
Abstract
Expression of viral proteins causes important epigenetic changes leading to abnormal cell growth. Whether viral proteins directly target histone methyltransferases (HMTs), a key family enzyme for epigenetic regulation, and modulate their enzymatic activities remains elusive. Here we show that the E6 proteins of both low-risk and high-risk human papillomavirus (HPV) interact with three coactivator HMTs, CARM1, PRMT1 and SET7, and downregulate their enzymatic activities in vitro and in HPV-transformed HeLa cells. Furthermore, these three HMTs are required for E6 to attenuate p53 transactivation function. Mechanistically, E6 hampers CARM1- and PRMT1-catalyzed histone methylation at p53-responsive promoters, and suppresses the binding of p53 to chromatinized DNA independently of E6-mediated p53 degradation. p53 pre-methylated at lysine-372 (p53K372 mono-methylation) by SET7 protects p53 from E6-induced degradation. Consistently, E6 downregulates p53K372 mono-methylation and thus reduces p53 protein stability. As a result of the E6-mediated inhibition of HMT activity, expression of p53 downstream genes is suppressed. Together, our results not only reveal a clever approach for the virus to interfere with p53 function, but also demonstrate the modulation of HMT activity as a novel mechanism of epigenetic regulation by a viral oncoprotein.
Similar content being viewed by others


Introduction
Human papillomaviruses (HPVs) cause cervical carcinoma and are associated with several other human cancers such as lung cancer (Chen et al., 2004; Ganguly and Parihar, 2009). They are small viruses with a double-stranded circular DNA, which replicates in the nucleus of infected cells. Among the HPV proteins, the early proteins E6 and E7 attract most attention as they transform and immortalize cells (Ganguly and Parihar, 2009; McLaughlin-Drubin and Munger, 2009). One of the well-documented mechanisms by which HPV causes cervical carcinoma is E6-mediated degradation of the tumor suppressor p53 (Scheffner et al., 1990; Munger and Howley, 2002; Howley and Livingston, 2009). E6, complexed with the E6-associated protein (E6AP), functions as a ubiquitin ligase to perform p53 ubiquitination for destruction (Scheffner et al., 1990; Scheffner et al., 1993). Nevertheless, this is unlikely the only mechanism by which E6 transforms cells. Additional p53-independent functions/targets of E6 have been described (Howley, 2006; Howie et al., 2009). Several groups reported that E6 inhibits the histone acetyltransferase activity of the important coactivator p300 (Patel et al., 1999; Zimmermann et al., 1999; Thomas and Chiang, 2005).
To activate gene transcription from the condensed chromatin with DNA wrapped by histone and non-histone chromosomal proteins, transcriptional activators with sequence-specific DNA-binding activity will recruit coactivators to the regulatory sequence of a particular gene. These coactivators usually contain at least one of the following activities: as a scaffold protein, histone modification, ATP-dependent chromatin remodeling or the activity to regulate DNA methylation (Trojer and Reinberg, 2007). Histone modifiers can post-translationally modify histones as well as a growing list of non-histone proteins (Lee and Stallcup, 2009; Pradhan et al., 2009). The modifications on histones, alone or in combination, constitute an epigenetic language, which is believed to alter gene expression by changing the DNA–histone interaction, histone–protein interaction or by serving as signals to recruit other regulators (Strahl and Allis, 2000).
Many sites on histone tails can be modified by various histone modifiers. Up to date, the reported modifications include acetylation, methylation, phosphorylation, ubiquitination, sumoylation, ADP-ribosylation, proline isomerization, and so on (Kouzarides, 2007; Campos and Reinberg, 2009). Unlike histone acetylation, a hallmark of gene activation occurring exclusively on lysine, histone methylation is involved in both gene activation and repression, and takes place on both lysine and arginine. Arginine could be di-methylated either asymmetrically by type-I arginine histone methyltransferases (HMTs) or symmetrically by type-II HMTs (Wolf, 2009). An et al. (2004) showed that p53 recruits the type-I arginine HMTs CARM1 and PRMT1 to methylate histones at p53-responsive promoters and activate p53 downstream genes. Notably, CARM1 and PRMT1 coactivate and methylate many other proteins (Lee and Stallcup, 2009). By contrast, lysine can be mono-, di- or tri-methylated (Shukla et al., 2009). SET7 performs mono-methylation on lysine-4 of H3 to exert its coactivation function (Wang et al., 2001; Nishioka et al., 2002). Likewise, SET7 methylates non-histone substrates, which include p53 (Pradhan et al., 2009). SET7-mediated mono-methylation of p53 at K372 (p53K372me1) increases p53 stability, likely through recruitment of the histone acetyltransferase Tip60 for p53 acetylation (Kurash et al., 2008). SET7-mediated p53 methylation also antagonizes the transcriptional repression by Smyd2-mediated p53 methylation (Huang et al., 2006).
Whether functions of HMTs can be modulated by viral proteins remains mysterious. In the current study, we take p53, a well-known target of E6, as an example to illustrate how E6 modulates the activities of HMTs CARM1, PRMT1 and SET7 for controlling gene-specific transcription. We found that E6 at the physiological concentration downregulated p53 transcriptional activity and stability through inhibition of the arginine-specific HMTs CARM1 and PRMT1, and the lysine-specific HMT SET7. As these HMTs are important coactivators and enzymes not only for p53, the study strongly suggests that the oncogenic function of E6 is contributed by more factors than previously appreciated. Our study provides the first evidence to show HMT inhibition by a viral oncoprotein.
Results
E6 interacts with endogenous CARM1, PRMT1 and SET7 in cells
To investigate whether E6 functions through modulation of HMTs, CARM1, PRMT1 and SET7 were analyzed as these HMTs facilitate the function of p53, a well-known target of E6 (An et al., 2004; Chuikov et al., 2004). First we asked whether E6 interacts with these HMTs in cells. The high-risk HPV type-18 E6 (18E6)-expressing cervical carcinoma cell HeLa was harvested and subjected to immunoprecipitation (IP) using IgG or antibody (Ab) against 18E6, followed by western blotting with Ab against CARM1, PRMT1 or SET7. As shown in Figure 1a, the E6 Ab only co-immunoprecipitated CARM1, PRMT1 and SET7 in HeLa cells with a scramble RNA, but failed to do so when 18E6 was knocked down by si-18E6 (compare lane 6 with lane 5, lane 12 with lane 11, and lane 18 with lane 17). Note that depleting 18E6 from HeLa cells only increased the p53 protein level, but did not affect the level of CARM1, PRMT1 or SET7 (Figure 1 and Supplementary Figure S1). Next we investigated whether E6 of the low-risk HPV type-11 (11E6) is also capable of binding to these HMTs. To do so, U2OS cells ectopically expressing Flag-tagged 11E6 or 18E6 were subjected to IP and western blot analysis. Figure 1b shows that CARM1, PRMT1 and SET7 Abs pulled down both Flag-11E6 and Flag-18E6, but not actin. These results indicate that both high- and low-risk HPV E6 specifically associate with endogenous CARM1, PRMT1 and SET7.

In vivo interaction of E6 and HMTs. HeLa cells (a) or E6-transfected U2OS cells (b) were harvested for IP with anti-18E6, anti-CARM1, anti-PRMT1, anti-SET7 or IgG, followed by western blotting using Ab against the indicated proteins. The asterisk indicates the immunoprecipitated HMTs in panel a. The input control represents 5% (a) or 10% (b) volume of cell lysates used for IP.
E6 inhibits the methyltransferase activity of CARM1, PRMT1 and SET7 in vitro
In vitro methyltransferase assays were then applied to test whether E6 directly affects the enzymatic activities of CARM1, PRMT1 and SET7. To this end, western blotting using Ab against CARM1-mediated asymmetric di-methylation of histone H3 at R17 (Asy-H3R17me2) or PRMT1-induced asymmetric di-methylation of H4 at R3 (Asy-H4R3me2) was performed. As shown in Figures 2a and b, CARM1 and PRMT1 methylated histones H3 and H4, respectively (compare lane 2 with lane 1). Increasing amounts of 11E6, 16E6 (E6 of high-risk HPV 16) or 18E6 markedly reduced histone methylation in an E6 dose-dependent manner (lanes 3–8). As control, glutathione-S-transferase did not affect CARM1- or PRMT1-mediated histone methylation (lane 9). The phenomenon was further confirmed by using a radiolabeled methionine donor (Supplementary Figures S2A and B). Similar results were obtained when SET7 was examined on histones (Supplementary Figure S2C) or p53 was used as the substrate for SET7 (Figure 2c). Note that neither CARM1 nor PRMT1 methylated p53 (data not shown). Stoichiometry analysis of the proteins included in the above in vitro methyltransferase assays (shown as molar rations in the figure legends) indicates that less than three-fold molar excess of E6 to HMT greatly downregulated HMT function. These results show that E6 directly inhibits HMT activity.

