
Abstract
The HIV-1 protein Nef inhibits antigen presentation by class I major histocompatibility complex (MHC-I). We determined the mechanism of this activity by solving the crystal structure of a protein complex comprising Nef, the MHC-I cytoplasmic domain (MHC-I CD) and the μ1 subunit of the clathrin adaptor protein complex 1. A ternary, cooperative interaction clamps the MHC-I CD into a narrow binding groove at the Nef-μ1 interface, which encompasses the cargo-recognition site of μ1 and the proline-rich strand of Nef. The Nef C terminus induces a previously unobserved conformational change in μ1, whereas the N terminus binds the Nef core to position it optimally for complex formation. Positively charged patches on μ1 recognize acidic clusters in Nef and MHC-I. The structure shows how Nef functions as a clathrin-associated sorting protein to alter the specificity of host membrane trafficking and enable viral evasion of adaptive immunity.
Similar content being viewed by others

Main
Evasion of immune surveillance is a strategy commonly used by viruses to persist in the host. The human immunodeficiency virus type 1 (HIV-1) protein Nef enables evasion of cellular adaptive immunity by antagonizing the antiviral activity of cytotoxic T lymphocytes1. These cells recognize viral peptides displayed on the cell surface by MHC-I and limit viral replication by killing the infected cells. Nef antagonizes cytotoxic T cells by decreasing surface expression of MHC-I, thereby reducing viral antigen presentation on infected cells1,2. Nef has a critical role in the pathogenesis of HIV-1 infection3: defective nef genes are associated with substantially delayed progression to AIDS4,5.
Nef decreases surface expression of MHC-I by associating the MHC-I CD with the clathrin adaptor protein complex 1 (AP1) at the trans-Golgi network6,7,8,9,10. Adaptor protein complexes are a family of heterotetramers that recognize cytoplasmic sorting motifs in transmembrane proteins and package them into clathrin-coated vesicles for intracellular transport11,12. AP1 transports transmembrane proteins between endosomes and the trans-Golgi network. Nef co-opts this pathway to retain MHC-I in the trans-Golgi network before diverting it to lysosomes for degradation7,13. Sequences recognized by adaptor protein complexes include the tyrosine-based Yxxφ motif (φ, bulky hydrophobic residue; x, any amino acid) and the leucine-based [ED]xxxL[LI] motif (bracketed residues represent alternative choices at the position). Yxxφ sequences bind the medium-sized (μ) subunit of adaptor protein complexes at a well-defined binding pocket14. A tyrosine in MHC-I CD and the tyrosine-binding pocket in the μ1 subunit are critical for modulation of MHC-I by Nef6,7,8,9,10. However, the context of this tyrosine, 320-YSQA-323, lacks a hydrophobic residue at the Y+3 position, rendering the sequence intrinsically inactive as an adaptor protein–binding signal. Nef has the crucial role of enabling AP1 to recognize and bind the MHC-I CD.
Nef is a 23- to 35-kDa myristoylated peripheral membrane protein (~206 residues in most HIV-1 strains) expressed by primate lentiviruses. Structural studies on Nef and its interaction complex with Src-family kinases15 show that Nef comprises a folded core domain and two long, flexible loops, one at the N terminus (residues 24–68) and one near the C terminus (residues 148–178)15,16,17,18. The regions of Nef important for MHC-I downregulation include an N-terminal α-helix, an acidic cluster (62-EEEE-65), a (PxxP)3 repeat at the junction of the N-terminal loop and the folded core, and residue Asp123 (refs. 19,20). The acidic dileucine motif in the C-terminal loop is not required for downregulation of MHC-I, although it is crucial for binding of Nef to AP2 and downregulation of CD4, the cellular receptor for the virus7,8,9,10,19,21. Despite this progress, most Nef-binding events are incompletely characterized. In particular, little is known about the formation of the Nef–MHC-I–AP1 complex at the structural level.
Results
To clarify how Nef enables recognition of MHC-I by AP1, we determined the crystal structure of HIV-1 Nef in complex with the MHC-I CD (residues 314–341) and the C-terminal domain (residues 158–423) of the μ1 subunit. We assembled the complex using a construct of the MHC-I CD fused to the N terminus of Nef8,10. This fusion protein binds μ1 directly, and the effects of various mutations on binding in vitro largely recapitulate the behavior of MHC-I downregulation by Nef in vivo10. The potential artifact caused by the fusion design concerned us initially; however, the crystal structure shows that the linkage is in a flexible region of Nef and does not affect the structure (see below). We obtained three crystal forms diffracting to resolutions of 2.6 Å, 2.9 Å and 3.3 Å (Table 1) and observed six independent Nef–MHC-I CD–μ1 complexes of the same structure in the asymmetric units of the crystals. The structure provides a detailed molecular description of how Nef hijacks the host membrane–trafficking machinery to decrease the expression of MHC-I at the cell surface.
Structure of Nef–MHC-I CD–μ1 complex
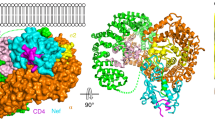
The Nef–MHC-I CD–μ1 complex adopts a clamp-like structure with the MHC-I cytoplasmic tail (residues 314–332) held tightly in a long, narrow groove at the Nef and μ1 interface (Fig. 1). The membrane-proximal end of MHC-I CD forms an extended strand that augments the μ1 β-sheets at the side of the cargo recognition site, with Tyr320 of the MHC-I CD in the tyrosine-binding pocket of μ1. The binding is secured by the proline-rich strand (PxxP repeats) of Nef, which runs along the MHC-I CD to form the other arm of the clamp. Nef is further affixed to μ1 by extensive contacts at the core domain and by crucial electrostatic interactions involving its acidic cluster (62-EEEE-65). The C terminus of Nef, together with a region of μ1 that is disordered in an earlier structure22, forms a snug pocket at one end of the binding groove to cradle a tight turn of the MHC-I CD (residues 327–332; Fig. 1c). The two membrane-anchoring points, the N termini of the MHC-I CD and Nef, are located on the same side of the complex, consistent with their membrane attachment roles in vivo.

