
Abstract
Autophagy is a highly conserved self-degradative process that has a key role in cellular stress responses and survival. Recent work has begun to explore the function of autophagy in cancer metastasis, which is of particular interest given the dearth of effective therapeutic options for metastatic disease. Autophagy is induced upon progression of various human cancers to metastasis and together with data from genetically engineered mice and experimental metastasis models, a role for autophagy at nearly every phase of the metastatic cascade has been identified. Specifically, autophagy has been shown to be involved in modulating tumor cell motility and invasion, cancer stem cell viability and differentiation, resistance to anoikis, epithelial-to-mesenchymal transition, tumor cell dormancy and escape from immune surveillance, with emerging functions in establishing the pre-metastatic niche and other aspects of metastasis. In this review, we provide a general overview of how autophagy modulates cancer metastasis and discuss the significance of new findings for disease management.
Similar content being viewed by others

Introduction
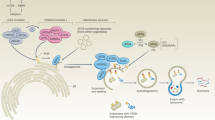
Macro-autophagy (hereafter autophagy) is a highly conserved catabolic process that targets cellular contents to the lysosomal compartment for degradation. Because autophagy has the ability to degrade very large structures, cells depend on this pathway to turnover damaged organelles, pathogens and large protein aggregates.1 Autophagic degradation serves as an important source of amino acids, nucleotides and fatty acids, especially for cells unable to acquire sufficient nutrients from the extracellular milieu to sustain ATP production and biosynthesis.2 Autophagy has a complex and highly context-dependent role in tumorigenesis3 with work from genetically engineered mouse models demonstrating that autophagy suppresses primary tumor growth on the one hand4, 5, 6 but is required for tumor maintenance and progression to advanced disease on the other.7, 8, 9, 10, 11, 12, 13 More recently, investigation of the role of autophagy in metastatic progression has suggested that autophagy promotes multiple steps in the metastatic cascade (Figure 1).

Schematic illustrating roles of autophagy in the metastatic cascade. Autophagy increases as tumor cells progress to invasiveness and this in turn is linked to increased cell motility, EMT, a stem cell phenotype, secretion of pro-migratory factors, release of MMPs, drug resistance and escape from immune surveillance at the primary site in some tumors. Many aspects of these autophagy-dependent changes during acquisition of invasiveness also likely contribute to the ability of disseminating tumor cells to intravasate, survive and migrate in the circulation before extravasating at secondary site. At the secondary site, autophagy is required to maintain tumor cells in a dormant state, possibly through its ability to promote quiescence and a stem cell phenotype, that in turn is linked to tumor cell survival and drug resistance. Emerging functions for autophagy in metastasis include a role in establishing the pre-metastatic niche as well as promoting tumor cell survival, escape from immune surveillance and other aspects required to ultimately grow out an overt metastasis.
The metastatic cascade can be divided into a series of stages: local invasion, intravasation, survival in the circulation, extravasation, survival at a second site and finally outgrowth at a second site14, 15 (Figure 1). All of these steps involve the physical translocation of cancer cells to new microenvironments, where they must survive altered nutrient, growth factor and physical support in order to colonize successfully.16 During local invasion, epithelial cancer cells break through the basement membrane and acquire a motile phenotype through induction of the epithelial–mesenchymal transition (EMT), a process that is active during mammalian embryonic development and wound healing in the adult but then co-opted by the tumor as a means to escape and migrate.17 The now-motile cancer cells then cross pericyte and endothelial cell barriers to enter the circulation by utilizing some of the same matrix-degrading enzymes upregulated during EMT and facilitated by the inherently leaky and disordered organization of the tumor vasculature.18 Once in the circulation, tumor cells face additional stresses including cell death signals triggered by the absence of anchorage to extracellular matrix (ECM) (that is, anoikis)19 in addition to the mechanical injury inherent in transit through narrowing blood vessels.16 As tumor cells arrive at secondary sites in other organs, they either extravasate from the vessel or grow intraluminally until the new lesion ruptures vessel walls.15, 16 The factors determining the target organ at which the tumor cell arrests and potentially grows out has been the subject of historical debate between the ‘seed and soil’ theory, wherein certain tumors (the ‘seed’) exhibit tropism for select secondary sites above others (the ‘soil’),20 and the theory that circulatory patterns are sufficient to dictate sites of tumor cell arrest.21 It is likely that both patterns of metastasis contribute to determining the success of colonization although this may vary from tissue to tissue.22, 23 The colonization process itself during the last stages of metastasis is multi-step with tumor dormancy, micrometastasis and macrometastasis defining how rapidly colonization takes place.14, 15 Basically, once in a new location, tumor cells need to adapt rapidly to new and unfamiliar stromal interactions; whether and how quickly tumor cells form micrometastases may be related to the similarities or differences between the stroma at the tumor origin versus the stroma at the site of metastasis.15, 16
Emerging evidence shows that autophagy promotes not only the survival of dormant tumor cells24, 25 and disseminating tumor cells in the circulation26, 27 but also specifically promotes the survival of stem-like subpopulations of tumor cells that drive invasion and treatment resistance.28, 29, 30 In addition, several recently identified autophagy substrates indicate novel functions for autophagy early in the metastatic cascade in the direct regulation of the EMT, tumor cell migration and invasion31, 32, 33, 34 (Figure 2). In this review, we will survey what is known about functions of autophagy in metastasis and discuss some of the remaining open questions in this area, with the goal of elucidating whether autophagy might be a useful target in clinical efforts to prevent the most deadly aspect of cancer: metastasis.

Autophagy promotes tumor cell motility. Autophagy promotes tumor cell motility through various mechanisms including: (a) promoting a stem cell phenotype that may be linked to as association of increased autophagy in response to stresses such as hypoxia and TGF-β, with EMT; (b) promoting survival in response to matrix detachment; (c) modulating levels of RhoA and Rho signaling while conversely being induced by Rho signaling; (d) through coordinated control of autophagy and FAK via FIP200; (e) a role in promoting focal adhesion turnover both directly and via regulation of SRC activity; (f) autophagy-dependent production of secreted factors that promote invasion, such as IL-6 and MMP2.
Autophagy is upregulated during metastasis
Given the numerous challenges that metastatic tumor cells overcome to successfully establish distant colonies (for example, invasion, anoikis resistance and colonization) and the critical role of autophagy as a response to cellular stress, various roles for autophagy in the metastatic cascade have been postulated.35 Indeed, autophagic flux is induced by many of the environmental stresses that are known to promote metastasis, such as hypoxia, as well as those that are experienced by disseminating tumor cells, including nutrient deprivation36 and detachment from the ECM.15, 16, 37, 38 Although at present it is not possible to directly assess autophagic flux in primary human tumor samples, various studies using surrogate markers have identified an association between increased autophagy and metastasis. For example, increased punctate staining for microtubule associated light chain B (LC3B) was associated with lymph node metastasis and reduced survival in human breast cancer39, 40 whereas melanoma metastases exhibited increased LC3B staining compared with matched primary tumor samples.39, 41, 42 LC3B expression was also correlated with metastasis in hepatocellular carcinoma, with higher LC3B staining in metastases relative to primary tumors and in early metastatic colonies relative to late metastatic colonies.26, 27 Meanwhile in human glioblastoma, increased expression of an autophagy gene signature was associated with a more aggressive and invasive phenotype.43
Autophagy is regulated at both the transcriptional and post-translational level in response to nutrient signaling pathways.36, 44, 45, 46 However, it remains to be fully elucidated whether increased autophagy associated with progression to invasive cancer is a downstream effect of reduced nutrient availability in the changing microenvironment of the growing tumor, or whether, as seems more likely, control of autophagy by nutrient sensing mechanisms becomes uncoupled during tumor progression. Tumors have evolved to scavenge nutrients from their environment by unconventional means, such as macropinocytosis,47 and have frequently upregulated cellular nutrient transporters and rewired their metabolism to make them less sensitive to loss of nutrient support from the environment.48 In human pancreatic cancer, for example, elevated expression of autophagy genes was found to occur due to constitutive activation of the microphthalmia/transcription factor E (MiT/TFE) transcription factors that removed them from negative regulation by target of rapamycin complex-I (mTORC1).44 TFE transcription factors are phosphorylated by and co-localize with mTORC1 at the lysosome when nutrients are present.49 Inactivation of mTORC1 or nutrient deprivation promotes nuclear translocation of TFEB and expression of target genes involved in lysosomal biogenesis thereby providing a mechanistic link between lysosomal activity and nuclear gene expression in response to changes in nutrient availability.49 In contrast to tight nutrient control of MiT/TFE factors in untransformed cells, nuclear translocation of MiT/TFE factors in pancreatic cancer was promoted by Importin-8 in a manner that overrides suppression by mTORC1.44 Deregulation of MiT/TFE transcription factors has also been identified in other tumor types, including human melanoma.50 Evidence addressing how post-translational negative control of autophagy by target of rapamycin (mTOR) signaling is surmounted in tumors came with work showing that protein phosphatase 2A has elevated phosphatase activity against Unc51-like autophagy activating kinase-1 (ULK1) in pancreatic cancer compared with normal cells.51 ULK1 activity is inhibited by mTORC1 phosphorylation on S637 amongst other sites but protein phosphatase 2A acts against this by de-phosphorylating phosphor-S637 resulting in elevated ULK1 activity in tumor cells despite mTORC1 activity.51 Thus, there are both transcriptional and post-translational mechanisms that may explain the conundrum of how robust autophagic flux and mTOR pathway activity can co-exist in tumor cells. In addition, it is possible that elevated autophagy during cancer progression to metastasis is genetically determined by additional oncogenic lesions or metastasis-specific mutations that remain to be characterized.
