to preserve blood flow in the face of low CO and/or BP during STREP infusion, then it is possible no reserve would be available for further vasodilation in response to hypercarbia. To test this hypothesis, we compared cerebral vascular reactivity in piglets receiving STREP with cerebral vascular reactivity in a group of nonseptic piglets, in which CO and BP were reduced comparably to septic piglets by inflation of a LAB.
Finally, we assessed the potential role of arachidonic acid metabolites in the reduction of cerebral vascular responsiveness during STREP infusion. Excess arachidonic acid metabolites, especially thromboxanes, have been demonstrated to be responsible for sepsis-induced pulmonary vasoconstriction(7–9) as well as perturbations of the matching of pulmonary blood flow to ventilation(10). Further, in nonseptic piglets, it is known that both vasoconstricting and vasodilating prostaglandins contribute to the regulation of CBF(11–14). We hypothesized that arachidonic acid metabolites, formed in excess quantities during STREP infusion, might be responsible for the observed loss of cerebral vascular responsiveness in septic piglets. We tested this hypothesis by inhibiting the formation of prostaglandins with indomethacin before the onset of STREP infusion. We asked whether indomethacin pretreatment would preserve cerebral vascular reactivity to acute hypercarbia during STREP sepsis in piglets.
METHODS
Anesthesia and Ventilation
Twenty-one piglets (2-3 wk old, 4.6 ± 0.4 kg) received ketamine intraperitoneally (20 mg/kg) and were intubated endotracheally. An ear vein was cannulated for venous access. The piglets were anesthetized with sodium pentobarbital (20 mg/kg, i.v.). Throughout the experiment, the level of anesthesia was assessed by response of heart rate and BP to noxious stimuli, and adjusted by intermittent bolus of pentobarbital. approximately 2 mg/kg-h. Muscle relaxation was achieved with D-tubocurarine (1 mg/kg) and was maintained with hourly doses of D-tubocurarine (0.3 mg/kg). The endotracheal tube was replaced by a tracheostomy tube; air leak around the tracheostomy tube was prevented by a snug circumferential tie at the membrane between tracheal rings. Mechanical ventilation (Harvard Small Animal Ventilator, Harvard Medical Supplies, Dover, MA) was used with initial ventilator settings of tidal volume = 15 mL/kg, rate = 10 breaths/min, positive end-expiratory pressure = 3 cm H2O. Excluding the periods of hyperventilation (see below), a 50% O2, 50% N2 gas mixture was used, and Paco2 was maintained at approximately 35 torr. Warming blankets and overhead heating lamps were used to maintain body core temperature at 37-38°C. A femoral artery catheter was placed to monitor systemic BP, and a suprapubic cystostomy catheter was placed to establish urinary drainage.
Thoracic Surgery
Polyethylene catheters were surgically introduced into the right atrium(via left external jugular vein), and PA via left sternotomy. Following our previously described methodology(6, 15), an external electromagnetic flow probe (no. EP 425; Carolina Medical Electronics, King, NC) was placed around the PA, proximal to the tip of the PA catheter. During each experiment, the PA flow probe was zeroed frequently using the approximation that diastolic blood flow in the PA is zero. In the documented absence of systemic to pulmonary vascular shunts, PA blood flow was taken as equivalent to total body CO. The existence of vascular shunts between the systemic and pulmonary circulations through either the foramen ovale or ductus arteriosus was excluded by comparison of oxygen content in blood samples obtained simultaneously from the right atrium, PA, and aorta. The PA and right atrium catheters were connected to pressure transducers to determine PAP and CVP.
In addition to the surgery listed above, in six of the piglets a 5 French Foley catheter was placed directly into the left atrial appendage, in a modification of the technique of Fahey and Lister(16). The catheter was placed through a small incision in the left atrial appendage, and was secured in place with a circumferential tie.
CBF Monitoring Surgery
We have previously described a technique for assessing CBF, which is sensitive to rapid changes in CBF(17, 18). This procedure represents a modification of the methods of Buckley et al.(19) and Scremin et al.(20, 21), and provides a continuous measure of phasic ICBF. Two separate dissections were made in the right neck region. Through the first dissection, at a level midway between the root of the aorta and the angle of the jaw, the right common carotid artery was identified. The vagus nerve was carefully freed from the vessel at this level, and an external electromagnetic flow probe (no. EP 406; Carolina Medical Electronics) was placed around the right common carotid artery. A separate dissection was then made at the level of the angle of the jaw, leaving approximately 3 cm of tissue intact between dissections to isolate the carotid flow probe from any physical manipulation during the “mechanical zero” procedure. Through the distal dissection, the right external carotid artery was identified, immediately cephalad to the carotid bifurcation, and ligated. All residual common carotid blood flow was then taken to reflect unilateral ICBF.
ICBF was recorded continuously on a polygraph recorder, while digital values were displayed simultaneously on the electromagnetic flow meter(Carolina Medical Electronics). A mechanical “zero flow” signal was produced without disturbing the carotid flow probe by briefly occluding the right common carotid artery with a vascular clamp through the distal dissection. This procedure was performed at frequent intervals throughout the experiment. In addition, at the end of each experiment the accuracy of the mechanical zero procedure was verified by comparing the flow probe signal during distal occlusion (mechanical zero) with both the signal produced when the probe was turned off (electrical zero) and the flow probe signal after the animal had been killed (absolute zero). Verification of the zero flow signal is particularly important in this model, as it allows changes in CBF to be expressed as a percentage of initial values.