(a) Ribbon representation. (b) Surface representation. Previously unobserved region of μ1 is yellow. Key interaction motifs are shown as sticks and labeled. Spheres, membrane-attachment sites. Dashed lines, disordered linker region and Nef N-terminal loop. (c) Experimentally phased (SAD) electron density map (1 σ level) of the MHC-I CD (sticks) in a close-up view rotated 90° from that in a,b.
Both Nef and μ1 show differences from their previously reported structures15,22. The C-terminal domain of μ1 (residues 158–423) remains as a banana-shaped structure of mostly β-strands22, with a slight twist (~10°) along its long axis upon formation of complex (Supplementary Fig. 1). An unstructured region of μ1 (residues 218–231) becomes ordered to interact with both Nef and the MHC-I CD. The core domain of Nef has the same structure as seen earlier, whereas the functionally important acidic cluster (62-EEEE-65) and the PxxP repeats (residues 68–78) swing to a different trajectory to interact with μ1 (Supplementary Fig. 1). The N-terminal helix of Nef is observed for the first time to be attached to the protein's core, on the side opposite the MHC-I-binding site (Fig. 1). Much of the Nef N- and C-terminal loops (residues 27–56 and 162–174, respectively) are disordered and not seen in the structure. In addition, the connecting region of the MHC-I CD–Nef fusion protein, including the last nine residues of MHC-I, a two-residue linker (TS) and the first four residues of Nef, is also disordered. This flexible region of 15 residues can span up to 55 Å in distance, whereas its two ends observed in the structure are only 28 Å apart, suggesting that the fusion linker probably does not cause distortion of the structure.
A cooperative, ternary interaction interface
The three-protein binding is cooperative in nature. A calculation of the buried interface areas predicts weak or no binding between any two components23. The largest binary interface, 1,058 Å2, occurs between Nef and μ1 and falls into the category of weak, transient binding24. The MHC-I–μ1 and MHC-I–Nef interfaces bury 771 Å2 and 437 Å2, respectively, predicting that no binary complexes would form with MHC-I. In contrast, all three components together generate a total of 2,266 Å2 buried area in a strong three-protein complex. This analysis also suggests a possible sequence of binding events. The association between Nef and AP1 may take place first, as their buried surface area predicts the only probable binary binding event. The binding groove would then be created at the Nef-μ1 interface to sequester the MHC-I CD. Consistent with this scenario, we found that the Nef-μ1 binary complex can form at high protein concentrations (~30 μM; Supplementary Fig. 2), although this interaction is not strong enough to be detected by in vitro pull-down assay10. This is corroborated by the weak, binary interaction between Nef and μ1 (refs. 6,25). Moreover, the binding of Nef to μ1 is enhanced by the presence of the MHC-I CD7,10; this further supports the cooperative nature of the three-protein interaction.
Nef compensates for incomplete trafficking motif in MHC-I
The tyrosine-containing sequence of MHC-I (YSQA) binds the canonical Yxxφ recognition site on μ1 (Fig. 2), with Tyr320 of MHC-I fitting snugly into the tyrosine-binding pocket6,8,9,10. The binding geometry closely resembles that of the Yxxφ peptide binding to μ2 of AP2 (Fig. 2a)14. However, the lack of a bulky hydrophobic side chain at the Y+3 position (Fig. 2b) in the MHC-I CD (320-YSQA-323) leads to an empty hydrophobic pocket on μ1 (Fig. 2c); this explains why MHC-I is not recognized by AP1 in the absence of Nef. Nef therefore functions to compensate for this intrinsically defective binding and forces a tight association of the MHC-I CD with AP1.

(a) Superposition of μ1 (green) and μ2 (gray) showing the MHC-I YSQA (magenta) at the same site as the canonical YNRL peptide (yellow). (b) Canonical bulky hydrophobic residue at Y+3 position binds in a hydrophobic pocket on μ1. (c) MHC-I Ala323 in Y+3 position leaves the pocket on μ1 empty. (d) Nef (cyan) PxxP motifs secure bound MHC-I CD by forming a side wall of the binding groove and stacking onto μ1 aromatic residues.
The structure shows that the Nef PxxP repeats lock the MHC-I CD onto μ1 by forming a side wall of the binding groove and by shaping a binding path between Nef and μ1 (Fig. 2d). The Nef proline-rich region has a crucial role in modulating MHC-I8,10,19,20,26. The last two PxxP repeats (72-PQVPLRP-78) form the major component of the side wall of the MHC-I CD–binding groove. This region of Nef probably has a confinement role to prevent the MHC-I CD from diffusing away, as only weak van der Waals interactions occur between these two proteins along the groove, with all interatomic distances >3.5 Å. The Nef chain makes a 90° turn immediately upstream of the binding groove. This sharp turn is pivoted at the first PxxP repeat (69-PVTP-72) and is stabilized by stacking interactions with μ1 (Fig. 2d). The turn changes the binding trajectory of Nef on μ1 and leads to an electrostatic interaction involving the Nef 62-EEEE-65 residues (described below).