Links between autophagy, EMT and cancer stem cells
Tumor cells undergo EMT resulting in adherent epithelial cells morphing into highly motile mesenchymal cells, as part of the early stages of cancer progression to invasiveness and metastasis.17 This involves reorganization of the cytoskeleton, downregulation of proteins important for maintaining epithelial cell–cell junctions, particularly E-cadherin and upregulation of proteins that confer mesenchymal characteristics, leading to loss of cell polarity, dissolution of cell–cell junctions, and secretion of matrix metalloproteinases (MMPs) and other proteases involved in ECM degradation.17 Significantly, various inducers of EMT, including hypoxia and transforming growth factor beta (TGFβ), also potently activate autophagy.52, 53
However, it is not clear to what extent EMT is dependent on autophagy and thus whether EMT and autophagy act in sequence or in parallel to promote tumor cell invasiveness (Figure 2a). One study reported that autophagy is required for TGF-β-induced EMT and invasion of hepatocellular carcinoma cell lines, in part through a dependence on autophagy for TGF-β signaling.53 It was also shown that ULK2, which promotes autophagy through phosphorylation of the Beclin1-containing initiation complex, stimulates EMT, downregulation of E-cadherin and increased invasiveness in vitro.54 Increased autophagy was also linked to a more mesenchymal stem-like phenotype and to be required for invasion and migration of glioblastoma stem cell lines43 although a more recent study has argued that autophagy limits glioblastoma tumor cell migration through downregulation of Snail and Slug.55
Interestingly, the p62/Sqstm1 autophagy cargo adapter was shown to bind to the EMT regulator, Twist and prevent its proteasomal degradation leading to increased EMT, increased invasiveness in vitro and metastasis in vivo.31 A similar study validated these effects of p62/Sqstm1 on EMT through p62-dependent stabilization of both Twist and SMAD4, a TGF-β signal transducer.56 By inference this suggests that autophagy, by limiting p62/Sqstm1 accumulation indirectly inhibits EMT and thus acts to reduce tumor cell migration which contradicts the aforementioned findings showing a role of autophagy in promoting tumor cell motility.13, 32, 33, 34, 35, 39 These apparently disparate conclusions in the field highlight the need for increased effort to determine whether autophagy and EMT are directly or indirectly linked and if so, whether this varies as a function of tumor sub-type and underlying genetics, and also whether p62/Sqstm1 can act independently of autophagy to promote cell migration and metastasis.
Both EMT and autophagy promote the cancer stem cell state28, 57, 58 making the link between EMT and autophagy particularly intriguing (Figure 2a). Cancer stem cells (CSCs) have been postulated to drive tumor metastasis due to both their more motile and plastic phenotype and their ability to propagate de novo CSCs and tumor heterogeneity at secondary sites,14, 15, 59 with numerous reports of a clinical correlation between expression of CSC markers and increased invasiveness and metastasis.17, 60, 61, 62, 63 Induction of EMT promotes the cancer stem cell phenotype through the activity of transcription factors, such as Slug that are part of the so-called EMT-TF, that activates self-renewal gene expression programs, upregulation of CD44 and tumor-propagating properties in breast cancer.15, 57, 58, 59, 64, 65 Intriguingly, components of the EMT-TF modulate MITF activity during melanomagenesis66 that, as mentioned, activates autophagy gene expression.44
Autophagy is similarly required to maintain CD44+CD24−/low breast cancer stem cells28, 29 and the central autophagy initiator, Beclin1 emerged from an shRNA screen for genes that modulate cancer stem cell plasticity.67 Autophagy is required for normal tissue stem cell maintenance and differentiation68, 69, 70 with hematopoietic stem cells dependent on autophagy for survival18, 71 and muscle stem cells dependent on autophagy to prevent senescence.72 In human breast ductal carcinoma in situ (DCIS), subpopulations of cells exhibiting increased tumor-initiating capacity and migration/invasion capabilities were shown to have increased autophagy and were dependent on autophagic flux for their survival and stem cell phenotype.28, 29 The requirement for autophagy in CD44+/CD24−/low breast cancer cells was further supported by studies identifying ATG4 (autophagy related gene 4) as a regulator of this cell population and their ability to form mammospheres in vitro and form tumors in vivo.20, 30 Thus, a model emerges in which cancer stem cells couple increased autophagy to induction of EMT to promote self-renewal, motility, survival and drug resistance in a hypoxic or otherwise stressful microenvironment (Figure 2a).
Autophagy and resistance to anoikis
Epithelial cells rely on attachment to the ECM via integrins to maintain survival, and prolonged detachment from the ECM results in a form of apoptotic cell death called anoikis (Figure 2b).73 Autophagy is induced in epithelial cells by matrix detachment or by direct inhibition of integrins, while inhibition of autophagy increases epithelial cell death upon detachment.38 Thus, it has been proposed that autophagy has a key role in preventing anoikis and supporting the survival of detaching tumor cells during metastatic dissemination.74 In mammary tumor models, autophagy induction in response to matrix detachment or integrin blockade was associated with reactive oxygen species (ROS)-dependent activation of the ER-stress responsive kinase, protein kinase R-like ER kinase (PERK1).37 Consistently, inhibition of PERK or autophagy itself during matrix detachment or integrin signaling blockade promoted cell death and reduced clonogenic recovery following detachment, supporting a role for PERK-induced autophagy in mammary tumor cell survival during matrix detachment.37, 38
However, the mechanism through which PERK activates autophagy downstream of integrin blockade during matrix detachment is less well understood. Activated PERK activates eIF-2α to suppress general protein translation but permit selective translation of the ATF4 transcription factor,37 which is known to induce ATG5 (autophagy related gene 5) and LC3B expression.75 Active PERK also induces the NF-E2 related factor 2 (NRF2) de-toxification pathway to activate LKB1-AMPK signaling downstream of integrin blockade. This in turn inhibits mTOR signaling76 to theoretically relieve mTORC1-mediated inhibition of autophagy. However, a subsequent study showed that while mTORC1 signaling is indeed reduced during matrix detachment in mammary epithelial cells, enforced re-activation of mTORC1 during detachment does not inhibit autophagy, indicating that autophagy induction during detachment does not require mTORC1 inhibition in contrast to settings such as nutrient deprivation.77 Instead, this study found that inhibitor of kappa B kinase activity which stimulates autophagy independent of NF-κB in response to nutrient deprivation78 was required for autophagy induction in response to integrin blockade or matrix detachment in mammary epithelial cells.77 Elevated ROS induced by matrix detachment also likely contributes to autophagy induction via activation of Atg4 directly.79 Thus, it appears that matrix detachment activates autophagy downstream of integrin blockade through multiple signaling pathways (Figure 2b). Intriguingly, some preliminary work has also suggested that specific components of the ECM, including collagen VI and laminin α2,80 can modulate intracellular autophagy through as-yet unknown mechanisms.
In addition to the studies described above in mammary tumor systems,37, 38, 77 autophagy has also been shown to promote survival following matrix detachment of hepatocellular carcinoma and melanoma cells leading to increased lung colonization during metastasis.26, 27, 81 Together, this work points to a critical role for autophagy in promoting the survival of detached epithelial cells when tumor cells are initially escaping the primary site, circulating in the periphery or following arrival at secondary sites prior to re-attachment.
Autophagy and cell motility
Metastasis depends on the increased motility of tumor cells to both escape the primary tumor site and to successfully colonize secondary sites.14, 22, 58 Cell motility requires the execution of a series of mechanical acts: protrusion of the plasma membrane, adhesion to the ECM at the front end of the cell, contraction of the cell body and detachment from the ECM at the rear of the cell.82 Protrusion of the plasma membrane and contraction of the cell body are driven by actin polymerization83 whereas adhesion to the ECM is mediated by integrin signaling to focal adhesion complexes.82, 84 Although it is now understood that cells migrate in different ways depending on the cell type and microenvironment,82 all cell migration still requires some combination of these basic components.
In addition to modulating tumor cell invasiveness through regulation of an EMT-like program as described above, evidence is accumulating that autophagy also has a direct role in key aspects of tumor cell motility and invasion,35, 85, 86, 87 including through modulation of the tumor cell secretome,32 turnover of components of the cell migration machinery33, 34 and ECM proteins88 amongst other roles as discussed below.
Control of RhoA and downstream signaling by autophagy
The Rho family of small GTPases, that include RhoA, Rac1 and CDC42, are key regulators of cell motility89 through effects on membrane protrusion and cytoskeletal remodeling, with different cell types using different patterns of migration depending on which Rho family members are active.82, 90 Several reports link autophagy to the function of Rho family members including an initial study in Drosophila showing that autophagy is required for hemocyte migration during wound healing.85 Specifically, the production of Rho1-induced cell protrusions and cell spreading of hemocytes, but not cortical actin dynamics, was dependent on both Atg1 (autophagy related gene 1) and the Drosophila homolog of cargo adapter p62/sqstm1 and inactivation of autophagy prevented blood cell migration to larval wound sites.85 Similarly, knockdown of ULK1 or Beclin1 prevented cell spreading in mouse macrophages,85 although the specific targets of autophagic degradation underlying the cell spreading defect in either cell type were not identified. Subsequently, a role for p62/Sqstm1 in targeting active RhoA, the mammalian homolog of Drosophila Rho1, to the autophagosome for degradation was shown.91 Aberrant accumulation of RhoA at the cell mid-body, when autophagy was inhibited through ATG5 knockdown, resulted in cytokinesis defects, multinucleation and aneuploidy91 demonstrating novel consequences of autophagy deficiency in cancer cells.
