
Key Points
-
This article presents the currently available mucosal vaccines and their principle mechanisms of action. The concepts of live attenuated and non-living subcomponent vaccines are explained.
-
Principles for mucosal vaccine design and development are discussed, with special reference to vaccine formulations based on soluble or particulate forms. The strengths and weaknesses of various routes of vaccine administration — including intranasal, oral, sublingual, aerosol and rectal — are also considered.
-
Mucosal adjuvants and their mechanisms of action, especially toxin-based adjuvants and derivatives of these, are explored.
-
Long-term B and T cell memory development following mucosal vaccination is discussed.
-
Concepts and regulatory mechanisms governing mucosal IgA responses and the synchronization of gut IgA immunity, in particular, are explained.
-
Finally, future directions, new technologies and new candidate mucosal vaccines that are in the pipeline are summarized.
Abstract
Most pathogens access the body through the mucosal membranes. Therefore, effective vaccines that protect at these sites are much needed. However, despite early success with the live attenuated oral polio vaccine over 50 years ago, only a few new mucosal vaccines have been subsequently launched. This is partly due to problems with developing safe and effective mucosal adjuvants. In the past decade, however, the successful development of live attenuated mucosal vaccines against influenza virus and rotavirus infections has boosted interest in this field, and great expectations for new mucosal vaccines lie ahead. Here, I discuss the expanding knowledge and strategies used in the development of mucosal vaccines.
Similar content being viewed by others
Main
There is general agreement that effective mucosal vaccines — that is, oral, nasal, sublingual and genital tract vaccines — could dramatically contribute to the improvement of global health by stimulating protective immune responses not only against mucosal infections but also against HIV, Mycobacterium tuberculosis and many other infections1,2.
Mucosal vaccines are advantageous compared with systemic vaccines from a production and regulatory perspective3,4. For example, vaccines for oral use do not require extensive purification from bacterial by-products, as the gut is already heavily populated by bacteria, whereas the same vaccine formulation injected parenterally, for example, would have unacceptable endotoxin levels. In addition, mucosal vaccines are practical for mass vaccination and do not involve the risk of spreading blood-borne infections, as can occur with contaminated injection needles. The ease of administration, better compliance and the possibility that they can be delivered by personnel without medical training are also viewed as benefits of mucosal vaccine strategies5,6, especially for preventing the pandemic spread of infections, such as influenza virus infections7,8,9,10.
Both live attenuated and non-living mucosal vaccines are being developed to meet the need for better protection against pathogens that gain access to the body through mucosal membranes. Mucosal vaccination can trigger both humoral and cell-mediated immune protection not only at mucosal sites but also systemically1,11. Indeed, mucosal immunizations strongly induce long-term B and T cell memory12,13. Protection against pathogens can, thus, be effectively achieved by directing memory and effector immune cells to the mucosal membranes through tissue-specific homing receptors. B and T cells acquire mucosal homing properties only in the draining lymph nodes from specialized dendritic cells (DCs) that migrate from the mucosal tissue to these lymph nodes14 (Box 1). Hence, vaccination via the intramuscular or subcutaneous routes poorly promotes immune protection at mucosal membranes. Following mucosal immunization, antigen-triggered B and T cells leave the draining lymph nodes, transit through the lymph, enter the blood circulation and then 'seed' the mucosal tissues11,15.
The size of the mucosal tissues provides a formidable challenge to inducing protective immunity at the natural portals of pathogen entry12. In general, the strongest immune response is usually induced at the site of vaccine exposure and in anatomically adjacent mucosal sites (for example, an immune response can be induced in both the rectal and genital tract in response to a rectal vaccine). Of note, intranasal immunization efficiently stimulates a protective immune response in the lungs and upper respiratory tract and at distant sites, such as the gastric and genital tract mucosa, but it is rather poor at stimulating intestinal immune responses.
Mucosal vaccine-induced immune responses are initiated in specific mucosa-associated lymphoid tissue (MALT) structures1,12,16, such as the Peyer's patches in the small intestine in response to oral immunization or the tonsils and adenoids in the nasal cavity in response to nasal or sublingual immunization (Fig. 1). In addition, the colon patches, the appendix, the abundant small isolated lymphoid follicles in the small intestine, large intestine and respiratory tract, and the mesenteric lymph nodes are all viewed as inductive sites. The immune effector sites, to which protective B and T cells home following priming in the MALT, are restricted to the lamina propria and intra-epithelial compartments of the mucosal membranes13,15,17.

Within the mucosa-associated lymphoid tissue (MALT), subcompartments can be identified, such as the nasopharynx-associated lymphoid tissue (NALT), bronchus-associated lymphoid tissue (BALT), gut-associated lymphoid tissue (GALT) and genital tract-associated lymphoid tissue. Certain immunization routes are more effective at stimulating immunity within specific, most often closely located, subcompartments of the MALT. Intranasal vaccination is preferred for targeting the respiratory, gastric and genital tracts; oral vaccination is effective for immunity in the gut and for the induction of mammary gland antibodies (which are secreted in milk); rectal immunization is best for the induction of colon and rectal immunity and to some extent genital tract immunity; and intravaginal vaccination is the most effective for antibody and T cell immunity in the genital tract.
Following mucosal vaccination, the mucosal barriers to infection are reinforced, mainly through the induction of antigen-specific secretory IgA (SIgA) antibody production, which prevents pathogens and toxins from adhering to or infecting the epithelial cells and breaching the mucosal barrier1,11. In addition, specific effector T cells are distributed to mucosal sites to reinforce their barrier functions13,18 (Box 2). Mucosal vaccination may also alter the growth conditions for pathogens and thereby reduce susceptibility to infection. For example, by changing the secretion of host factors involved in mucosal resistance to infection (such as antimicrobial peptides, chemokines and cytokines), the mucosal membrane can better resist attacks from intruding pathogens19,20.
Although the route of vaccination is important for a successful result, the selection of an appropriate formulation and adjuvant is also crucial for effective mucosal immunization using non-living vaccines. The reason for this is that tolerance is the natural immune response induced in response to a soluble antigen at mucosal sites. Mucosal tolerance prevents harmful inflammatory responses, for example to food antigens16,20, and has a role in the establishment of homeostasis between the microbiota and the host at mucosal membranes21. A particular problem with mucosal antigen exposure is, thus, the risk of inducing mucosal tolerance rather than protective immunity11,16. Adjuvants trigger innate immunity to support the induction of effector rather than regulatory T cell responses and, therefore, adjuvants are crucially needed to stimulate strong mucosal immune responses.
Here, I review some of the main strategies that have been used for successful mucosal vaccination and discuss the importance of the formulation, route of administration and adjuvant choice for achieving protective immunity at mucosal membranes. In addition, I comment on the recent progress in mucosal vaccine and adjuvant development and the novel strategies for finding correlates of protection through systems biology approaches.
Strategies for mucosal vaccination
Only a few commercially available mucosal vaccines currently exist. Although many mucosal vaccine candidates have been found to be effective in animal models, clinical trials have often failed. However, live attenuated mucosal vaccines against influenza virus, rotavirus and Salmonella enterica subsp. enterica serovar Typhi (the causative agent of typhoid fever) have been successfully launched. Moreover, two non-living whole-cell oral vaccines against Vibrio cholerae (marketed as Dukoral (Crucell) and Shanchol (Shantha Biotechnics)) provide proof-of-concept that non-living vaccines can also be effective via the mucosal route6,22. Unfortunately, the stability of live attenuated vaccines is still a problem, whereas non-living whole-cell or subcomponent vaccines are usually poorly immunogenic and require effective mucosal adjuvants. Therefore, many laboratories are focused on developing new and effective mucosal adjuvants (see below).
Live attenuated vaccines. Live attenuated vaccines are composed of live bacteria or viruses that are made much less virulent than the pathogenic parental bacterial or viral strains. The most important feature of live attenuated vaccines is that they provide a high level of antigen exposure and to some extent also have in-built adjuvanticity23. Live bacteria and viruses can also be engineered to carry recombinantly expressed antigens from other pathogens and, thus, can act as vaccine vectors. However, vaccine vectors that express recombinant antigens may be less effective than live attenuated vaccines owing to the presence of pre-existing vector-specific immunity that could lower vaccine efficacy, as has been reported with adenovirus vectors24.
Ideally, live attenuated vaccines establish a mild infection at the site of administration. They can also be engineered to have a limited capacity for replication and to deliver a sufficiently high antigen load at the site of immunization to be effective, while avoiding unwanted local inflammatory responses25. These attributes are important because prolonged antigen exposure and a large amount of antigen are usually required for the induction of mucosal immune responses, especially following oral vaccination26.
A major challenge for the development of live attenuated vaccines is the balance between sufficient attenuation and vaccine immunogenicity. For example, the effective live attenuated vaccines against rotavirus infection have been made less virulent (that is, less infectious) through serial passage of the virus in host cell cultures25,27. These vaccines result in only a mild subclinical infection and are, thus, sufficiently attenuated, while still being highly immunogenic. By contrast, the live oral Ty21a typhoid vaccine is only moderately immunogenic, but it is safe because of extensive attenuation. However, the molecular basis for the attenuation of these vaccines is poorly understood25,28. The production of a new generation of live S. Typhi vaccines, designed to be administered in a single dose, was achieved by attenuation through selected gene deletion, giving rise to a safe and highly immunogenic vaccine25,29. Thus, precise gene deletions represent a more controlled strategy for attenuation than serial passage, and this approach is highly attractive for developing stable and safe live vaccines. A better understanding of the genetic basis for attenuation will be crucial for the next generation of live vaccines, and will allow for vaccines with increased safety and stability.