Sagittal Sinus Catheterization
As described previously(17), a 2-cm diameter burr hole was made in the midline of the cranium halfway between the nasion and inion, and the sagittal sinus was identified. A 16-gauge plastic catheter(Angiocath, Deseret Medical, Sandy, UT) was placed in the sagittal sinus and advanced to the confluence of sinuses. Passive blood return was verified and the catheter was secured to the scalp. The catheter was connected to a pressure transducer to determine SSP. We used this measurement to assess intracranial pressure.
Blood Gas Monitoring
Blood specimens were taken from the aorta after surgery (during the stabilization period), during hyperventilation with 5% CO2 and 10% CO2, and during STREP infusion or LAB inflation when Paco2 was normal. From these blood samples, pH, Po2, Pco2, and base excess were determined using a blood gas analyzer (ABL II, Radiometer, Copenhagen, Denmark). Hb content and oxygen saturation were determined using a cooximeter(IL 282, Instrumentation Laboratories, Lexington, MA), previously calibrated in our laboratory for piglet Hb using the carbon monoxide scrubbing technique(22).
Hyperventilation Protocol
To measure the phasic response of ICBF to step changes in Paco2, we designed a technique to produce extremely rapid alteration in Paco2 without changing mean airway pressure or Pao2. Piglets were hyperventilated (40 breaths/min; 25 mL/kg tidal volume; 1000 mL/kg-min ventilation) with an inspired gas mixture of 5% CO2, 50% O2, 45% N2 to fix their Paco2 at approximately 40 torr. Mechanical zero signals from both the PA and carotid flow probes were confirmed, and hemodynamic values and a blood gas sample from the arterial catheter were obtained. Without any other change of ventilator settings, the inspired gas circuit was instantly switched to 10% CO2, 50% O2, 40% N2 to fix their Paco2 at approximately 70 torr. The time required to switch inspired gas circuits was <1 s. Continuous observations of CO, ICBF, BP, PAP, CVP, and SSP were determined for 7.5 min, a time previously determined by us to produce stable responses in cerebral hemodynamics. Mechanical zero signals from both the PA and carotid flow probes were confirmed in the hypercarbia condition, pressure and flow readings were determined, and another arterial blood gas was obtained. At the conclusion of each hyperventilation trial, the ventilator and the gas mixture were switched back to the settings before hyperventilation.
Preparation of Bacteria
STREP, serotype Ib, previously isolated from an infected human newborn, were grown in 250 mL of Todd-Hewitt broth to late log phase (≈3 × 107 organisms/mL). The bacteria were then centrifuged, the supernatant decanted, and the organisms resuspended to one-quarter of their original concentration in 0.9% NaCl. Previously, quantitative cultures of the bacterial inoculum have been performed by serial dilution to allow calculation of the rate of bacterial infusion.
After all surgery had been completed, a 1-h stabilization was allowed each piglet before the onset of any experimental protocol.
For us to test the role of prostaglandin production during 4 h of STREP sepsis in reducing cerebral vascular reactivity to hypercarbia, we first demonstrated that the effects of indomethacin on cerebral reactivity in nonseptic piglets were short-lived. Five piglets, designated group A, were studied. These piglets were initially subjected to the hyperventilation protocol (baseline). They then received indomethacin, 5 mg/kg diluted in normal saline, administered i.v. over 5 min. At 1 and 2 h after indomethacin administration, the piglets were subjected to the hyperventilation protocol(total of three hyperventilation episodes). These animals did not receive STREP and were not used in any other protocol.
Group 1. To investigate whether a 4-h STREP infusion attenuated the response of CBF to hypercarbia, five piglets designated group 1 were studied. The piglets were subjected to the hyperventilation protocol(baseline). They then received a 4-h, continuous infusion of live, washed, resuspended STREP organisms at approximately 2.0 × 107 STREP/kg/min × 60 min, followed by 4.0 × 107 STREP/kg/min× 60 min, and 8.0 × 107 STREP/kg/min × 120 min. At hourly intervals during the STREP infusion, the piglets were subjected to the hyperventilation protocol. Animals were thus subjected to a total of five hyperventilation episodes.
Group 2. To investigate the effect of the cyclooxygenase inhibitor indomethacin on the STREP-induced attenuation of the CBF response to hypercarbia, five other piglets designated group 2 were studied. The piglets were initially subjected to the hyperventilation protocol (baseline). They then received indomethacin, 5 mg/kg diluted in normal saline, administered i.v. over 5 min. Thirty minutes were allowed for the piglets to recover from any acute effects of the indomethacin infusion. These piglets then received the 4-h, continuous infusion of live, washed, resuspended STREP exactly as did group 1. At hourly intervals during the STREP infusion, the piglets were subjected to the hyperventilation protocol (five hyperventilation episodes).
Group 3. Six other piglets, designated group 3, did not receive STREP but rather received a 5 French Foley catheter placed in the left atrial appendage. These piglets were initially subjected to the hyperventilation protocol (baseline). Then, at hourly intervals for 4 h, the LAB was incrementally inflated to reduce CO to levels approximately equal to those of the piglets in group 1. At each steady-state after balloon inflation, the piglets were subjected to the hyperventilation protocol (five hyperventilation episodes).
