
Key Points
-
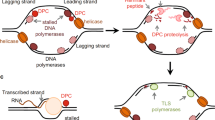
Covalent DNA–protein crosslinks (DPCs) are highly toxic DNA lesions that are induced by widely used classes of chemotherapeutics and also by various external and endogenous agents.
-
DPCs consist of three distinct components, which are harnessed by distinct repair mechanisms as a starting point for repair.
-
Tyrosyl-DNA phosphodiesterases directly hydrolyse the covalent bond between protein and DNA at DPCs.
-
Nuclease-dependent repair by the MRE11–RAD50–NBS1 (MRN) complex targets the DNA component of DPCs.
-
Proteolytic repair by the spartan (SPRTN)/weak suppressor of SMT3 protein 1 (Wss1) protease family degrades the protein component of DPCs.
-
Inhibition of DPC repair pathways offers novel therapeutic opportunities for anticancer combination therapies.
Abstract
Covalent DNA–protein crosslinks (DPCs, also known as protein adducts) of topoisomerases and other proteins with DNA are highly toxic DNA lesions. Of note, chemical agents that induce DPCs include widely used classes of chemotherapeutics. Their bulkiness blocks virtually every chromatin-based process and makes them intractable for repair by canonical repair pathways. Distinct DPC repair pathways employ unique points of attack and are crucial for the maintenance of genome stability. Tyrosyl-DNA phosphodiesterases (TDPs) directly hydrolyse the covalent linkage between protein and DNA. The MRE11–RAD50–NBS1 (MRN) nuclease complex targets the DNA component of DPCs, excising the fragment affected by the lesion, whereas proteases of the spartan (SPRTN)/weak suppressor of SMT3 protein 1 (Wss1) family target the protein component. Loss of these pathways renders cells sensitive to DPC-inducing chemotherapeutics, and DPC repair pathways are thus attractive targets for combination cancer therapy.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

References
Lindahl, T. Instability and decay of the primary structure of DNA. Nature 362, 709–715 (1993).
Jackson, S. P. & Bartek, J. The DNA-damage response in human biology and disease. Nature 461, 1071–1078 (2009).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Bryant, H. E. et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 (2005).
Farmer, H. et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (2005).
Oza, A. M. et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol. 16, 87–97 (2015).
Nakano, T. et al. T7 RNA polymerases backed up by covalently trapped proteins catalyze highly error prone transcription. J. Biol. Chem. 287, 6562–6572 (2012).
Nakano, T. et al. Translocation and stability of replicative DNA helicases upon encountering DNA-protein cross-links. J. Biol. Chem. 288, 4649–4658 (2013).
Fu, Y. V. et al. Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell 146, 931–941 (2011).
Kuo, H. K., Griffith, J. D. & Kreuzer, K. N. 5-Azacytidine induced methyltransferase-DNA adducts block DNA replication in vivo. Cancer Res. 67, 8248–8254 (2007).
Stingele, J. & Jentsch, S. DNA-protein crosslink repair. Nat. Rev. Mol. Cell Biol. 16, 455–460 (2015).
Chvalova, K., Brabec, V. & Kasparkova, J. Mechanism of the formation of DNA-protein cross-links by antitumor cisplatin. Nucleic Acids Res. 35, 1812–1821 (2007).
Pommier, Y. & Marchand, C. Interfacial inhibitors: targeting macromolecular complexes. Nat. Rev. Drug Discov. 11, 25–36 (2011).
Nitiss, J. L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 9, 338–350 (2009).
Fenaux, P. et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 10, 223–232 (2009).
Fenaux, P. et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J. Clin. Oncol. 28, 562–569 (2010).
Maslov, A. Y. et al. 5-Aza-2′-deoxycytidine-induced genome rearrangements are mediated by DNMT1. Oncogene 31, 5172–5179 (2012).
O'Connor, M. J. Targeting the DNA damage response in cancer. Mol. Cell 60, 547–560 (2015).
Mateo, J. et al. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 373, 1697–1708 (2015).
Murai, J. et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 72, 5588–5599 (2012).
Barker, S., Weinfeld, M. & Murray, D. DNA-protein crosslinks: their induction, repair, and biological consequences. Mutat. Res. 589, 111–135 (2005).