Conversely, Rho signaling has also been implicated in the regulation of autophagy.92, 93 Chan et al.92 identified Rho-associated kinase-1 (ROCK1), a downstream effector of Rho, as a regulator of starvation induced but not basal autophagy. Inhibition of ROCK1 elevated autophagic flux and led to the accumulation of enlarged, early autophagosomes that were highly enriched for ULK1 kinase (the catalytic component of the pre-initiation complex) but not WIPI2 or other later stage autophagosome markers. This led the authors to speculate that ROCK1 limits autophagosome size by reducing time spent in the phagophore elongation phase of early autophagy.93 Subsequent work showed ROCK1 activation by amino acid deprivation promoting autophagy through direct phosphorylation of Beclin1.94 ROCK1 interacted directly with and phosphorylated Beclin1 on Thr119 leading to disruption of the autophagy inhibitory complex of Bcl-2 with Beclin1.94 Furthermore, conditional deletion of Rock1 in murine cardiac muscle impaired starvation-induced autophagy, attesting to an important role for Rock1 in autophagy in vivo.94 These findings, that Rho signaling regulates autophagy, are in agreement with a systems wide approach in which a Rho GTPase signaling module including RhoA, Rac1 and CDC42, was identified within an autophagy-centered human gene interaction network.95 The authors further experimentally validated CDC42 as a protein that fine-tunes autophagic responses to environmental conditions.95
The interplay between autophagic flux and control of cell migration was further highlighted by work identifying coordinated control of Rab7, a small GTPase involved in the maturation of late stage autophagosomes and lysosomal fusion,96 with Rac1, a Rho family GTPase required for formation of lamellipodia and cell motility.97 Armus, a known RabGAP, was shown to localize to autophagosomes through direct interaction with LC3 thereby promoting efficient cycling of the Rab7 GTPase activity required for autophagolysosome maturation in response to amino acid starvation in primary keratinocytes.98, 99 This was coordinated with inactivation of Rac1 that also interacts with Armus and shown in previous studies to promote autophagic degradation and recycling of E-Cadherin during EGF-stimulated cell scattering.98 Failure to inactivate Rac1 during amino acid starvation blocked autophagy as Rac1 competed with LC3 for binding to Armus thereby preventing Rab7 localization to maturing autophagolysosomes resulting in a block to autophagic flux.99 However, the mechanism of Rac1 inhibition during starvation was not described and how differential control of Rac1 during EGF-induced scattering98 versus starvation-induced autophagy is achieved,99 remains unclear. Nevertheless, these studies highlight how control of autophagy is closely coordinated with control of cell migration (Figure 2c). In summary, there appears to be reciprocal regulation of autophagy by Rho GTPase signaling components, and of Rho family activity and cell migration by autophagy. Our understanding of how and in what physiological contexts these processes interact will likely increase as further studies emerge.
Autophagic control of focal adhesion dynamics
In addition to the interplay with Rho GTPases, autophagy also has a direct role in focal adhesion dynamics.33, 34, 87 Early evidence for a close functional relationship between autophagy and focal adhesions came with the identification of FIP200 (focal adhesion kinase (FAK)-interacting protein of 200 kD). FIP200 was initially isolated as a protein that binds to and inhibits the kinase activity of FAK,100 a key regulator of focal adhesion dynamics, but was subsequently also found to be the mammalian homolog of yeast autophagy gene Atg17 (autophagy related gene 17) and a critical component of the autophagy pre-initiation/Ulk1 complex in mammals101 (Figure 2d).
FIP200 inhibits FAK autophosphorylation when overexpressed, leading to decreased cell spreading, decreased migration and inhibition of cell cycle progression100 although it is now clear that aspects of the ability of FIP200 to modulate cell cycle are dependent on its interaction with and positive regulation of p53 leading to activation of p21Cip1, p16/Ink4A and pRB1 expression.102 Intriguingly, it has been proposed that p53 negatively modulates autophagy through its interaction with FIP200.103 In autophagy, FIP200 activity lies downstream of both starvation-induced AMPK-dependent activation and cell growth induced mTORC1 inhibition of the ULK1 pre-initiation complex.46, 104, 105, 106 In one study, limiting AMPK and ULK1 activity through the activity of specific HSP90s induced release of FAK from FIP200-mediated inhibition enhancing cellular invasion and metastatic dissemination of tumor cells in experimental metastasis models.107 This suggested that FIP200 mediates an inverse relationship between autophagic flux and FAK activity/cell migration. However, the mechanism by which reduced AMPK/ULK1 activity titrated FIP200 away from FAK was not elucidated and AMPK has other known functions in cell migration, for example in control of microtubule polymerization, that were not discussed.108 Thus, the extent to which FIP200 mediates cross-talk between autophagy and cell migration remains unclear. Numerous factors affecting FIP200 levels and post-translational modification likely have into which functions of FIP200 are dominant or indeed exclusive in any given cell type or cellular stress condition. Recent studies have begun to separate out the functions of FIP200 in autophagy from those in other cellular processes.109 By mutating amino acids 582–585 to alanine (FIP200-4A) and preventing the interaction of FIP200 with Atg13 (autophagy related gene 13), Chen et al.109 were able to block the function of FIP200 in autophagy. Interestingly, knock-in of the FIP200-4A mutant allele into mice extended the lifespan of Fip200−/− mice from E16.5110 to birth when other Atg gene knockout mice are known to die.111 However, the extended lifespan was attributed to the non-autophagy functions of FIP200 in protecting against TNF-α-induced apoptosis109 as opposed to any role in cell migration. Nevertheless, reagents from these mice and future structure–function analyses with FIP200 may ultimately allow us to determine how important FIP200 is to coordinated control of autophagy and cell migration.
Further insight to the role played by autophagy in focal adhesion dynamics (Figure 2e) came from work demonstrating that active SRC kinase is targeted for autophagic degradation in cells in which FAK activity is inhibited through substrate detachment, expression of non-phosphorylatable FAK, or FAK deletion.87 These results suggest that autophagy has a role in regulating the function of the FAK-Src pathway through direct degradation of active SRC by autophagosomes downstream of FAK signaling. Targeting of SRC to autophagosomes was mediated by c-CBL, an E3 Ubiquitin ligase that possesses a LC3-interacting region (LIR) motif and interacts directly with processed LC3B.87 The authors showed that c-CBL functioned as a cargo adapter for active SRC, targeting it for autophagic degradation through interaction with LC3 in a manner not requiring its E3 Ub-ligase activity. Inhibition of autophagy restored SRC expression at focal adhesions but was associated with cell death87 suggesting that autophagic turnover of SRC was an adaptive response to cell detachment or FAK inactivation. Levels of autophagic flux were increased when FAK was inhibited in cells suggesting that FAK activity can inhibit autophagy, possibly by sequestering or otherwise interfering with FIP200, although this was not examined. Alternatively, FAK and FIP200 have both been reported to positively regulate mTOR activity through inhibitory interactions with tuberous schlerosis complex 2 (TSC2)112, 113 and mTOR is a potent autophagy inhibitor.104 It will be interesting to further determine how FAK modulates rates of autophagy and whether this requires FIP200, TSC2 or other signaling mechanisms.