Data Collection and Statistical Analysis
Hemodynamic data (both steady state and response to hypercarbia) were obtained at hourly intervals during the experimental protocols. Within each experimental protocol, changes in each analyzed variable were compared as a function of time using RMANOVA. At each hourly interval each analyzed variable was compared across groups 1, 2, and 3 using three-group analysis of variance(23). For both of these analyses, multiple pairwise comparisons were accounted for using the Newman-Keuls test. Statistical significance was accepted at p < 0.05.
RESULTS
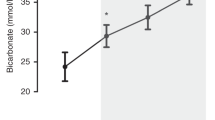
Figure 1 presents the percent change in unilateral CBF during 10% CO2 hyperventilation for five nonseptic group A piglets before (time 0, baseline) and after prostaglandin inhibition induced by infusion of indomethacin (5 mg/kg). At baseline, ICBF rose during hypercarbia by 81.1 ± 18.4% (16.0 ± 3.5 mL/min). One hour after indomethacin the cerebral vascular response to hypercarbia was significantly inhibited, as ICBF increased during hypercarbia by only 20.7 ± 12.9% (3.0 ± 2.1 mL/min) (p < 0.01 versus the value at baseline). By 2 h after indomethacin administration, however, the cerebral vascular response to hypercarbia was restored. At 2 h, ICBF increased during hypercarbia by 61.8± 27.1% (12.3 ± 6.6 mL/min), significantly greater than the value at 1 h after indomethacin, and not significantly different from the value at baseline (F = 11.4, p = 0.004).

Time course of the effects of indomethacin on cerebral vascular reactivity to hypercarbia. Data during 10% CO2 hyperventilation (Paco2 ≅ 70 torr) expressed as percent change in unilateral internal carotid blood flow in nonseptic group A piglets.n = 5. Indomethacin (5 mg/kg) was administered after the baseline values were obtained. The asterisk (*) indicates a significant difference at 1 h vs both time 0 and 2 h (RMANOVA). p ≤ 0.05. At 1 h postadministration, indomethacin significantly inhibited the cerebral vascular response to hypercarbia. By 2 h after indomethacin administration, however, the cerebral vascular response to hypercarbia was restored.%ΔICBF at 2 h was significantly greater than%ΔICBF at 1 h, but not significantly different from the value at baseline.
Figure 2 presents the effects of 4 h of STREP infusion or LAB inflation in groups 1, 2, and 3. The baseline values which are displayed in the graphs are comparable to those reported previously for piglets by us and others(6, 15).Figure 2a reveals that for all three groups, CO decreased steadily during STREP infusion or LAB inflation. After 4 h of STREP infusion in group 1, CO had fallen from 634 ± 121 (SD) mL/min at baseline to 324± 172 mL/min (p < 0.001). The decrease in CO during STREP infusion is consistent with previous findings reported in piglets by ourselves and others(6, 7, 15, 24). Despite pretreatment with indomethacin, CO also decreased significantly during STREP infusion in group 2 (600 ± 68 to 291 ± 80 mL/min). By design, CO in group 3 was also reduced significantly to levels approximately equal to those of group 1. At no time did CO differ significantly between the groups(F = 0.199; p = 0.821).

Steady state values. Mean ± SEM data for group 1(STREP infusion, n = 5, open triangles); group 2(indomethacin + STREP, n = 5, open squares); and nonseptic group 3 (LAB, n = 6, closed circles) at baseline (time 0) and at each hour of STREP infusion or LAB inflation. In group 2, indomethacin was administered after the baseline values, but before the start of STREP infusion. The numer sign (#) indicates a significant difference between group 3 vs groups 1 and 2; the asterisk (*) indicates a significant difference vs time 0 (RMANOVA); the section sign (§) indicates a significant difference between group 1 vs groups 2 and 3.p ≤ 0.05. (a) CO fell significantly in both septic groups 1 and 2 by 4 h. By design, CO was reduced as well as in group 3 by LAB inflation. At no time did the groups differ significantly. (b) Systemic BP. Aortic pressure decreased significantly in all experimental groups by 4 h. Aortic pressure did not differ significantly at any time comparing the three experimental groups. (c) Unilateral internal carotid blood flow. In group 1, 4 h of STREP infusion caused ICBF to decrease significantly. In group 2, despite indomethacin pretreatment, STREP infusion again caused ICBF to fall significantly by 4 h. In nonseptic group 3, despite the reduction in CO (cf. a) ICBF was unchanged at 4 h. (d) PAP. STREP infusion caused PAP to rise significantly in Group 1 at 1 h. Indomethacin pretreatment in Group 2 delayed the STREP-induced rise in PAP. By 3 h, PAP in both groups 1 and 2 was significantly greater than that in group 3, which did not change significantly.
Figure 2b reveals BP decreased significantly in all three groups, from approximately 100 mm Hg at baseline to approximately 55 mm Hg at 4 h. There were no significant differences in aortic pressure between the groups at any time (F = 0.239; p = 0.790).