Pommier, Y. et al. Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2). DNA Repair (Amst.) 19, 114–129 (2014).
DeMott, M. S. et al. Covalent trapping of human DNA polymerase beta by the oxidative DNA lesion 2-deoxyribonolactone. J. Biol. Chem. 277, 7637–7640 (2002).
Sczepanski, J. T., Wong, R. S., McKnight, J. N., Bowman, G. D. & Greenberg, M. M. Rapid DNA–protein cross-linking and strand scission by an abasic site in a nucleosome core particle. Proc. Natl Acad. Sci. USA 107, 22475–22480 (2010).
Shi, Y. et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953 (2004).
Yang, S. W. et al. A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc. Natl Acad. Sci. USA 93, 11534–11539 (1996).
Pouliot, J. J., Yao, K. C., Robertson, C. A. & Nash, H. A. Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science 286, 552–555 (1999). References 26 and 27 identify Tdp1 in yeast as an enzyme that is capable of releasing covalent Top1 adducts.
Lin, C. P., Ban, Y., Lyu, Y. L., Desai, S. D. & Liu, L. F. A ubiquitin-proteasome pathway for the repair of topoisomerase I-DNA covalent complexes. J. Biol. Chem. 283, 21074–21083 (2008).
Interthal, H. & Champoux, J. J. Effects of DNA and protein size on substrate cleavage by human tyrosyl-DNA phosphodiesterase 1. Biochem. J. 436, 559 (2011).
Debethune, L., Kohlhagen, G., Grandas, A. & Pommier, Y. Processing of nucleopeptides mimicking the topoisomerase I-DNA covalent complex by tyrosyl-DNA phosphodiesterase. Nucleic Acids Res. 30, 1198–1204 (2002).
El-Khamisy, S. F. To live or to die: a matter of processing damaged DNA termini in neurons. EMBO Mol. Med. 3, 78–88 (2011).
Hudson, J. J., Chiang, S. C., Wells, O. S., Rookyard, C. & El-Khamisy, S. F. SUMO modification of the neuroprotective protein TDP1 facilitates chromosomal single-strand break repair. Nat. Commun. 3, 733 (2012).
Das, B. B. et al. PARP1-TDP1 coupling for the repair of topoisomerase I-induced DNA damage. Nucleic Acids Res. 42, 4435–4449 (2014).
Das, B. B. et al. Optimal function of the DNA repair enzyme TDP1 requires its phosphorylation by ATM and/or DNA-PK. EMBO J. 28, 3667–3680 (2009).
Chiang, S. C., Carroll, J. & El-Khamisy, S. F. TDP1 serine 81 promotes interaction with DNA ligase IIIalpha and facilitates cell survival following DNA damage. Cell Cycle 9, 588–595 (2010).
Shiloh, Y. & Ziv, Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 14, 197–210 (2013).
Cortes Ledesma, F., El Khamisy, S. F., Zuma, M. C., Osborn, K. & Caldecott, K. W. A human 5′-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 461, 674–678 (2009). Identified TDP2 as the first-characterized human enzyme with 5′-tyrosyl DNA phosphodiesterase activity, which is important for releasing covalent TOP2 adducts.
Gao, R. et al. Proteolytic degradation of topoisomerase II (Top2) enables the processing of Top2·DNA and Top2·RNA covalent complexes by tyrosyl-DNA-phosphodiesterase 2 (TDP2). J. Biol. Chem. 289, 17960–17969 (2014).
Mao, Y., Desai, S. D., Ting, C. Y., Hwang, J. & Liu, L. F. 26 S proteasome-mediated degradation of topoisomerase II cleavable complexes. J. Biol. Chem. 276, 40652–40658 (2001).
Gomez-Herreros, F. et al. TDP2 protects transcription from abortive topoisomerase activity and is required for normal neural function. Nat. Genet. 46, 516–521 (2014).
Gomez-Herreros, F. et al. TDP2-dependent non-homologous end-joining protects against topoisomerase II-induced DNA breaks and genome instability in cells and in vivo. PLoS Genet. 9, e1003226 (2013).
Rao, T. et al. Novel TDP2-ubiquitin interactions and their importance for the repair of topoisomerase II-mediated DNA damage. Nucleic Acids Res. 44, 10201–10215 (2016).