Both FAK and SRC are key regulators of focal adhesion dynamics114, 115, 116, 117, 118 and intriguingly, several recent reports have identified a novel role for autophagy in focal adhesion turnover33, 34 (Figure 2e). Kenific et al.33 reported that autophagy was required for both focal adhesion assembly and disassembly in HRasV12-transformed MCF10A cells although autophagosomes were only associated with focal adhesions during disassembly. This work directly implicated the autophagy cargo adapter, Near BRCA1 (NBR1) in delivery of multiple focal adhesion proteins to the autophagosome.33 In a separate study, Sharifi et al.34 showed that inhibition of autophagy in metastatic cancer cell lines inhibited tumor cell migration and invasion in vitro and suppressed metastasis to lung and liver in vivo in orthotopic mouse models. This defect in cell motility and metastasis was associated with reduced focal adhesion disassembly but in contrast to Kenific et al., this was mediated by a direct interaction between LC3B at the autophagosome and the focal adhesion protein Paxillin (PXN).34 The targeting of PXN for autophagic degradation was independent of NBR1, and also independent of p62/Sqstm1, but required a conserved LIR motif in PXN.34 PXN is a critical scaffolding and signaling component of focal adhesions that is phosphorylated on multiple tyrosine residues by SRC kinase.119 Interestingly, the interaction between PXN and LC3B was strongly stimulated by oncogenic SRC and required the Y40 residue at position +1 of the LIR motif in PXN,34 a site previously identified as a target of SRC phosphorylation but with unknown function.119
Conversely, PXN has also been implicated in the regulation of autophagic flux. Atg1 (the Drosophila homolog of ULK1) was identified using a transposon-based mis-expression screen as a gene whose inactivation could suppress defects in wing development in dPax (the Drosophila homolog of PXN) mutant flies.120 Follow-up analyses showed that Atg1/Ulk1 could phosphorylate Pxn in vitro and that, in response to nutrient deprivation, Pxn and vinculin, a different focal adhesion protein, relocated from the plasma membrane to cytosolic puncta resembling autophagosomes, with relocation of vinculin to autophagosomes dependent on functional Pxn.120 Intriguingly, Pxn was required for efficient autophagosome formation in nutrient-deprived mouse embryonic fibroblasts (MEFs)120 again suggesting reciprocal control of autophagy by the processes and molecules it controls. Given the significance of Rho-dependent signaling for effective autophagy, as discussed above, it will be informative in future studies to examine whether PXN modulates autophagosome formation via its critical activity in the proper localization of Rho GTPases in cells.119
Autophagy and tumor cell invasion
One of the most fascinating findings linking autophagy to increased tumor cell migration and invasion has been work demonstrating a requirement for autophagy in the production of pro-migratory cytokines, such as IL-6, during tumor cell invasion.32 This study found that inhibition of autophagy-blocked cellular protrusions and invasion of Ras-transformed MCF10A cells through a laminin-rich ECM, whereas conditioned media from autophagy competent tumor cells rescued protrusion and invasion defects in autophagy-deficient cells.32 The authors identified interleukin-6 (IL-6), MMP2 and WNT-5A as the specific factors that were dependent on autophagy for their secretion and required for effective protrusion formation and tumor cell invasion.32 Although autophagy is known to transcriptionally regulate cytokine expression during cellular senescence,121 the effect of autophagy on IL-6 production in this study occurred at a post-transcriptional level, although further work is needed to elucidate exactly how autophagy promotes secretion of pro-migratory cytokines in Ras-transformed epithelial cells.32 Certainly, this study raises several provocative questions, including whether autophagy modulates the secretion of other pro-invasive factors beyond those examined here and also whether the same autophagy machinery is involved in promoting both secretion and degradation. Autophagy has been shown in other studies to promote secretion of factors, such as IL-1β122 where it was proposed that the ‘secretory’ and ‘degradative’ forms of autophagy are distinct.123, 124 However, the relationship between autophagy and secretory phenotypes is likely more complex (Figure 2f), as illustrated by a recent study showing an inhibitory function for autophagy in senescence-associated secretory phenotype associated with DNA damage induced senescence, via p62-dependent degradation of GATA-4 upstream of NFκB signaling.121 How these different functions of autophagy in control of secreted factors are integrated or deregulated in cancer metastasis is another area of future endeavor.
Autophagy in tumor cell dormancy and therapy resistance
Metastatic dormancy has emerged over the past 10 years as a significant clinical challenge, but also as a potential therapeutic target to prevent the outgrowth of macrometastatic disease.14, 25, 125 The realization that a lag period of up to 20 years between ‘successful’ treatment of primary breast cancer126 and the emergence of metastatic disease in patients led to investigation of this phenomenon in breast, prostate and other cancers.14 Dormant tumor cells are generally considered to be growth arrested, although there is some debate about whether sites of micrometastatic disease instead exist in a balance of proliferation and death that only appears as an arrested state.25 By analogy with tissue stem cells, it has been proposed that dormant tumor cells are in fact tumor stem cells that exist in a quiescent state but occasionally expand to propagate tumor outgrowth.25 Indeed, there is significant evidence that stem cell markers are upregulated on disseminated tumor cells found in the bone marrow of breast cancer patients.127 Given the role of autophagy in cancer stem cells and in promoting tumor cell survival, several groups have proposed that dormant tumor cells depend on autophagy to survive at secondary sites over extended periods of time to grow out later as macrometastases.24, 25
As already discussed, autophagy may simply support the metabolic needs and survival of disseminated tumor cells deprived of key growth factors by maintaining amino acid levels, ATP production and preventing energetic catastrophe.2, 25, 128 Alternatively, autophagy could facilitate tumor cell dormancy by promoting quiescence, a process that is also required for the cancer stem cell state,72 which as we have described above, has also been linked to autophagy induction. Indeed, the induction of autophagy, G1 arrest and cell survival have been shown to be coordinated downstream of LKB1-AMPK activation.129 Liang et al.129 showed that, in addition to stimulating ULK1-dependent autophagy,46 LKB1-AMPK activates a p27kip1-dependent growth arrest in G1 of cell cycle. In the absence of p27/KIP1, LKB1-AMPK signaling under nutrient stress resulted in rapid apoptotic cell death129 suggesting a mechanism by which autophagy induction is linked to growth arrest to prevent programmed cell death.
The ARHI protein (aplasia Ras homolog member I), a tumor suppressor that is decreased in over 60% of ovarian cancers130 has also been proposed to have a role in cancer cell dormancy through induction of autophagy.24 ARHI induces autophagy in part by inhibiting the phosphatidyl inositol 3-kinase (PI3K)-protein kinase B (AKT) growth factor signaling pathway and this has been suggested to underlie aspects of its tumor suppressor function131 while re-expression of ARHI in ARHI-deficient SKOv3 ovarian cancer cells in vitro induces autophagy.24 When ARHI was re-introduced into ARHI-deficient SKOv3 ovarian cancer cells and implanted under the skin of nude mice, the tumors failed to grow but when ARHI was subsequently knocked down again, tumors grew out suggesting that ARHI was maintaining tumor dormancy.24 Inhibition of autophagy, during the period of non-proliferation in tumors re-expressing ARHI, prevented tumor regrowth when ARHI was subsequently repressed, indicating that the dormancy observed was autophagy dependent.24 Taken together, these studies linking coordination of autophagy induction with growth arrest and cell survival during growth arrest, support a function for autophagy in tumor cell dormancy as part of a growth arrest/quiescence program, possibly one determined by the stem-like state of these cells.
If autophagy is required for this state, either to maintain quiescence or other aspects of stemness, then inhibition of autophagy comes into focus as a therapeutic option, as dormancy has been postulated to underlie the persistence of minimal residual disease.25 Indeed, Gupta et al.132 showed that autophagy induction was associated with Imatinib-induced reversible growth arrest (quiescence) in gastrointestinal stromal tumor cells. Inhibition of autophagy through either knockdown of key autophagy genes or treatment with lysosomotrophic agents, such as chloroquine, caused rapid cell death.132 This is consistent with a role for autophagy in promoting the survival of cells undergoing stress-induced growth arrest and suggests that therapies combining Imatinib or other TKIs with autophagy inhibitors may increase the likelihood of durable treatment responses.
The quiescent stem-like state of dormant tumor cells has also been suggested to lead to tumor cell resistance to genotoxic therapies that target proliferating cells. Indeed, radiation treatment has been shown to induce autophagy in vitro, whereas conversely inhibition of autophagy reduced clonogenic survival of breast, lung and cervical cancer cell lines following irradiation.133 Significantly, reduced clonogenic survival was noted for radioresistant but not radiosensitive sub-clones after irradiation.133 Similar observations have been made with several cytotoxic chemotherapies. For example, doxorubicin resistance in the HEp3 liver cancer dormancy model was attributed to p38-induced upregulation of ER chaperones and PERK134 that stimulates autophagy via induction of ATF4 and its downstream targets, ATG5 and LC3B.75 Also, autophagy inhibition in a Myc-driven model of lymphoma suppressed tumor recurrence in mice treated with alkylating agents consistent again with a role for autophagy in promoting both tumor dormancy and drug resistance.135 Taken together, these studies suggest that autophagy promotes survival of dormant tumor cells and contributes to therapy resistance. This argues that the combination of genotoxic therapy with autophagy inhibition would preferentially eliminate dormant tumor cells and thereby limit metastatic dormancy and minimal residual disease.