Figure 2c displays the effects of 4 h of STREP infusion or LAB inflation on the absolute value of ICBF (unilateral CBF) in the three experimental groups. At baseline, ICBF was 32.0 ± 10.8 mL/min in group 1, 22.9 ± 9.9 mL/min in group 2, and 23.1 ± 7.4 mL/min in group 3, values which did not differ significantly from each other. By the 4th h of STREP infusion, ICBF fell significantly in both groups of septic piglets, to 21.0 ± 7.3 mL/min in group 1, and to 13.1 ± 4.3 mL/min despite indomethacin in group 2. Thus in the groups receiving STREP for 4 h ICBF fell markedly, by almost 40%. In contrast, after 4 h of LAB inflation (group 3), ICBF was 19.6 ± 6.3 mL/min, not significantly different from baseline. Not shown in the graph was the transient reduction in ICBF, by ≈10 mL/min(≈33%), seen acutely after indomethacin administration, before the onset of STREP infusion. This phasic reduction and recovery in CBF after indomethacin has been described previously (cf.Ref. 25).
Figure 2d reveals that in group 1, PAP increased significantly from 19.5 ± 4.0 mm Hg at baseline to 38.6 ± 14.0 mm Hg at 1 h and 32.8 ± 10.0 mm Hg at 4 h of STREP infusion. This response to STREP infusion in piglets has been well described by us and others previously(6, 7, 15, 24). In contrast, in group 2 piglets which received indomethacin, PAP was not significantly increased despite 1 h of STREP infusion: 16.8 ± 3.9 mm Hg at time 0versus 18.6 ± 4.4 mm Hg at 1 h of STREP. By the 2nd h of STREP infusion in group 2, however, PAP had risen significantly to 28.8± 8.7 mm Hg and remained elevated for the duration of the protocol. PAP did not change significantly from baseline at any point during the LAB inflation protocol, and at the 3rd and 4th h was significantly less than either of the septic groups.
At baseline, SSP for the piglets averaged 7.5 ± 3.0 mm Hg(n = 16). There were no significant changes in SSP in any group comparing baseline values with those at 4 h. Despite a significant reduction in Pao2 in groups 1 and 2 during 4 h of STREP infusion (from 260± 15 to 176 ± 59 torr in group 1 and 277 ± 23 to 166± 88 torr in group 2) the use of 50% O2 ensured that there was no significant fall in arterial oxygen saturation over time for any experimental group.
Paco2 rose during 10% CO2 ventilation from 43.5 ± 3.2 to 71.6 ± 4.2 torr (80 hypercarbia trials). By design, Pao2 did not change significantly during 10% CO2 hyperventilation compared with 5% CO2 hyperventilation (ΔPao2 = 5.4 ± 29.0 torr). Systemic BP decreased slightly, but significantly, during 10% CO2 hyperventilation compared with 5% CO2 hyperventilation (ΔBP =-5.5 ± 10.9 mm Hg; 80 hypercarbia trials). Importantly, the effect of 10% CO2 hyperventilation on BP did not change significantly for any group over the 4 h of STREP infusion or LAB inflation, nor were the groups significantly different in the change in BP during hyperventilation at any time.
Figure 3 presents the percent changes in unilateral CBF during 10% CO2 hyperventilation for groups 1, 2, and 3. At baseline, ICBF rose during hypercarbia by 68.7 ± 13.0% (22.1 ± 7.8 mL/min) in group 1, 62.2 ± 15.6% (13.1 ± 2.2 mL/min) in group 2, and 87.7 ± 34.0% (20.7 ± 9.2 mL/min) in group 3, values which did not differ significantly (p = 0.137). By the 4th h of STREP infusion,%ΔICBF was significantly less than baseline value in each septic group. In the nonseptic group 3 piglets, however,%ΔICBF was not significantly different compared with baseline even after 4 h of LAB inflation. At the 4th h, in the nonseptic group 3 ICBF rose during hypercarbia by 61.8 ± 23.3% (12.2 ± 5.2 mL/min), significantly greater than the values in the two groups which received STREP. In marked contrast, for both septic groups, ICBF actually tended to fall during hypercarbia by the fourth hour of STREP infusion.%ΔICBF was -7.8 ± 23.2%(-1.5 ± 4.5 mL/min) in group 1, and -10.9 ± 17.7% (-1.5 ± 2.4 mL/min) in group 2 at 4 h. At no time did%ΔICBF differ significantly comparing groups 1 and 2. The differences in%ΔICBF at 4 h compared with baseline were unrelated to alteration in the magnitude of the change in Paco2 or SSP, as neither ΔSSP or ΔPco2 was not different at 4 h compared with baseline for any of the three groups. Over all trials, during hypercarbia SSP rose by only 2.2 ± 2.0 mm Hg.

Percent change in unilateral internal carotid blood flow during hypercarbia. Data during 10% CO2 hyperventilation(Paco2 ≅ 70 torr) for group 1 (STREP infusion, n = 5,open triangles); group 2 (indomethacin + STREP, n = 5,open squares), and nonseptic group 3 (LAB, n = 6,closed circles) at baseline (time 0) and at each hour of STREP infusion or LAB inflation. In group 2, indomethacin was administered after the baseline values, but before the start of STREP infusion. Mean ± SEM. The number sign (#) indicates a significant difference between group 3vs groups 1 and 2; the asterisk (*) indicates a significant difference vs time 0 (RMANOVA). p ≤ 0.05. At baseline, ICBF increased significantly during hypercarbia in each experimental group, by approximately ≈73%. At 4 h, in septic groups 1 and 2,%ΔICBF fell during hypercarbia, significantly different from baseline, and significantly different from the rise in%ΔICBF in nonseptic group 3. At no time did the indomethacin pretreated group 1 differ significantly from group 2.%ΔICBF did not change significantly over 4 h of LAB inflation in group 3.