Stracker, T. H. & Petrini, J. H. The MRE11 complex: starting from the ends. Nat. Rev. Mol. Cell Biol. 12, 90–103 (2011).
Woodworth, D. L. & Kreuzer, K. N. Bacteriophage T4 mutants hypersensitive to an antitumor agent that induces topoisomerase-DNA cleavage complexes. Genetics 143, 1081–1090 (1996).
Stohr, B. A. & Kreuzer, K. N. Repair of topoisomerase-mediated DNA damage in bacteriophage T4. Genetics 158, 19–28 (2001).
Connelly, J. C., de Leau, E. S. & Leach, D. R. Nucleolytic processing of a protein-bound DNA end by the E. coli SbcCD (MR) complex. DNA Repair (Amst.) 2, 795–807 (2003).
Keeney, S., Giroux, C. N. & Kleckner, N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88, 375–384 (1997).
Neale, M. J., Pan, J. & Keeney, S. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 436, 1053–1057 (2005). Provides the first direct evidence that the nuclease MRN (MRX) can release covalent protein adducts from DSB ends.
Hartsuiker, E. et al. Ctp1CtIP and Rad32Mre11 nuclease activity are required for Rec12Spo11 removal, but Rec12Spo11 removal is dispensable for other MRN-dependent meiotic functions. Mol. Cell. Biol. 29, 1671–1681 (2009).
Hartsuiker, E., Neale, M. J. & Carr, A. M. Distinct requirements for the Rad32Mre11 nuclease and Ctp1CtIP in the removal of covalently bound topoisomerase I and II from DNA. Mol. Cell 33, 117–123 (2009).
Malik, M. & Nitiss, J. L. DNA repair functions that control sensitivity to topoisomerase-targeting drugs. Eukaryot. Cell 3, 82–90 (2004).
Lee, K. C. et al. MRE11 facilitates the removal of human topoisomerase II complexes from genomic DNA. Biol. Open 1, 863–873 (2012).
Hoa, N. N. et al. Mre11 is essential for the removal of lethal topoisomerase 2 covalent cleavage complexes. Mol. Cell 64, 580–592 (2016).
Aparicio, T., Baer, R., Gottesman, M. & Gautier, J. MRN, CtIP, and BRCA1 mediate repair of topoisomerase II-DNA adducts. J. Cell Biol. 212, 399–408 (2016).
Deshpande, R. A., Lee, J. H., Arora, S. & Paull, T. T. Nbs1 converts the human Mre11/Rad50 nuclease complex into an endo/exonuclease machine specific for protein-DNA adducts. Mol. Cell 64, 593–606 (2016).
Cannavo, E. & Cejka, P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 514, 122–125 (2014).
Sartori, A. A. et al. Human CtIP promotes DNA end resection. Nature 450, 509–514 (2007).
Sacho, E. J. & Maizels, N. DNA repair factor MRE11/RAD50 cleaves 3′-phosphotyrosyl bonds and resects DNA to repair damage caused by topoisomerase 1 poisons. J. Biol. Chem. 286, 44945–44951 (2011).
de Graaf, B., Clore, A. & McCullough, A. K. Cellular pathways for DNA repair and damage tolerance of formaldehyde-induced DNA-protein crosslinks. DNA Repair (Amst.) 8, 1207–1214 (2009).
Stingele, J., Schwarz, M. S., Bloemeke, N., Wolf, P. G. & Jentsch, S. A. DNA-dependent protease involved in DNA-protein crosslink repair. Cell 158, 327–338 (2014). Identifies Wss1 as a DNA-dependent protease that repairs covalent DPCs.
Duxin, J. P., Dewar, J. M., Yardimci, H. & Walter, J. C. Repair of a DNA-protein crosslink by replication-coupled proteolysis. Cell 159, 346–357 (2014). Describes the detailed mechanisms of replication-coupled proteolytic DPC repair.
Stingele, J. et al. Mechanism and regulation of DNA-protein crosslink repair by the DNA-dependent metalloprotease SPRTN. Mol. Cell 64, 688–703 (2016).
Vaz, B. et al. Metalloprotease SPRTN/DVC1 orchestrates replication-coupled DNA-protein crosslink repair. Mol. Cell 64, 704–719 (2016).
Lopez-Mosqueda, J. et al. SPRTN is a mammalian DNA-binding metalloprotease that resolves DNA-protein crosslinks. eLife 5, e21491 (2016).