Several live attenuated mucosal vaccines are currently being developed. Most of the vaccines of this type that are being evaluated in clinical trials are oral vaccines, typified by vaccines against Shigella spp. (the causative agents of shigellosis) and Salmonella infections, but some are intranasal and sublingual vaccines against Bordetella pertussis (the causative agent of whooping cough), influenza virus, rotavirus, norovirus and measles virus25,30,31,32.
Non-living whole-cell or subcomponent vaccines. Although attenuated vaccines are immunogenic and can deliver high levels of antigen to the mucosal target site, their development is a multistep process that often fails to generate safe and stable vaccines. Therefore, the development of non-living whole-cell or subcomponent mucosal vaccines is still much warranted. A large number of candidate vaccines are being developed, including inactivated influenza virus, norovirus and Shigella spp. vaccines10,32,33,34. Various delivery strategies are being adopted, with intranasal and oral administrations being the most used immunization routes in clinical trials. Particulate vaccine formulations (see below) are generally more effective at mucosal surfaces than soluble antigens, but it is generally thought that both types of vaccine require adjuvants that are more efficacious than those currently available to improve their immunogenicity.
Existing mucosal vaccines. The mucosal vaccines that are licensed and available25,27,35,36,37 (Table 1) have been used to vaccinate populations in both developing countries and industrialized countries, and they have achieved considerable success in disease prevention3. For example, the recently developed rotavirus vaccines have proven effective in infants, although their efficacy is 30% lower in developing countries than it is in developed countries, probably owing to differences in nutritional status, gut microflora and/or pre-existing natural or maternal antibodies38,39. This highlights a crucial problem that has hampered the global success of mucosal vaccines, namely the poor outcome of these vaccines in developing countries, referred to as a tropical barrier3. Indeed, other oral vaccines — including vaccines against V. cholerae and prototypic vaccines against enterotoxigenic Escherichia coli (ETEC) — have been found to be less effective in developing countries than in developed countries3,25. The mechanisms involved in the tropical barrier are still incompletely understood, but chronic environmental enteropathy is a cardinal feature associated with this vaccine inefficacy. Chronic environmental enteropathy is marked by intestinal inflammation and blunting of the small intestinal villi, which leads to malabsorption of nutrients and a lack of vitamin A (and its derivative retinoic acid) and/or zinc. These deficiencies hamper the development of mucosal immune responses4,14,40; for example, a lack of retinoic acid directly impairs DC function in the gut mucosa41. Furthermore, persistent infections with helminths or other parasites, which are common in developing countries, may skew or suppress mucosal immune responses42. Therefore, the treatment of such infections could help to increase mucosal vaccine efficacy in developing countries. In addition, a more controlled strategy for pathogen attenuation through specific gene deletions is being investigated. Indeed, the use of precise and appropriate gene deletions to control the safety and stability of live attenuated vaccines and to improve their immunogenicity is another approach that might enhance vaccine efficacy in developing countries.
The only licensed non-living mucosal vaccine available for human use is the oral cholera vaccine. This vaccine, named Dukoral, was developed in the 1970s and early 1980s and consists of killed whole V. cholerae bacteria admixed with recombinant cholera toxin subunit B (CTB)22,43. It conveys protection even 2–3 years after vaccination, and the resulting protective immune response is dominated by antibacterial SIgA. The SIgA is mainly specific for V. cholerae lipopolysaccharide, but CTB-specific antibodies are also produced following vaccination44,45. These CTB-specific antibodies also convey short-term cross-protection against ETEC, owing to the similarity between CTB and the B subunit of the heat-labile toxin from ETEC1,46. These gut SIgA antibodies inhibit bacterial colonization and the binding of the toxins to intestinal epithelial cells22,46.
An important feature of the cholera vaccine is its ability to stimulate herd immunity47. This is a hallmark of mucosal vaccines, as pathogen-specific SIgA antibodies prevent infection and therefore reduce the virulence and spread of infection. Hence, even if the efficacy of a mucosal vaccine is lower than that of most parenteral vaccines, it might effectively prevent the spread of infection in a given population. Therefore, future research should be devoted to better understanding SIgA-dependent herd immunity and how it can be exploited for the prevention of mucosal infections48.
In sum, there are many strategies — such as the use of targeted gene deletions and the improvement of adjuvant functions — currently being used to refine the next generation of mucosal vaccines. These developments will result in mucosal vaccines that are more stable and more immunogenic, and that can better promote strong long-term memory responses and herd immunity.
Mucosal vaccine delivery routes
Mucosal vaccination was initially dominated by oral vaccines. The development of intranasal vaccines followed, and today we are exploring many different routes for the delivery of mucosal vaccines, including aerosol inhalation and intravaginal, rectal and sublingual routes (Fig. 1). Vaccine delivery via most of these routes works well in experimental animal models, but only the oral and intranasal routes have so far been used for licensed human vaccines49.
Oral vaccination. Oral vaccines represent the biggest challenge for mucosal vaccine development. This is not only because of the harsh gut environment, which degrades most antigenic epitopes that are delivered in soluble form, but also due to mucosal tolerance, which protects against unwanted immune responses to digested antigens. A plethora of studies have reported success in experimental animals with oral vaccine delivery, although results in ensuing clinical trials have mostly been disappointing25,49. Hence, it appears that more sophisticated formulations using both cell-specific targeting approaches and immunomodulation will be required. Attempts to develop live attenuated oral vaccines are continuing, but stronger immunogenicity and better stability and safety profiles are required to launch new live mucosal vaccines, and this appears to be difficult to achieve3. Therefore, non-living vaccines containing strong mucosal adjuvants are much warranted. Although we know that particulate antigens are more immunogenic than soluble vaccine formulations at mucosal sites, we still lack a licensed oral vaccine based on, for example, biodegradable microparticles and nanoparticles adjuvanted by a mutated enterotoxin or Toll-like receptor (TLR) agonist50,51 (see below). Notwithstanding these hurdles, it should be pointed out that effective oral vaccines do exist, with rotavirus and cholera vaccines as representative examples. However, at present we lack a good understanding of why these oral vaccines work when others do not. Hence, it has not been possible to mimic these successful vaccines by simply using the same formulations or protocols. It could be speculated that these oral vaccines can deliver sufficient antigen for sustained time periods to effectively prime the gut immune system, by gaining access to follicle-associated epithelium (FAE) and the Peyer's patches (discussed below).
Intranasal vaccination. Intranasal vaccination stimulates immune responses in the nasopharynx-associated lymphoid tissue (NALT) and is effective at inducing systemic and mucosal immunity in the gastric mucosa and the respiratory and genital tracts52,53Fig. 2. For example, intranasal administration of a live attenuated influenza virus vaccine (FluMist; MedImmune) has proven effective at protecting against seasonal infection, and it even provides cross-protection against drifted influenza virus strains (which arise through the continuous process of genetic and antigenic change in influenza virus)9,54. In addition, recent attempts at stimulating protective immunity against gastrointestinal infections, such as Shigella flexneri infections, via the intranasal route have been successful25,32. In general, intranasal vaccination is an attractive approach, as much lower antigen and adjuvant doses are required compared with oral vaccination. Furthermore, vaccine delivery via aerosol spray and droplets is an attractive possibility owing to the development of new delivery devices53,55,56.

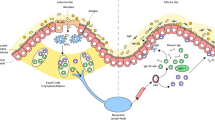
The nasopharynx-associated lymphoid tissue (NALT) is composed of the lymphoid tissues in the Waldeyer's ring, including the adenoids and the lingual and palatine tonsils. It is an attractive inductive site for stimulating immune responses against airborne and alimentary antigens. Antigens are taken up by the follicle-associated epithelium (FAE) overlaying the NALT, where dendritic cells (DCs) prime naive CD4+ or CD8+ T cells. Activated CD4+ T cells differentiate into T helper 1 (TH1), TH2 and TH17 cells, regulatory T cells and, importantly, T follicular helper (TFH) cells. TFH cells help the development of long-lived plasma cells and memory B cells through the germinal centre reaction. Intranasally administered adjuvants enhance immune responses mainly by augmenting the innate immune response through the upregulation of co-stimulatory molecule, chemokine and cytokine expression. Such molecules include CD80, CD86, CD40, interferon-α (IFNα), B cell-activating factor (BAFF) and a proliferating-inducing ligand (APRIL). DCs are the main targets of most adjuvants, but follicular dendritic cells (FDCs) may also be activated, as seen with the adjuvant CTA1-DD. Memory B and T cells migrate to draining lymph nodes and the non-organized effector tissues, such as the lungs and the genital tract. Crucial cytokines for germinal centre development in the NALT are shown. Nasal vaccines are formulated as sprays or droplets. CXCL13, CXC-chemokine ligand 13; ICAM1, intercellular adhesion molecule 1; IL, interleukin; TGFβ, transforming growth factor-β; VCAM1, vascular cell adhesion molecule 1.
Moreover, intranasal immunization is effective at inducing antibody responses in the genital tract57 (Fig. 2). This has obvious practical implications for the development of mucosal vaccines against sexually transmitted diseases. A promising strategy in HIV-1 vaccine development has been suggested by a study in which rhesus macaques were intranasally vaccinated with a virosome-coupled trimeric gp41 protein, which elicited strong protective IgA antibody responses in the genital tract and also prevented the transmission of infection58. Phase I clinical trials are now underway with intranasal delivery of this HIV-1 candidate vaccine59. Although intranasal immunization in females results in strong genital tract antibody responses, the stimulation of genital tract T cells appears to require local vaginal vaccination. Thus, combining intranasal priming and intravaginal boosting immunizations could be an effective strategy to achieve strong immune protection in the genital tract60,61. Indeed, the poor priming ability of the vaginal route, at least in the absence of an effective adjuvant, was highlighted in a recent clinical trial by the failure of an HIV-1 gp140 vaccine delivered vaginally to induce significant immune responses in healthy women62.