DISCUSSION
Under normal physiologic conditions hypercarbia increases CBF. The experimental protocols described here were designed to address two questions. We first hypothesized that STREP sepsis would impair the responsiveness of the cerebral circulation to hypercarbia in piglets. That hypothesis was confirmed. We demonstrated that the cerebral vasodilator response to hypercarbia was completely ablated after four hours of STREP sepsis in piglets.
Next, we addressed a question of mechanism-we hypothesized that sepsis-induced prostanoids mediated the loss of cerebral reactivity to hypercarbia. We also hypothesized that the reduction of cerebral reactivity to hypercarbia resulted from the reduced systemic BP or CO. We found no support for either of these hypothesized mechanisms. The reduction in the cerebral vascular response to hypercarbia was not altered by pretreatment with a dose of indomethacin that has been previously demonstrated to inhibit prostaglandin synthesis for a time period approximately equal to that of the entire STREP protocol used here(7). Consequently, we conclude that the synthesis of abnormal arachidonic acid metabolites is not likely to be the mechanism by which STREP reduces CBF reactivity to hypercarbia.
We considered the possibility that the effect of STREP infusion on CBF reactivity was simply a response to significant reduction of CO and BP noted during the sepsis protocols, and not related to the STREP organisms per se. It has long been held that CBF may fall if cerebral perfusion pressure is reduced below a critical threshold of autoregulation. We have previously demonstrated in piglets, for example, that under certain conditions, CBF may fall as CO falls despite a constant or even elevated BP(17). To assess these possibilities, we matched the reductions in CO and BP noted during STREP infusions (groups 1 and 2) with reductions in CO and BP produced by LAB inflation (group 3). At every stage of the LAB protocol cerebral vascular reactivity was preserved, and after 4 h of observations%ΔICBF was significantly greater in group 3 than in either septic group. We conclude that nonspecific effects of reductions of CO or BP do not to account for the obliteration of cerebral reactivity to hypercarbia during STREP infusion. Further, the preservation of cerebral vascular reactivity in the nonseptic group argues against the introduction of artifacts by anesthesia or surgical preparation (all of which were matched in the three experimental protocols).
Arachidonic acid metabolites modulate vasomotion in a number of vascular beds in piglets, both in health and disease. For example, when given i.v. to piglets, indomethacin reduces CBF and cerebral metabolic rate for oxygen, presumably through its inhibition of cyclooxygenase(26). When piglets were ventilated with a 9% CO2 + 10% O2 mixture, cerebral spinal fluid concentrations of prostaglandin F1a and E2, as well as thromboxane B2, rose in conjunction with pial artery dilation (observed through cranial windows)(12). Direct application of prostaglandin E2 to the pial surface of piglets also caused pial artery vasodilation. Further, Chemtob et al.(14) have concluded that prostaglandins and thromboxane contribute significantly in the autoregulation of CBF in piglets.
In the pulmonary circulation, thromboxane A2 has been shown to be responsible for sepsis-induced pulmonary hypertension. Within minutes after infusion of either live(7, 10, 27) or heat-killed STREP organisms(28), both plasma thromboxane levels and PAP rise in concert. If the rise in thromboxane is blocked by either indomethacin(7) or the thromboxane inhibitor dazemgrel(10, 27), the rise in PAP disappears. This relationship holds true even when thromboxane is blocked after STREP sepsis has been established.
If, however, prostaglandin metabolites do not mediate the loss of cerebral vascular reactivity during sepsis, and the phenomenon cannot be accounted for by a reduction in CO or BP, what is it about sepsis that“paralyzes” the cerebral vascular bed? Certainly, the most likely explanation is an indomethacin-insensitive toxic metabolite, and several possible candidates can be suggested. The role of EDRF (nitric oxide) in controlling the cerebral vascular circulation, either under baseline physiologic conditions or during disease states, remains unclear. Investigators have shown in piglets that EDRF-like substances dilate pial arterioles(29), and competitive inhibition of EDRF formation results in increased cerebral vascular resistance(17) and reduced forebrain blood flow(30). EDRF has also been postulated to mediate many abnormal responses in models of sepsis using other circulations, organisms or age spectrums(31, 32). Other potential mediators of the loss of cerebral vascular reactivity include cytokines induced by sepsis (i.e. platelet-activating factor, tumor necrosis factor, and IL-1). Platelet-activating factor is known to be a potent vasoconstrictor in piglets(33), and tumor necrosis factor-α and IL-1 are believed to mediate the septic state, in part, by influencing vascular resistance(34).
Our in vivo observations of the loss of cerebral vascular reactivity in neonatal piglets during STREP bacteremia are in accord with the findings of others using different assessments of vascular reactivity and different models of sepsis. McKenna et al.(3) demonstrated a diminished maximal contractile response to norepinephrine in aortic rigns from rats made septic by cecal ligation. Wylam et al.(1) demonstrated that arterial rings from several different vascular beds of endotoxin-treated dogs contracted normally to phenylephrine, but had markedly impaired relaxation when exposed to acetylcholine. In confirmation of these findings, sera from septic humans and septic dogs have been shown to alter rat aortic vascular reactivity(35). To the extent that diminution in vascular reactivity during sepsis is a robust phenomenon which can be demonstratedin vivo and in vitro, in adult and newborn animals, induced by Gram-negative endotoxin and live STREP organisms, this phenomenon is likely to be an increasingly physiologically relevant feature of sepsis, and its amelioration may be an important marker of physiologic improvement during sepsis. We note, however, that these observations differ from the findings of Bowton et al.(5), who reported normal cerebral vascular reactivity to hypercarbia in septic human adults, assessed by the xenon clearance technique. At present, we cannot reconcile these disparate observations.