In vitro inhibition of HMT activities by E6. E6 inhibits the methyltransferase activities of CRAM1 (a), PRMT1 (b) and SET7 (c). (a, b) The indicated proteins (2 μg of each HMT, 1 or 2 μg of each E6, and 10 μg of core histone proteins) were mixed with SAM. The reaction products were separated by 15% SDS–PAGE. The gels were analyzed by western blotting using Abs against H3 asymmetrically di-methylated at R17 (Asy-H3R17me2) and histone H3 in panel a or H4 asymmetrically di-methylated at R3 (Asy-H4R3me2) and histone H4 in panel b. The molar ratios of histone H3 to CARM1 to E6 are 1:0.2:0.37 (lanes 3, 5 and 7) and 1:0.2:0.72 (lanes 4, 6 and 8) in panel a, and of histone H4 to PRMT1 to E6 are 1:0.21:0.24 (lanes 3, 5 and 7) and 1:0.21:0.48 (lanes 4, 6 and 8) in panel b. (c) The indicated proteins (2 μg of each HMTs, 2 or 5 μg of each E6, and 5 μg of p53) were mixed with [3H]-SAM (S-adenosyl-L-[methyl-3H]-methionine). The reaction products were separated by 15% SDS–PAGE. The gels were stained with Coomassie blue (upper panel) and analyzed by phosphoimaging (lower panel). The molar ratios of p53 to SET7 to E6 are 1:0.44:1.18 (lanes 3, 5 and 7) and 1:0.44:2.94 (lanes 4, 6 and 8).
E6 inhibits CARM1- and PRMT1-mediated p53 transactivation function in a p53 degradation-independent manner
As E6 interacted with CARM1, PRMT1 and SET7 (Figure 1), and directly downregulated their enzymatic activities (Figure 2), it is expected that gene transcription modulated through this pathway is affected. To test this possibility, we used the p53-target gene p21 as an example. As expected, exogenous expression of p53 in p53-null H1299 cells increased the activity of the luciferase reporter driven by the p21 promoter (Figure 3a, column 2), which was further boosted by CARM1 or PRMT1, or both (columns 3–5). In the presence of 18E6, the transactivation function of p53 was reduced (compare column 7 with column 2), presumably because of the degradation of a certain portion of p53 (western blot in Supplementary Figure S3, compare lane 4 with lane 3). Interestingly, neither CARM1 nor PRMT1 further enhanced the transcriptional activity of the remaining p53 under this condition (Figure 3a, compare columns 8, 9 and 10 with column 7). As the protein levels of CARM1 and PRMT1 were same regardless of the presence of E6 (Supplementary Figure S3, compare lane 6 with lane 5, lane 8 with lane 7, and lane 10 with lane 9), the result indicates that the coactivation function of both CARM1 and PRMT1 was abolished by E6. The reverse transcription (RT)–PCR experiment in Figure 3b further confirms that (i) the endogenous p21 mRNA level in H1299 cells was stimulated by p53, CARM1 and PRMT1 (compare column 1 with column 3); (ii) E6-mediated p53 degradation resulted in a great loss of p21 mRNA level (compare column 4 with column 2) and (iii) in the presence of E6, the exogenous CARM1 and PRMT1 no longer coactivated the remaining p53 in stimulating p21 mRNA synthesis (compare column 5 with column 4).

E6 inhibits the CARM1- and PRMT1-stimulated p53-dependent transcription. (a) CARM1 and PRTM1 fail to stimulate the p53 transactivation function in the presence of E6. (b) E6 suppresses CARM1- and PRMT1-induced p21 expression. (a, b) H1299 cells were transfected with the indicated plasmids, followed by luciferase assay (a), RT–PCR and western blot analysis (b). (c) 11E6 and 16E66C/6S F47R do not interfere with p53 protein level but inhibit p21 expression. H1299 cells were transfected with the indicated plasmids for 48 h and then treated with Adr for another 6 h, followed by RT–PCR and western blot analysis. (d) Knocking down CARM1 and PRMT1 relieves the E6-mediated inhibition of endogenous p21 expression. After 24 h of siRNA transfection, U2OS cells were transfected with a plasmid encoding E6 for another 48 h. (e) Deletion of E6 from HeLa cells restores CARM1- and PRMT1-enhanced p21 expression. After 24 h of siRNA transfection, HeLa cells were transfected with plasmids encoding CARM1 and PRMT1 for 48 h, or treated with Adr for 6 h. (d, e) After transfection, quantitative RT–PCR and western blotting were performed. sc-RNA indicates si-scramble control.
To further demonstrate that E6 is able to inhibit p53-target gene expression through HMT inhibition but not p53 degradation, an experiment similar to Figure 3a was conducted, with H1299 treated with the proteasome inhibitor MG132 to ensure that the p53 protein level is equal regardless of the presence of E6. Consistently, E6 still suppressed the HMT-stimulated p53 activity under this condition (Supplementary Figure S4, compare columns 6, 8 and 10 with columns 5, 7 and 9), indicating that HMT inhibition likely contributes to E6-mediated downregulation of p53 downstream gene expression.
The hypothesis was further supported by using a 16E6-6C/6S/F47R mutant or 11E6, both of which cannot induce p53 degradation (Crook et al., 1991; Nomine et al., 2006). As shown in Figure 3c, these two E6 were still capable of inhibiting p53-dependent p21 mRNA synthesis in H1299 treated with Adriamycin (Adr), a DNA damage inducer known to stimulate p53 activity (Tewey et al., 1984). Similar results were obtained in U2OS cells (Supplementary Figure S5). Using a short interfering RNA (siRNA) against CARM1 or PRMT1, we further show that the E6-mediated reduction of Adr-stimulated p21 mRNA level was partly relieved upon knockdown of CARM1 and PRMT1 (Figure 3d, columns 1–4). As control, a scramble RNA did not release the E6-dependent reduction of p21 mRNA level (Figure 3d, columns 5–8). The data also indicated that, in the absence of E6, depleting CARM1 and PRMT1 reduced Adr-mediated p21 expression (Supplementary Figure S6, compare column 4 with column 2), indicating that both HMTs are essential in coactivating p53 function in vivo. These data together argue that inhibition of endogenous CARM1 and PRMT1 might contribute to the E6-mediated repression of p53 function. Consistent results were obtained from HeLa cells, which stably express 18E6. As shown in Figure 3e, Adr and exogenous CARM1/PRMT1 significantly enhanced endogenous p21 mRNA expression only in cells depleted with 18E6 (lanes 7–9). The effect was not observed in mock or scramble RNA-transfected HeLa cells (lanes 1–6). These studies demonstrate that E6 at the physiological level is sufficient to inhibit CARM1- and PRMT1-stimulated p53 function.