Sublingual vaccination. The sublingual route for vaccine administration is gaining interest63. Unexpectedly, vaccine delivery via this route stimulates broad and disseminated mucosal and systemic immune responses, similarly to the intranasal route64. Sublingual vaccination with soluble or particulate antigens (including live or killed bacteria and viruses) evokes strong mucosal IgA and systemic IgG antibody responses as well as cytotoxic CD8+ T cell responses65,66,67. Moreover, sublingual immunization with human papillomavirus (HPV) or Chlamydia muridarum antigens provides protection against genital infection by these pathogens65,66. In contrast to intranasal immunization, this route avoids the perturbation of central nervous system functions (which can result in Bell's palsy) by adjuvants such as cholera toxin and E. coli heat-labile toxin (see below). This route also requires substantially lower amounts of antigen compared with oral immunization63.
Protection against lung infection appears to be stimulated by sublingual immunization, as seen in the case of influenza virus vaccination68. Furthermore, experimental sublingual immunization with Helicobacter pylori antigens or with ETEC or V. cholerae antigens effectively induces protective immunity in the stomach and intestine, respectively. This finding indicates that the tropical barrier may be circumvented by this route, as the antigen would not have to pass down the intestinal canal to prime B and T cells with gut-homing properties63,69. Clinical trials using sublingual vaccination are underway70.
Mucosal vaccine formulations
No vaccine is effective unless it is formulated in a way that promotes its immunogenicity. This means that it should preferably include components that enable the effective uptake of the antigens across the mucosal membrane. It has been found that antigens delivered in particles are better recognized by the innate immune system and, hence, are stronger inducers of mucosal immune responses than soluble antigens.
Soluble formulations. There are various soluble formulations for non-living mucosal vaccines. The access of vaccine antigens to the underlying tissues, where they activate immune cells, is impeded by a constantly renewing mucus layer that covers mucosal tissues. Thus, to increase the chances of effective antigen uptake, mucosal vaccines have been developed to include components — such as the mucoadhesives chitosan and starch — that improve adhesion to mucosal membranes71. The aim is to create close contact between the soluble vaccine antigens and the mucosal membrane. Use of these soluble formulations has been partly successful, although the choice of adjuvant rather than the mucoadhesive appears, in many cases, to be key for vaccine efficacy. Nevertheless, chitosan has been shown to have both mucoadhesive and adjuvant properties and in this way can fulfil two functions that enhance vaccine efficacy (Table 2). Notably, mucoadhesive formulations have been shown to effectively enhance intranasal and intravaginal immunizations, probably owing to prolonged antigen retention72,73.
Particulate formulations. Vaccines based on particulate formulations have generally been more effective than soluble antigens at stimulating protective immunity50,51,74,75,76. Examples of such formulations include virus-like particles (VLPs), bacterial ghosts, biodegradable microparticles and nanoparticles (such as PLG (poly(lactide-co-glycolide))-containing microparticles and liposomes) and immunostimulating complexes. Particulate vaccines can also establish a close contact with the mucosal epithelium through the inclusion of anchoring devices that have adhesive properties (such as lectins and specific antibodies) or through the inclusion of immunomodulating molecules (such as TLR ligands and CTB) that bind to distinct receptors50,51. Thus, the particles can be developed to have both bioadhesive and immunomodulating properties.
Targeting a particle to the mucosal membrane also facilitates antigen uptake across the epithelial barrier through the specialized FAE, which overlies the MALT77. This allows the particle to be targeted to antigen-presenting cells (APCs) and to directly modulate the release of chemokines and cytokines to enhance T cell priming through increased co-stimulation. A particular type of FAE that is composed of microfold (M) cells and found over the NALT and the Peyer's patches has special properties that enable the engulfment and transcellular transport of antigens to the subepithelial dome, where the antigens can stimulate specific CD4+ T cells in the interfollicular region78. The targeting of mucosal vaccines to M cells has been successfully explored using TLR agonists, specific lectins (such as Ulex europaeus agglutinin 1) and antibodies specific for M cell surface markers (such as glycoprotein 2)79.
Although work with mucosal vaccine formulations has been ongoing for more than two decades, we still lack licensed vaccines based on these approaches. Thus, the future challenge is to develop novel mucosal vaccine formulations that meet the demand for safe and effective delivery of vaccine antigens. Perhaps nanotechnology-based vaccines will prove to be the most effective delivery vehicles for non-living subcomponent mucosal vaccines, as their size, contents and cell-targeting properties can be engineered. However, we are only beginning to understand the optimal chemical composition of nanoparticles for the effective stimulation of mucosal immune responses50,51.
Mucosal vaccine adjuvants
The selection of adjuvants for a mucosal vaccine is as crucial as the selection of the antigen (or combination of antigens). Recent data from humans and experimental models have shown that the choice of adjuvant can dramatically affect not only the immediate immune response but also the long-term protective effect of a vaccine23,80. Also, the quality of the immune response — especially the development of high-affinity B cell clones, long-lived memory B cells and plasma cells — can be influenced by the choice of adjuvant80. Furthermore, by using whole-genome phage-display libraries to test antibody recognition, recent studies with influenza vaccines have demonstrated that adjuvants may dramatically augment epitope spreading following vaccination, which could lead to the production of higher avidity antibodies with improved neutralizing properties81.
From a mechanistic point of view, mucosal adjuvants modulate innate immune responses in the same way as adjuvants in injectable vaccines26,82,83 (Table 2). Thus, TLR agonists constitute a major category of mucosal adjuvants84. These adjuvants are based on pathogen-associated molecular patterns (PAMPs), which are often formulated in oil-in-water emulsions. For example, the commercial adjuvant IC31 and other CpG oligodeoxynucleotide (ODN)-containing formulations, which act through TLR9, have been used successfully in experimental vaccine models and in clinical trials85. Monophosphoryl lipid A, which signals through TLR4, and the TLR5 ligand flagellin have also been used as adjuvants in mucosal immunizations and have shown intermediate potency86. Cationic liposomes (composed of the synthetic amphiphile dimethyldioctadecylammonium (DDA), for example) and the saponin derivative QS21 have been found to be effective for mucosal immunization, although the mechanism for their action is poorly understood87,88,89. The combination of a particulate vaccine formulation with an adjuvant has been found to be superior in many cases; for example, coating a CpG ODN onto microparticles enhances the immunostimulatory capacity of this TLR9 agonist in mucosal vaccination90.
The second category of mucosal adjuvants includes the most effective immunomodulators known today: the bacterial enterotoxins cholera toxin and E. coli heat-labile toxin, which have considerable sequence similarity91,92. These enterotoxins are AB5 complexes, in which the A subunit is comprised of an ADP-ribosylating A1 portion and an A2 chain that interacts with the pentamer of B subunits1,93. Mucosal immune responses using these holotoxin adjuvants are probably dependent on DCs94; however, current evidence suggests that they do not activate the innate immune system through TLRs or NOD-like receptors (NLRs)26. Instead, they bind via their B subunits to gangliosides (preferentially GM1) that are present on the cell membrane of most nucleated cells95. Following binding, the A1 portion mediates the ADP-ribosylation of the membrane-bound heterotrimeric guanine nucleotide-binding protein subunit Gsα, resulting in increased cytoplasmic levels of cyclic AMP96. The binding of these toxins to gangliosides facilitates their uptake across mucosal membranes and, perhaps, by the FAE97. However, gangliosides are ubiquitously expressed, and therefore these adjuvants can be associated with unwanted side effects. For example, intranasal administration of a mucosal VLP-based influenza vaccine containing E. coli heat-labile toxin as an adjuvant has resulted in the development of facial paralysis (Bell's palsy) in a few individuals, owing to the presence of gangliosides on nerve cells98,99,100. Furthermore, oral vaccination with cholera toxin or heat-labile toxin as the adjuvant has resulted in overt diarrhoea in some vaccine recipients101.
Mutant enterotoxins are safer adjuvants. Enterotoxins can be engineered into less toxic and safer mucosal adjuvants through site-directed mutations of the enzymatically active A1–A2 subunit102. In the case of E. coli heat-labile toxin, this has resulted in the generation of a single-mutant toxin (LTR192G) and a double-mutant toxin (LTR192G/L211A; known as dmLT), both of which appear to be effective for enhancing mucosal immune responses91,103. dmLT in particular is trypsin resistant and, hence, is stable after oral administration, and because it exerts no enterotoxicity it holds promise as an oral vaccine adjuvant. dmLT is currently being tested experimentally in various models and will soon be assessed in clinical trials. Promising results from these studies could convey optimism for future use of mutated holotoxins in mucosal vaccines. This would pave the way for a renewed confidence from regulatory authorities in mucosal adjuvants based on cholera toxin and E. coli heat-labile toxin.
LTK63 is another genetically detoxified E. coli heat-labile toxin mutant that has been developed and has shown efficacy in humans. Although this mutant completely lacks enzymatic activity102,104, it was associated with the development of Bell's palsy in a few vaccine recipients in two recent intranasal vaccine clinical trials, thereby cautioning against the use of GM1-binding adjuvants in intranasal vaccines105. However, for oral or sublingual vaccination the neurotoxicity problem may not exist.