Of interest is the observation that, in the piglets reported here, cerebral vascular reactivity decreased significantly only after 4 h of STREP infusion. The dogs in the study of Wylam et al.(1) received endotoxin i.v. 5 h before sacrifice, whereas the rats in the work of McKenna et al.(3) “were obviously” ill at sacrifice, which took place up to 29 h after cecal ligation. Similarly, a latency of pathologic vascular response has been observed in other studies involving STREP. Using the cranial window technique, McKnight et al.(36) reported in rats that dilation of pial cerebral arterioles occurred only after 2.5 h of exposure to STREP(36). Runkle et al.(7) reported a terminal decline in CO beginning at the 3rd h of STREP infusion in piglets, despite a stable CO in the previous 150 min; we also have observed this phenomenon during STREP infusion(37). Finally, we have observed the development of significant base deficit and lactic acidemia after 3.5 h of STREP infusion in piglets(15). These data suggest that the altered vascular reactivity in bacteremic and endotoxin-treated animals becomes manifest only after a “latent” period of at least several hours.
Quantitatively, the increases in ICBF during hypercarbia noted in our piglets at baseline (before STREP or LAB inflation) agree well with previously reported findings for comparable elevations of Paco2, whether expressed as milliliters of increase in flow per min per mm Hg Paco2 [2.0 mL/100 gm-min-torr in lambs(38), 1.3 mL/100 gm-min-torr in humans(5), compared with 1.7 mL/100 gm-min-torr in our piglets], or as percentage rise of CBF/mm Hg Paco2 [2-3.6%/torr in lambs(39); 3%/torr in human infants(40); 2.9-3.6%/torr in baboons(41); 1.5%/torr in rabbits(20, 21), compared with approximately 2.7%/torr at baseline in the piglets which we report here]. In a separate series of experiments using this methodology, we have determined the half-time of change of Paco2 as it rose from approximately 40-70 torr to be <19 s(18).
Considering steady-state effects of STREP infusion, we found that ICBF fell significantly after 4 h of STREP infusion in group 1 (by approximately 34%). These data are consistent with the report of Bressack et al.(24), who demonstrated a significant fall in microsphere-determined CBF in piglets 3.5 h after a 45-min infusion of STREP. The mechanism of the reduction in steady-state ICBF induced by STREP is not readily apparent. Reduction in systemic BP or CO during STREP infusion do not completely account for the fall in ICBF, as both BP and CO fell equivalently during the LAB protocol, yet ICBF did not fall significantly. Sepsis-induced alterations in prostaglandin metabolism are also unlikely to underlie this effect, as pretreatment with indomethacin did not prevent the decrease in ICBF in the group 2 piglets.
Wagerle et al.(13) as well as other investigators(12) have demonstrated in piglets the central role of arachidonic acid metabolites in the response of the cerebral circulation to acute hypercarbia. The cerebral vascular response to hypercarbia is inhibited by the systemic administration of 5 mg/kg indomethacin. This inhibition of cerebral vascular reactivity by indomethacin, however, appears to be of relatively short duration. Leffler et al.(42) have shown in piglets that, at 1 h after infusion of 5 mg/kg indomethacin, pial arterioles observed through cranial windows exhibited very little dilation in response to acute hypercarbia compared with that found in pre-indomethacin controls. By the 2nd h after indomethacin, the capacity of pial arterioles to dilate in response to hypercarbia was significantly restored. Our series of experiments reveal very similar findings. As shown in Figure 1, 5 mg/kg indomethacin virtually ablated the response to acute hypercarbia at 1 h. By 2 h after indomethacin, however, the increase in%ΔICBF in response to hypercarbia was not significantly different from time 0 (before indomethacin). Indeed, as suggested by the data in Figure 1, for piglets which received indomethacin and STREP (group 2), the duration of indomethacin's inhibition of the CBF response to hypercarbia may be as short as only 90 min. That is, at 1 h of STREP infusion (actually 90 min after administration of indomethacin) (cf. “Methods”),%ΔICBF in group 2 was not significantly different from the value of%ΔICBF at time 0 or the value of%ΔICBF at 1 h in groups 1 and 3.
Some authors have suggested that acute effects of anti-inflammatory agents such as indomethacin on CBF may not be prostanoid related(43). In the context of either the steady-state effects of sepsis on ICBF or the sepsis-induced impairment of vascular responsiveness to hypercarbia, such non-prostaglandin-related effects, should they exist, would be irrelevant to our conclusions, as we could not detect a beneficial effect of indomethacin on ICBF or cerebral reactivity to hypercarbia at any time in group 2. Indomethacin has been shown by us and others(18, 42, 44, 45) to temporarily reduce both baseline CBF. In the septic piglets reported here, CBF had returned to baseline within approximately 90 min of indomethacin administration.