Morocz, M. et al. DNA-dependent protease activity of human Spartan facilitates replication of DNA-protein crosslink-containing DNA. Nucleic Acids Res. 45, 3172–3188 (2017).
Maskey, R. S. et al. Spartan deficiency causes accumulation of Topoisomerase 1 cleavage complexes and tumorigenesis. Nucleic Acids Res. 45, 4564–4576 (2017). References 62–66 identify SPRTN as the DPC protease that operates in higher eukaryotes.
Stingele, J., Habermann, B. & Jentsch, S. DNA-protein crosslink repair: proteases as DNA repair enzymes. Trends Biochem. Sci. 40, 67–71 (2015).
Ruijs, M. W. G. et al. Atypical progeroid syndrome: an unknown helicase gene defect? Am. J. Med. Genet. A 116A, 295–299 (2003).
Lessel, D. et al. Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat. Genet. 46, 1239–1244 (2014).
Machida, Y., Kim, M. S. & Machida, Y. J. Spartan/C1orf124 is important to prevent UV-induced mutagenesis. Cell Cycle 11, 3395–3402 (2012).
Nakano, T. et al. Homologous recombination but not nucleotide excision repair plays a pivotal role in tolerance of DNA-protein cross-links in mammalian cells. J. Biol. Chem. 284, 27065–27076 (2009).
Nakano, T. et al. Nucleotide excision repair and homologous recombination systems commit differentially to the repair of DNA-protein crosslinks. Mol. Cell 28, 147–158 (2007).
Baker, D. J. et al. Nucleotide excision repair eliminates unique DNA-protein cross-links from mammalian cells. J. Biol. Chem. 282, 22592–22604 (2007).
Vance, J. R. & Wilson, T. E. Yeast Tdp1 and Rad1-Rad10 function as redundant pathways for repairing Top1 replicative damage. Proc. Natl Acad. Sci. USA 99, 13669–13674 (2002).
Zhang, Y. W. et al. Poly(ADP-ribose) polymerase and XPF-ERCC1 participate in distinct pathways for the repair of topoisomerase I-induced DNA damage in mammalian cells. Nucleic Acids Res. 39, 3607–3620 (2011).
Ridpath, J. R. et al. Cells deficient in the FANC/BRCA pathway are hypersensitive to plasma levels of formaldehyde. Cancer Res. 67, 11117–11122 (2007).
Garaycoechea, J. I. et al. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 489, 571–575 (2012).
Langevin, F., Crossan, G. P., Rosado, I. V., Arends, M. J. & Patel, K. J. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475, 53–58 (2011).
Rosado, I. V., Langevin, F., Crossan, G. P., Takata, M. & Patel, K. J. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat. Struct. Mol. Biol. 18, 1432–1434 (2011).
Kottemann, M. C. & Smogorzewska, A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 493, 356–363 (2013).
Duxin, J. P. & Walter, J. C. What is the DNA repair defect underlying Fanconi anemia? Curr. Opin. Cell Biol. 37, 49–60 (2015).
Orta, M. L. et al. 5-Aza-2′-deoxycytidine causes replication lesions that require Fanconi anemia-dependent homologous recombination for repair. Nucleic Acids Res. 41, 5827–5836 (2013).
Deng, C., Brown, J. A., You, D. & Brown, J. M. Multiple endonucleases function to repair covalent topoisomerase I complexes in Saccharomyces cerevisiae. Genetics 170, 591–600 (2005).
Delabaere, L. et al. The Spartan ortholog maternal haploid is required for paternal chromosome integrity in the Drosophila zygote. Curr. Biol. 24, 2281–2287 (2014).
Huang, S. N., Pommier, Y. & Marchand, C. Tyrosyl-DNA phosphodiesterase 1 (Tdp1) inhibitors. Expert Opin. Ther. Pat. 21, 1285–1292 (2011).
Takemura, H. et al. Defective Mre11-dependent activation of Chk2 by ataxia telangiectasia mutated in colorectal carcinoma cells in response to replication-dependent DNA double strand breaks. J. Biol. Chem. 281, 30814–30823 (2006).
Giannini, G. et al. Mutations of an intronic repeat induce impaired MRE11 expression in primary human cancer with microsatellite instability. Oncogene 23, 2640–2647 (2004).