Mutants of cholera toxin that retain strong adjuvant activity have also been generated98,102,106. The mutations have mostly affected the enzymatically active cleft of cholera toxin subunit A (CTA), similarly to those used for E. coli heat-labile toxin, rendering the molecule enzymatically inactive. As an example, a double mutant of cholera toxin (CTA112/KDEV) was found to be safe and did not accumulate in the central nervous system, and retained good adjuvant function when administered intranasally106. It is presently not known whether the cholera toxin and heat-labile toxin mutants have similar adjuvant activities. However, it is likely that there would be minimal, if any, significant differences between the two families of mutant holotoxins with regard to adjuvant impact.
Taking toxin adjuvants one step further. To circumvent the problem with toxicity, we have developed a non-toxic derivative of cholera toxin, termed CTA1-DD107, which has been shown to be a highly effective adjuvant in mice, guinea pigs and non-human primates following admixing with numerous vaccine candidates108. CTA1-DD is a fusion protein of CTA1, which hosts the full enzymatic activity of the toxin, and a dimer of the D-fragment from Staphylococcus aureus protein A. Unlike cholera toxin and E. coli heat-labile toxin, CTA1-DD cannot bind to the ganglioside GM1, as it lacks the B subunit, and it has been shown to be non-toxic and safe in experimental animal models, including non-human primates109,110. Furthermore, it does not bind to nervous tissues and no side effects were observed following intranasal administration109. CTA1-DD has adjuvant effects comparable to those of cholera toxin and heat-labile toxin, and it enhances a wide range of immune responses following intranasal immunizations, including antibody, CD4+ T cell and cytotoxic CD8+ T cell responses26 (Box 3). Candidate vaccines containing CTA1-DD have been shown to enhance immunity to HIV-1, Chlamydia trachomatis, H. pylori, M. tuberculosis, rotavirusandinfluenza A virus26,110. By contrast, intranasal administration of an enzymatically inactive mutant of CTA1-DD (CTA1R7K-DD) with an incorporated collagen peptide induced tolerance via interleukin-10 (IL-10)-producing regulatory T cells and protected against collagen-induced arthritis in mice111. This suggests that the ADP-ribosylating activity of CTA1 has a central role in determining whether immunity or tolerance is induced following mucosal immunization112. The use of the mutant CTA1R7K-DD for the treatment of autoimmune diseases is now being explored in our laboratory in animal models of rheumatoid arthritis, multiple sclerosis and type 1 diabetes.
Vaccine-induced long-term memory
B cell memory. Field trials of mucosal vaccines have shown that overall protection against infection can last for several years, even though intestinal IgA responses vanish after 6–9 months12,22,43. The longer duration of protection is thought to reflect the ability of IgA+ memory B cells to elicit a rapid recall response to a renewed exposure to the pathogen and thereby to improve the capacity for preventing infection12,113. These and other observations have demonstrated that mucosal vaccination can induce both memory IgA+ and memory IgG+ B cells, not only after oral immunization but also after intranasal and intravaginal vaccinations31,44,114,115,116,117.
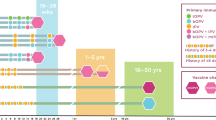
The development of memory IgA+ B cells following mucosal immunization has been studied in detail in mice12,26,108. The constitutive presence of germinal centres in Peyer's patches implies that these are important sites for generating long-lived plasma cells and memory B cells, although oral immunizations have not been shown to influence the overall number of germinal centres in the gut-associated lymphoid tissue (GALT)12. Cholera toxin-specific IgA+ plasma cells were found in the gut lamina propria for up to 6 months following oral immunization with this toxin118,119. Although at later time points toxin-specific IgA+ plasma cells had disappeared, memory B cells remained in the GALT and could be triggered by a single re-exposure to oral cholera toxin, resulting in a vigorous IgA+ plasma cell response in the lamina propria within 3 days18. Recently, we found evidence for extensive clonal relationships between antigen-specific secondary IgA+ B cells in multiple Peyer's patches, suggesting that B cells can migrate from one Peyer's patch to another120. Therefore, it appears that, following an oral priming immunization, activated IgA+ B cells can enter already existing germinal centres in multiple Peyer's patches, where they can continue proliferating and undergo affinity maturation (Fig. 3). By using already established germinal centres in multiple Peyer's patches, the IgA response can be synchronized, leading to the selection of only high-affinity B cell clones, provided that the antigen is present in the germinal centre. This explains why IgA+ plasma cells in the lamina propria and memory B cells in the GALT are clonally related and oligoclonal. Accordingly, and contrary to the trend in parenteral vaccination, mucosal vaccine strategies need to consider repeated immunizations — for example, using live vectors, slow-release systems or repeated administrations — to ensure sustained antigen stimulation and to thereby drive high-quality IgA+ B cell responses in the GALT.

In the gut, antigens are taken up by specialized microfold (M) cells overlaying the Peyer's patches. Subepithelial dome (SED)-resident dendritic cells (DCs) then take up and process these antigens, and naive CD4+ T cells are primed in the interfollicular space. T follicular helper (TFH) cells expressing B cell lymphoma 6 and CXC-chemokine receptor 5 (not shown) interact with cognate B cells at the B cell follicular border. Next, TFH cells colocalize with B cells in the B cell follicle in close proximity to a follicular dendritic cell (FDC) network, and this allows the formation of a germinal centre. In the germinal centre, the antigen-specific B cells undergo class-switching to IgA and somatic hypermutation to generate higher affinity antibodies. The resulting IgA+ long-lived plasma cells and memory B cells generated within the germinal centre leave the Peyer's patch through the efferent lymph and migrate to the mesenteric lymph nodes and then to the blood, from where plasma cells home to bone marrow and to effector sites in the lamina propria of the small and large intestine. The gut IgA response to oral vaccines is highly synchronized and strongly oligoclonal. Specific IgA+ B cells and plasma cells in both the gut-associated lymphoid tissue and non-organized lamina propria are clonally related, suggesting that multiple Peyer's patches are involved in the build-up of protective immunity. This can only occur if already existing germinal centres in multiple Peyer's patches are involved in the process of expanding and selecting high-affinity antigen-specific IgA+ B cells. In particular, the distribution of clonally related B cells to multiple Peyer's patches explains why repeated oral immunizations are needed to successfully drive high-affinity IgA responses in the gut lamina propria.
T cell memory. Memory CD4+ and CD8+ T cells are also generated by mucosal vaccination. Indeed, most T cells in the lamina propria are effector memory T cells121, and only low numbers of naive T cells are found there13. Although the function of these memory T cells in mucosal tissues is not fully understood, all the major effector and regulatory CD4+ T cell subsets, as well as CD8+ T cells, are present. In particular, T helper 17 (TH17) cells are frequent in the gut lamina propria, reflecting colonization by certain bacterial strains122,123. However, the exact protective functions of mucosal vaccine-induced memory TH1 and TH17 cells still await better definition (Box 2). Indeed, both protective effector functions and immunosuppressive functions of TH17 cells in the intestine have recently been documented124. Resistance against H. pylori or C. trachomatis infection relies on the presence of memory CD4+ T cells in the gastric and genital tracts, respectively, but the mechanisms of protection are much debated125,126. It appears, however, that tissue-resident memory T cell responses at mucosal sites can be activated in both an antigen-dependent and an antigen-independent manner, and in both cases this may limit pathogen replication127. A better understanding of the triggering factors and the function of these memory T cells for protection against pathogens is needed to allow for a more rational design of mucosal vaccines.
Hurdles in mucosal vaccine development
A major goal in mucosal vaccine research is the development of efficiently attenuated live vaccines with improved stability and safety. Directed gene mutagenesis is likely to successfully generate more stable vaccine vectors than random mutagenesis, which has been used previously. Good examples are the recent genetic engineering of the single-dose S. Typhi vaccines and the well-tolerated live S. flexneri 2a attenuated oral vaccine CVD1208 (Ref. 31). Indeed, CVD1208 was shown to be as effective as the more reactogenic and less mutated CVD1204 vaccine at stimulating lipopolysaccharide-specific memory B cells, indicating that safety may not have to be compromised to achieve an efficacious mucosal vaccine.
Furthermore, effective targeting of particulate vaccine formulations combined with strong immunomodulation should improve the efficacy of non-living subcomponent mucosal vaccines. New strategies to target M cells using lectins and antibodies, as well as further exploration of the use of ligands for TLRs and complement receptors, will undoubtedly be given special attention. In addition, the development of more effective mucosal adjuvants is of key importance. Prototype vaccines have successfully targeted DCs by using antibodies specific for cell-surface receptors such as DEC205 (also known as LY75) and DC-SIGN, or by using the natural ligands of these receptors, such as mannan and mannosylated liposomes128. Whether these targeting components will be effective in mucosal vaccines awaits full investigation. Moreover, a promising DC-specific target that was recently identified is C-type lectin domain family 9 member A (CLEC9A), activation of which stimulates antibody production129. Targeting derivatives of mutant holotoxins specifically to DCs could be an attractive way forward. For example, we are currently testing the effect of targeting CTA1 to DC-specific cell-surface receptors. Targeting adjuvants to cells expressing Fc receptors for IgG (FcγRs) could be an alternative strategy. Interestingly, intranasally administered CTA1-DD–IgG immune complexes were shown to activate mast cells and to enhance antibody production via FcγRIIIA130, highlighting the role of mast cells in augmenting mucosal immune responses.