E6 suppresses CARM1- and PRMT1-mediated histone methylation on p53-responsive promoters in cells
Given that E6 was demonstrated to inhibit CARM1- and PRMT1-mediated p53 transactivation on p21 gene expression (Figure 3), we continued to investigate if E6 suppresses the corresponding histone methylation at p53-responsive promoters. To this end, the levels of Asy-H3R17me2 and Asy-H4R3me2 known to be mediated by CARM1 and PRMT1, respectively, were analyzed in U2OS cells transiently expressing hemagglutinin (HA)-tagged wild-type (wt) or catalytically dead CARM1 or PRMT1 with or without E6. As observed, only low levels of histone methylation and acetylation were detected without overexpressing HMTs (Figure 4a, lane 1). Adding wt CARM1 or PRMT1, alone or together, increased the levels of Asy-H3R17me2 and Asy-H4R3me2, respectively, H4 acetylation and p53 binding to chromatin (lanes 2–4). By contrast, the catalytic mutant of CARM1 or PRMT1 failed to induce the same effect (lanes 5 and 6), indicating that the CARM1- and PRMT1-catalyzed histone modifications were critical for binding of p53 to chromatin. These experiments demonstrate for the first time that the enzymatic activities of these two HMTs were important for binding of p53 to its target promoters in vivo. Importantly, in the presence of MG132 (lanes 7–9), 18E6 repressed the CARM1- and PRMT1-stimulated histone methylation and acetylation, as well as p53 binding (compare lane 9 with lane 8). Quantification of these chromatin IP (ChIP) results by real-time PCR is shown in Supplementary Figure S7. The western blots below the ChIP results (Figure 4a, bottom panel) further confirm that the above effects were not due to variation of the protein levels of p53, CARM1 and PRMT1. Subsequently we examined whether E6 downregulates the histone methylation caused by endogenous CARM1 and PRMT1. In the presence of MG132 and Adr, U2OS cells with or without 18E6 were subjected to ChIP assays. Similarly, expression of E6 suppressed Asy-H4R3me2 and Asy-H3R17me2 on the promoters of p21 and another p53-target gene, GADD45. Only the p53-binding region, but not on the distal p53-unrelated region, was affected (Supplementary Figure S8). As control, the western blot shows that, in the presence of MG132, the p53 level was unchanged with or without E6 (bottom panel). Thus, E6 downregulates endogenous CARM1 and PRMT1 activities on p53-target genes.

E6 represses CRAM1 and PRMT1 activity-dependent p53 binding and histone modifications on p53-responsive promoters. U2OS cells transfected with the indicated expression vectors in the absence (lanes 1–6) or presence (lanes 7–9) of MG132 (a) and H1299 cells transfected with the indicated expression vectors in the presence of MG132 (b) were subjected to ChIP assays on the p21 or the GADD45 promoter, and western blotting was performed by using Abs against the indicated proteins. (c) HeLa cells treated with Adr and MG132 in the presence of scramble RNA (open square) or si-18E6 (filled square) were subjected to ChIP using Abs against the indicated proteins.
Whether the HMT- and E6-mediated alteration of histone modification on p21 and GADD45 genes was indeed through p53 was analyzed. The ChIP assays shown in Figure 4b indicate that ectopically expressed p53 bound to both p21 and GADD45 promoters in H1299 cells. p53 recruited CARM1 and PRMT1 to the p21 and GADD45 promoters, and enhanced the corresponding histone methylation as well as histone acetylation. As observed, all these effects were abolished in the presence of 18E6 (lanes 3 and 6). As a negative control, symmetric di-methylation of H3R26 (Sym-H3R26me2), a mark mediated by neither CARM1 nor PRMT1, was unaffected by expression of p53 or E6. Again, the western blot results show no decrease of p53 protein level by E6 in the presence of MG132 (compare lane 9 with lane 8).
Furthermore, we showed that depleting E6 from HeLa cells enhanced Asy-H4R3me2, Asy-H3R17me2 and H4 acetylation on both p21 and GADD45 promoters (Figure 4c), indicating that E6 at the physiological level suppresses endogenous CARM1- and PRMT1-mediated histone methylation on p53-responsive promoters. Similar to 18E6, 11E6, which does not degrade p53, also repressed CARM1 and PRMT1 activities on p53-responsive promoters (Figure 5). Collectively, these results suggest that both high-risk and low-risk E6 are capable of downregulating p53-dependent promoter activity through suppression of HMT function in vivo.

Low-risk HPV type-11 E6 downregulates CARM1 and PRMT1 activities in vivo. (a) 11E6 represses the CARM1- and PRMT1-induced GADD45 mRNA level and the corresponding histone modifications on the GADD45 promoter. U2OS cells ectopically expressing HA-CARM1 and HA-PRMT1 with or without 11E6 were subjected to RT–PCR, western blotting and ChIP assays. As shown, overexpression of CARM1 and PRMT1 increased the GADD45 mRNA level (compare lane 2 with lane 1) as well as the corresponding histone modifications on the GADD45 promoter (compare lane 5 with lane 4). In the presence of 11E6, all these CARM1- and PRMT1-stimulated effects were attenuated (compare lane 3 with lane 2 and lane 6 with lane 5), indicating that 11E6, which does not degrade p53 (as seen in the western blotting), could antagonize the CARM1- and PRMT1-mediated expression of the p53-target gene. (b) 11E6 downregulates the Adr-induced GADD45 mRNA level and the corresponding histone methylation and acetylation on the GADD45 promoter. U2OS cells with or without 11E6 and Adr were subjected to RT–PCR for GADD45 mRNA analysis and ChIP assays using Ab against Asy-H4R3me2 or Ace-H4.
E6 represses p53 binding to chromatinized DNA by interacting with p53
The ChIP experiments in Figure 4 have identified that 18E6 binding to p53-target gene promoters accompanied the loss of p53 binding. To further confirm this phenomenon, ChIP and DNA affinity protein assay (DAPA) were applied in H1299 cells, with exogenously added p53. The ChIP assay indicates that 18E6 alone, even at a high concentration, failed to bind to the p21 or the GADD45 promoter (Figure 6a, lane 1). Association of E6 with both p21 and GADD45 promoters was only observed when p53 was present (lane 3), indicating that E6 targets p53-responsive promoters by interacting with p53. Interestingly, binding of p53 and E6 were both diminished when more E6 was added (lane 4). Quantification of the results by real-time PCR is shown in Supplementary Figure S9.

E6 inhibits p53 binding to chromatin. H1299 cells were transfected with the indicated expression vectors and treated with MG132 followed by ChIP (a, top and middle panels), western (a, bottom panel) and DAPA (b) assays. (a) E6 suppresses p53 binding to endogenous p21 (upper panel) and GADD45 (middle panel) promoters. (b) E6 does not affect p53 binding to the non-chromatinized DNA containing the p53-binding site of the p21 promoter. (c) Knocking down E6 increases p53 binding to the p21 promoter. (d) Knocking down p53 reduces E6 binding to the p21 promoter. (c, d) HeLa cells were treated with MG132 or transfected with the indicated siRNAs, followed by ChIP, RT–PCR and western blot analysis.
We hypothesized that the E6-reduced p53 binding to chromatin was a result of alteration in histone modification, but not due to a conformational change in p53 that impairs the binding of p53 to free DNA. The idea was confirmed by the following DAPA assay. A biotin-labeled DNA fragment containing the p53-binding element of the p21 promoter or a random sequence (Figure 6b, illustration at the top) was linked to streptavidin beads and incubated with H1299 cell lysates under the same transfection conditions as in Figure 6a. p53 was precipitated by beads containing the p53-binding element of the p21 promoter, but not by beads conjugated with a random probe, indicating that p53 only binds to a specific DNA fragment (lane 3). Whereas E6 alone did not associate with the p53-binding element or the random sequence (lane 2), it bound to the p53-responsive element when p53 was expressed in cells (lanes 4 and 5). As a negative control, β-tubulin could not be precipitated by the p21 probe, suggesting that E6 recruitment by p53 to the p53-binding element was specific. Importantly, E6 did not interfere with the DNA-binding ability of p53 (compare lanes 4 and 5 with lane 3). A similar DAPA result was obtained by using a 214-bp DNA fragment containing the same sequence and length as that examined by the ChIP in Figure 6a (Supplementary Figure S10A). Note that, although the 214-bp DNA fragment theoretically could hold a nucleosome, which occupies approximately 146 bp, the DNA was unlikely assembled into the nucleosome-like structure as successful reconstitution of chromatin requires additional chromatin assembly factors/histone chaperons, which were not included in this case. To further confirm this, we show that the 214-bp probe in the DAPA condition failed to pull down histone H3 (Supplementary Figure S10B). Thus, it is concluded that E6 only affects p53 binding to histone-bound DNA, but not free DNA, consistent with the idea that histone methylation determines the binding of p53 to chromatin (Figure 4a).