Crucial electrostatic interactions
The Nef–MHC-I CD–μ1 interaction has strong electrostatic characteristics, highlighted by two large, complementarily charged surface areas at the binding interface (Fig. 3a). Two prominent positively charged patches on μ1 are matched by two negatively charged patches on Nef and the MHC-I CD. Concentrated in these regions are many residues important for Nef-mediated MHC-I downregulation, although the mechanisms have been unclear. The structure establishes a framework for understanding the functions of these residues and reveals their previously unidentified interaction partners.

(a) Complementary electrostatic surface areas (ovals) between μ1 (top) and MHC-I CD-Nef (bottom). Blue, positively charged; red, negatively charged. Nef, cyan; MHC-I, magenta; μ1, green. (b) Electrostatic interactions between Nef acidic cluster and positive residues of μ1. (c) Glutathione S-transferase (GST) pull-down assay detecting interaction in vitro between μ1, MHC-I CD-Nef and their mutants. (d) Three-way interaction network at the three-protein interface. (e) Nef C terminus induces a conformational change (yellow) in μ1 to form MHC-I CD-binding pocket. (f) Mutagenesis verifications in vivo (fluorescence-activated cell sorting, FACS) of key residues involved in interactions. FACS experiments were carried out using human T lymphocytes of cell line SupT1 transfected to express Nef and green fluorescent protein (GFP, as a transfection marker), followed by staining for surface MHC-I. Relative fluorescence intensity of MHC-I versus GFP. Nef, red contour lines; negative control (GFP only), blue contour lines. Right, western blots of Nef proteins during FACS experiments.
A key electrostatic interaction occurs at the Nef acidic cluster, 62-EEEE-65, which is attracted to a basic patch on μ1 (Fig. 3b). This acidic cluster is functionally important19,26,27. The interaction is long range, where Glu65 is the only residue within hydrogen-bonding distance of μ1 (Arg303). This long-range electrostatic interaction is supported by the earlier finding that mutation of glutamic acid residues to uncharged residues abolishes the Nef-mediated modulation of MHC-I, whereas mutation to aspartic acid residues has no effect28. A quadruple mutation of μ1 (K274E K298E K302E R303D) that reverses the positive charge at the interface abolished binding to the MHC I CD–Nef fusion protein in our in vitro pull-down assay (Fig. 3c). Furthermore, interaction with the mutant μ1 was completely restored by a complementary mutation that reverses the negative charge in the acidic cluster in Nef (EEEE→KKKK; Fig. 3c), validating the role of this positively charged patch on μ1.
The second major charged interface (Fig. 3a,d) involves a crucial three-way electrostatic network. This interaction involves Asp327 of MHC-I, Asp123 of Nef and an elongated basic patch on μ1 (Arg225, Arg393, Lys396, Arg211 and Arg246). These interactions shape one end of the binding groove and provide a second anchoring point at Asp327, in addition to Tyr320, for the MHC-I CD to latch into the groove. The D327A mutation in MHC-I or D123G in HIV-1 Nef abolishes downregulation of MHC-I9,26,29. However, in contrast with the proposed role of Asp123 in Nef dimerization29, the crystal structure shows that this residue is essential because of the three-way electrostatic interaction. Moreover, the R225A R393A double mutation in μ1 abolished binding with the MHC I–CD–Nef fusion protein in vitro (Fig. 3c), as predicted from the structure.
The key roles of the two anchoring residues of the MHC-I CD, Tyr320 and Asp327, are reflected by selective downregulation of MHC-I subtypes by Nef30,31, in which their presence is correlated with susceptibility to Nef (Supplementary Fig. 3). Tyr320 and Asp327 are present in human leukocyte antigen A (HLA-A) and HLA-B, but not in HLA-C, accounting for the selective downregulation of HLA-A and HLA-B, but not HLA-C, by Nef6. The presence of Asn327 in HLA-C indicates that the interactions involving Asp327 in HLA-A and HLA-B are electrostatic rather than hydrogen bonding.
A binding pocket induced by the Nef C terminus
The Nef C terminus has an indispensable role in establishing the three-way interaction network by stabilizing a structural change in μ1 to complete the MHC-I CD binding groove (Fig. 3e). Residues 215–233 of μ1 rearrange from a disordered loop in the crystal structure of the AP1 core into a helix-turn motif when in complex with the MHC-I CD and Nef. The Nef C terminus and the helix-turn motif of μ1 constitute one end of the MHC-I CD binding groove, which cradles the tight turn of MHC-I residues 327–332 (Figs. 1c and 3e). The stabilization of the helix-turn motif also places Arg225 of μ1 in position to participate in the important three-way electrostatic interaction described above.
We verified the essential function of the Nef C terminus in biochemical and functional assays (Fig. 3c,f). A double mutation (Y202A F203A) or a five-residue C-terminal truncation (residues 202–206) of Nef abolished formation of complex in vitro. These mutations also abolished downregulation of cell surface MHC-I detected by flow cytometry when Nef was expressed in human T lymphocytes, the primary host cells of HIV-1 in vivo. Either Y202A or F203A mutation also markedly disrupted complex formation in vitro.
Membrane-positioning role of Nef N terminus–core interaction
The structure also suggests that the Nef N-terminal helix9,20,26 (residues 6–23) has a previously unknown role in positioning the Nef core at an optimal distance from the lipid membrane for efficient interaction with the MHC-I CD and AP1 (Fig. 4a). This helix, as part of the flexible N terminus of Nef, has been predicted to be detached from the core32. It is nonetheless important, as deletion of part of it, or mutation of Met20 within it, abolishes Nef-mediated downregulation of MHC-I9,20,26. Our structure shows that the helix instead is anchored via two hydrophobic residues, Trp13 and Met20, to the surface of the Nef core opposite the MHC-I binding site. Rather than directly participating in intermolecular binding, the N-terminal helix–core association is predicted to bring the Nef core close to the membrane, facilitating interaction with the MHC-I CD and the AP1 complex. Consistent with an important helix-core anchoring interaction, either W13A or M20A mutation in Nef abolished downregulation of MHC-I in human T lymphocytes (Fig. 4b). Mutations of the two residues also inhibited complex formation in our in vitro pull-down assays (Fig. 4c). The identification of two adjacent mutations with the same phenotype supports the role of the anchoring interaction in Nef function.