Raoof, A. et al. Toxoflavins and deazaflavins as the first reported selective small molecule inhibitors of tyrosyl-DNA phosphodiesterase II. J. Med. Chem. 56, 6352–6370 (2013).
Dupre, A. et al. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat. Chem. Biol. 4, 119–125 (2008).
Shibata, A. et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell 53, 7–18 (2014).
Takashima, H. et al. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat. Genet. 32, 267–272 (2002).
Interthal, H. et al. SCAN1 mutant Tdp1 accumulates the enzyme—DNA intermediate and causes camptothecin hypersensitivity. EMBO J. 24, 2224–2233 (2005).
Hernandez, D. et al. A family showing no evidence of linkage between the ataxia telangiectasia gene and chromosome 11q22-23. J. Med. Genet. 30, 135–140 (1993).
Stewart, G. S. et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 99, 577–587 (1999).
Saar, K. et al. The gene for the ataxia-telangiectasia variant, Nijmegen breakage syndrome, maps to a 1-cM interval on chromosome 8q21. Am. J. Hum. Genet. 60, 605–610 (1997).
Varon, R. et al. Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 93, 467–476 (1998).
Weemaes, C. M. et al. A new chromosomal instability disorder: the Nijmegen breakage syndrome. Acta Paediatr. Scand. 70, 557–564 (1981).
Carney, J. P. et al. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell 93, 477–486 (1998).
Chrzanowska, K. H., Gregorek, H., Dembowska-Baginska, B., Kalina, M. A. & Digweed, M. Nijmegen breakage syndrome (NBS). Orphanet J. Rare Dis. 7, 13 (2012).
Waltes, R. et al. Human RAD50 deficiency in a Nijmegen breakage syndrome-like disorder. Am. J. Hum. Genet. 84, 605–616 (2009).
Barbi, G. et al. Chromosome instability and X-ray hypersensitivity in a microcephalic and growth-retarded child. Am. J. Med. Genet. 40, 44–50 (1991).
Acknowledgements
The authors apologize to those whose important findings could not be mentioned as primary literature and/or cited owing to space constraints. The authors thank S. Panier and G. Hewitt for comments and discussions. J.S. is supported by an EMBO Long-term Fellowship (ALTF 470–2015). S.J.B.'s laboratory is supported by the Francis Crick Institute, which receives its core funding from Cancer Research UK (FC0010048), the UK Medical Research Council (FC0010048), and the Wellcome Trust (FC0010048); European Research Council (ERC) Advanced Investigator Grants (RecMitMei; TelMetab); and a Wellcome Trust Senior Investigator Grant.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Glossary
- Abasic sites
-
Positions within DNA that lack a DNA base; also known as apurinic or apyrimidinic (AP) sites. They can occur spontaneously or through base excision by DNA glycosylases.
- Intrastrand and interstrand DNA crosslinks
-
Chemical crosslinking of DNA can occur between positions on the same DNA strand (intrastrand) or between opposing strands (interstrand).
- Poly(ADP-ribose) polymerase 1
-
(PARP1). A member of a family of enzymes that catalyse the formation of poly(ADP-ribose) chains, thereby regulating various cellular processes, most notably DNA repair.
- Pseudosubstrate
-
Any molecule that inhibits an enzyme by mimicking a substrate.
- Homologous recombination
-
(HR). A process resulting in the exchange of identical or highly similar DNA sequences between two DNA molecules; it is involved in meiosis, DNA repair and DNA replication.
- DNA polymerase β
-
The DNA polymerase required for gap filling during base excision repair and DNA single-strand break repair.
- Ataxia
-
A medical condition that is characterized by a lack of coordination of voluntary muscle movements; it is often caused by inherited or acquired cerebellum diseases (cerebellar ataxia).
- Areflexia
-
The absence of neurological reflexes.
- Telangiectasias
-
Small dilated blood vessels in the outer layer of the skin or in mucosae. Usually a benign condition, which can be associated with serious genetic or acquired diseases.
- Apraxia
-
A neurological motor disorder that is caused by partial brain damage; affected individuals experience difficulty with motor planning to carry out motor tasks or movements.
- Dysarthria
-
A motor speech disorder characterized by poor articulation of phonemes. It is caused by injuries to the motor component of the motor–speech system of the brain.
