
Abstract
Mucosal vaccines offer the potential to trigger robust protective immune responses at the predominant sites of pathogen infection. In principle, the induction of adaptive immunity at mucosal sites, involving secretory antibody responses and tissue-resident T cells, has the capacity to prevent an infection from becoming established in the first place, rather than only curtailing infection and protecting against the development of disease symptoms. Although numerous effective mucosal vaccines are in use, the major advances seen with injectable vaccines (including adjuvanted subunit antigens, RNA and DNA vaccines) have not yet been translated into licensed mucosal vaccines, which currently comprise solely live attenuated and inactivated whole-cell preparations. The identification of safe and effective mucosal adjuvants allied to innovative antigen discovery and delivery strategies is key to advancing mucosal vaccines. Significant progress has been made in resolving the mechanisms that regulate innate and adaptive mucosal immunity and in understanding the crosstalk between mucosal sites, and this provides valuable pointers to inform mucosal adjuvant design. In particular, increased knowledge on mucosal antigen-presenting cells, innate lymphoid cell populations and resident memory cells at mucosal sites highlights attractive targets for vaccine design. Exploiting these insights will allow new vaccine technologies to be leveraged to facilitate rational mucosal vaccine design for pathogens including severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and for cancer.
Similar content being viewed by others

Introduction
The global burden of mortality and morbidity associated with infectious diseases caused by mucosal pathogens remains unacceptably high. Indeed, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic provides a brutal reminder of the continual threat of novel mucosal infectious challenges, in addition to the threat posed by many widespread mucosal infections for which no or only suboptimal vaccines exist. Now more than ever, there is a clear focus on vaccine requirements for respiratory pathogens but, importantly, new and improved vaccines are also urgently needed for numerous enteric pathogens and other agents such as those causing sexually transmitted diseases (STDs) and oncogenic viruses that gain access through the mucosae.
Respiratory pathogens remain a prominent cause of global mortality, with lower respiratory tract infections constituting the fourth leading cause of death worldwide1. Lower respiratory tract infections are responsible for approximately 2.4 million deaths per annum, with Streptococcus pneumoniae, respiratory syncytial virus (RSV), Haemophilus influenzae B and influenza virus taking a particularly high toll on the young (<5 years old) and older people2. There is currently no approved vaccine for RSV infection, which is particularly prevalent in children and infants2,3,4, and although there are licensed vaccines targeting respiratory pathogens such as Mycobacterium tuberculosis, S. pneumoniae, Bordetella pertussis and influenza virus, improved vaccines for these pathogens are required to enhance suboptimal protection, particularly at the site of infection, and to increase coverage (Fig. 1). There are indications that innovative mucosal vaccine approaches offer promise for these infections. For example, live attenuated influenza vaccines given intranasally are now an integral part of influenza vaccination strategies with particular application to children5,6, intranasally administered B. pertussis vaccines have entered phase II trials7,8 (Supplementary Table 1) following successful phase I completion, and preclinical data investigating the intranasal delivery of Bacillus Calmette–Guérin for M. tuberculosis have yielded promising results9. The emergence of SARS-CoV-2 has firmly demonstrated how deadly and disruptive respiratory pathogens can be, with approximately 2.6 million deaths attributed to this pathogen at the time of writing10 and estimates that the SARS-CoV-2 pandemic will continue to stunt global economic growth in 2021, particularly in low-income countries11. Although an array of effective SARS-CoV-2 vaccines have been designed and implemented, challenges in mass production and deployment still provide an unmet need for global coverage (Fig. 1). New vaccines could help to circumvent these issues. In particular, orally delivered SARS-CoV-2 vaccines would be suited to global vaccination attempts, especially in lower-income countries, as these vaccines will not only allow for enhanced convenience and compliance but also the intestine may represent a viral target organ12. Indeed, the development of ‘universal’ mucosal vaccines targeting conserved antigens on coronaviruses13 and influenza viruses, although challenging, may be a viable option for prevention of future pandemics.