(a) Nef N-terminal helix (blue) is anchored to the core (cyan) via two hydrophobic residues, Trp13 and Met20. (b,c) Mutagenesis verification in vivo (FACS) and in vitro (GST pull-down assay), respectively, of the importance of the two residues. FACS experiments were carried out using human T lymphocytes of cell line SupT1 transfected to express Nef and GFP (as a transfection marker), followed by staining for surface MHC-I. Relative fluorescence intensity of MHC-I versus GFP. Nef, red; negative control (GFP only), blue. Lower left in b, western blots of Nef proteins.
Functional distinction between AP1 (μ1) and AP2 (μ2) by Nef
Specific interaction between μ1 and Nef may also determine the selective utilization of AP1 rather than AP2 by Nef for MHC-I downregulation (Fig. 5 and Supplementary Fig. 4). The sequences of μ1 and μ2 differ substantially in regions that enable μ1 to interact with Nef. In the region corresponding to the Nef-induced μ1 helix-turn structure, a much longer loop exists in μ2, which probably does not adopt the same conformation (Supplementary Fig. 4). μ2 also lacks the important tyrosine residue (Tyr374 in μ1) that stacks onto Nef Pro72 to stabilize the sharp Nef turn (Figs. 2 and 5). Furthermore, μ2 does not have the essential basic patch that attracts the Nef acidic cluster to secure formation of complex. Indeed, the Nef-MHC-I CD construct did not interact with μ2 in our in vitro pull-down experiments (Fig. 5c). These differences highlight an important distinction between the two otherwise highly homologous μ subunits in AP1 and AP2, which may contribute to different interactions with distinct cellular sorting proteins and cargos. The lack of interaction between Nef and μ2 also could explain why Nef uses a distinct mode of interaction—involving its acidic dileucine motif and the α and σ2 subunits of AP2—to downregulate CD4 (refs. 33,34).

(a,b) μ2 lacks the positively charged surface patch that interacts with the Nef acidic cluster. It also lacks the tyrosine residue that stacks onto Nef Pro72 to stabilize the sharp Nef turn (oval). Model in b was generated by superposing the μ2 structure35 onto μ1 in the structure of the MHC-I CD–Nef–μ1 complex. (c) GST-tagged MHC-I CD-Nef does not pull down μ2 in vitro.
Discussion
A picture of the Nef-mediated modulation of MHC-I emerged when we overlaid our structure with the crystal structure of the 'open' form of the AP2 core by superposition of the μ1 and μ2 C-terminal domains (Fig. 6)35. There is a striking coplanar arrangement of the Nef N-terminal myristoylation site, the MHC-I CD membrane-proximal end and multiple phosphatidylinositol-4,5-bisphosphate (PIP2) binding sites on AP2 (ref. 35), consistent with formation of complex along the lipid bilayer in vivo. The coplanar arrangement of all the membrane attachment sites indicates that this model is likely to be accurate. Through myristoylation and anchoring of its N-terminal helix to the core, Nef is positioned at an optimal distance from the lipid membrane to bind the AP1 complex; this probably occurs after recruitment of AP1 to the membrane by the small GTPase ADP-ribosylation factor 1 (refs. 36,37). The Nef-μ1 interaction may also contribute to inducing the active, open conformation of AP1 through electrostatic attractions resembling those involving the PIP2-containing membranes35. This interaction probably further stabilizes the membrane association of AP1, as we have reported earlier38. The association of Nef and AP1 then creates a snug binding groove for recruitment of the MHC-I CD and ultimately leads to packaging of MHC-I into the clathrin-coated vesicle.

Circled crosses, PIP2-binding sites35; arrows, membrane-anchoring sites.
Nef uses a wide range of structural features throughout its length to hijack the AP-1 complex and modulate cell surface expression of MHC-I. Perhaps owing to its highly cooperative nature, the interaction is very delicate: mutation of any of its key elements disrupts the complex and causes loss of Nef activity. This explains the observation that virtually every domain in Nef required for modulation of MHC-I is also required for coimmunoprecipitation of the two proteins from human cells26. Moreover, our model explains why the dileucine motif in the C terminus of native Nef is not active during downregulation of MHC-I, yet is functional as a trafficking signal when Nef is fused to the CD of MHC-I39. Our structure shows that the cooperative Nef–MHC-I CD–μ1 ternary interaction is key to recruitment of MHC-I to AP1. This conformation, however, precludes the involvement of the dileucine motif of Nef, as the binding site for [ED]xxxL[LI] motifs on the γ and σ1 subunits of AP1 (ref. 40) is too distant (Fig. 6). In contrast, when MHC-I is covalently linked to Nef, the critical MHC-I recruitment step is bypassed, and any trafficking signal in Nef, including the dileucine motif, can contribute to trafficking of the MHC-I–Nef fusion protein. Nevertheless, the principle of cooperative binding probably also applies to the mechanism by which Nef modulates CD4 via clathrin-mediated endocytosis and AP2 (ref. 41), even though that interaction involves the dileucine motif of Nef and the α and σ2 subunits of AP2 rather than μ2.