We currently have only imprecise correlates of protection for most mucosal vaccines113. For example, toxin-specific salivary IgA antibody titres and serum vibriocidal antibodies are considered markers of protection against V. cholerae, although we do not know why serum vibriocidal antibodies can protect against a strictly intestinal mucosal infection1,25. A systems biology approach could help to identify biological markers for mucosal vaccine efficacy. Systems biology offers a new strategy for vaccine design that is based on understanding the molecular network that is activated by vaccination. Using functional genomics, specific molecular signatures of individual vaccines can be identified and used as predictors of vaccine efficacy131,132,133,134,135. In addition to complementing the conventional approaches to vaccine assessments, this approach investigates more general correlates of protection (beyond antigen-specific responses), including additional protective measures in the target tissues. To this end, complex tissue-specific interplay between epithelial cells, effector lymphocytes, inflammatory cells and nerve cells may be revealed, and such interactions could represent key protective measures that are involved, for example, in antitoxin immune responses in the intestine46,49. With this information, vaccine developers also hope to identify the very early immune events that occur after vaccination, in particular those crucial for memory development. Ultimately, this information will help our understanding of the mechanisms that underlie successful mucosal vaccination. By incorporating genomics, transcriptomics, proteomics, metabolomics and mathematical modelling with studies of immune protection in humans and in experimental animal models, we could gain a better understanding of the efficiency of mucosal vaccination136. Finally, a continuous exchange of information between preclinical research and clinical vaccine development is much needed to secure improved formulations, adjuvants and delivery protocols for the next generation of effective mucosal vaccines.
References
Holmgren, J. & Czerkinsky, C. Mucosal immunity and vaccines. Nature Med. 11, S45–S53 (2005). An excellent overview of the field with a special focus on oral vaccines.
Neutra, M. R. & Kozlowski, P. A. Mucosal vaccines: the promise and the challenge. Nature Rev. Immunol. 6, 148–158 (2006).
Levine, M. M. Immunogenicity and efficacy of oral vaccines in developing countries: lessons from a live cholera vaccine. BMC Biol. 8, 129 (2010).
Walker, R. I. Considerations for development of whole cell bacterial vaccines to prevent diarrheal diseases in children in developing countries. Vaccine 23, 3369–3385 (2005).
Levine, M. M. & Dougan, G. Optimism over vaccines administered via mucosal surfaces. Lancet 351, 1375–1376 (1998).
Yuki, Y. & Kiyono, H. Mucosal vaccines: novel advances in technology and delivery. Expert Rev. Vaccines 8, 1083–1097 (2009).
Amorij, J. P., Hinrichs, W., Frijlink, H. W., Wilschut, J. C. & Huckriede, A. Needle-free influenza vaccination. Lancet Infect. Dis. 10, 699–711 (2010).
Burt, D. et al. Proteosome-adjuvanted intranasal influenza vaccines: advantages, progress and future considerations. Expert Rev. Vaccines 10, 365–375 (2011).
Carter, N. J. & Curran, M. P. Live attenuated influenza vaccine (FluMist; Fluenz): a review of its use in the prevention of seasonal influenza in children and adults. Drugs 71, 1591–1622 (2011). An updated review of the current status of the FluMist vaccine.
Langley, J. M. et al. A nasally administered trivalent inactivated influenza vaccine is well tolerated, stimulates both mucosal and systemic immunity, and potentially protects against influenza illness. Vaccine 29, 1921–1928 (2011).
Brandtzaeg, P. Function of mucosa-associated lymphoid tissue in antibody formation. Immunol. Invest. 39, 303–355 (2010). An excellent overview of the mucosal immune system and its regulatory mechanisms.
Brandtzaeg, P. Induction of secretory immunity and memory at mucosal surfaces. Vaccine 25, 5467–5484 (2007).
Sheridan, B. S. & Lefrancois, L. Regional and mucosal memory T cells. Nature Immunol. 12, 485–491 (2011).
Mora, J. R. & von Andrian, U. H. Role of retinoic acid in the imprinting of gut-homing IgA-secreting cells. Semin. Immunol. 21, 28–35 (2009). This review details the role of retinoic acid in the gut-homing ability of IgA+ B cells.
Kiyono, H. & Fukuyama, S. NALT- versus Peyer's-patch-mediated mucosal immunity. Nature Rev. Immunol. 4, 699–710 (2004).
Mowat, A. M., Millington, O. R. & Chirdo, F. G. Anatomical and cellular basis of immunity and tolerance in the intestine. J. Pediatr. Gastroenterol. Nutr. 39, S723–S724 (2004).
Brandtzaeg, P. Update on mucosal immunoglobulin A in gastrointestinal disease. Curr. Opin. Gastroenterol. 26, 554–563 (2010).
Bevan, M. J. Memory T cells as an occupying force. Eur. J. Immunol. 41, 1192–1195 (2011).
Blaschitz, C. & Raffatellu, M. Th17 cytokines and the gut mucosal barrier. J. Clin. Immunol. 30, 196–203 (2010).
Feng, T. & Elson, C. O. Adaptive immunity in the host–microbiota dialog. Mucosal Immunol. 4, 15–21 (2011). This paper highlights the various elements in the interaction between the microbiota and host T cells in gut homeostasis.
Slack, E. et al. Innate and adaptive immunity cooperate flexibly to maintain host–microbiota mutualism. Science 325, 617–620 (2009). An elegant study on the mutualism between the microbiota and the host with special reference to the role of TLR signalling.
Czerkinsky, C. & Holmgren, J. Enteric vaccines for the developing world: a challenge for mucosal immunology. Mucosal Immunol. 2, 284–287 (2009).
Manicassamy, S. & Pulendran, B. Modulation of adaptive immunity with Toll-like receptors. Semin. Immunol. 21, 185–193 (2009).
Tucker, S. N., Tingley, D. W. & Scallan, C. D. Oral adenoviral-based vaccines: historical perspective and future opportunity. Expert Rev. Vaccines 7, 25–31 (2008).
Pasetti, M. F., Simon, J. K., Sztein, M. B. & Levine, M. M. Immunology of gut mucosal vaccines. Immunol. Rev. 239, 125–148 (2011). An outstanding review of the field of oral vaccines.
Lycke, N. in Vaccine adjuvants and delivery systems (ed. Singh, M.) 53–79 (John Wiley & Sons, 2007).
Greenberg, H. B. & Estes, M. K. Rotaviruses: from pathogenesis to vaccination. Gastroenterology 136, 1939–1951 (2009). A very comprehensive report on the background and efforts to develop rotavirus vaccines.
Ruiz-Palacios, G. M. et al. Safety and efficacy of an attenuated vaccine against severe rotavirus gastroenteritis. N. Engl. J. Med. 354, 11–22 (2006).
Kirkpatrick, B. D. et al. Evaluation of Salmonella enterica serovar Typhi (Ty2 aroC-ssaV-) M01ZH09, with a defined mutation in the Salmonella pathogenicity island 2, as a live, oral typhoid vaccine in human volunteers. Vaccine 24, 116–123 (2006).
Li, R., Lim, A. & Alonso, S. Attenuated Bordetella pertussis BPZE1 as a live vehicle for heterologous vaccine antigens delivery through the nasal route. Bioeng. Bugs 2, 315–319 (2011).
Simon, J. K. et al. Antigen-specific IgA B memory cell responses to Shigella antigens elicited in volunteers immunized with live attenuated Shigella flexneri 2a oral vaccine candidates. Clin. Immunol. 139, 185–192 (2011).
Tribble, D. et al. Safety and immunogenicity of a Shigella flexneri 2a Invaplex 50 intranasal vaccine in adult volunteers. Vaccine 28, 6076–6085 (2010).
El-Kamary, S. S. et al. Adjuvanted intranasal Norwalk virus-like particle vaccine elicits antibodies and antibody-secreting cells that express homing receptors for mucosal and peripheral lymphoid tissues. J. Infect. Dis. 202, 1649–1658 (2010).
Heinonen, S. et al. Effectiveness of inactivated influenza vaccine in children aged 9 months to 3 years: an observational cohort study. Lancet Infect. Dis. 11, 23–29 (2011).
DeRoeck, D. et al. Typhoid vaccination: the Asian experience. Expert Rev. Vaccines 7, 547–560 (2008).
Shin, S., Desai, S. N., Sah, B. K. & Clemens, J. D. Oral vaccines against cholera. Clin. Infect. Dis. 52, 1343–1349 (2011).
Zakay-Rones, Z. Human influenza vaccines and assessment of immunogenicity. Expert Rev. Vaccines 9, 1423–1439 (2010).
Madhi, S. A. et al. Effect of human rotavirus vaccine on severe diarrhea in African infants. N. Engl. J. Med. 362, 289–298 (2010).
Vesikari, T. et al. Safety and efficacy of a pentavalent human–bovine (WC3) reassortant rotavirus vaccine. N. Engl. J. Med. 354, 23–33 (2006). A classical study on the protective efficacy of a rotavirus vaccine.
Grimwood, K. & Forbes, D. A. Acute and persistent diarrhea. Pediatr. Clin. North Am. 56, 1343–1361 (2009).
Jaensson-Gyllenback, E. et al. Bile retinoids imprint intestinal CD103+ dendritic cells with the ability to generate gut-tropic T cells. Mucosal Immunol. 4, 438–447 (2011).
Cooper, P. J. et al. Impact of early life exposures to geohelminth infections on the development of vaccine immunity, allergic sensitization, and allergic inflammatory diseases in children living in tropical Ecuador: the ECUAVIDA birth cohort study. BMC Infect. Dis. 11, 184 (2011).
Holmgren, J. Actions of cholera toxin and the prevention and treatment of cholera. Nature 292, 413–417 (1981).
Quiding, M. et al. Intestinal immune responses in humans. Oral cholera vaccination induces strong intestinal antibody responses and interferon-γ production and evokes local immunological memory. J. Clin. Invest. 88, 143–148 (1991). One of the first convincing studies demonstrating the ability to generate immunological memory through oral vaccination.