Autophagy in tumor immune surveillance
Autophagy has a major role in the immune response to infection and disease, as has been reviewed eloquently elsewhere.136, 137 These functions include intracellular degradation of pathogens, secretion of immune modulatory cytokines and proteases, generation of antigen peptides for MHC-II presentation and modulation of pro-inflammatory signaling.136, 137, 138 The importance of these autophagy functions in immunity is illustrated by the susceptibility of autophagy-deficient animals to infection13 and by the linkage of certain autoimmune conditions to autophagy defects.139
Tumor cells both at the primary site and throughout metastatic dissemination and colonization evolve to evade immune surveillance by cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells in order to survive and proliferate.140 Significantly, autophagy acts within the tumor cell itself to modulate recruitment and interaction with components of both the adaptive and innate immune system.136 Recruitment of both dendritic cells and T cells to subcutaneous CT26 murine colon carcinoma tumors in response to chemotherapy was dependent on autophagy, such that inhibition of autophagy significantly inhibited immune cell recruitment to these tumors in vivo.141 Furthermore, strong punctate LC3B staining in human breast cancers correlated with increased tumor infiltration by CD8+ CTLs and reduced infiltration by FoxP3+ T-regulatory cells (Tregs) consistent with tumor cell autophagy promoting increased immune surveillance.142 Similar conclusions were drawn from studies in which genetic targeting of autophagy in a K-Ras driven GEM model of lung cancer, showed increased infiltration of pro-tumorigenic FoxP3+CD25+ Tregs into autophagy-deficient tumors compared with controls.9 Increased lung tumor initiation in this autophagy-deficient mouse model was reversed by depletion of CD25+ Treg cells supporting a role for autophagy in preventing recruitment of Tregs to the tumor and in promoting tumor immune surveillance.9 Intriguingly, the ability to promote tumor immune surveillance with caloric restriction mimetics was dependent on functional autophagy.143 Mechanistically, autophagy has been shown to promote release of ATP and other danger-associated molecular patterns from dying tumor cells, in addition to its role in trafficking tumor epitopes to the lysosome for processing and presentation on antigen-presenting dendritic cells.136, 138, 141
However, autophagy has also been reported to block tumor immune surveillance in other settings. In MCF-7 breast cancer cells induced to undergo EMT, acquisition of the mesenchymal phenotype was associated with both increased autophagic flux and Beclin1-dependent resistance to CTL-mediated cell lysis, suggesting a model in which the acquisition of pro-metastatic behavior is linked via autophagy with resistance to immune surveillance.144 Similarly, autophagy has been reported to suppress immune surveillance in other tumor contexts, such as in the MMTV-PyMT mouse mammary tumor model in which deletion of FIP200 increased CTL infiltration into the primary tumors, most likely secondary to increased CXCL10 production by autophagy-deficient tumor cells, leading to reduced primary tumor growth and decreased metastastic dissemination,145 although, in this scenario, autophagy was deficient in both the tumor and the immune system. Autophagy also promotes hypoxic tumor cell resistance to both CTL and NK cell-mediated through autophagy-dependent granzyme B degradation in breast cancer cells146 and destabilization of the immune synapse and other functions in melanoma cells.147, 148 A feedback loop has also been described wherein inhibition of autophagy sensitizes renal cell carcinoma cells to NK cell-mediated lysis, whereas NK cells themselves induce autophagy in renal cell carcinoma cells via the protein, Inositol 1, 4, 5-triphosphate receptor type 1.149
The identification of both pro- and anti-immune surveillance functions of autophagy in different tumor models may reflect tissue- or stage-specific differences in responses to stresses such as hypoxia or chemotherapy that modulate tumor cell interactions with the immune system. Cancer cells are likely to have evaded anticancer immune surveillance in order to become metastatic but whether autophagy plays a positive or negative role in this aspect of the metastatic cascade requires further analysis.
Other functions of autophagy in the tumor microenvironment
The tumor stroma, particularly fibroblasts and tumor-infiltrating immune cells, are a major source of pro-migratory growth factors and chemokines in the tumor microenvironment150 and thus have an important role in modulating the metastatic potential of tumor cells. For example, M2 polarized tumor-associated macrophages (TAMs) are key contributors to tumor cell motility and invasiveness.151 The extent of TAM recruitment is correlated with poor prognosis in a number of different solid tumor types and loss of macrophage recruitment to primary tumors inhibits metastasis in mouse models of breast and ovarian cancer.152, 153 Defective autophagy in tumor cells promoted macrophage recruitment to tumors in vivo as a result of increased pro-inflammatory necrotic cell death in hypoxic tumor cells4 that in turn stimulated pro-invasive signaling in the tumor. Meanwhile, stromal cells, particularly macrophages150 and fibroblasts154 are a major source of matrix-degrading enzymes required in the tumor microenvironment to promote invasion and intra-vital imaging has suggested that perivascular macrophages are required for tumor cell intravasation into blood vessels.155, 156 As described above, autophagy functions in both secretion and immune cell function32, 137, 157 making it likely that autophagic flux within the stroma, as distinct from autophagy in tumor cells, alters the metastatic phenotype. Put simply, there is growing evidence that autophagy has multiple significant functions in modulating paracrine signaling between tumor cells and stromal cells in the tumor microenvironment to regulate tumor cell escape.
Autophagy has a particularly important role in macrophage activation by modulating inflammasome activation and release of pro-inflammatory cytokines IL-1β and IL-18.122, 137, 138, 158 For example, several groups have shown that autophagic degradation of damaged mitochondria inhibits initial activation of inflammosomes by limiting levels of ROS and free mitochondrial DNA in the cell, both of which stimulate inflammasome activation.138, 159, 160 Autophagy also acts downstream to degrade active inflammasomes and thus limit the duration of the inflammatory response.158 The immune response generated by inflammasome activation may be considered anti-tumorigenic as it opposes the pro-invasive and immune-suppressive microenvironment generated by M2 macrophages and other cell types.137 Indeed, mice deficient in Atg8 family member GABARAP are resistant to chemically induced carcinogenesis due to increased macrophage and lymphocyte secretion of chemokines and cytokines, such as IL-1β, IL-6, IL-2 and IFN-γ that augment the anti-tumor immune response.161 Thus, autophagy in TAMs appears to limit anti-tumor responses, allowing them to switch their secretome towards a pro-invasive, pro-angiogenic, pro-metastatic phenotype. However, it has also been suggested that failure to dampen inflammasome activation in a timely manner as a result of autophagy deficiencies could promote cancer as a result of macrophage cell death and the uncontrolled recruitment of other immune cell types to the tumor.138 Again, further work is required to fully resolve these questions in metastatic cancer models in vivo.
Cancer associated fibroblasts (CAFs) also make up a key part of the tumor stroma that promotes both transformation of normal epithelium and progression to carcinoma and metastasis through release of matrix-degrading enzyme and pro-migratory cytokines and chemokines.162 It has been proposed that release of hydrogen peroxide by tumor cells induces senescence in CAFs that forces an autophagy-dependent switch to aerobic glycolysis in CAFs and the production of lactic acid, ketone bodies and free fatty acids that fuel tumor cell growth when secreted by the CAFs.163 This role for autophagy in promoting the pro-tumorigenic and pro-metastatic functions of CAFs was also dependent on autophagic degradation of caveolin-1, a putative tumor suppressor protein found in lipid rafts at the plasma membrane that negatively regulates Ras signaling.164 Loss of stromal caveolin-1 in human tumor-associated stroma has been associated with early progression from DCIS to invasive cancer,165 lymph node metastasis in breast cancer and metastatic disease in prostate cancer.166 Increased autophagy in CAFs may explain reduced stromal expression of caveolin and increased CAF proliferation that would in turn support tumor cell progression to metastasis. Beyond caveolin, autophagy may directly promote release of MMPs and pro-migratory cytokines by CAFs, as was previously reported for tumor cells.32
Finally, it is important to consider that autophagy may be regulated differently in the tumor stroma from in tumor cells in a way that has significance for the use of autophagy inhibitors in cancer therapy. The use of lineage-specific knockout of autophagy in GEM models may be needed to properly separate out distinct functions of autophagy in the tumor versus the tumor stroma and the implications of inhibiting autophagy to limit metastasis.
Autophagy and emerging functions in establishing the pre-metastatic niche
Through co-evolution with the tumor stroma, metastatic cancer cells acquire the ability to modulate the tumor microenvironment both at the primary site and at secondary sites.15 The re-programming of the new microenvironment to which the tumor cell will metastasize is achieved through secretion of circulating chemokines and other factors, such as lysyl oxidase (LOX), a matrix remodeling factor that accumulates at distant sites where it is required to cross-link collagen and trap bone marrow-derived myeloid cells.167 Such bone marrow-derived myeloid cells promote MMP release and vascular endothelial growth factor bio-availability amongst other activities to establish the so-called ‘pre-metastatic niche’ resulting in suppression of anti-tumor immunity, as well as provision of growth factors and pro-angiogenic factors to support tumor cell proliferation.168 Given the critical role of autophagy in release of secreted factors by cells,32 it will be interesting to assess in future studies whether autophagy is required to establish the pre-metastatic niche in tumor models and in human metastases.
The production of exosomes by tumor cells also contributes to the remodeling of distant sites to promote metastatic outgrowth.169, 170, 171, 172 Exosomes are small membrane vesicles derived from the endosomal system, primarily late endosomes and multivesicular bodies, and subsequently secreted through fusion with the plasma membrane.173 They contain soluble factors such as cytokines, integrins and growth factors but are also capable of delivering other proteins, mRNAs, microRNAs and lipids at distant sites.174, 175 Exosomes released by metastatic melanoma cells reprogram bone marrow progenitor cells to be pro-vasculogenic and pro-metastatic, promoting vascular leakiness at pre-metastatic sites.170 Similarly, exosomes released by renal carcinoma CSCs have been shown to promote an activated angiogenic phenotype in normal endothelial cells in vitro and tumor cell colonization of the lung and angiogenesis in vivo.176 As autophagy itself is also part of the endolysosomal membrane system,177 it is unsurprising that the autophagy machinery has been linked to exosome production178 and it is tempting to speculate that autophagic regulation of exosome release by tumor cells might participate in pre-metastatic niche conditioning. However, at this time, the mechanisms that might underlie such regulation and whether this becomes functionally relevant during tumor progression and metastasis remain an important area of on-going investigation.