Our structure supports the notion that Nef is a virally encoded clathrin-associated sorting protein (CLASP) that alters the breadth and specificity of cargo recognition by adaptor protein complexes12. The cooperative binding mechanism by which Nef enables an adaptor protein complex to recognize a suboptimal sorting sequence could be used by cellular CLASPs. For example, in mouse MHC-I, the tyrosine residue corresponding to Tyr320 is required for 'cross-presentation' of exogenous antigens by dendritic cells, an event required for priming of antiviral cytotoxic T lymphocytes42. This observation, together with the role of Tyr320 in Nef-mediated modulation of MHC-I via AP1, has led us and others to speculate that cross-presentation might involve AP-1 and a cellular CLASP that is reminiscent of HIV-1 Nef8,9. Moreover, the positively charged clusters on μ1 that contribute to the interaction between Nef and the MHC-I CD may function as binding sites for the acidic trafficking motifs of cellular proteins. For example, the cytoplasmic domain of the cation-independent mannose 6-phosphate receptor contains acidic clusters that include glutamic acid, aspartic acid and phosphoserine residues, and these sequences contribute to interaction with AP1 (ref. 43).
Finally, the narrow binding groove for MHC-I formed by Nef and μ1 is a potential target for a small molecule that would inhibit the Nef-mediated MHC-I downregulation pathway. Inhibition of this Nef activity would sensitize infected cells to killing by cytotoxic T lymphocytes and potentially facilitate control of infection by the host.
Methods
Cloning, Escherichia coli expression and purification.
The C-terminal domain of the mouse μ1 subunit (residues 158–423) of AP1 (a gift from J. Bonifacino, NIH) was cloned into multiple cloning site 1 of the pCDFDuet vector (Novagen). A severe acute respiratory syndrome-coronavirus (SARS-CoV) main protease (Mpro) cleavage site44,45 was introduced between the N-terminal His6 tag and μ1158–423. The MHC-I CD-Nef fusion was created as described earlier10. The fusion gene was cloned into multiple cloning site 2 of the pCDFDuet vector. The pCDFDuet plasmid encoding both μ1158–423 and CD-Nef genes was expressed in Rosetta cells in Terrific Broth. Cells were induced with 0.1 mM IPTG at A600 of 0.6 and grown at 16 °C overnight. Proteins were first purified by Ni-NTA resin using the gravity-flow batch method. A subsequent purification step through a HiTrap Q anion exchange column produced the CD-Nef–μ1 complex in good purity. The His6 tag on μ1158–423 was then cleaved off using the SARS-CoV Mpro protease44,45. A final gel filtration purification using a Sephadex 200 column yielded homogeneous monomeric CD-Nef–μ1 complex. Selenomethionine-derivatized (SeMet) proteins were expressed from the same vector using the methionine biosynthesis inhibition method46. Purification of the SeMet derivative of the protein complex was accomplished as described above for the native proteins.
For GST pull-down experiments, μ1158–423 was cloned into a pMAT9s expression vector47 containing a N-terminal His6 tag followed by maltose-binding protein (MBP) and a SARS-CoV Mpro cleavage site. Mouse μ2159–435 of AP2 was cloned into the pMAT9s expression vector in a similar fashion. The pMAT9s-μ1 plasmid was coexpressed with the pGro7 vector (Takara Bio), which encodes the groES and groEL chaperone proteins, in E. coli BL21(DE3) cells in Terrific Broth. L-(+)-Arabinose was added to 2 mg ml−1 to induce chaperone expression. Expression of MBP-μ1 was later induced by 0.1 mM IPTG at A600 of 0.6 and continued at 16 °C overnight. The protein was purified to homogeneity by using Ni-NTA gravity flow, HiTrap SP cation exchange column and Sephadex 200 gel filtration column. All MBP-μ1 mutants (and the GST–CD-Nef mutants below) were generated by site-directed mutagenesis using the QuikChange method. The mutants of MBP-μ1158–423 and wild-type MBP-μ2159–435 were expressed and purified similarly as for the wild-type MBP-μ1.
The expression vector for the GST-tagged CD-Nef (pGEX-4T-1-CD-Nef) was created earlier10. GST–CD-Nef was expressed in Rosetta cells using conditions similar to those described above for the CD-Nef–μ1 complex. GST–CD-Nef and its mutants were purified the same way. The protein was first bound to a GSTrap column, washed and eluted with buffer containing 10 mM glutathione. A final anion-exchange chromatography step (HiTrap Q column) yielded homogeneous protein.
Crystallization and data collection.
Crystallization of the protein complex was carried out using the microbatch under-oil method48. Equal volumes (1.5 μl each) of the protein solution (3.5 mg ml−1 in 25 mM Tris, pH 8, 100 mM NaCl, 0.1 mM Tris-(2-carboxyethyl)phosphine) and the precipitant solution (0.1 M HEPES, pH 6.5, 3% PEG8000) were mixed. The drop was sealed using a mixture of paraffin and silicon oil at a 2:1 ratio. Crystals formed within 24 h at room temperature and grew to full size in 5–6 d. SeMet crystals were obtained similarly except that streak seeding was carried out to obtain good-sized crystals.
Crystals were cryoprotected using the precipitant solution containing 20% glycerol and then frozen in liquid nitrogen. Diffraction data were collected at the NE-CAT beamline 24ID-E at the Advanced Photon Source, Argonne National Laboratory, and beamline X29 at the National Synchrotron Light Source, Brookhaven National Laboratory. Two forms of the native crystals were obtained in P21 and P212121 space groups and diffracted to resolution of 3.3 Å and 2.6 Å, respectively. The SeMet crystals are in P212121 space group and diffracted to a resolution of 2.9 Å. The statistics are summarized in Table 1.
Structure determination and refinement.