Shamsuzzaman, S. et al. Robust gut associated vaccine-specific antibody-secreting cell responses are detected at the mucosal surface of Bangladeshi subjects after immunization with an oral killed bivalent V. cholerae O1/O139 whole cell cholera vaccine: comparison with other mucosal and systemic responses. Vaccine 27, 1386–1392 (2009).
Svennerholm, A. M. & Holmgren, J. Oral vaccines against cholera and enterotoxigenic Escherichia coli diarrhea. Adv. Exp. Med. Biol. 371B, 1623–1628 (1995).
Ali, M. et al. Herd immunity conferred by killed oral cholera vaccines in Bangladesh: a reanalysis. Lancet 366, 44–49 (2005). An excellent description of herd immunity and its consequences for vaccine-induced protection.
Wijburg, O. L. et al. Innate secretory antibodies protect against natural Salmonella typhimurium infection. J. Exp. Med. 203, 21–26 (2006).
Czerkinsky, C. & Holmgren, J. Mucosal delivery routes for optimal immunization: targeting immunity to the right tissues. Curr. Top. Microbiol. Immunol. 354, 1–18 (2010).
Chadwick, S., Kriegel, C. & Amiji, M. Nanotechnology solutions for mucosal immunization. Adv. Drug Deliv. Rev. 62, 394–407 (2010). A comprehensive and detailed description of the novel nanotechnologies that may change mucosal vaccine development.
Peek, L. J., Middaugh, C. R. & Berkland, C. Nanotechnology in vaccine delivery. Adv. Drug Deliv. Rev. 60, 915–928 (2008).
Brandtzaeg, P. Potential of nasopharynx-associated lymphoid tissue for vaccine responses in the airways. Am. J. Respir. Crit. Care Med. 183, 1595–1604 (2011).
Jabbal-Gill, I. Nasal vaccine innovation. J. Drug Target. 18, 771–786 (2010). A review of current nasal vaccine strategies.
Kreijtz, J. H., Fouchier, R. A. & Rimmelzwaan, G. F. Immune responses to influenza virus infection. Virus Res. 162, 19–30 (2011).
Bakke, H. et al. Oral spray immunization may be an alternative to intranasal vaccine delivery to induce systemic antibodies but not nasal mucosal or cellular immunity. Scand. J. Immunol. 63, 223–231 (2006).
Djupesland, P. G. & Skretting, A. Nasal deposition and clearance in man: comparison of a bidirectional powder device and a traditional liquid spray pump. J. Aerosol Med. Pulm. Drug Deliv. 17 Jan 2012 (doi:10.1089/jamp.2011.0924). This paper reports on the development of nasal vaccine devices.
Mestecky, J., Alexander, R. C., Wei, Q. & Moldoveanu, Z. Methods for evaluation of humoral immune responses in human genital tract secretions. Am. J. Reprod. Immunol. 65, 361–367 (2011).
Bomsel, M. et al. Immunization with HIV-1 gp41 subunit virosomes induces mucosal antibodies protecting nonhuman primates against vaginal SHIV challenges. Immunity 34, 269–280 (2011).
Lopalco, L. & Bomsel, M. Protecting the initial site of viral entry: an alternative HIV vaccine target. Expert Rev. Vaccines 10, 1253–1256 (2011). This article outlines successful approaches to mucosal HIV vaccination.
Marks, E., Helgeby, A., Andersson, J. O., Schon, K. & Lycke, N. Y. Elicitation of CD4+ T cell immunity in the female genital tract is critically dependent on local mucosal immunization. Eur. J. Immunol. 41, 2642–2653 (2011). The study shows that CTA1-DD cannot bind to the cells of the central nervous tissues following intranasal administration.
Wegmann, F. Mucosally-targeted HIV-1 vaccines. Hum. Vaccin. 7, 982–985 (2011).
Lewis, D. J. et al. Phase I randomised clinical trial of an HIV-1(CN54), clade C, trimeric envelope vaccine candidate delivered vaginally. PLoS ONE 6, e25165 (2011).
Czerkinsky, C., Cuburu, N., Kweon, M. N., Anjuere, F. & Holmgren, J. Sublingual vaccination. Hum. Vaccin. 7, 110–114 (2011).
Kweon, M. N. Sublingual mucosa: a new vaccination route for systemic and mucosal immunity. Cytokine 54, 1–5 (2011). This article critically reviews various aspects of sublingual vaccination.
Carmichael, J. R., Pal, S., Tifrea, D. & de la Maza, L. M. Induction of protection against vaginal shedding and infertility by a recombinant Chlamydia vaccine. Vaccine 29, 5276–5283 (2011).
Cho, H. J. et al. Enhanced humoral and cellular immune responses after sublingual immunization against human papillomavirus 16 L1 protein with adjuvants. Vaccine 28, 2598–2606 (2010).
Domm, W. et al. Robust antigen-specific humoral immune responses to sublingually delivered adenoviral vectors encoding HIV-1 Env: association with mucoadhesion and efficient penetration of the sublingual barrier. Vaccine 29, 7080–7089 (2011).
Song, J. H. et al. Sublingual vaccination with influenza virus protects mice against lethal viral infection. Proc. Natl Acad. Sci. USA 105, 1644–1649 (2008).
Flach, C. F. et al. A truncated form of HpaA is a promising antigen for use in a vaccine against Helicobacter pylori. Vaccine 29, 1235–1241 (2011).
Huo, Z. et al. Systemic and mucosal immune responses to sublingual or intramuscular human papilloma virus antigens in healthy female volunteers. PLoS ONE 7, e33736 (2012).
van der Lubben, I. M., Verhoef, J. C., Borchard, G. & Junginger, H. E. Chitosan for mucosal vaccination. Adv. Drug Deliv. Rev. 52, 139–144 (2001).
Garg, N. K., Mangal, S., Khambete, H. & Tyagi, R. K. Mucosal delivery of vaccines: role of mucoadhesive/biodegradable polymers. Recent Pat. Drug Deliv. Formul. 4, 114–128 (2010).
Wassen, L., Schon, K., Holmgren, J., Jertborn, M. & Lycke, N. Local intravaginal vaccination of the female genital tract. Scand. J. Immunol. 44, 408–414 (1996). This study demonstrates the efficiency of intravaginal vaccination.
Eliasson, D. G. et al. A novel non-toxic combined CTA1-DD and ISCOMS adjuvant vector for effective mucosal immunization against influenza virus. Vaccine 29, 3951–3961 (2011). This study describes in detail the benefits of combined vectors for mucosal vaccination.
Ishii, M. & Kojima, N. Mucosal adjuvant activity of oligomannose-coated liposomes for nasal immunization. Glycoconj. J. 27, 115–123 (2010).
Schneider-Ohrum, K. & Ross, T. M. Virus-like particles for antigen delivery at mucosal surfaces. Curr. Top. Microbiol. Immunol. 354, 53–73 (2011).
Kuolee, R. & Chen, W. M cell-targeted delivery of vaccines and therapeutics. Expert Opin. Drug Deliv. 5, 693–702 (2008).
Kelsall, B. Recent progress in understanding the phenotype and function of intestinal dendritic cells and macrophages. Mucosal Immunol. 1, 460–469 (2008). An excellent description of DC subsets in the gut immune system.
Azizi, A., Kumar, A., Diaz-Mitoma, F. & Mestecky, J. Enhancing oral vaccine potency by targeting intestinal M cells. PLoS Pathog. 6, e1001147 (2010).
Galli, G. et al. Fast rise of broadly cross-reactive antibodies after boosting long-lived human memory B cells primed by an MF59 adjuvanted prepandemic vaccine. Proc. Natl Acad. Sci. USA 106, 7962–7967 (2009).
Dormitzer, P. R. et al. Influenza vaccine immunology. Immunol. Rev. 239, 167–177 (2011).
Elson, C. O. & Dertzbaugh, M. T. in Mucosal Immunology (eds Mestecky, J. et al.) 967–986 (Elsevier Academic Press, 2005).
Lambrecht, B. N., Kool, M., Willart, M. A. & Hammad, H. Mechanism of action of clinically approved adjuvants. Curr. Opin. Immunol. 21, 23–29 (2009). A clear and concise description of current vaccine adjuvants and their mechanisms of action.
Harandi, A. M. & Medaglini, D. Mucosal adjuvants. Curr. HIV Res. 8, 330–335 (2010).
Blaas, S. H., Stieber-Gunckel, M., Falk, W., Obermeier, F. & Rogler, G. CpG-oligodeoxynucleotides stimulate immunoglobulin A secretion in intestinal mucosal B cells. Clin. Exp. Immunol. 155, 534–540 (2009).
Uematsu, S. et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nature Immunol. 9, 769–776 (2008). An important study demonstrating the role of TLR5 signalling in gut IgA-mediated immunity.
Boyaka, P. N. et al. Oral QS-21 requires early IL-4 help for induction of mucosal and systemic immunity. J. Immunol. 166, 2283–2290 (2001).
Christensen, D. et al. Liposome-based cationic adjuvant formulations (CAF): past, present, and future. J. Liposome Res. 19, 2–11 (2009).
Kool, M. et al. Cutting edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J. Immunol. 181, 3755–3759 (2008).
Malyala, P. et al. The potency of the adjuvant, CpG oligos, is enhanced by encapsulation in PLG microparticles. J. Pharm. Sci. 97, 1155–1164 (2008).