- Microcephaly
-
A neurological condition in which affected individuals present with a smaller than normal head owing to defective brain development.
- Micrognathia
-
A medical condition that is characterized by underdevelopment of the jaw.
- Proteasome
-
A protein complex that degrades unneeded or damaged proteins. Proteins are commonly marked for degradation by modification with polyubiquitin chains.
- PARylation
-
A post-translational modification, also known as polyADP-ribosylation, by which polymers of ADP-ribose are attached to substrate proteins by poly(ADP-ribose) polymerases (PARPs).
- X-ray repair cross-complementing protein 1
-
(XRCC1). A molecular scaffold protein that orchestrates the repair of DNA single-strand breaks by recruiting and stabilizing multiple other repair factors.
- Ataxia telangiectasia mutated
-
(ATM). A protein kinase best known for its function in signalling the presence of DNA double-strand breaks, thereby activating a highly sophisticated cellular response.
- DNA-dependent protein kinase
-
(DNA-PK). Heterotrimeric DNA-dependent kinase that comprises KU70, KU80 and the catalytic subunit DNA-PKcs; best known for its central function in non-homologous end joining repair of DNA double-strand breaks.
- Replication fork run-off
-
If a replication fork encounters a single-strand break, the replicative helicase runs off the template strand, resulting in the formation of a highly toxic single-ended DNA double-strand break.
- Non-homologous end joining
-
(NHEJ). The repair of DNA double-strand breaks by directly ligating two DNA ends, irrespective of DNA sequence.
- SPO11
-
A type II DNA topoisomerase that is required for the generation of DNA double-strand breaks during meiosis, which is crucial for genetic recombination.
- Synthetic lethality screen
-
A genetic screening method, which aims to reveal genes that when knocked out cause lethality in combination with a knockout of a gene of interest.
- DNA damage checkpoint
-
A cellular signalling mechanism that pauses the cell cycle in response to the presence of DNA damage, thereby providing enough time to ensure that repair can occur before the next cell division.
- AAA-ATPase
-
A diverse class of enzymes that are involved in various cellular processes. AAA-ATPases use energy obtained from ATP hydrolysis to generate mechanical forces through conformational changes.
- Hypomorphic mutations
-
Mutations that cause a partial loss of function; for example, by reduced gene expression or decreased protein stability. Hypomorphic mutations mostly display less severe phenotypes than a complete gene loss.
- Hepatocellular carcinomas
-
The most common type of liver cancer; often caused by hepatitis virus (B or C) and substances such as aflatoxin and ethanol.
- Translesion synthesis polymerases
-
DNA polymerases with the ability to synthesize past DNA lesions due to a flexible active site; they often display low fidelity and thus tend to incorporate mutations.
- Nucleotide excision repair
-
(NER). A DNA repair pathway that repairs primarily bulky adducts, such as those induced by ultraviolet (UV) light. Repair is achieved by excision of a short stretch of nucleotides that contains the DNA lesion.
- Template switch
-
A recombination process that allows the replication of damaged DNA strands by using the newly replicated sister strand as a template for DNA synthesis.
- Mismatch repair
-
A conserved DNA repair pathway that detects and repairs base–base mismatches and insertion–deletion mispairs, which can be generated by mistakes during replication.
Rights and permissions
About this article

Cite this article
Stingele, J., Bellelli, R. & Boulton, S. Mechanisms of DNA–protein crosslink repair. Nat Rev Mol Cell Biol 18, 563–573 (2017). https://doi.org/10.1038/nrm.2017.56
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrm.2017.56
This article is cited by
-
MRE11A: a novel negative regulator of human DNA mismatch repair
Cellular & Molecular Biology Letters (2024)
-
MDM2 provides TOP2 poison resistance by promoting proteolysis of TOP2βcc in a p53-independent manner
Cell Death & Disease (2024)
-
Transcription-coupled DNA–protein crosslink repair by CSB and CRL4CSA-mediated degradation
Nature Cell Biology (2024)
-
Transcription-coupled repair of DNA–protein cross-links depends on CSA and CSB
Nature Cell Biology (2024)
-
MUS81 cleaves TOP1-derived lesions and other DNA–protein cross-links
BMC Biology (2023)