Initial phases for the SeMet derivative data were obtained using SAD. Thirteen selenium sites were found using the program Shelx49 and subsequent SAD phasing was done in SOLVE50. A two-fold noncrystallographic symmetry (NCS) was identified using the Se sites. The phases were improved by a twofold NCS averaging using RESOLVE50. Two copies of both μ1 (model PDB 1W63)22 and Nef (model PDB 1EFN)15 were located in the electron density map by the real-space molecular replacement method using the CCP4 program MOLREP51,52. Multidomain multicrystal averaging was carried out between the nonisomorphous native and the SeMet crystals using the program DMMulti53, which greatly improved the phases. Iterative rounds of model building in COOT54 and refinement with REFMAC5 (ref. 55) and PHENIX56 were carried out. Strong NCS restraint was applied initially and released gradually at the final stages of refinement. The final model has an Rwork / Rfree of 0.208 / 0.258. A Ramachandran plot showed that 96.1% of the residues are in the favored region and the remaining 3.9% are in the allowed region. The refinement statistics are summarized in Table 1.
In vitro GST pull-down assay.
The purified proteins GST–CD-Nef (0.16 mg) and MBP-μ1158–423 (0.25 mg) or their mutants were mixed in a final volume of 100 μl and incubated at 4 °C for 1.5 h. The protein solution was then loaded onto a small gravity-flow column containing 0.2 ml GST resin. Flow through was collected and the resin was extensively washed with 5 × 0.9 ml GST binding buffer (50 mM Tris, pH 8, 100 mM NaCl, 0.1 mM Tris-(2-carboxyethyl)phosphine). The bound proteins were eluted with 5 × 0.1 ml GST elution buffer containing 10 mM reduced glutathione. The eluted proteins were analyzed by SDS-PAGE stained with Coomassie blue.
MHC-I downregulation assays.
We used the human T cell lines SupT1, which expresses HLA-A2 endogenously; the stably transfected CEM derivative line CEM 4B, which expresses Nef from a replication-defective provirus; and 4BNS, which is similar to 4B except that the provirus is nef-negative57. Nef-mediated downregulation of MHC-I A2 (or related mutants) was measured by flow cytometric analysis of CD4+ T cells of the CEM line, which express MHC-A1, with or without constitutive expression of Nef; cells were transfected with a plasmid encoding the α-chain of MHC-I A2. For transient expression experiments, 4 × 106 CEM cells per sample were transfected using Amaxa program X-001 or 3 × 106 SupT1 cells per sample were transfected using program O-017. All cellular samples were washed in medium without antibiotics and then resuspended in 100 μl of Amaxa Kit V nucleofection reagent (Lonza). For the SupT1 cells, the plasmid DNA was 2 μg pCG-GFP (a gift from J. Skowronski, Case Western Reserve University, Cleveland, Ohio, USA) plus 20 μg pCI-Neo (Promega) encoding Nef or the indicated Nef-mutants. For the CEM 4B and 4BNS cells, the plasmid DNA was 2 μg pCG-GFP plus 20 μg pcDNA3 (Invitrogen) encoding HLA-A2 (a gift from O. Schwartz, Pasteur Institute, Paris) or the indicated HLA-A2 mutants. After incubation at 37 °C for 16 h, the cells were surface stained while alive for HLA-A2, and the cell lysates were analyzed by western blot using a goat antiserum to HIV-1 Nef (a gift from C. Spina, UCSD). For the flow cytometry, each sample was resuspended in 100 μl PBS and stained using 10 μg ml−1 MA2.1 (a murine monoclonal antibody to HLA-A2, a gift from D. Camerini, University of California Irvine, Irvine, California, USA) for 30 min on ice, followed by incubation with goat anti-mouse IgG conjugated to allophycocyanin, before fixation with 2% paraformaldehyde and two-color analysis using a Becton Dickinson FACSCanto flow cytometer.
Accession numbers.
Protein Data Bank: coordinates and structure factors have been deposited with accession codes 4EMZ (selenomethionine derivative) and 4EN2 (native).
References
Collins, K.L., Chen, B.K., Kalams, S.A., Walker, B.D. & Baltimore, D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391, 397–401 (1998).
Schwartz, O., Marechal, V., Le Gall, S., Lemonnier, F. & Heard, J.M. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 2, 338–342 (1996).
Kirchhoff, F., Schindler, M., Specht, A., Arhel, N. & Munch, J. Role of Nef in primate lentiviral immunopathogenesis. Cell Mol. Life Sci. 65, 2621–2636 (2008).
Deacon, N.J. et al. Genomic structure of an attenuated quasi species of HIV-1 from a blood transfusion donor and recipients. Science 270, 988–991 (1995).
Kirchhoff, F., Greenough, T.C., Brettler, D.B., Sullivan, J.L. & Desrosiers, R.C. Absence of intact Nef sequences in a long-term survivor with nonprogressive Hiv-1 infection. N. Engl. J. Med. 332, 228–232 (1995).
Le Gall, S. et al. Nef interacts with the mu subunit of clathrin adaptor complexes and reveals a cryptic sorting signal in MHC I molecules. Immunity 8, 483–495 (1998).
Roeth, J.F., Williams, M., Kasper, M.R., Filzen, T.M. & Collins, K.L. HIV-1 Nef disrupts MHC-1 trafficking by recruiting AP-1 to the MHC-1 cytoplasmic tail. J. Cell Biol. 167, 903–913 (2004).
Noviello, C.M., Benichou, S. & Guatelli, J.C. Cooperative binding of the class I major histocompatibility complex cytoplasmic domain and human immunodeficiency virus type 1 Nef to the endosomal AP-1 complex via its mu subunit. J. Virol. 82, 1249–1258 (2008).