We continue to investigate whether E6 represses p53 binding to chromatin in HeLa cells. As shown in Figure 6c, both MG132 treatment and E6 depletion increased the p53 protein level (compare lanes 2 and 3 with lane 1). Nevertheless, p53 binding to p21 promoter was largely increased only in cells deficient in E6 (compare lane 3 with lane 2), indicating that the stably expressed E6 is sufficient and necessary to repress p53 binding to its target promoter. In HeLa cells, we also showed that knockdown of p53 not only reduced the chromatin-bound p53, but also diminished E6 binding to the same promoter region (Figure 6d), further supporting our hypothesis that E6 targets p53-resposive promoters by associating with p53.
E6 downregulates p53 stability by inhibiting SET7-catalyzed p53 methylation
So far our data demonstrate that the E6-mediated downregulation of the HMT activities of CARM1 and PRMT1 has important function in vivo. Whether inhibition of SET7 by E6 observed in vitro (Figure 2) is also physiologically relevant was investigated. Again, p53 was used as a working model as p53 is one of the SET7 substrates and E6 targets. First we confirmed whether SET7, and presumably the resulting mono-methylation of p53 at lysine-372 (p53K372me1), stabilizes p53 protein level as reported earlier (Chuikov et al., 2004; Ivanov et al., 2007; Kurash et al., 2008). As observed in Supplementary Figure S11, without Adr treatment, knocking down SET7 (lane 3) or expressing methylase-dead SET7 (lane 4) affected neither p53 nor p21 protein level in U2OS cells, indicating that SET7 unlikely controls p53 stability and activity under normal conditions. By contrast, Adr induced p53 and p21 levels (compare lane 5 with lane 1), but failed to do so when SET7 was knocked down (lane 7) or the catalytically dead SET7 was included (lane 8), indicating that SET7 activity on p53 (p53K372me1) is required for p53 stabilization and downstream gene expression in response to genotoxic stresses. Subsequently, we investigated whether SET7 indeed contributes to the E6-mdiated repression of p53 function under DNA damage conditions. As shown in Figure 7a, the 16E6-mediated inhibition of p21 expression (compare lane 2 with lane 1) was greatly lost when SET7 was depleted from U2OS cells (compare lane 4 with lane 3), indicating that E6-mediated repression of p53 function depends on SET7. Consistently, E6 reduced SET7-mediated p53 methylation in DNA damage-induced cells. As shown in Figure 7b, p53K372me1 signal was abolished in U2OS cells expressing Flag-18E6 (lanes 1 and 2) and stimulated in HeLa cells depleted of 18E6 (lanes 3 and 4). p21 protein level was induced as well in HeLa cells without 18E6 (lane 4). We further addressed that SET7 was required to maintain Adr-induced p53 protein level in both U2OS and HeLa cells as p53 protein level decreased upon SET7 knockdown (Figure 7c, lanes 3 and 6). As expected, Adr only slightly enhanced the p53 level in HeLa cells, compared with that in U2OS cells (compare lane 5 with lane 2). SET7 depletion almost totally abolished the p53 protein level in HeLa cells (compare lane 6 with lane 4), whereas a certain portion of p53 was left upon SET7 depletion in U2OS cells (compare lane 3 with lane 1). These differential effects are likely due to the stable E6 expression in HeLa cells. In agreement with this idea, E6 knockdown in HeLa cells greatly recovered the p53 protein level (compare lane 7 with lane 5). These in vivo results strongly suggest that endogenous SET7 protects p53 from E6-mediated p53 degradation.

E6 downregulates SET7-mediated p53 methylation and stability. (a) Knocking down SET7 relieves the E6-mediated repression of p21 expression. U2OS cells transfected with 16E6 in the absence (lanes 1 and 2) or presence (lanes 3 and 4) of si-SET7 were treated with Adr for 6 h, followed by RT–PCR for p21 mRNA level and western blotting. (b) E6 inhibits p53 methylation at K372 in vivo. 18E6-transfected U2OS cells (lanes 1 and 2) or si-18E6-transfected HeLa cells (lanes 3 and 4) were treated by UV irradiation/MG132 or Adr/MG132, respectively, followed by western blotting. (c) 18E6 depletion from HeLa cells increases Adr-dependent p53 accumulation. The siRNA-transfected U2OS cells (lanes 1–3) and HeLa cells (lanes 4–7) were treated with or without Adr, followed by western blotting. The protein level of p53 was quantified and is shown in the bar chart below the western blots. (d) E6 preferentially degrades K372-unmethylated p53. Equal amounts of p53 from 40 μl of mock-treated cells or 5 μl of Adr-treated cells were subjected to IP using Ab against p53 mono-methylated at K372 (p53K372me1; see Materials and methods for Ab information) (lanes 1 and 2) or Ab against total p53 (FL-393; Santa Cruz), followed by E6-mediated in vitro degradation assay (lanes 3 and 4). The protein level of total p53 of each sample was then analyzed by western blotting using anti-p53 (DO-1) (sc-126; Santa Cruz). (e) SET7 and SAM together partially prevent E6-mediated p53 degradation. (f) E6 does not degrade p53 pre-methylated by SET7. (g) SET7 does not protect the p53K372R mutant from E6-mediated degradation. (e–g) Both p53 and 16E6 were in vitro transcribed and translated. p53 was incubated with or without 16E6, purified SET7 or SAM in the order shown in the scheme, followed by western blotting to show the p53 protein level at the indicated stage.
An IP-coupled degradation assay was next used to address whether p53 mono-methylated at K372 is preferentially protected from E6-mediated degradation. As shown in Figure 7d, equal amounts of p53 from U2OS cells with or without Adr treatment were immunoprecipitated with Ab against p53K372me1 (lanes 1 and 2) or Ab against total p53, and then subjected to E6-mediated in vitro degradation assays (lanes 3 and 4) (see section Materials and methods). p53 from cells stimulated with Adr was preferentially mono-methylated at K372 (compare lane 2 with lane 1) and prevented from degradation (compare lane 4 with lane 3), indicating that E6 preferentially degrades the endogenous p53 unmethylated by SET7.
The interrelationship between E6-dependent SET7 inhibition and E6-mediated p53 degradation was further clarified with a more purified system, the in vitro transcription–translation-coupled degradation assay. p53, SET7 and E6 were in vitro transcribed and translated from rabbit reticular lysate, mixed in different combination with the methyl group donor S-adenosyl-L-methionine (SAM) and incubated as illustrated in Figure 7e. The protein levels of p53 at Stage 1 show equal input. E6 reduced the protein level of p53, presumably unmethylated in the reaction lysates (compare lane 2 with lane 1). Interestingly, the p53 protein level was partly recovered when the reaction mixture also contained both exogenous SET7 and SAM (lane 5). SAM or SET7 alone did not prevent the E6-mediated p53 reduction (lanes 3 and 4). These data support our hypothesis that the methyltransferase activity of SET7 protects p53 from E6-mediated degradation. We continued to investigate whether p53 pre-methylated by SET7 resists E6-mediated degradation. To this end, we examined whether SAM added later than E6 fails to work with SET7 to prevent p53 from degradation by E6. The in vitro transcribed and translated p53 and SET7 were mixed at Stage 1, followed by incubation with E6 at Stage 2. SAM was added at Stage 1 or Stage 2. As shown in Figure 7f, the p53 protein level remained unchanged at all conditions at Stage 1 or Stage 2. Remarkably, only SAM added at Stage 1 successfully protected p53 from E6-mediated degradation, as determined by the p53 protein level at the stop stage (lane 3). By contrast, SAM added at Stage 2 failed to work with SET7 to recover the p53 protein level (lane 6). These data indicate that E6 only degrades p53, which is not previously methylated by SET7. To further support this notion, we examined the effect of E6 on the p53K372R mutant, which lacks the residue to be methylated by SET7. SET7, SAM, p53K372R and E6 were incubated in the order illustrated in Figure 7g. Consistently, the protein level of p53K372R was reduced by E6 (lane 2) and could not be recovered by SET7 and SAM (lane 3). The results strongly indicate that SET7 fails to protect the methylation-site mutant p53K372R from E6-mediated degradation. In other words, p53 requires to be methylated by SET7 to escape from E6-mediated degradation. Overall, these experiments demonstrate that E6-mediated inhibition of SET7 activity is upstream from E6-induced p53 degradation.