In sum, we have shown that cerebral vascular reactivity to a physiologic stimulus (acute hypercarbia) is attenuated during STREP sepsis. This attenuation cannot be accounted for by the fall in CO or BP due to STREP infusion, nor is it likely that cyclooxygenase products are responsible for this effect. The mechanism(s) responsible for the attenuation in cerebral vascular reactivity to hypercarbia during sepsis remains to be elucidated.
Abbreviations
- STREP:
-
group B Streptococcus
- CBF:
-
cerebral blood flow
- LAB:
-
left atrial ballon
- ICBF:
-
internal carotid artery blood flow
- CO:
-
cardiac output
- PA:
-
pulmonary artery
- PAP:
-
pulmonary artery pressure
- BP:
-
systemic blood pressure
- EDRF:
-
endothelium-derived relaxation factor
- CVP:
-
central venous pressure
- SSP:
-
sagittal sinus pressure
- RMANOVA:
-
repeated measures analysis of variance
- Pao2:
-
partial pressure of arterial O2
- Paco2:
-
partial pressure of arterial CO2
References
Wylam ME, Samsel RW, Umans JG, Mitchell RW, Leff AR, Schumacker PT 1990 Endotoxin in vivo impairs endothelium-dependent relaxation of canine arteries in vivo. Am Rev Respir Dis 142: 1263–1267
Nelson DP, Beyer C, Samsel RW, Wood LDH, Schumacker PT 1987 Pathological supply dependence of O2 uptake during bacteremia in dogs. J Appl Physiol 63: 1487–1492
McKenna TM, Martin FM, Chernow B, Briglia FA 1986 Vascular endothelium contributes to decreased aortic contractility in the adult respiratory distress syndrome. Circ Shock 19: 267–73
Bone RC 1994 Sepsis and its complications: the clinical problem. Crit Care Med 22:S8–S11
Bowton DL, Bertels NH, Prough DS, Stump DA 1989 Cerebral blood flow is reduced in patients with sepsis syndrome. Crit Care Med 17: 399–403
Meadow WL, Rudinsky BF, Strates E 1987 Oxygen delivery, oxygen consumption, and metabolic acidosis during GBS sepsis in piglets. Pediatr Res 22: 509–512
Runkle B, Goldberg RN, Streitfeld MM, Clark MR, Buron E, Setzer ES, Bancalari E 1984 Cardiovascular changes in group B streptococcal sepsis in the piglet: response to indomethacin and relationship to prostacyclin and thromboxane A2 . Pediatr Res 18: 874–878
Gibson RL, Truog WE, Redding GJ 1988 Thromboxane-associated pulmonary hypertension during three types of Gram-positive bacteremia in piglets. Pediatr Res 23: 553–556
Tarpey MN, Graybar GB, Lyrene RK, Gofoy G, Oliver J, Gray BM, Philips JB 1987 Thromboxane synthesis inhibition reverses group B Streptococcus-induced pulmonary hypertension. Crit Care Med 15: 644–647
Troug WE, Sorenson GK, Standaert A, Redding GJ 1986 Effects of the thromboxane inhibitor, dazemgrel (UK 38,485), on pulmonary gas exchange and hemodynamics in neonatal sepsis. Pediatr Res 20: 481–486
Leffler CW, Busija DW, Fletcher AM, Beasley DG, Hessler JR, Green RS 1985 Effects of indomethacin upon cerebral hemodynamics of newborn pigs. Pediatr Res 19: 1160–1164
Leffler CW, Busija DW 1985 Prostanoids in cortical subarachnoid cerebrospinal fluid and pial arterial diameter in newborn pigs. Circ Res 57: 689–694
Wagerle LC, Mishra OP 1988 Mechanism of CO2 response in cerebral arteries of the newborn pig: role of phospholipase, cyclooxygenase, and lipoxygenase pathways. Circ Res 62: 1019–1026
Chemtob S, Beharry K, Rex J, Varma DR, Aranda JV 1990 Prostanoids determine the range of cerebral blood flow autoregulation of newborn piglets. Stroke 21: 777–784
Rudinsky BF, Meadow WL 1992 Relationship between oxygen delivery and metabolic acidosis during sepsis in piglets. Crit Care Med 20: 831–39
Fahey JT, Lister G 1987 A simple method for reducing cardiac output in the conscious lamb. Am J Physiol 249:H188–H192
Meadow W, Rudinsky B, Bell A, Lozon M, Randle C, Hipps R 1994 The role of prostaglandins and endothelium derived relaxation factor in the regulation of cerebral blood flow and cerebral oxygen utilization in the piglet: operationalizing the concept of an essential circulation. Pediatr Res 35: 649–656
Lozon M, Rudinsky B, Randle C, Hipps R, Meadow W 1991 Effects of indomethacin on cerebral O2 regulation and cerebrovascular reactivity: in situ measurements in neonatal piglets. Pediatr Res 29: 223
Buckley NM, Gootman PM, Gootman N, Reddy GD, Weaver LC, Crane LA 1976 Age-dependent cardiovascular effects of afferent stimulation in neonatal pigs. Biol Neonate 30: 268–279
Scremin OU, Sonnenschein RR, Rubinstein EH 1982 Cerebrovascular anatomy and blood flow measurements in the rabbit. J Cereb Blood Flow Metab 2: 55–66