Wonderlich, E.R., Williams, M. & Collins, K.L. The tyrosine binding pocket in the adaptor protein 1 (AP-1) μ1 subunit is necessary for nef to recruit AP-1 to the major histocompatibility complex class I cytoplasmic tail. J. Biol. Chem. 283, 3011–3022 (2008).
Singh, R.K., Lau, D., Noviello, C.M., Ghosh, P. & Guatelli, J.C. An MHC-I cytoplasmic domain/HIV-1 Nef fusion protein binds directly to the musubunit of the AP-1 endosomal coat complex. PLoS ONE 4, e8364 (2009).
Ohno, H. Clathrin-associated adaptor protein complexes. J. Cell Sci. 119, 3719–3721 (2006).
Traub, L.M. Tickets to ride: selecting cargo for clathrin-regulated internalization. Nat. Rev. Mol. Cell Biol. 10, 583–596 (2009).
Schaefer, M.R., Wonderlich, E.R., Roeth, J.F., Leonard, J.A. & Collins, K.L. HIV-1 Nef targets MHC-I and CD4 for degradation via a final common beta-COP-dependent pathway in T cells. PLoS Pathog. 4, e1000131 (2008).
Owen, D.J. & Evans, P.R. A structural explanation for the recognition of tyrosine-based endocytotic signals. Science 282, 1327–1332 (1998).
Lee, C.H., Saksela, K., Mirza, U.A., Chait, B.T. & Kuriyan, J. Crystal structure of the conserved core of HIV-1 Nef complexed with a Src family SH3 domain. Cell 85, 931–942 (1996).
Arold, S. et al. The crystal structure of HIV-1 Nef protein bound to the Fyn kinase SH3 domain suggests a role for this complex in altered T cell receptor signaling. Structure 5, 1361–1372 (1997).
Grzesiek, S. et al. Refined solution structure and backbone dynamics of HIV-1 Nef. Protein Sci. 6, 1248–1263 (1997).
Horenkamp, F.A. et al. Conformation of the dileucine-based sorting motif in HIV-1 Nef revealed by intermolecular domain assembly. Traffic 12, 867–877 (2011).
Greenberg, M.E., Iafrate, A.J. & Skowronski, J. The SH3 domain-binding surface and an acidic motif in HIV-1 Nef regulate trafficking of class I MHC complexes. EMBO J. 17, 2777–2789 (1998).
Mangasarian, A., Piguet, V., Wang, J.K., Chen, Y.L. & Trono, D. Nef-induced CD4 and major histocompatibility complex class I (MHC-I) down-regulation are governed by distinct determinants: N-terminal alpha helix and proline repeat of Nef selectively regulate MHC-I trafficking. J. Virol. 73, 1964–1973 (1999).
Guy, B. et al. HIV F/3′ orf encodes a phosphorylated GTP-binding protein resembling an oncogene product. Nature 330, 266–269 (1987).
Heldwein, E.E. et al. Crystal structure of the clathrin adaptor protein 1 core. Proc. Natl. Acad. Sci. USA 101, 14108–14113 (2004).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
Nooren, I.M. & Thornton, J.M. Structural characterisation and functional significance of transient protein-protein interactions. J. Mol. Biol. 325, 991–1018 (2003).
Craig, H.M., Reddy, T.R., Riggs, N.L., Dao, P.P. & Guatelli, J.C. Interactions of HIV-1 Nef with the mu subunits of adaptor protein complexes 1, 2, and 3: Role of the dileucine-based sorting motif. Virology 271, 9–17 (2000).
Williams, M., Roeth, J.F., Kasper, M.R., Filzen, T.M. & Collins, K.L. Human immunodeficiency virus type 1 Nef domains required for disruption of major histocompatibility complex class I trafficking are also necessary for coprecipitation of Nef with HLA-A2. J. Virol. 79, 632–636 (2005).
Riggs, N.L., Craig, H.M., Pandori, M.W. & Guatelli, J.C. The dileucine-based sorting motif in HIV-1 Nef is not required for down-regulation of class I MHC. Virology 258, 203–207 (1999).
Baugh, L.L., Garcia, J.V. & Foster, J.L. Functional characterization of the human immunodeficiency virus type 1 Nef acidic domain. J. Virol. 82, 9657–9667 (2008).
Liu, L.X. et al. Mutation of a conserved residue (D123) required for oligomerization of human immunodeficiency virus type 1 Nef protein abolishes interaction with human thioesterase and results in impairment of Nef biological functions. J. Virol. 74, 5310–5319 (2000).
Cohen, G.B. et al. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity 10, 661–671 (1999).
Specht, A. et al. Selective downmodulation of HLA-A and -B by Nef alleles from different groups of primate lentiviruses. Virology 373, 229–237 (2008).
Geyer, M., Fackler, O.T. & Peterlin, B.M. Structure-function relationships in HIV-1 Nef. EMBO Rep. 2, 580–585 (2001).
Craig, H.M., Pandori, M.W. & Guatelli, J.C. Interaction of HIV-1 Nef with the cellular dileucine-based sorting pathway is required for CD4 downregulation and optimal viral infectivity. Proc. Natl. Acad. Sci. USA 95, 11229–11234 (1998).
Chaudhuri, R., Lindwasser, O.W., Smith, W.J., Hurley, J.H. & Bonifacino, J.S. Downregulation of CD4 by human immunodeficiency virus type 1 Nef is dependent on clathrin and involves direct interaction of Nef with the AP2 clathrin adaptor. J. Virol. 81, 3877–3890 (2007).
Jackson, L.P. et al. A large-scale conformational change couples membrane recruitment to cargo binding in the AP2 clathrin adaptor complex. Cell 141, 1220–1229 (2010).