Respiratory, enteric and sexually transmitted infections constitute prominent causes of death worldwide, and this is exacerbated in low-income regions. Aetiological agents shown are vaccine targetable, but there remains an unmet need for new or improved vaccination approaches to address global vaccine coverage. Mucosal vaccination strategies hold promise to address this unmet need, providing more robust mucosal immunity and an alternative to parenteral vaccination. In addition to their centrality in the pathogenesis of infectious disease, mucosal tissues are frequent sites of tumour development and mucosal vaccination strategies may play a role in the prophylactic and therapeutic targeting of these malignancies. CRC, colorectal cancer; ETEC, enterotoxigenic Escherichia coli; p.a., per annum; RSV, respiratory syncytial disease; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Enteric pathogens causing diarrhoeal disease are the eighth leading cause of death worldwide, with children, in particular, at risk14. Among these, Shigella and enterotoxigenic Escherichia coli (ETEC) have an urgent vaccine requirement (Fig. 1). Enteric pathogens and associated acute and chronic infections have a stark impact on the livelihoods of at-risk individuals in lower-income countries. Aside from diarrhoeal disease, the impact of such infections on physical and cognitive development is becoming more apparent14, not only highlighting the need for vaccine development but also impacting how we determine vaccine efficacy. Lack of moderate-to-severe symptoms may not be an adequate correlate of protection — prevention of colonization and/or low-grade infection may be the crucial determinant. The World Health Organization (WHO) has endeavoured to end cholera by 2030 through implementation of widespread preventive measures, including vaccination15, providing a challenge to oral cholera vaccine manufacturers globally. This may be addressed through successful development of lower-cost alternative oral cholera vaccines such as Hillchol, which is currently under clinical evaluation16.
Mucosal vaccines targeting the genital tract have the potential to combat STDs and local tumours, which is important given that cervical cancer represents the fourth most common cancer in women17. Clearly, there is an enormous need for an effective HIV vaccine and given the intestinal tropism of the virus18, mucosal vaccine strategies are warranted19. Additionally, the emergence of multidrug-resistant STDs is of concern and could be combatted through preventive mucosal vaccine strategies.
Whereas most licensed vaccines are currently administered by injection, mucosal vaccines can outperform parenteral vaccination strategies in eliciting protective mucosal immune responses that block infection or transmission20,21. The nature of the infection must be considered from the outset in designing mucosal vaccines or, indeed, in assessing whether targeting the mucosal route is necessary. For example, in the case of enteric pathogens, the infections may be invasive (as is seen in typhoid and polio), partially/locally invasive (as seen in shigellosis) or strictly mucosal (as seen for cholera and ETEC infections)22,23. This will impact on whether a systemic immune response is an appropriate objective, or whether predominantly mucosal or both mucosal and systemic immunity would be more effective. In this context, the nature of the mucosal surface (for example, the uninflamed small intestine versus the lower respiratory tract) will influence the accessibility of circulating antibodies, the nature of the dominant antibody isotype and the transport mechanism governing access of antibodies to mucosal infectious pathogens24,25.
Strong mucosal cellular and humoral immune responses have the potential to induce sterilizing immunity by impeding pathogen binding to and uptake across epithelial surfaces. However, there are significant hurdles to mucosal vaccine development, including incomplete knowledge of the nature of protective mucosal immune responses. Advancing new mucosal vaccines and improving existing vaccines requires innovative adjuvant approaches and delivery strategies, which is the focus of this Review. Given the specific architecture of the mucosal surfaces and their unique cellular composition, vaccine strategies should be specifically tailored for the target site rather than redeploying effective injectable vaccines. In any case, many adjuvants that are effective by injection are not optimal for mucosal delivery.
Lessons from licensed mucosal vaccines
Over recent decades, there has been a broad shift from injectable whole-cell killed and attenuated vaccines towards adjuvanted subunit and, more recently, viral vectored, RNA and DNA vaccines26,27. This can reduce the potential for excessive reactogenicity and is facilitated by advances in antigen discovery, adjuvants and delivery systems. However, the landscape for mucosal vaccines is very different. Of the nine mucosal vaccines approved for use in humans — eight oral and one intranasal — all are either live attenuated or whole-cell inactivated vaccine formulations (Fig. 2). This current dichotomy in approaches is, in part, due to greater tolerability of orally administered whole-cell killed antigens, the susceptibility of unprotected subunit antigens to degradation and clearance, and, crucially, a lack of proven mucosal adjuvants.