Scremin OU, Sonnenschein RR, Rubinstein EH 1983 Cholinergic cerebral vasodilatation: lack of involvement of cranial parasympathetic nerves. J Cereb Blood Flow Metab 3: 362–368
Kirk BW, Raber MB 1973 A practical apparatus for rapid determination of blood oxygen content. J Appl Physiol 34: 724–725
Glanz SA 1987 Primer of Biostatistics, 2nd Ed. McGraw-Hill, New York, pp 30–63
Bressack MA, Morton NS, Hortop J 1987 Group B streptococcal sepsis in the piglet: effects of fluid therapy on venous return, organ edema, and organ blood flow. Circ Res 61: 659–669
Mirro R, Leffler CW, Armstead W, Beasley DG, Busija DW 1988 Indomethacin restricts cerebral blood flow during pressure ventilation of newborn piglets. Pediatr Res 24: 59–62
Pourcyrous M, Leffler CW, Bada HS, Korones SB, Busija DW 1994 Cerebral blood flow responses to indomethacin in awake newborn piglets. Pediatr Res 35: 565–570
Hammerman C, Komar K, Meadow W, Strates E 1988 Selective inhibition of thromboxane synthase reduces group B β-hemolytic streptococci-induced pulmonary hypertension in piglets. Dev Pharmacol Ther 11: 306–312
Barefield ES, Hicks TP, Philips JB 1994 Thromboxane and pulmonary morphometry in the development of the pulmonary hypertensive response to group B Streptococcus. Crit Care Med 22: 506–514
Busija DW, Leffler CW, Wagerle LC 1990 Mono-L-arginine-containing compounds dilate piglet pial arterioles via an endothelium-derived relaxing factor-like substance. Circ Res 67: 1374–1380
Greenberg RS, Helfaer MA, Kirsch JR, Moore LE, Traystman RJ 1994 Nitric oxide synthase inhibition with N-monomethyl-L-arginine reversibly decreases cerebral blood flow in piglets. Crit Care Med 22: 384–392
Julou-Schaeffer G, Gray GA, Fleming I, Schott C, Parratt JR, Stoclet JC 1990 Loss of vascular responsiveness induced by endotoxin involves L-arginine pathway. Am J Physiol 259:H1038–H1043
Thiemermann C, Vane J 1990 Inhibition of nitric oxide synthesis reduces the hypotension induced by bacterial lipopolysaccharides in the rat in vivo. Eur J Pharmacol 182: 591–595
Armstead WM, Pourcyrous M, Mirro R, Leffler CW, Busija DW 1988 Platelet activating factor: a potent constrictor of cerebral arterioles in newborn pigs. Circ Res 62: 1–7
Moldawer LL 1994 Biology of proinflammatory cytokines and their antagonists. Crit Care Med 22:S3–S8
Hollenberg SM, Cunion RE, Parrillo JE 1992 Effect of septic serum on vascular smooth muscle: in vitro studies using rat aorta. Crit Care Med 20: 993–998
McKnight AA, Keyes WG, Hudak ML, Jones DM 1992 Oxygen free radicals and the cerebral arteriolar response to group B Streptococcus. Pediatr Res 31: 640–644
Meadow WL, Rudinsky BF, Strates E 1987 Oxygen delivery, oxygen consumption, and metabolic acidosis during GBS sepsis in piglets. Pediatr Res 22: 509–512
Rosenberg AA 1986 Cerebral blood flow and O2 metabolism after asphyxia in neonatal lambs. Pediatr Res 20: 778–782
Sonesson S, Herin P 1988 Intracranial arterial blood flow velocity and brain blood flow during hypocarbia and hypercarbia in newborn lambs. Pediatr Res 24: 423–426
Ashwal S, Stringer W, Tomasi L, Schneider S, Thompson J, Perkin R 1990 Cerebral blood flow and carbon dioxide reactivity in children with bacterial meningitis. J Pediatr 117: 523–530
Raju TNK, Kim SY 1991 The effect of hematocrit alteration on cerebral vascular reactivity in newborn baboons. Pediatr Res 20: 778–782
Leffler CW, Mirro R, Shibata M, Parfenova H, Armstead WM, Zuckerman S 1993 Effects of indomethacin on cerebral vasodilator responses to arachidonic acid and hypercapnia in newborn pigs. Pediatr Res 33: 609–614
Chemtob S, Beharry K, Barna T, Varma DR, Aranda JV 1991 Differences in the effects in the newborn piglet of various nonsteroidal antiinflammatory drugs on cerebral blood flow but not on cerebrovascular prostaglandins. Pediatr Res 30: 106–111
Pourcyrous M, Leffler CW, Bada HS, Korones SB, Busija DW 1994 Cerebral blood flow responses to indomethacin in awake newborn pigs. Pediatr Res 35: 565–570
Rudinsky BF, Bell A, Hipps R, Meadow WL 1994 Group B Streptococcal sepsis disrupts, and indomethacin restores, hemodynamic tolerance to acute hypoxia in piglets. Pediatr Res 35: 58( abstr)
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Rudinsky, B., Lozon, M., Bell, A. et al. Group B Streptococcal Sepsis Impairs Cerebral Vascular Reactivity to Acute Hypercarbia in Piglets. Pediatr Res 39, 55–63 (1996). https://doi.org/10.1203/00006450-199601000-00008
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199601000-00008