Discussion
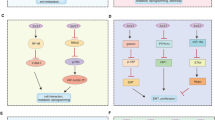
The current work presents the first evidence to show that HPV E6 is able to suppress the activities of both the arginine-specific HMTs, CARM1 and PRMT1, and the lysine-specific HMT, SET7. The inhibition is functionally significant in HPV-transformed cells as through this E6 downregulates the stability and transactivation function of p53. Figure 8 provides a model based on our current findings and reports by others. In HPV-non-infected normal cells (Figure 8a), DNA damage induces SET7-mediated p53 methylation and stabilization (Chuikov et al., 2004; Ivanov et al., 2007; Kurash et al., 2008), as well as p53-dependent recruitment of CARM1 and PRMT1 to p53-target gene promoters for transcriptional activation (An et al., 2004). In HPV E6-expressing cells (Figure 8b), E6 attenuates stress-induced p53 function at least through the following two distinct pathways: (1) E6 downregulates p53 protein stability depending on E6AP (Scheffner et al., 1990, 1993) or by inhibiting SET7-mediated p53 methylation at K372 (Figure 7). Given that p53 pre-methylated by SET7 resists E6-triggered degradation (Figure 7), inhibition of SET7 by E6 might prime E6-mediated p53 degradation. This also in part explains the fact that not all p53 is degraded in E6-expressing cells (Howie et al., 2009); (2) E6 can further target p53-responsive promoters (Figure 6) and downregulate the corresponding histone methylation mediated by the p53 coactivators CARM1 and PRMT1 (Figure 4), leading to reduced binding of p53 to chromatin (Figures 4 and 6) and loss of p53 transactivation (Figure 3). Together, a novel model is provided to show the dual roles of E6 in regulating p53 function through modulation of distinct HMT activity. We not only demonstrate the upstream regulatory mechanism leading to E6-mediated p53 degradation, but also reveal an alternative approach for E6 to shut down the function of the remaining p53 not degraded by E6. As CARM1, PRMT1, and SET7 are critical co-regulators not only for p53 (Lee and Stallcup, 2009; Pradhan et al., 2009) and E6 associates with these HMTs in p53-null H1299 cells (Supplementary Figure S12), E6-mediated HMT inhibition is expected to have a broader impact.

A model: E6 downregulates p53 function through HMT repression. (a) Previous reports indicate that, in HPV-non-infected cells, DNA-damage stress induces SET7-mediated p53 methylation and stabilization, as well as recruitment of p53 and its coactivators CARM1 and PRMT1 to p53-regulated genes for gene activation. (b) We demonstrate that, in HPV E6-expressing cells, in addition to directly targeting p53 for degradation through the E6AP pathway, E6 is able to inhibit SET7-mediated p53 methylation and thus reduce p53 protein stability. E6 can also target chromatin-bound p53 and repress the activities of the p53 coactivators CARM1 and PRMT1, leading to reduction of local histone methylation, which, likely, in turn dissociates p53 from chromatin. p53-dependent gene transcription is thus downregulated.
The remaining p53 escaping from E6-mediated degradation is still active in transcription
As shown in Figure 3, the remaining p53 escaping from E6-mediated degradation was still able to activate transcription to 5.7-fold (compare lane 7 with lane 6). Results published by two other laboratories also support the same conclusion. First, Zimmermann et al. (1999) showed that the remaining p53 still retained 1/3 of the total p53 activity (Figure 7a in Zimmermann et al., 1999, compare bar 4 with bar 2). Second, Patel et al. (1999) demonstrated that the remaining p53 regained activity in the presence of the histone acetyltransferase p300 (Figure 8a in Patel et al., 1999, compare bar 5 with bar 4). Therefore, we believe that the remaining p53 is still active in transcription.
Distinct roles of CARM1, PRMT1 and SET7 in mediating p53 function
Although CARM1, PRMT1 and SET7 all interacted with p53, unlike PRMT1 and CARM1, SET7 did not enhance p53 transactivation function without DNA-damage insult (compare Figure 3 with Supplementary Figure S13). This is not surprising as SET7 activity is minimal unless being induced by DNA damage (Ivanov et al., 2007). In addition, only SET7 among these three HMTs methylates p53 under stress (Chuikov et al., 2004; Ivanov et al., 2007; Kurash et al., 2008; Figure 2, and data not shown). In agreement with previous reports (Chuikov et al., 2004; Ivanov et al., 2007; Kurash et al., 2008), we found that the enzymatic activity of SET7 was important for p53 stabilization and downstream gene expression in response to DNA-damage stress (Supplementary Figure S11). It is likely that SET7 methylates p53 and directly prevents p53 from being ubiquitylated as K372 is one of p53 ubiquitylation sites (Kruse and Gu, 2008). Alternatively, p53 methylation by SET7 might indirectly influence p53 protein stability by facilitating p53 acetylation. Indeed, SET7-mediated p53K372 methylation primes p53 acetylation on several sites in the C-terminus (Ivanov et al., 2007; Kurash et al., 2008), and p53 acetylation increases p53 stability by blocking the ubiquitination sites of p53 (Li et al., 2002). These mechanisms likely explain how p53 methylation by SET7 resists E6-dependent p53 degradation through E6AP/ubiquitin (Scheffner et al., 1990, 1993). Nevertheless, E6-triggered p53 degradation can rely on neither E6AP (Massimi et al., 2008) nor ubiquitin (Camus et al., 2007). Whether E6-mediated SET7 inhibition also contributes to the ubiquitin-independent pathway remains unknown.
Low-risk E6, which does not degrade p53, downregulates p53 function through HMTs
Although low-risk HPVs have no detectable cell transformation or immortalization activity, they are major causative agents of anogenital warts and recurrent respiratory papillomatosis (Lacey et al., 2006; Hu and Goldie, 2008; Pim and Banks, 2010). For example, almost 100% of anogenital warts are caused by HPV type-6 or type-11 (Pim and Banks, 2010). The mechanism by which low-risk HPVs cause abnormal cell growth is largely unknown. Consistent with the previous report in which low-risk E6 is shown to affect p53 function through inhibition of p300 (Thomas and Chiang, 2005) or destabilization of TIP60 (Jha et al., 2010), in the current study we further observe that, similar to the high-risk HPV E6, the low-risk E6 from HPV type-11 suppressed p53 transactivation function likely by targeting HMTs (Figures 2, 3c and 5, and Supplementary Figures S2 and S5). This strongly suggests that both low-risk and high-risk E6 proteins may induce abnormal cell growth by interfering with the function of p53 and HMTs.
Global impact of E6-mediated HMT inhibition
Given that (i) E6 interacts with CARM1, PRMT1 and SET7 regardless of the presence of p53 (Supplementary Figure S12); (ii) E6 directly inhibits the enzymatic activities of these HMTs (Figure 2); and (iii) these HMTs are involved in a variety of cellular functions by methylating a growing list of substrates, the p53-independent impact of E6-mediated HMT inhibition is expected. CARM1 and PRMT1 belong to the protein arginine methyltransferase family, which at least contains 10 members with homology within the active site (Lee and Stallcup, 2009). They participate in various cellular processes involved in signal transduction, cell proliferation, transcriptional regulation, chromatin remodeling, DNA repair, RNA processing, protein stability and nucleo-cytoplasmic trafficking (Pahlich et al., 2006; Pal and Sif, 2007; Lee and Stallcup, 2009; Wolf, 2009). Importantly, preventing PRMT1-mediated methylation disturbs the DNA-repair function of MRE11 and 53BP1 (Boisvert et al., 2005a, 2005b). PRMT1 is also critical in maintaining genome integrity (Yu et al., 2009). The lysine-specific HMT SET7 methylates a list of non-histone proteins besides p53. These include TAF10, p65 of nuclear factor-κB, DNA methyltransferase-1 (DNMT1) and estrogen receptor (ER) (Kouskouti et al., 2004; Subramanian et al., 2008; Ea and Baltimore, 2009; Esteve et al., 2009; Yang et al., 2009). SET7-mediated DNMT1 methylation decreases DNMT1 protein stability (Esteve et al., 2009). Note that defects in DNA repair and genome integrity are signs for cancer, and that DNMT1 is generally induced and stabilized in cancer cells to methylate the tumor-suppressor gene promoter for subsequent transcriptional suppression of the gene (Miremadi et al., 2007). Thus, loss of CARM1, PRMT1 or SET7 function by E6 might contribute to the oncogenic activity of E6.