Eight oral vaccines are currently licensed for use against cholera, salmonella, poliovirus and rotavirus. Live attenuated influenza vaccines remain the sole licenced intranasal vaccines. To date, live attenuated and inactivated vaccines have proved the most successful platforms for mucosal vaccine design. CTB, cholera toxin B subunit; LPS, lipopolysaccharide.
Currently, the only subunit antigen included in a licensed mucosal vaccine is cholera toxin B subunit (CTB), included as an additional component of the killed whole-cell Vibrio cholerae vaccine Dukoral. CTB cannot be regarded as a ‘model’ subunit antigen as this is the binding component of cholera toxin — it binds with high affinity to GM1 on epithelial cells and is highly immunogenic28,29,30 (Box 1). Nevertheless, this does indicate that potent immune responses can be induced against an orally administered purified protein even if this is in the presence of whole bacteria. The tolerability of oral whole-cell antigens is instructive as, although adjuvanted subunits are preferable for parenteral formulations, leveraging potent mucosal adjuvants with whole-cell antigens may be a more productive approach for mucosal vaccination, especially via oral routes.
Developing whole-cell antigens as a platform offers potential for combination with purified subunits but also, perhaps more encouragingly, for overexpression of antigens on whole cells — ETVAX provides a stellar example of this31. Developed at the University of Gothenburg in collaboration with Scandinavian Biopharma, ETVAX comprises four E. coli strains engineered to overexpress colonization factor antigens on the bacterial surface, namely CFA/I, CS3, CS5 and CS6, in combination with LCTBA31,32 (a CTB and E. coli heat-labile enterotoxin B subunit (LTB) hybrid; see Box 1). Overexpression of antigens on inactivated whole bacteria is thus a promising approach to increase immunogenicity, leveraging the adjuvanticity of inactivated bacteria while helping to minimize the vaccine dose required. This approach may also be applied to inactivated viruses or virus-like particles, taking advantage of their inherent immunogenicity, particulate antigen presentation and well-established expression systems33,34.
Aside from Dukoral, Euvichol and Shanchol, the other licensed mucosal vaccines are all live attenuated bacteria (Salmonella enterica subsp. enterica serovar Typhimurium) or attenuated and/or reassortant viruses delivered orally (polio vaccine, rotavirus) or nasally (influenza A and influenza B viruses). Overall, this highlights the tolerability and effectiveness of mucosal attenuated and whole-cell vaccines but also points to the key question of why the successful shift to more modern vaccine strategies has not yet occurred for mucosal vaccines.
Vaccine lessons from mucosal tissues
Unique aspects of mucosal immune responses
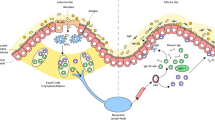
There are many distinctive features of mucosal immune responses that impact on mucosal vaccine design, ranging from the structure and location of immune inductive sites to the type of effector and memory cells induced and their longevity and location. The mucosal immune system can be broadly categorized into inductive sites where antigen-specific B cell and T cell responses are initiated and effector sites (such as the lamina propria and epithelium) where the adaptive immune responses are mediated. The nature of the inductive sites varies between species and also between different mucosae. In the case of the intestine, the inductive sites are the gut-associated lymphoid tissue and the intestine-draining mesenteric lymph nodes. Gut-associated lymphoid tissue in humans and mice comprises Peyer’s patches and isolated lymphoid follicles35. The connection between inductive and effector sites is facilitated by selective expression of integrins and chemokine receptors on B cells36 and T cells37. For example, in the case of the small intestine, imprinting of α4β7 integrin and CC-chemokine receptor 9 (CCR9) expression on lymphocytes is key for tissue-specific homing of cells. Although mucosal immune responses are compartmentalized, there is crosstalk between different mucosae, and it is thus possible to vaccinate at a single mucosal site but also promote immune responses at distant mucosal sites38. Understanding the nature of the signals regulating such homing in a human context is critical to allow design of novel mucosal vaccines that can potentially target mucosae distant from the site of vaccination.