Freytag, L. C., Clements, J. D., Grdic Eliasson, D. & Lycke, N. in New Generation Vaccines (ed. Levine, M.M.) 273–283 (Informa, 2010). This article reviews the role of the bacterial enterotoxins cholera toxin and E. coli heat-labile enterotoxin in mucosal vaccine efficacy.
Liang, S. & Hajishengallis, G. Heat-labile enterotoxins as adjuvants or anti-inflammatory agents. Immunol. Invest. 39, 449–467 (2010).
Spangler, B. D. Structure and function of cholera toxin and the related Escherichia coli heat-labile enterotoxin. Microbiol. Rev. 56, 622–647 (1992).
Fahlen-Yrlid, L. et al. CD11chigh dendritic cells are essential for activation of CD4+ T cells and generation of specific antibodies following mucosal immunization. J. Immunol. 183, 5032–5041 (2009).
Connell, T. D. Cholera toxin, LT-I, LT-IIa and LT-IIb: the critical role of ganglioside binding in immunomodulation by type I and type II heat-labile enterotoxins. Expert Rev. Vaccines 6, 821–834 (2007).
Snider, D. P. The mucosal adjuvant activities of ADP-ribosylating bacterial enterotoxins. Crit. Rev. Immunol. 15, 317–348 (1995).
Anosova, N. G. et al. Cholera toxin, E. coli heat-labile toxin, and non-toxic derivatives induce dendritic cell migration into the follicle-associated epithelium of Peyer's patches. Mucosal Immunol. 1, 59–67 (2008).
Fujihashi, K., Koga, T., van Ginkel, F. W., Hagiwara, Y. & McGhee, J. R. A dilemma for mucosal vaccination: efficacy versus toxicity using enterotoxin-based adjuvants. Vaccine 20, 2431–2438 (2002).
Glueck, R. Pre-clinical and clinical investigation of the safety of a novel adjuvant for intranasal immunization. Vaccine 20, S42–S44 (2001).
Mutsch, M. et al. Use of the inactivated intranasal influenza vaccine and the risk of Bell's palsy in Switzerland. N. Engl. J. Med. 350, 896–903 (2004). An informative example of holotoxin-mediated side effects after nasal vaccination.
Levine, M. M. et al. Evaluation in humans of attenuated Vibrio cholerae El Tor Ogawa strain Texas Star-SR as a live oral vaccine. Infect. Immun. 43, 515–522 (1984).
Pizza, M. et al. Mucosal vaccines: non toxic derivatives of LT and CT as mucosal adjuvants. Vaccine 19, 2534–2541 (2001). A classical review of mucosal vaccine strategies based on holotoxins or derivatives thereof.
Summerton, N. A. et al. Toward the development of a stable, freeze-dried formulation of Helicobacter pylori killed whole cell vaccine adjuvanted with a novel mutant of Escherichia coli heat-labile toxin. Vaccine 28, 1404–1411 (2010).
Brereton, C. F. et al. Escherichia coli heat-labile enterotoxin promotes protective Th17 responses against infection by driving innate IL-1 and IL-23 production. J. Immunol. 186, 5896–5906 (2011). Mechanistic studies of E. coli heat-labile enterotoxin and LTK63 and their role for augmenting T H 17 cell responses.
Lewis, D. J. et al. Transient facial nerve paralysis (Bell's palsy) following intranasal delivery of a genetically detoxified mutant of Escherichia coli heat labile toxin. PLoS ONE 4, e6999 (2009).
Hagiwara, Y. et al. A second generation of double mutant cholera toxin adjuvants: enhanced immunity without intracellular trafficking. J. Immunol. 177, 3045–3054 (2006).
Agren, L. C., Ekman, L., Lowenadler, B. & Lycke, N. Y. Genetically engineered nontoxic vaccine adjuvant that combines B cell targeting with immunomodulation by cholera toxin A1 subunit. J. Immunol. 158, 3936–3946 (1997). The first description of the CTA1-DD adjuvant.
Lycke, N. & Bemark, M. Mucosal adjuvants and long-term memory development with special focus on CTA1-DD and other ADP-ribosylating toxins. Mucosal Immunol. 3, 556–566 (2010).
Eriksson, A. M., Schon, K. M. & Lycke, N. Y. The cholera toxin-derived CTA1-DD vaccine adjuvant administered intranasally does not cause inflammation or accumulate in the nervous tissues. J. Immunol. 173, 3310–3319 (2004).
Sundling, C. et al. CTA1-DD adjuvant promotes strong immunity against human immunodeficiency virus type 1 envelope glycoproteins following mucosal immunization. J. Gen. Virol. 89, 2954–2964 (2008).
Hasselberg, A., Schon, K., Tarkowski, A. & Lycke, N. Role of CTA1R7K-COL-DD as a novel therapeutic mucosal tolerance-inducing vector for treatment of collagen-induced arthritis. Arthritis Rheum. 60, 1672–1682 (2009). The first demonstration that mutated CTA1-DD could work to tolerize CD4+ T cells in the context of autoimmune conditions.
Hasselberg, A., Ekman, L., Yrlid, L. F., Schon, K. & Lycke, N. Y. ADP-ribosylation controls the outcome of tolerance or enhanced priming following mucosal immunization. J. Immunol. 184, 2776–2784 (2010).
Plotkin, S. A. Correlates of protection induced by vaccination. Clin. Vaccine Immunol. 17, 1055–1065 (2010). A critical review of the correlates of protection in mucosal vaccines.
Alam, M. M. et al. Antigen-specific memory B-cell responses in Bangladeshi adults after one- or two-dose oral killed cholera vaccination and comparison with responses in patients with naturally acquired cholera. Clin. Vaccine Immunol. 18, 844–850 (2011).
Di Fabio, S. et al. Vaginal immunization of Cynomolgus monkeys with Streptococcus gordonii expressing HIV-1 and HPV 16 antigens. Vaccine 16, 485–492 (1998).
Rudenko, L. et al. Live attenuated pandemic influenza vaccine: clinical studies on A/17/California/2009/38 (H1N1) and licensing of the Russian-developed technology to WHO for pandemic influenza preparedness in developing countries. Vaccine 29, A40–A44 (2011).
Tengvall, S., Lundgren, A., Quiding-Jarbrink, M. & Svennerholm, A. M. BAFF, stimulatory DNA and IL-15 stimulates IgA+ memory B cells and provides a novel approach for analysis of memory responses to mucosal vaccines. Vaccine 28, 5445–5450 (2010).
Lycke, N. & Holmgren, J. Long-term cholera antitoxin memory in the gut can be triggered to antibody formation associated with protection within hours of an oral challenge immunization. Scand. J. Immunol. 25, 407–412 (1987).
Lycke, N. & Holmgren, J. Adoptive transfer of gut mucosal antitoxin memory by isolated B cells 1 year after oral immunization with cholera toxin. Infect. Immun. 57, 1137–1141 (1989).
Bergqvist, P. et al. Re-utilization of germinal centers in multiple Peyer's patches results in highly synchronized, oligoclonal, and affinity-matured gut IgA responses. Mucosal Immunol. 11 Jul 2012 (doi:10.1038/mi.2012.56). The first study to provide evidence that antigen-specific gut IgA responses expand in multiple Peyer's patches by re-using already existing germinal centres.
Sallusto, F., Geginat, J. & Lanzavecchia, A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu. Rev. Immunol. 22, 745–763 (2004).
McAleer, J. P. & Kolls, J. K. Mechanisms controlling Th17 cytokine expression and host defense. J. Leukoc. Biol. 90, 263–270 (2011).
Ohnmacht, C. et al. Intestinal microbiota, evolution of the immune system and the bad reputation of pro-inflammatory immunity. Cell. Microbiol. 13, 653–659 (2011).
Esplugues, E. et al. Control of TH17 cells occurs in the small intestine. Nature 475, 514–518 (2011).
Howie, S. E., Horner, P. J., Horne, A. W. & Entrican, G. Immunity and vaccines against sexually transmitted Chlamydia trachomatis infection. Curr. Opin. Infect. Dis. 24, 56–61 (2011).
Velin, D. & Michetti, P. Advances in vaccination against Helicobacter pylori. Expert Rev. Gastroenterol. Hepatol. 4, 157–166 (2010). This article discusses various aspects of H. pylori vaccine development that may point to more successful approaches.
Woodland, D. L. & Kohlmeier, J. E. Migration, maintenance and recall of memory T cells in peripheral tissues. Nature Rev. Immunol. 9, 153–161 (2009).
Shortman, K., Lahoud, M. H. & Caminschi, I. Improving vaccines by targeting antigens to dendritic cells. Exp. Mol. Med. 41, 61–66 (2009). This article describes in detail DC-targeting strategies.
Caminschi, I. et al. The dendritic cell subtype-restricted C-type lectin Clec9A is a target for vaccine enhancement. Blood 112, 3264–3273 (2008).
Fang, Y., Larsson, L., Mattsson, J., Lycke, N. & Xiang, Z. Mast cells contribute to the mucosal adjuvant effect of CTA1-DD after IgG-complex formation. J. Immunol. 185, 2935–2941 (2010).
Nakaya, H. I. & Pulendran, B. Systems vaccinology: its promise and challenge for HIV vaccine development. Curr. Opin. HIV AIDS 7, 24–31 (2012).
Six, A., Bellier, B., Thomas-Vaslin, V. & Klatzmann, D. Systems biology in vaccine design. Microb. Biotechnol. 5, 295–304 (2011).
White, O. J. et al. A genomics-based approach to assessment of vaccine safety and immunogenicity in children. Vaccine 30, 1865–1874 (2012).
Pulendran, B. & Ahmed, R. Translating innate immunity into immunological memory: implications for vaccine development. Cell 124, 849–863 (2006).