Viral regulation of histone methylation
Several viral mechanisms to affect gene transcription involve modulation of histone methylation. These include recruitment of G9a, Suv39h1 to the viral major immediate-early promoter by the immediate-early-2 protein of the human cytomegalovirus (Reeves et al., 2006); removal of the HMT-associated cellular transcriptional regulator from DNA by adenovirus E1A (Ghosh and Harter, 2003); recruitment of the histone demethylase LSD1 by host cell factor-1 to immediate-early promoters of herpes simplex virus and varicella zoster virus (Liang et al., 2009); and direct expression of viral protein with HMT activity such as the vSET protein of paramecium bursaria chlorella virus-1 (Mujtaba et al., 2008). Together with our current study, the importance of HMT regulation in viral survival is highlighted.
Materials and methods
Cell line, DNA transfection and luciferase assay
The human cervix adenocarcinoma cell line, HeLa, the human osteosarcoma cell line, U2OS, and the human lung carcinoma cell line, H1299, were obtained from, and maintained as instructed by, the ATCC (Manassas, VA, USA). DNA transfections were performed by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. For ectopic protein expression, cells were harvested at 36∼72 h after transfection. For knockdown of endogenous proteins, cells were collected 72 h after transfection with 10 nM specific siRNA (Ambion, Austin, TX, USA). The sense sequence of si-SET7 is 5′-GGACCGCACUUUAUGGGAAtt-3′; of si-CARM1 is 5′-AGACAGAGCUACGACAUCAtt-3′; of si-PRMT1 is 5′-GCAACUCCAUGUUUCAUAAtt-3′; of si-18E6 is 5′-GAAAAACUUAGACACCUUAtt-3′; and of si-p53 is 5′-GUAAUCUACUGGGACGGAAtt-3′. Luciferase assays were performed as described previously (Juan et al., 2000). Briefly, transfections were performed at 80% confluence for H1299 and U2OS cells. Cells grown in six-well dishes were harvested 48 h after transfection. A 2- to 10-μl volume of each lysate was mixed with 50 μl of buffer (dual-luciferase reporter assay system; Promega, Madison, WI, USA) and measured for luciferase activities.
Plasmids
The following plasmids have been described previously: pcDNA3-F16E6 (Patel et al., 1999; Thomas and Chiang, 2005) and p513-16E66C/6S F47R (Nomine et al., 2006), which encode 16E66C/6S F47R and Flag-18E6, respectively; pcDNA 3.1-p53, which encodes wt p53 for mammalian expression (Ou et al., 2005); pRL-SV40 (Dual-Luciferase Reporter Assay System; Promega); p21-Luc (el-Deiry et al., 1993); pSG5HA-CARM1, pSG5HA-PRMT1, pSG5HA-CARM1 E267Q and pSG5HA-PRMT1 E153Q, encoding wt HA-CARM1, HA-PRMT1 and their methylase-dead mutants, respectively (Chen et al., 1999; Strahl et al., 2001; Lee et al., 2002, 2007); and pcDNA3.1-SET7 and SET7 H297A, encoding, respectively, wt SET7 and the methylase-dead mutant (Chuikov et al., 2004). Plasmids pRK5-11E6 and pRK5-18E6, which express Flag-HPV-11E6 and Flag-HPV-18E6, respectively, were constructed by inserting the corresponding cDNA fragments into the EcoRI and SalI sites of pRK5-Flag. Plasmids pET-29a11E6, pET-29a16E6 and pET29a18E6, which express His-tagged 11E6, 16E6 and 18E6, respectively, in Escherichia coli were constructed by inserting the corresponding cDNA fragments into the EcoRI and SalI sites of the pET-29a(+) vector (Novagen, Darmstadt, Germany). The plasmid encoding the p53 point mutant p53K372R was generated by using pcDNA 3.1-p53 as the template for in vitro site-directed mutagenesis (Stratagene, Santa Clara, CA, USA) according to the manufacturer's instructions.
In vitro methyltransferase assay
A 10-μg weight of core histones (Millipore, Basel, Switzerland or Roche, Basel, Switzerland) or a 5-μg weight of p53 made from baculovirus (Wang et al., 2003) was mixed with recombinant E6 protein purified from bacteria and recombinant CARM1, PRMT1 or SET7 (Upstate, Billerica, MA, USA, 14-575, 14-474 or 14-469, respectively), followed by methylation reactions at 30 °C for 30 min using 1 μl of S-adenosyl-L-[methyl-3H]-methionine ([3H]-SAM (1 mCi/ml; Amersham, Piscataway, NJ, USA) in HMT buffer (50 mM Tris–HCl (pH 8.0) and 0.5 mM dithiothreitol). The reaction mixtures were then separated by 15% sodium dodecyl sulfate (SDS)–PAGE and the gels were stained with Coomassie blue, dried and subjected to autoradiography. The non-isotope methyltransferase assays were performed by using unlabeled SAM to replace 3H-SAM. After in vitro methylation reaction, proteins were separated by 15% SDS–PAGE, followed by western blot analysis.
Western blotting, IP and DAPA
Cells were lysed on ice in ice-cold IP buffer (137 mM NaCl, 2.7 mM KCl, 7.7 mM NaH2PO4, 1.5 mM KH2PO4, 0.5% NP-40, 1 mM EDTA, 10% glycerol, 1 mM dithiothreitol) for IP or in DAPA buffer (137 mM NaCl, 2.7 mM KCl, 7.7 mM NaH2PO4, 1.5 mM KH2PO4, 0.1% NP-40, 1 mM EDTA, 10% glycerol, 1 mM dithiothreitol) for DAPA. Both buffers contain a mixture of protease inhibitors (Complete, EDTA-free, Protease Inhibitor Cocktail Tablets; Roche Molecular Biochemicals, Basel, Switzerland). Western blot and IP assays were performed as described by Hsu et al. (2004). Briefly, 4 mg of cell extracts from HeLa cells or 1 mg from U2OS cells were pre-cleaned with protein-A or protein-G beads for 30 min, and then incubated with the specific Ab at 4 °C overnight. Protein-A or protein-G-conjugated beads were added for an additional 2 h incubation. The beads were then collected by centrifugation and washed six times with IP buffer. For DAPA, the biotinylated DNA fragments were incubated with streptavidin beads (S-1638; Sigma, St Louis, MO, USA) at 4 °C overnight and washed three times with DAPA buffer. Subsequently, the DNA-conjugated beads were incubated with 2 mg of cell lysates and 10 μg of Poly dI–dC (Sigma; P4929) at 4 °C overnight. The beads were then collected by centrifugation and washed six times with DAPA buffer containing 0.5% NP-40. The pulled down complexes were then resolved by 15% SDS–PAGE and analyzed by western blotting.
Chromatin IP
ChIP assays were performed as described by Hsu et al. (2004), with modifications. The immunoprecipitated DNA fragments were extracted by using a PCR purification kit (Qiagen, Hilden, Germany; no. 28106) and analyzed by real-time PCR (Light Cycler 480; Roche). The amplifications were performed in a reaction volume of 20 μl containing 2 μl of immunoprecipitated material. The sequences of the p21 promoter-specific primers are 5′-GGCTGTGGCTCTGATTG-3′ (forward) and 5′-GAGGTCTCCTGTCTCCTAC-3′ (reverse). The sequences of the GADD45 promoter-specific primers are 5′-TAGAGTGTGGCTGGACTTT-3′ (forward) and 5′-TCACAGGGATCTCTTCCG-3′ (reverse). The sequences of primers recognizing the p53-unrelated region, approximately 2 kb downstream from the p53-response element on the GADD45 promoter (An et al., 2004), are 5′-GGAGTTGGAGTTGTCAGGAAAAAGGG-3′ (forward) and 5′-GGTTGTGGTCTTTCAGGCCTCCACACC-3′ (reverse).