Mucosal sites cover a surface area of 30–40 m2 in humans39 predominantly in the gastrointestinal tract, respiratory tract, urogenital tract and ocular cavities, playing a crucial role in homeostasis and interactions with the microbiome, dietary antigens and other environmental material. As a result, they constitute prominent points of pathogen entry and are often sites of tumour development. This high antigenic load and constant exposure to microbes necessitates mucosal immunoregulatory mechanisms that are vital to maintain homeostasis and prevent damaging chronic inflammatory responses40. This has significant implications with regard to vaccine development, for example, many Toll-like receptor (TLR) agonists that are effective adjuvants when injected parenterally have poor efficacy when administered orally. This results, at least in part, from tolerization of intestinal antigen-presenting cells (APCs) to pathogen-associated molecular patterns, particularly TLR ligands to which they are continuously exposed via the microbiome41, and also as a result of the basolateral rather than luminal expression of TLRs at epithelial surfaces. Detailed knowledge on mucosal responsiveness to pathogen-associated molecular patterns, responsive target cells and their location is critical so that productive target pathways can be identified for adjuvant discovery and optimization.
A recent report demonstrated that proximal intestinal gut-draining lymph nodes preferentially supported regulatory T cell responses whereas distal gut-draining lymph nodes supported the induction of effector T helper cells42. These insights into the balance of regulatory and effector responses can inform vaccine design — if antigen uptake in proximal regions of the small intestine preferentially enhances tolerogenic responses, delivery of oral vaccines in solution may not be optimal and targeting of antigens to the distal small intestine or large intestine may be more effective. This preferential induction of regulatory T cells in the proximal intestine could also be affected by the presence of adjuvants or the nature of the orally administered antigen. Vaccines could thus potentially over-ride the tolerogenic environment in the proximal intestine by inducing an inflammatory signature to allow the induction of effector T cell responses, although overall there may be an advantage in targeting the distal intestine.
One such large intestine-targeted oral delivery system was described where vaccine nanoparticles were delivered within pH-dependent microparticles. This oral construct induced protective antiviral immunity in the rectum and vagina comparable with levels seen with colorectal vaccination and protected against rectal and vaginal viral challenge43, providing a potentially productive route for mucosal vaccination for STDs. Although compartmentalization of mucosal immune responses is well established, confirming that the same subdivisions and connections apply from rodents to humans can be a challenge. However, it was shown that oral Dukoral vaccination increased the numbers of circulating IgA+ memory B cells with a surface marker expression profile indicative of homing to the large intestine and respiratory tract38. Furthermore, translation of vaccine delivery strategies from small animal models to humans can be challenging owing to differences in parameters including gastrointestinal pH, gastrointestinal tract residence times and intestinal surface area. Some of these factors have been addressed in the case of oral drug delivery, but it is quite clear that in the absence of immunogenic antigens and effective adjuvants, addressing delivery challenges in isolation offers modest benefits and the vaccine components must be optimized for the targeted mucosal pathogen and its site of infection. The nature of the antigen is also a major determinant of responses, whether living or non-living, soluble or particulate. This can dictate the nature of antigen uptake pathways at mucosal sites and should be a principal design feature in the development of mucosal vaccines (Fig. 3). Particulate antigens — whether as whole bacterial cells, attenuated or inactivated viruses, virus-like particles or synthetic particulate formulations — are more immunogenic than purified proteins33 and, in addition to their greater inherent immunogenicity, when delivered mucosally their particulate nature can impact on the site of uptake and APC targeting.