Conclusions
In sum, a growing body of work has identified a number of critical functions for autophagy throughout cancer metastasis, offering the opportunity to define specific points in the metastatic cascade where autophagy inhibitors may be directed to reduce cancer mortality rates. Various studies have highlighted the key role of autophagy in modulating tumor cell motility and invasion, the cancer stem cell phenotype, drug resistance, tumor dormancy and tumor immune surveillance with emerging roles in other processes determining the metastatic success of cancer cells. Several themes emerge from this review, including the key role of autophagy in the induction of EMT and the stem cell state in response to hypoxia, TGF-β and other signals, although there are clearly gaps in our current understanding of how this occurs mechanistically. Investigation into autophagic degradation of key cell fate regulators, such as Twist, Smad4 and GATA-4 suggests one mechanistic paradigm, but as with many of the higher functions of autophagy, it is most likely that additional as-yet-unidentified mechanisms are involved. Another recurring theme that emerges is the existence of reciprocal regulatory mechanisms between autophagy and the cellular processes in which it participates. For example, during tumor cell migration, autophagy modulates Rho activity and conversely Rho signaling regulates autophagy; also, autophagy promotes PXN degradation while PXN is required for autophagosome formation, and so on. Finally, at the cellular level, it will be important to determine how the functions of autophagy in tumor cells are coordinated with those of autophagy in the tumor microenvironment, to properly predict the outcome of autophagy inhibitors used in the clinic to treat or prevent metastatic disease.
References
Mizushima N, Komatsu M . Autophagy: renovation of cells and tissues. Cell 2011; 147: 728–741.
Galluzzi L, Pietrocola F, Levine B, Kroemer G . Metabolic control of autophagy. Cell 2014; 159: 1263–1276.
Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F et al. Autophagy in malignant transformation and cancer progression. EMBO J 2015; 34: 856–880.
Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson KL, Chen G et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation and tumorigenesis. Cancer Cell 2006; 10: 51–64.
Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011; 25: 717–729.
Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzumbas G et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 2011; 25: 460–470.
Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev 2013; 27: 1447–1461.
Rosenfeldt MT, O'Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013; 504: 296–300.
Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun 2014; 5: 3056–3070.
Strohecker AM, Guo JY, Karsli-Uzunbas G, Price SM, Chen GJ, Mathew R et al. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov 2013; 3: 1272–1285.
Xie X, Koh JY, Price S, White E, Mehnert JM . Atg7 overcomes senescence and promotes growth of BrafV600E-driven melanoma. Cancer Discov 2015; 5: 410–423.
Santanam U, Banach-Petrosky W, Abate-Shen C, Shen MM, White E, DiPaola RS . Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev 2016; 30: 399–407.
Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov 2014; 4: 914–927.
McGowan PM, Kirstein JM, Chambers AF . Micrometastatic disease and metastatic outgrowth: clinical issues and experimental approaches. Future Oncol 2009; 5: 1083–1098.
Chaffer CL, Weinberg RA . A perspective on cancer cell metastasis. Science 2011; 331: 1559–1564.
Valastyan S, Weinberg RA . Tumor metastasis: molecular insights and evolving paradigms. Cell 2011; 147: 275–292.
Nieto MA, Huang RY, Jackson RA, Thiery JP . EMT: 2016. Cell 2016; 166: 21–45.
Carmeliet P, Jain RK . Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov 2011; 10: 417–427.
Guo W, Giancotti FG . Integrin signaling during tumor progression. Nat Rev Cancer 2004; 5: 816–826.
Paget S . The distribution of secondary growths in cancer of the breast. The Lancet 1889; 133: 571–573.
Ewing J . Neoplastic Diseases; A Treatise on Tumors. W.B. Saunders: Philadelphia, London, 1928.
Langley RR, Fidler IJ . The seed and soil hypothesis revisited—-the role of tumor-stroma interactions in metastasis to different organs. Int J Cancer 2011; 128: 2527–2535.
Brabletz T . To differentiate or not—-routes towards metastasis. Nat Rev Cancer 2012; 12: 425–436.
Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare S et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cells. J Clin Invest 2008; 118: 3917–3929.
Sosa MS, Bragado P, Aguirre-Ghiso JA . Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 2014; 14: 611–622.
Peng YF, Shi YH, Ding ZB, Ke AW, Gu CY, Hui B et al. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy 2013; 9: 2056–2068.
Peng YF, Shi YH, Shen YH, Ding ZB, Ke AW, Zhou J et al. Promoting colonization in metastatic HCC cells by modulation of autophagy. PLoS One 2013; 8: e74407.
Espina V, Mariani BD, Gallagher RI, Tran K, Banks S, Wiedemann J et al. Malignant precursor cells pre-exist in human breast DCIS and require autophagy for survival. PLoS One 2010; 5: e10240.
Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Vellon L, Menendez JA . Autophagy positively regulates the CD44(+) CD24(-/low) breast cancer stem-like phenotype. Cell Cycle 2011; 10: 3871–3885.
Wolf J, Dewi DL, Fredebohm J, Muller-Decker K, Flechtenmacher C, Hoheisel JD et al. A mammosphere formation RNAi screen reveals that ATG4A promotes a breast cancer stem-like phenotype. Breast Cancer Res 2013; 15: R109.
Qiang L, Zhao B, Ming M, Wang N, He TC, Hwang S et al. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc Natl Acad Sci USA 2014; 111: 9241–9246.
Lock R, Kenific CM, Leidal AM, Salas E, Debnath J . Autophagy dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov 2014; 4: 466–479.
Kenific CM, Stehbens SJ, Goldsmith J, Leidal AM, Faure N, Ye J et al. NBR1 enables autophagy-dependent focal adhesion turnover. J Cell Biol 2016; 212: 577–590.
Sharifi MN, Mowers EE, Drake LE, Collier C, Chen H, Zamora M et al. Autophagy promotes focal adhesion disassembly and cell motility of metastatic tumor cells through the direct interaction of paxillin with LC3. Cell Rep 2016; 15: 1660–1672.
Kenific CM, Thorburn A, Debnath J . Autophagy and metastasis: another double-edged sword. Curr Opin Cell Biol 2010; 22: 1–5.
Kroemer G, Marino G, Levine B . Autophagy and the integrated stress response. Mol Cell 2010; 40: 280–293.
Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J et al. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol Cell Biol 2011; 31: 3616–3629.
Fung C, Lock R, Gao S, Salas E, Debnath J . Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell 2008; 19: 797–806.
Lazova R, Camp RL, Klump V, Siddiqui SF, Amaravadi RK, Pawelek JM . Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin Cancer Res 2012; 18: 370–379.
Zhao H, Yang M, Zhao J, Wang J, Zhang Y, Zhang Q . High expression of LC3B is associated with progression and poor outcome in triple-negative breast cancer. Med Oncol 2013; 30: 475.
Lazova R, Klump V, Pawelek J . Autophagy in cutaneous malignant melanoma. J Cutan Pathol 2010; 37: 256–268.
Han C, Sun B, Wang W, Cai W, Lou D, Sun Y et al. Overexpression of microtubule-associated protein-1 light chain 3 is associated with melanoma metastasis and vasculogenic mimicry. Tohoku J Exp Med 2011; 223: 243–251.
Galavotti S, Bartesaghi S, Faccenda D, Shaked-Rabi M, Sanzone S, McEvoy A et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene 2013; 32: 699–712.
Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M et al. Transcriptional control of the autophagy-lysosome system in pancreatic cancer. Nature 2015; 524: 361–365.
Pattingre S, Tassa A, Qu X, Garuti R, Linag XH, Mizushima N et al. Bcl-2 anti-apoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122: 927–939.
Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331: 456–461.
Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013; 497: 633–637.
Deberardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB . The biology of cancer: metabolic programming fuels cell growth and proliferation. Cell Metab 2008; 7: 11–20.
Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 2012; 31: 1095–1108.
Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005; 436: 117–122.
Wong PM, Feng Y, Wang J, Shi R, Jiang X . Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat Commun 2015; 6: 8048.
Kiyono K, Suzuki HI, Matsuyama H, Morishita Y, Komuro A, Kano MR et al. Autophagy is activated by TGF-b and potentiates TGF-b-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res 2009; 69: 8844–8852.
Li J, Yang B, Zhou Q, Wu Y, Shang D, Guo Y et al. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis 2013; 34: 1343–1351.
Kim YH, Baek SH, Kim EK, Ha JM, Jin SY, Lee HS et al. Uncoordinated 51-like kinase 2 signaling pathway regulates epithelial-mesenchymal transition in A549 lung cancer cells. FEBS Lett 2016; 590: 1365–1374.
Catalano M, D'Alessandro G, Lepore F, Corazzari M, Caldarola S, Valacca C et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol 2015; 9: 1612–1625.
Bertrand M, Petit V, Jain A, Amsellem R, Johansen T, Larue L et al. SQSTM1/p62 regulates the expression of junctional proteins through epithelial-mesenchymal transition factors. Cell Cycle 2015; 14: 364–374.
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133: 704–715.
May CD, Sphyris N, Evans KW, Werden SJ, Guo W, Mani SA . Epithelial-mesenchymal transition and cancer stem cells: a dangerously dynamic duo in breast cancer progression. Breast Cancer Res 2011; 13: 202.
Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci USA 2011; 108: 7950–7955.
Comen E, Norton L, Massague J . Clinical implications of cancer self-seeding. Nature Rev Clin Oncol 2011; 8: 369–377.
Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res 2009; 69: 1302–1313.
Pang R, Law WL, Chu AC, Poon JT, Lam CS, Chow AK et al. A subpopulation of CD26+ cancer stem cells with metastatic capacity in human colorectal cancer. Cell Stem Cell 2010; 6: 603–615.
Chen C, Wei Y, Hummel M, Hoffmann TK, Gross M, Kaufmann AM et al. Evidence for epithelial-mesenchymal transition in cancer stem cells of head and neck squamous cell carcinoma. PLoS One 2011; 6: e16466.
Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 2012; 148: 1015–1028.
Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, Ng Eaton E et al. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015; 525: 256–260.
Caramel J, Papadogeorgakis E, Hill L, Browne GJ, Richard G, Wierinckx A et al. A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell 2013; 24: 466–480.
Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell 2011; 146: 633–644.
Oliver L, Hue E, Priault M, Vallette FM . Basal autophagy decreased during the differentiation of human adult mesenchymal stem cells. Stem Cells Dev 2012; 21: 2779–2788.
Tra T, Gong L, Kao LP, Li XL, Grandela C, Devenish RJ et al. Autophagy in human embryonic stem cells. PLoS One 2011; 6: e27485.
Vazquez P, Arroba AI, Cecconi F, de la Rosa EJ, Boya P, de Pablo F . Atg5 and Ambra1 differentially modulate neurogenesis in neural stem cells. Autophagy 2012; 8: 187–199.
Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R et al. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013; 494: 323–327.
Garcia-Prat L, Martinez-Vicente M, Perdiguero E, Ortet L, Rodriguez-Ubreva J, Rebollo E et al. Autophagy maintains stemness by preventing senescence. Nature 2016; 529: 37–42.
Guadamillas MC, Cerezo A, Del Pozo MA . Overcoming anoikis—-pathways to anchorage-independent growth in cancer. J Cell Sci 2011; 124: 3189–3197.
Lock R, Debnath J . Extracellular matrix regulation of autophagy. Curr Opin Cell Biol 2008; 20: 583–588.
Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest 2010; 120: 127–141.
Avivar-Valderas A, Bobrovnikova-Marjon E, Alan Diehl J, Bardeesy N, Debnath J, Aguirre-Ghiso JA . Regulation of autophagy during ECM detachment is linked to a selective inhibition of mTORC1 by PERK. Oncogene 2013; 32: 4932–4940.
Chen N, Debnath J . IkappaB kinase complex (IKK) triggers detachment-induced autophagy in mammary epithelial cells independently of the PI3K-AKT-MTORC1 pathway. Autophagy 2013; 9: 1214–1227.
Criollo A, Senovilla L, Authier H, Maiuri MC, Morselli E, Vitale I et al. The IKK complex contributes to the induction of autophagy. EMBO J 2010; 29: 619–631.
Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z . Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007; 26: 1749–1760.
Neill T, Schaefer L, Iozzo RV . Instructive roles of extracellular matrix on autophagy. Am J Pathol 2014; 184: 2146–2153.
Maes H, Kuchnio A, Peric A, Moens S, Nys K, De Bock K et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 2014; 26: 190–206.
Ridley AJ . Life at the leading edge. Cell 2011; 145: 1012–1022.
Olson MF, Sahai E . The actin cytoskeleton in cancer cell motility. Clin Exp Metastasis 2009; 26: 273–287.
Parsons JT, Horwitz AF, Schwartz MA . Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Biol 2010; 11: 633–643.
Kadandale P, Stender JD, Glass CK, Kiger AA . Conserved role for autophagy in Rho1-mediated coritcal remodeling and blood cell recruitment. Proc Natl Acad Sci USA 2010; 107: 10502–10507.
Mackintosh RL, Timpson P, Thorburn J, Anderson KI, Thornburn A, Ryan KM . Inhibition of autophagy impairs tumor cell invasion in an organotypic model. Cell Cycle 2012; 11: 2022–2029.
Sandilands E, Serrels B, McEwan DG, Morton JP, Macagno JP, McLeod K et al. Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat Cell Biol 2011; 14: 51–60.
Kim SI, Na HJ, Ding Y, Wang Z, Lee SJ, Choi ME . Autophagy promotes intracellular degradation of type I collagen induced by transforming growth factor (TGF)-beta1. J Biol Chem 2012; 287: 11677–11688.
Sahai E, Marshall CJ . RHO-GTPases and cancer. Nat Rev Cancer 2002; 2: 133–142.
Sahai E, Marshall CJ . Differing modes of tumor cell invasion have distinct requirements for Rho/ROCK signaling and extracellular proteolysis. Nat Cell Biol 2003; 5: 711–719.
Belaid A, Cerezo M, Chargui A, Corcelle-Termeau E, Pedeutour F, Giuliano S et al. Autophagy plays a critical role in the degradation of active RHOA, the control of cell cytokinesis, and genomic stability. Cancer Res 2013; 73: 4311–4322.
Chan EY, Kir S, Tooze SA . siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem 2007; 282: 25464–25474.
Mleczak A, Millar S, Tooze SA, Olson MF, Chan EY . Regulation of autophagosome formation by Rho kinase. Cell Signal 2013; 25: 1–11.
Gurkar AU, Chu K, Raj L, Bouley R, Lee SH, Kim YB et al. Identification of ROCK1 kinase as a critical regulator of Beclin1-mediated autophagy during metabolic stress. Nat Commun 2013; 4: 2189.
Till A, Saito R, Merkurjev D, Liu JJ, Syed GH, Kolnik M et al. Evolutionary trends and functional anatomy of the human expanded autophagy network. Autophagy 2015; 11: 1652–1667.
Gutierrez MG, Munafo DB, Beron W, Colombo MI . Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci 2004; 117: 2687–2697.
Mack NA, Whalley HJ, Castillo-Lluva S, Malliri A . The diverse roles of Rac signaling in tumorigenesis. Cell Cycle 2011; 10: 1571–1581.
Frasa MA, Maximiano FC, Smolarczyk K, Francis RE, Betson ME, Lozano E et al. Armus is a Rac1 effector that inactivates Rab7 and regulates E-cadherin degradation. Curr Biol 2010; 20: 198–208.
Carroll B, Mohd-Naim N, Maximiano F, Frasa MA, McCormack J, Finelli M et al. The TBC/RabGAP Armus coordinates Rac1 and Rab7 functions during autophagy. Dev Cell 2013; 25: 15–28.
Abbi S, Ueda H, Zheng C, Cooper LA, Zhao J, Christopher R et al. Regulation of focal adhesion kinase by a novel protein inhibitor FIP200. Mol Biol Cell 2002; 13: 3178–3191.
Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 2008; 181: 497–510.
Melkoumian ZK, Peng X, Gan B, Wu X, Guan JL . Mechanism of cell cycle regulation by FIP200 in human breast cancer cells. Cancer Res 2005; 65: 6676–6684.
Morselli E, Shen S, Ruckenstuhl C, Bauer MA, Marino G, Galluzzi L et al. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle 2011; 10: 2763–2769.
Kim J, Kundu M, Viollet B, Guan KL . AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13: 132–141.
Lee JW, Park S, Takahashi Y, Wang HG . The association of AMPK with ULK1 regulates autophagy. PLoS One 2011; 5: e15394.
Löffler AS, Alers S, Dieterle AM, Keppeler H, Franz-Wachtel M, Kundu M et al. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy 2011; 7: 696–706.
Caino MC, Chae YC, Vaira V, Ferrero S, Nosotti M, Martin NM et al. Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J Clin Invest 2013; 123: 2907–2920.
Nakano A, Kato H, Watanabe T, Min KD, Yamazaki S, Asano Y et al. AMPK controls the speed of microtubule polymerization and directional cell migration through CLIP-170 phosphorylation. Nat Cell Biol 2010; 12: 583–590.
Chen S, Wang C, Yeo S, Liang CC, Okamoto T, Sun S et al. Distinct roles of autophagy-dependent and -independent functions of FIP200 revealed by generation and analysis of a mutant knock-in mouse model. Genes Dev 2016; 30: 856–869.
Gan B, Peng X, Nagy T, Alcaraz A, Gu H, Guan JL . Role of FIP200 in cardiac and liver development and its regulation of TNFalpha and TSC-mTOR signaling pathways. J Cell Biol 2006; 175: 121–133.
Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T et al. The role of autophagy during the early neonatal starvation period. Nature 2004; 432: 1032–1036.
Gan B, Melkoumian ZK, Wu X, Guan KL, Guan JL . Identification of FIP200 interaction with the TSC1-TSC2 complex and its role in regulation of cell size control. J Cell Biol 2005; 170: 379–389.
Gan B, Yoo Y, Guan JL . Association of focal adhesion kinase with tuberous sclerosis complex 2 in the regulation of s6 kinase activation and cell growth. J Biol Chem 2006; 281: 37321–37329.
Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT et al. FAK-Src signaling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Rev Cancer 2004; 6: 154–161.
Mitra SK, Hanson DA, Schlaepfer DD . Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol 2005; 6: 56–68.
Ezratty EJ, Partridge MA, Gundersen GG . Microtubule-induced focal adhesion disassembly is mediated by dynamin and focal adhesion kinase. Nat Cell Biol 2005; 7: 581–592.
Klinghoffer RA, Sachsenmaier C, Cooper JA, Soriano P . Src family kinases are required for integrin but not PDGFR signal transduction. EMBO J 1999; 18: 2459–2471.
Fincham VJ, Frame MC . The catalytic activity of Src is dispensable for translocation to focal adhesions but controls the turnover of these structures during cell motility. EMBO J 1998; 17: 81–92.
Deakin NO, Turner CE . Paxillin comes of age. J Cell Sci 2008; 121: 2435–2444.
Chen GC, Lee JY, Tang HW, Debnath J, Thomas SM, Settleman J . Genetic interactions between Drosophila Atg1 and paxillin reveal a role for paxillin in autophagosome formation. Autophagy 2008; 4: 37–45.
Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015; 349 (:) aaa5612.
Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V . Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J 2011; 30: 4701–4711.
Jiang S, Dupont N, Castillo EF, Deretic V . Secretory versus degradative autophagy: unconventional secretion of inflammatory mediators. J Innate Immun 2013; 5: 471–479.
Ponpuak M, Mandell MA, Kimura T, Chauhan S, Cleyrat C, Deretic V . Secretory autophagy. Curr Opin Cell Biol 2015; 35: 106–116.
Giancotti FG . Mechanisms governing metastatic dormancy and reactivation. Cell 2013; 155: 750–764.
Karrison TG, Ferguson DJ, Meier P . Dormancy of mammary carcinoma after mastectomy. JNCI J Natl Cancer Inst 1999; 91: 80–85.
Balic M, Lin H, Young L, Hawes D, Giuliano A, McNamara G et al. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin Cancer Res 2006; 12: 5615–5621.
Lum JJ, Bauer DE, Kong M, Harris MH, Li CY, Lindsten T et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 2005; 120: 237–249.
Liang J, Saho SH, Xu ZX, Hennessy B, Ding Z, Larrea M et al. The energy sensing LKB1-AMPK pathway regulates p27kip1 phosphorylation mediatiing the decision to enter autophagy or apoptosis. Nat Cell Biol 2007; 9: 218–224.
Rosen DG, Wang L, Jain AN, Lu KH, Luo RZ, Yu Y et al. Expression of the tumor suppressor gene ARHI in epithelial ovarian cancer is associated with increased expression of p21WAF1/CIP1 and prolonged progression-free survival. Clin Cancer Res 2004; 10: 6559–6566.
Lu Z, Yang H, Sutton MN, Yang M, Clarke CH, Liao WS et al. ARHI (DIRAS3) induces autophagy in ovarian cancer cells by downregulating the epidermal growth factor receptor, inhibiting PI3K and Ras/MAP signaling and activating the FOXo3a-mediated induction of Rab7. Cell Death Differ 2014; 21: 1275–1289.
Gupta A, Roy S, Lazar AJF, Wang WL, McAuliffe JC, Reynoso D et al. Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST). Proc Natl Acad Sci USA 2010; 107: 14333–14338.
Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A . Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res 2008; 68: 1485–1494.
Ranganathan AC, Zhang L, Adam AP, Aguirre-Ghiso JA . Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Research 2006; 66: 1702–1711.
Amaravadi RK, Yu DS, Lum JJ, Bui T, Christophorou MA, Evan GI et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007; 117: 326–336.
Ma Y, Galluzzi L, Zitvogel L, Kroemer G . Autophagy and cellular immune responses. Immunity 2013; 39: 211–227.
Deretic V, Saitoh T, Akira S . Autophagy in infection, inflammation and immunity. Nature Rev Immunol 2013; 13: 722–737.
Zhong Z, Sanchez-Lopez E, Karin M . Autophagy, inflammation, and immunity: a Troika governing cancer and its treatment. Cell 2016; 166: 288–298.
Rioux JD, Xavier R, Taylor K, Silverberg M, Goyette P, Huett A et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet 2007; 39: 596–604.
Schreiber RD, Old LJ, Smyth MJ . Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 2011; 331: 1565–1570.
Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011; 334: 1573–1577.
Ladoire S, Enot D, Senovilla L, Ghiringhelli F, Poirier-Colame V, Chaba K et al. The presence of LC3B puncta and HMGB1 expression in malignant cells correlate with the immune infiltrate in breast cancer. Autophagy 2016; 12: 864–875.
Pietrocola F, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE et al. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell 2016; 30: 147–160.
Akalay I, Janji B, Hasmim M, Noman MZ, Andre F, De Cremoux P et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Res 2013; 73: 2418–2427.
Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL . Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev 2011; 25: 1510–1527.
Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci USA 2013; 110: 17450–17455.
Tittarelli A, Janji B, Van Moer K, Noman MZ, Chouaib S . The selective degradation of synaptic connexin 43 protein by hypoxia-induced autophagy impairs natural killer cell-mediated tumor cell killing. J Biol Chem. 2015; 290: 23670–23679.
Noman MZ, Janji B, Kaminska B, Van Moer K, Pierson S, Przanowski P et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res 2011; 71: 5976–5986.
Messai Y, Noman MZ, Hasmim M, Janji B, Tittarelli A, Boutet M et al. ITPR1 protects renal cancer cells against natural killer cells by inducing autophagy. Cancer Res 2014; 74: 6820–6832.
Joyce JA, Pollard JW . Microenvironmental regulation of metastasis. Nat Rev Cancer 2009; 9: 239–252.
Condeelis J, Pollard JW . Macrophages: obligate partners for tumor cell migration, invasion and metastasis. Cell 2006; 124: 263–266.
Lin EY, Nguyen AV, Russell RG, Pollard JW . Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med 2001; 193: 727–739.
Robinson-Smith TM, Isaacsohn I, Mercer CA, Zhou M, Van Rooijen N, Husseinzadeh N et al. Macrophages mediate inflammation-enhanced metastasis of ovarian tumors in mice. Cancer Res 2007; 67: 5708–5716.
Wels J, Kaplan RN, Rafii S, Lyden D . Migratory neighbors and distant invaders: tumor-associated niche cells. Genes Dev 2008; 22: 559–574.
Wyckoff JB, Wang Y, Lin EY, Li JF, Goswami S, Stanley ER et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res 2007; 67: 2649–2656.
Roh-Johnson M, Bravo-Cordero JJ, Patsialou A, Sharma VP, Guo P, Liu H et al. Macrophage contact induces RhoA GTPase signaling to trigger tumor cell intravasation. Oncogene 2014; 33: 4203–4212.
Deretic V, Jiang S, Dupont N . Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol 2012; 22: 397–406.
Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 2012; 13: 255–263.
Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011; 12: 222–230.
Zhou R, Yazdi AS, Menu P, Tschopp J . A role for mitochondria in NLRP3 inflammasome activation. Nature 2011; 469: 221–225.
Salah FS, Ebbinghaus M, Muley VY, Zhou Z, Al-Saadi KR, Pacyna-Gengelbach M et al. Tumor suppression in mice lacking GABARAP, an Atg8/LC3 family member implicated in autophagy, is associated with alterations in cytokine secretion and cell death. Cell Death Dis 2016; 7: e2205.
Drake LE, Macleod KF . Tumour suppressor gene function in carcinoma-associated fibroblasts: from tumour cells via EMT and back again? J Pathol 2014; 232: 283–288.
Capparelli C, Guido C, Whitaker-Menezes D, Bonuccelli G, Balliet R, Pestell TG et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle 2012; 11: 2285–2302.
Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010; 9: 2423–2433.
Witkiewicz AK, Dasgupta A, Nguyen KH, Liu C, Kovatich AJ, Schwartz GF et al. Stromal caveolin-1 levels predict early DCIS progression to invasive breast cancer. Cancer Biol Ther 2009; 8: 1071–1079.
Di Vizio D, Morello M, Sotgia F, Pestell RG, Freeman MR, Lisanti MP . An absence of stromal caveolin-1 is associated with advanced prostate cancer, metastatic disease and epithelial Akt activation. Cell Cycle 2009; 8: 2420–2424.
Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009; 15: 35–44.
McAllister SS, Weinberg RA . The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol 2014; 16: 717–727.
Sceneay J, Smyth MJ, Moller A . The pre-metastatic niche: finding common ground. Cancer Metastasis Rev 2013; 32: 449–464.
Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med 2012; 18: 883–891.
Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol 2015; 17: 816–826.
Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015; 527: 329–335.
Colombo M, Raposo G, Thery C . Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 2014; 30: 255–289.
Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO . Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 2007; 9: 654–659.
Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ . Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia 2006; 20: 1487–1495.
Grange C, Tapparo M, Collino F, Vitillo L, Damasco C, Deregibus MC et al. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res 2011; 71: 5346–5356.
Huotari J, Helenius A . Endosome maturation. EMBO J 2011; 30: 3481–3500.
Baixauli F, Lopez-Otin C, Mittelbrunn M . Exosomes and autophagy: coordinated mechanisms for the maintenance of cellular fitness. Front Immunol 2014; 5: 403.
Acknowledgements
Grant support for this work includes NIH RO1 CA 162405 to KFM, T32 GM007281 supported EEM and T32 CA009594 supported MNS.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Mowers, E., Sharifi, M. & Macleod, K. Autophagy in cancer metastasis. Oncogene 36, 1619–1630 (2017). https://doi.org/10.1038/onc.2016.333
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.333
This article is cited by
-
Mitochondrial protein C15ORF48 is a stress-independent inducer of autophagy that regulates oxidative stress and autoimmunity
Nature Communications (2024)
-
Impairing hydrolase transport machinery prevents human melanoma metastasis
Communications Biology (2024)
-
Canonical WNT/β-catenin signaling upregulates aerobic glycolysis in diverse cancer types
Molecular Biology Reports (2024)
-
The heterogeneity of breast cancer metastasis: a bioinformatics analysis utilizing single-cell RNA sequencing data
Breast Cancer Research and Treatment (2024)
-
PINK1-PTEN axis promotes metastasis and chemoresistance in ovarian cancer via non-canonical pathway
Journal of Experimental & Clinical Cancer Research (2023)