Antibodies
The commercial Abs used for western blotting were against the following: β-tubulin (MAB1637; Millipore), actin (MAB1501; Millipore), Flag (M2; Sigma), HA (MMS-101R; Covance, Princeton, NJ, USA), CARM1 (A300-421A; Bethyl, Montgomery, TX, USA), PRMT1 (sc-13392; Santa Cruz, Santa Cruz, CA, USA), SET7 (04-805; Millipore), histone H3 (ab1791; Abcam), Asy-H3R17me2 (07-214; Millipore), histone H4 (ab10158), Asy-H4R3me2 (07-213; Millipore), p53 (DO-1) (sc-126; Santa Cruz), p53K372me1 (ab16033; Abcam, Cambridge, UK) and p21 (C-19; Santa Cruz). The commercial Abs for IP and ChIP were against the following: CARM1 (A300-421A; Bethyl, Figure 1a; and sc-5421; Santa Cruz, Figure 4b), PRMT1 (sc-13392; Santa Cruz), SET7 (04-805; Millipore), rabbit normal IgG (PP64B; Millipore), Ace-H3 (06-599; Millipore), Asy-H3R17me2 (07-214; Millipore), Ace-H4 (06-866; Millipore), Asy-H4R3me2 (07-213; Millipore), HPV E6 (sc-460 and sc-57835; Santa Cruz) and p53 (FL-393) (sc-6243; Santa Cruz). The p53K372me1 Ab used in Figure 7d for IP was generated by immunizing rabbits with the peptide NH2-CSRAHSSHLKSK(me1)KG-COOH. The Abs against Asy-H3R26me2 and Sym-H3R26me2 for IP were generated by immunizing rabbits with KLH-conjugated NH2-KVAR(me2)KSAPC-COOH. The synthetic peptides corresponding to the symmetric and asymmetric H3R26me2 were obtained from Open Biosystems (Huntsville, AL, USA).
In vitro transcription/translation-coupled degradation assay
The assay was performed as described (Nomine et al., 2006) with modifications. In vitro transcribed and translated proteins were expressed form pcDNA3.1-SET7, pcDNA3-F16E6, pcDNA3.1-p53, and pcDNA3.1-p53K372R by TNT system (Promega). p53 was then directly incubated with E6 in degradation buffer (25 mM Tris–HCl, pH 7.5, 100 mM NaCl, 2 mM dithiothreitol) at 28°C for 2 h, or first with SET7 and SAM at 30°C for 30 min and then E6. Reactions were terminated by adding SDS sample buffer and samples analyzed by western.
Reverse transcription–PCR
RNAs were extracted by using an RNA extraction kit (Qiagen; no. 74134), reversed-transcribed and analyzed by real-time RT–PCR (Light Cycler 480; Roche) using the QuantiTect SYBR Green reagent (Qiagen; no. 204243). The expression levels of the examined genes were normalized to actin expression. The sequences of primers amplifying p21, GADD45, 18E6 and the β-actin gene are as follows: p21: 5′-CTGGAGACTCTCAGGGTCGAAA-3′ (forward) and 5′-GATTAGGGCTTCCTCTTGGAGAA-3′ (reverse); GADD45: 5′-CCCGGACCTGCACTGCG-3′ (forward) and 5′-TCAGATGCCATCACCGT-3′ (reverse); 18E6: 5′-ATTAATAAGGTGCCTGCGG-3′ (forward) and 5′-CTCTATAGTGCCCAGCTATGTT-3′(reverse); and β-actin: 5′-CCCTGGACTTCGAGCAAGAGAT-3′ (forward) and 5′-AAGGTAGTTTCGTGGATGCCACA-3′ (reverse).
References
An W, Kim J, Roeder RG . (2004). Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell 117: 735–748.
Boisvert FM, Dery U, Masson JY, Richard S . (2005a). Arginine methylation of MRE11 by PRMT1 is required for DNA damage checkpoint control. Gene Dev 19: 671–676.
Boisvert FM, Rhie A, Richard S, Doherty AJ . (2005b). The GAR motif of 53BP1 is arginine methylated by PRMT1 and is necessary for 53BP1 DNA binding activity. Cell Cycle 4: 1834–1841.
Campos EI, Reinberg D . (2009). Histones: annotating chromatin. Annu Rev Genet 43: 559–599.
Camus S, Menendez S, Cheok CF, Stevenson LF, Lain S, Lane DP . (2007). Ubiquitin-independent degradation of p53 mediated by high-risk human papillomavirus protein E6. Oncogene 26: 4059–4070.
Chen DG, Ma H, Hong H, Koh SS, Huang SM, Schurter BT et al. (1999). Regulation of transcription by a protein methyltransferase. Science 284: 2174–2177.
Chen YC, Chen JH, Richard K, Chen PY, Christiani DC . (2004). Lung adenocarcinoma and human papillomavirus infection. Cancer 101: 1428–1436.
Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS et al. (2004). Regulation of p53 activity through lysine methylation. Nature 432: 353–360.
Crook T, Tidy JA, Vousden KH . (1991). Degradation of p53 can be targeted by HPV E6 sequences distinct from those required for p53 binding and transactivation. Cell 67: 547–556.
Ea CK, Baltimore D . (2009). Regulation of NF-kappa B activity through lysine monomethylation of p65. Proc Natl Acad Sci USA 106: 18972–18977.
el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM et al. (1993). WAF1, a potential mediator of p53 tumor suppression. Cell 75: 817–825.
Esteve PO, Chin HG, Benner J, Feehery GR, Samaranayake M, Horwitz GA et al. (2009). Regulation of DNMT1 stability through SET7-mediated lysine methylation in mammalian cells. Proc Natl Acad Sci USA 106: 5076–5081.
Ganguly N, Parihar SP . (2009). Human papillomavirus E6 and E7 oncoproteins as risk factors for tumorigenesis. J Biosci 34: 113–123.
Ghosh MK, Harter ML . (2003). A viral mechanism for remodeling chromatin structure in G0 cells. Mol Cell 12: 255–260.
Howie HL, Katzenellenbogen RA, Galloway DA . (2009). Papillomavirus E6 proteins. Virology 384: 324–334.
Howley PM . (2006). Warts, cancer and ubiquitylation: lessons from the papillomaviruses. Trans Am Clin Climatol Assoc 117: 113–126; discussion 126–127.
Howley PM, Livingston DM . (2009). Small DNA tumor viruses: large contributors to biomedical sciences. Virology 384: 256–259.
Hsu CH, Chang MD, Tai KY, Yang YT, Wang PS, Chen CJ et al. (2004). HCMV IE2-mediated inhibition of HAT activity downregulates p53 function. EMBO J 23: 2269–2280.
Hu D, Goldie S . (2008). The economic burden of noncervical human papillomavirus disease in the United States. Am J Obstet Gynecol 198: 500.e1–500.e7.
Huang J, Perez-Burgos L, Placek BJ, Sengupta R, Richter M, Dorsey JA et al. (2006). Repression of p53 activity by Smyd2-mediated methylation. Nature 444: 629–632.
Ivanov GS, Ivanova T, Kurash J, Ivanov A, Chuikov S, Gizatullin F et al. (2007). Methylation–acetylation interplay activates p53 in response to DNA damage. Mol Cell Biol 27: 6756–6769.
Jha S, Pol SV, Banerjee NS, Dutta AB, Chow LT, Dutta A . (2010). Destabilization of TIP60 by human papillomavirus E6 results in attenuation of TIP60-dependent transcriptional regulation and apoptotic pathway. Mol Cell 38: 700–711.
Juan LJ, Shia WJ, Chen MH, Yang WM, Seto E, Lin YS et al. (2000). Histone deacetylases specifically downregulate p53-dependent gene activation. J Biol Chem 275: 20436–20443.
Kouskouti A, Scheer E, Staub A, Tora L, Talianidis I . (2004). Gene-specific modulation of TAF10 function by SET9-mediated methylation. Mol Cell 14: 175–182.
Kouzarides T . (2007). Chromatin modifications and their function. Cell 128: 693–705.
Kruse JP, Gu W . (2008). SnapShot: p53 posttranslational modifications. Cell 133: 930.e1.
Kurash JK, Lei H, Shen Q, Marston WL, Granda BW, Fan H et al. (2008). Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Mol Cell 29: 392–400.