Stamnes, M.A. & Rothman, J.E. The binding of AP-1 clathrin adaptor particles to Golgi membranes requires ADP-ribosylation factor, a small GTP-binding protein. Cell 73, 999–1005 (1993).
Traub, L.M., Ostrom, J.A. & Kornfeld, S. Biochemical dissection of AP-1 recruitment onto Golgi membranes. J. Cell Biol. 123, 561–573 (1993).
Janvier, K. et al. HIV-1 Nef stabilizes the association of adaptor protein complexes with membranes. J. Biol. Chem. 278, 8725–8732 (2003).
Wonderlich, E.R., Williams, M. & Collins, K.L. The tyrosine binding pocket in the adaptor protein 1 (AP-1) mu1 subunit is necessary for Nef to recruit AP-1 to the major histocompatibility complex class I cytoplasmic tail. J. Biol. Chem. 283, 3011–3022 (2008).
Janvier, K. et al. Recognition of dileucine-based sorting signals from HIV-1 Nef and LIMP-II by the AP-1 gamma-sigma1 and AP-3 delta-sigma3 hemicomplexes. J. Cell Biol. 163, 1281–1290 (2003).
Chaudhuri, R., Mattera, R., Lindwasser, O.W., Robinson, M.S. & Bonifacino, J.S. A basic patch on α-adaptin is required for binding of human immunodeficiency virus type 1 Nef and cooperative assembly of a CD4-Nef-AP-2 complex. J. Virol. 83, 2518–2530 (2009).
Lizée, G. et al. Control of dendritic cell cross-presentation by the major histocompatibility complex class I cytoplasmic domain. Nat. Immunol. 4, 1065–1073 (2003).
Ghosh, P. & Kornfeld, S. The cytoplasmic tail of the cation-independent mannose 6-phosphate receptor contains four binding sites for AP-1. Arch. Biochem. Biophys. 426, 225–230 (2004).
Xue, X.Y. et al. Production of authentic SARS-CoV M-pro with enhanced activity: Application as a novel tag-cleavage endopeptidase for protein overproduction. J. Mol. Biol. 366, 965–975 (2007).
Xue, X.Y. et al. Structures of two coronavirus main proteases: Implications for substrate binding and antiviral drug design. J. Virol. 82, 2515–2527 (2008).
Van Duyne, G.D., Standaert, R.F., Karplus, P.A., Schreiber, S.L. & Clardy, J. Atomic structures of the human immunophilin Fkbp-12 complexes with Fk506 and rapamycin. J. Mol. Biol. 229, 105–124 (1993).
Peränen, J., Rikkonen, M., Hyvonen, M. & Kaariainen, L. T7 vectors with a modified T7lac promoter for expression of proteins in Escherichia coli. Anal. Biochem. 236, 371–373 (1996).
Chayen, N.E., Stewart, P.D.S., Maeder, D.L. & Blow, D.M. An automated system for microbatch protein crystallization and screening. J. Appl. Crystallogr. 23, 297–302 (1990).
Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 64, 112–122 (2008).
Terwilliger, T. SOLVE and RESOLVE: automated structure solution, density modification, and model building. J. Synchrotron Radiat. 11, 49–52 (2004).
Bailey, S. The Ccp4 suite—programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 (1994).
Vagin, A. & Teplyakov, A. An approach to multi-copy search in molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 56, 1622–1624 (2000).
Cowtan, K. & Main, P. Miscellaneous algorithms for density modification. Acta Crystallogr. D Biol. Crystallogr. 54, 487–493 (1998).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Murshudov, G.N., Vagin, A.A. & Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 (1997).
Adams, P.D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Little, S.J. et al. Cell surface CD4 downregulation and resistance to superinfection induced by a defective provirus of HIV-1. Virology 205, 578–582 (1994).
Acknowledgements
We thank Y. Modis and L. Wolfe for assistance in data collection. We also thank the staff at the Advanced Photon Source beamline 24-ID and the National Synchrotron Light Source beamline X29. This work was supported by US National Institutes of Health (NIH) grants AI097064 (Y.X.), AI076040 and AI038201 (J.G.) as well as by The James B. Pendleton Charitable Trust. R.S. was supported by grants from the California HIV-AIDS Research Program and the UCSD Center for AIDS Research (CFAR) developmental program P30 AI36214.
Author information
Authors and Affiliations
Contributions
X.J., J.G. and Y.X. designed the research; X.J., R.S., S.H., H.Y. and Y.X. performed the research; X.J., R.S., S.H., J.G. and Y.X. analyzed data; J.G. and Y.X. supervised the project; and X.J., J.G. and Y.X. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–4 (PDF 1117 kb)
Rights and permissions
About this article
Cite this article
Jia, X., Singh, R., Homann, S. et al. Structural basis of evasion of cellular adaptive immunity by HIV-1 Nef. Nat Struct Mol Biol 19, 701–706 (2012). https://doi.org/10.1038/nsmb.2328
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nsmb.2328
This article is cited by
-
Exploring the conformational dynamics and flexibility of intrinsically disordered HIV-1 Nef protein using molecular dynamic network approaches
3 Biotech (2021)
-
Hijacking of endocytosis by HIV-1 Nef is becoming crystal clear
Nature Structural & Molecular Biology (2020)
-
The autophagy protein ATG9A promotes HIV-1 infectivity
Retrovirology (2019)
-
The HIV-1 accessory proteins Nef and Vpu downregulate total and cell surface CD28 in CD4+ T cells
Retrovirology (2018)
-
Brain-specific HIV Nef identified in multiple patients with neurological disease
Journal of NeuroVirology (2018)