Salmi, M. & Jalkanen, S. Lymphocyte homing to the gut: attraction, adhesion, and commitment. Immunol. Rev. 206, 100–113 (2005).
Gorfu, G., Rivera-Nieves, J. & Ley, K. Role of β7 integrins in intestinal lymphocyte homing and retention. Curr. Mol. Med. 9, 836–850 (2009).
Mora, J. R. & von Andrian, U. H. Differentiation and homing of IgA-secreting cells. Mucosal Immunol. 1, 96–109 (2008).
del Rio, M. L., Bernhardt, G., Rodriguez-Barbosa, J. I. & Forster, R. Development and functional specialization of CD103+ dendritic cells. Immunol. Rev. 234, 268–281 (2010).
Schulz, O. et al. Intestinal CD103+, but not CX3CR1+, antigen sampling cells migrate in lymph and serve classical dendritic cell functions. J. Exp. Med. 206, 3101–3114 (2009).
Scott, C. L., Aumeunier, A. M. & Mowat, A. M. Intestinal CD103+ dendritic cells: master regulators of tolerance? Trends Immunol. 32, 412–419 (2011). A current update on the role of an important DC subset for mucosal immunity and tolerance.
Semmrich, M. et al. Directed antigen targeting in vivo identifies a role for CD103+ dendritic cells in both tolerogenic and immunogenic T-cell responses. Mucosal Immunol. 5, 150–160 (2011). This study describes an effective strategy to target antigens to the CD103+ DC subset.
Cha, H. R. et al. Mucosa-associated epithelial chemokine/CCL28 expression in the uterus attracts CCR10+ IgA plasma cells following mucosal vaccination via estrogen control. J. Immunol. 187, 3044–3052 (2011).
Lin, Y., Slight, S. R. & Khader, S. A. Th17 cytokines and vaccine-induced immunity. Semin. Immunopathol. 32, 79–90 (2010).
Khader, S. A., Gaffen, S. L. & Kolls, J. K. Th17 cells at the crossroads of innate and adaptive immunity against infectious diseases at the mucosa. Mucosal Immunol. 2, 403–411 (2009).
Belyakov, I. M. & Ahlers, J. D. What role does the route of immunization play in the generation of protective immunity against mucosal pathogens? J. Immunol. 183, 6883–6892 (2009).
Mattsson, J. et al. Complement activation and complement receptors on follicular dendritic cells are critical for the function of a targeted adjuvant. J. Immunol. 187, 3641–3652 (2011). This study demonstrates that adjuvants can bind to and activate follicular DCs.
Bemark, M. et al. A unique role of the cholera toxin A1-DD adjuvant for long-term plasma and memory B cell development. J. Immunol. 186, 1399–1410 (2011).
Dhere, R. et al. A pandemic influenza vaccine in India: from strain to sale within 12 months. Vaccine 29, A16–A21 (2011).
Aylward, R. B. Eradicating polio: today's challenges and tomorrow's legacy. Ann. Trop. Med. Parasitol. 100, 401–413 (2006).
Sabin, A. B. Oral poliovirus vaccine: history of its development and use and current challenge to eliminate poliomyelitis from the world. J. Infect. Dis. 151, 420–436 (1985).
Ferreccio, C., Levine, M. M., Rodriguez, H. & Contreras, R. Comparative efficacy of two, three, or four doses of TY21a live oral typhoid vaccine in enteric-coated capsules: a field trial in an endemic area. J. Infect. Dis. 159, 766–769 (1989).
Shafique, M., Wilschut, J. & de Haan, A. Induction of mucosal and systemic immunity against respiratory syncytial virus by inactivated virus supplemented with TLR9 and NOD2 ligands. Vaccine 30, 597–606 (2012).
Elson, C. O. Cholera toxin and its subunits as potential oral adjuvants. Curr. Top. Microbiol. Immunol. 146, 29–33 (1989).
Agren, L. C., Ekman, L., Lowenadler, B., Nedrud, J. G. & Lycke, N. Y. Adjuvanticity of the cholera toxin A1-based gene fusion protein, CTA1-DD, is critically dependent on the ADP-ribosyltransferase and Ig-binding activity. J. Immunol. 162, 2432–2440 (1999).
Muhammad, A., Champeimont, J., Mayr, U. B., Lubitz, W. & Kudela, P. Bacterial ghosts as carriers of protein subunit and DNA-encoded antigens for vaccine applications. Expert Rev. Vaccines 11, 97–116 (2012).
Thompson, A. L. et al. Maximal adjuvant activity of nasally delivered IL-1α requires adjuvant-responsive CD11c+ cells and does not correlate with adjuvant-induced in vivo cytokine production. J. Immunol. 188, 2834–2846 (2012).
Winstone, N. et al. Enhanced control of pathogenic simian immunodeficiency virus SIVmac239 replication in macaques immunized with an interleukin-12 plasmid and a DNA prime–viral vector boost vaccine regimen. J. Virol. 85, 9578–9587 (2011).
Acknowledgements
I would like to thank all past and present members of my research group for their devotion and hard work. Special thanks to M. Bemark for collaborative work on gut IgA B cell immunity and to B. Löwenadler, co-inventor of the CTA1-DD adjuvant. Lastly, I would like to thank my mentor and friend W. Strober for always giving generous support and engaging in helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no competing financial interests.
Glossary
- Adjuvant
-
An agent that enhances the immunogenicity of an antigen. Etymologically, the term adjuvant comes from the latin verb adjuvare, which means 'to help'.
- Mucosal tolerance
-
Specific unresponsiveness to systemic challenges after a prior mucosal exposure to the antigen.
- Subcomponent vaccines
-
Non-living vaccines that consist of whole cell walls or complete protein structures.
- Tropical barrier
-
An inability to fully respond to mucosal vaccination, especially oral vaccination, in developing countries.
- Herd immunity
-
Protection against spread of infection in a population based on a critical mass of successfully vaccinated individuals.
- Biodegradable microparticles and nanoparticles
-
Particles that are 1–1000 nm in size and made from materials that self-disintegrate, such as poly(lactide-co-glycolide). Biodegradable microparticles and nanoparticles have been extensively used in different mucosal vaccine formulations and are gaining increased attention for their ability to serve as viable carriers for site-specific delivery of vaccines. They offer enhanced biocompatibility, superior vaccine encapsulation and convenient release profiles for mucosal vaccines.
- Follicle-associated epithelium
-
(FAE). The epithelium that overlies mucosal lymphoid tissues, such as the Peyer's patches and isolated lymphoid follicles in the intestine. Lymphoid tissues induce the differentiation of normal intestinal epithelium into FAE, which is specialized in antigen capture and transport.
- Virus-like particles
-
Virus envelopes without nucleic acid that are used for the delivery of vaccine antigens.
- Bacterial ghosts
-
Empty cell envelopes of Gram-negative bacteria devoid of cytoplasmic content and chromosomal and plasmid DNA that are used for the delivery of vaccine antigens.
- Immunostimulating complexes
-
(ISCOMs). Spherical open cage-like structures (typically 40 nm in diameter) that spontaneously form from a mixture of cholesterol, phospholipids and Quillaja saponins. ISCOMs are used as vaccine adjuvants to induce an enhanced immune response and protection.
- Epitope spreading
-
The process by which an antibody response to one epitope of an antigen leads to the production of antibodies specific for other epitopes of the same antigen, or for epitopes of entirely unrelated antigens. This results from the internalization of the whole antigen and the subsequent display of a range of peptides derived from that antigen, leading to the generation of T cells with different epitope specificities. Simultaneous processing of two unrelated antigens by an antigen-presenting cell can lead to the production of antibodies directed against both antigens.
- Follicular dendritic cells
-
(FDCs). Stromal cells that are crucial for the development of germinal centres in B cell follicles. The interaction between FDCs and B cells is thought to be essential for isotype switching and somatic hypermutation.
- Somatic hypermutation
-
The process by which point mutations occur in the heavy- or light-chain variable region gene segments, resulting in a change in the expressed protein, which may alter the affinity or specificity of the antibody for an antigen.
- Germinal centres
-
The structures that are formed by the expansion of populations of antigen-activated B cell blasts that have migrated into the follicles of lymph nodes. The B cells in these structures proliferate and their immunoglobulin genes undergo somatic hypermutation, before the cells leave as plasma cells or memory B cells.
- Affinity maturation
-
The mutation of antibody variable region genes followed by selection for higher affinity variants in the germinal centre leads to an increase in average antibody affinity as an immune response progresses.
Rights and permissions
About this article
Cite this article
Lycke, N. Recent progress in mucosal vaccine development: potential and limitations. Nat Rev Immunol 12, 592–605 (2012). https://doi.org/10.1038/nri3251
Published:
Issue Date:
DOI: https://doi.org/10.1038/nri3251
This article is cited by
-
Advances of nanoparticles in transmucosal drug delivery
Nano Research (2024)
-
Improving expression and assembly of difficult-to-express heterologous proteins in Saccharomyces cerevisiae by culturing at a sub-physiological temperature
Microbial Cell Factories (2023)
-
Recombinant lipidated FLIPr effectively enhances mucosal and systemic immune responses for various vaccine types
npj Vaccines (2023)
-
Novel intranasal vaccine targeting SARS-CoV-2 receptor binding domain to mucosal microfold cells and adjuvanted with TLR3 agonist Riboxxim™ elicits strong antibody and T-cell responses in mice
Scientific Reports (2023)
-
An updated review on oral protein-based antigen vaccines efficiency and delivery approaches: a special attention to infectious diseases
Archives of Microbiology (2023)