Lacey CJN, Lowndes CM, Shah KV . (2006). Burden and management of non-cancerous HPV-related conditions: HPV-6/11 disease. Vaccine 24: 35–41.
Lee DY, Ianculescu I, Purcell D, Zhang X, Cheng XD, Stallcup MR . (2007). Surface-scanning mutational analysis of protein arginine methyltransferase 1: roles of specific amino acids in methyltransferase substrate specificity, oligomerization, and coactivator function. Mol Endocrinol 21: 1381–1393.
Lee YH, Koh SS, Zhang X, Cheng XD, Stallcup MR . (2002). Synergy among nuclear receptor coactivators: selective requirement for protein methyltransferase and acetyltransferase activities. Mol Cell Biol 22: 3621–3632.
Lee YH, Stallcup MR . (2009). Minireview: protein arginine methylation of nonhistone proteins in transcriptional regulation. Mol Endocrinol 23: 425–433.
Li M, Luo J, Brooks CL, Gu W . (2002). Acetylation of p53 inhibits its ubiquitination by Mdm2. J Biol Chem 277: 50607–50611.
Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM . (2009). Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat Med 15: 1312–1317.
Massimi P, Shai A, Lambert P, Banks L . (2008). HPV E6 degradation of p53 and PDZ containing substrates in an E6AP null background. Oncogene 27: 1800–1804.
McLaughlin-Drubin ME, Munger K . (2009). Oncogenic activities of human papillomaviruses. Virus Res 143: 195–208.
Miremadi A, Oestergaard MZ, Pharoah PDP, Caldas C . (2007). Cancer genetics of epigenetic genes. Hum Mol Genet 16: R28–R49.
Mujtaba S, Manzur KL, Gurnon JR, Kang M, Van Etten JL, Zhou MM . (2008). Epigenetic transcriptional repression of cellular genes by a viral SET protein. Nat Cell Biol 10: 1114–1122.
Munger K, Howley PM . (2002). Human papillomavirus immortalization and transformation functions. Virus Res 89: 213–228.
Nishioka K, Chuikov S, Sarma K, Erdjument-Bromage H, Allis CD, Tempst P et al. (2002). Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Gene Dev 16: 479–489.
Nomine Y, Masson M, Charbonnier S, Zanier K, Ristriani T, Deryckere F et al. (2006). Structural and functional analysis of E6 oncoprotein: insights in the molecular pathways of human papillomavirus-mediated pathogenesis. Mol Cell 21: 665–678.
Ou YH, Chung PH, Sun TP, Shieh SY . (2005). p53 C-terminal phosphorylation by CHK1 and CHK2 participates in the regulation of DNA-damage-induced C-terminal acetylation. Mol Biol Cell 16: 1684–1695.
Pahlich S, Zakaryan RP, Gehring H . (2006). Protein arginine methylation: cellular functions and methods of analysis. Biochim Biophys Acta 1764: 1890–1903.
Pal S, Sif S . (2007). Interplay between chromatin remodelers and protein arginine methyltransferases. J Cell Physiol 213: 306–315.
Patel D, Huang SM, Baglia LA, McCance DJ . (1999). The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J 18: 5061–5072.
Pim D, Banks L . (2010). Interaction of viral oncoproteins with cellular target molecules: infection with high-risk vs low-risk human papillomaviruses. APMIS 118: 471–493.
Pradhan S, Chin HG, Esteve PO, Jacobsen SE . (2009). SET7/9 mediated methylation of non-histone proteins in mammalian cells. Epigenetics 4: 282–285.
Reeves M, Murphy J, Greaves R, Fairley J, Brehm A, Sinclair J . (2006). Autorepression of the human cytomegalovirus major immediate-early promoter/enhancer at late times of infection is mediated by the recruitment of chromatin remodeling enzymes by IE86. J Virol 80: 9998–10009.
Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM . (1990). The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63: 1129–1136.
Scheffner M, Huibregtse JM, Vierstra RD, Howley PM . (1993). The HPV-16 E6 and E6-AP complex functions as a ubiquitin–protein ligase in the ubiquitination of p53. Cell 75: 495–505.
Shukla A, Chaurasia P, Bhaumik SR . (2009). Histone methylation and ubiquitination with their cross-talk and roles in gene expression and stability. Cell Mol Life Sci 66: 1419–1433.
Strahl BD, Allis CD . (2000). The language of covalent histone modifications. Nature 403: 41–45.
Strahl BD, Briggs SD, Brame CJ, Caldwell JA, Koh SS, Ma H et al. (2001). Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Curr Biol 11: 996–1000.
Subramanian K, Jia D, Kapoor-Vazirani P, Powell DR, Collins RE, Sharma D et al. (2008). Regulation of estrogen receptor alpha by the SET7 lysine methyltransferase. Mol Cell 30: 336–347.
Tewey KM, Rowe TC, Yang L, Halligan BD, Liu LF . (1984). Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase-II. Science 226: 466–468.
Thomas MC, Chiang CM . (2005). E6 oncoprotein represses p53-dependent gene activation via inhibition of protein acetylation independently of inducing p53 degradation. Mol Cell 17: 251–264.
Trojer P, Reinberg D . (2007). Facultative heterochromatin: is there a distinctive molecular signature? Mol Cell 28: 1–13.
Wang H, Cao R, Xia L, Erdjument-Bromage H, Borchers C, Tempst P et al. (2001). Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol Cell 8: 1207–1217.
Wang YH, Tsay YG, Tan BC, Lo WY, Lee SC . (2003). Identification and characterization of a novel p300-mediated p53 acetylation site, lysine 305. J Biol Chem 278: 25568–25576.
Wolf SS . (2009). The protein arginine methyltransferase family: an update about function, new perspectives and the physiological role in humans. Cell Mol Life Sci 66: 2109–2121.
Yang XD, Huang B, Li MX, Lamb A, Kelleher NL, Chen LF . (2009). Negative regulation of NF-kappa B action by Set9-mediated lysine methylation of the RelA subunit. EMBO J 28: 1055–1066.
Yu ZB, Chen TP, Hebert J, Li E, Richard S . (2009). A mouse PRMT1 null allele defines an essential role for arginine methylation in genome maintenance and cell proliferation. Mol Cell Biol 29: 2982–2996.
Zimmermann H, Degenkolbe R, Bernard HU, O'Connor MJ . (1999). The human papillomavirus type 16 E6 oncoprotein can downregulate p53 activity by targeting the transcriptional coactivator CBP/p300. J Virol 73: 6209–6219.
Acknowledgements
We thank Drs WH Lee and Y Zhang for valuable suggestions; WH Lee for critical reading of the manuscript; MR Stallcup for providing pSG5HA, pSG5HA-CARM1, pSG5HA-CARM1 E267Q, pSG5HA-PRMT1 and pSG5HA-PRMT1 E153Q; D Reinberg for pcDNA3.1-SET7 and SET7H297A; G Trave for p513-16E66C/6S F47R; SY Shieh for p21-Luc and pcDNA3.1-p53; and CW Wu for partial funding support. We also thank the GRC Peptide Synthesis Core for providing the p53 peptide. This research was primarily supported by grants from Academia Sinica and NRPGM/DOH to L-JJ, and partly from NHRI to L-JJ, as well as Grants CA103867 and CA124760 from the National Institutes of Health to C-MC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Hsu, CH., Peng, KL., Jhang, HC. et al. The HPV E6 oncoprotein targets histone methyltransferases for modulating specific gene transcription. Oncogene 31, 2335–2349 (2012). https://doi.org/10.1038/onc.2011.415
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2011.415
Keywords
This article is cited by
-
Epigallocatechin gallate inhibits HeLa cells by modulation of epigenetics and signaling pathways
3 Biotech (2020)
-
RETRACTED ARTICLE: Molecular mechanisms in progression of HPV-associated cervical carcinogenesis
Journal of Biomedical Science (2019)
-
Characterization of a new mouse p53 variant: loss-of-function and gain-of-function
Journal of Biomedical Science (2014)
-
Expression of a recombinant Human papillomavirus 16 E6GT oncoprotein fused to N- and C-termini of Potato virus X coat protein in Nicotiana benthamiana
Plant Cell, Tissue and Organ Culture (PCTOC) (2013)