Nature of antigen uptake in the intestine is dependent on the type of vaccine components used, whether soluble or particulate, inert or live. Innovative encapsulation and targeting strategies have the potential to protect antigens while enhancing uptake and delivery to optimal intestinal regions. Inclusion of an effective mucosal vaccine adjuvant can confer multiple benefits from preventing tolerogenic responses to antigens, recruiting and activating antigen-presenting cells (APCs) and engaging other innate immune cells that contribute to protective immunity. Although there are currently few safe and effective adjuvants, cell-targeting adjuvants such as Escherichia coli double-mutant heat-labile toxin (dmLT), which binds to gangliosides on microfold (M) cells and dendritic cells, or α-galactosylceramide (α-GalCer), which can promote activation of invariant natural killer T (iNKT) cells locally and in draining lymph nodes via dendritic cell-mediated presentation, offer promise. Optimal formulations will address antigen design, adjuvanticity and antigen protection and targeting to address the unique challenges of intestinal delivery in the case of protein antigens (part a), whole cell vaccines (part b) and viral vector vaccines (part c). cDC, conventional dendritic cell; WCK, whole-cell killed.
Antigen-presenting cells in mucosal tissues
APC populations at mucosal sites are highly dynamic. In addition to tissue-resident dendritic cells and macrophages, during inflammatory responses or infection, further APCs are recruited that can engage with absorbed antigen and contribute to effector responses44. Indeed, there is evidence that during inflammation, monocytes in the gut and lungs can upregulate CCR7, migrate to lymph nodes and prime T cell responses45, and recruited monocytes or immature macrophages can produce inflammatory cytokines that contribute to T helper 1 (TH1) cell and TH17 cell responses44. Local inflammation triggered by mucosal vaccines and/or adjuvants could thus contribute to enhanced adaptive immunity by recruiting APCs. Although tissue-resident macrophages do not migrate to lymph nodes and are thus unlikely to directly prime T cells, antigen sampling CX3C-chemokine receptor 1 (CX3CR1+) mononuclear phagocytes can transfer antigen to dendritic cells46 (Fig. 3) and colonic CX3CR1+ mononuclear phagocytes were shown to be required for induction of TH17 cell and antibody responses to intestinal fungi47.
Two key dendritic cell populations in gut-draining lymph nodes — CD103+CD11b– dendritic cells and CD103+CD11b+ dendritic cells — have been associated with tolerogenic or pro-inflammatory immune responses, respectively48. Assigning specific roles to dendritic cell and macrophage populations in the intestine can be challenging as this is context-dependent. A recent study in a model of S. Typhimurium infection found that intestinal CX3CR1+ macrophages were superior to conventional type 1 dendritic cell (cDC1) and cDC2 populations in promoting the production of S. Typhimurium-specific IgA49. Furthermore, these broad categories of dendritic cell populations may not capture the true complexity of responses in the intestine, and subsets of these populations can contribute to the recruitment and activation of T cells and B cells at the site of infection. Differential gene expression profiles in cDC1 and cDC2 populations from different gut regions were reported42, indicating that not only the type of gut dendritic cell but also its precise tissue location may be key for the outcome of oral vaccination. The latter study also found that, compared with proximal gut-draining lymph nodes, distal gut-draining lymph nodes are more efficient in promoting the differentiation of TH17 cells. Given the importance of TH17 cells for defence against extracellular pathogens and for support of intestinal IgA secretion, this finding may be instructive for delivery of oral vaccines. It is of note that cDC1 and cDC2 frequencies remain relatively stable throughout life50 (Table 1), an important consideration for cDC-targeted vaccine strategies. In the nasal mucosa, resident cDCs are vital for maintaining non-responsiveness to harmless inhaled antigens but viral infection promotes recruitment of monocyte-derived dendritic cells (moDCs) that can mediate T cell priming51. This study utilized a nasally administered chitosan hydrogel vaccine platform to trigger the inflammatory recruitment of moDCs coupled to the sustained release of antigen, which successfully promoted antigen-specific CD8+ T cell activation and expansion. This suggests that in addition to potentially targeting specific populations of mucosal dendritic cells, vaccines can also aim to promote recruitment of monocytes and moDCs to mediate protective mucosal immunity. Indeed, with subunit antigens, adjuvants may be critical to overcome tolerance induction and may also contribute to recruitment of ‘unconditioned’ APCs that prime antigen-specific T cells and B cells. Assessing the capacity of mucosal adjuvants to alter the activation states of tissue-associated or lymph node dendritic cells and to recruit monocytes and other APCs may be a useful indicator of efficacy, certainly more so than in vitro screening of cultured dendritic cells or macrophages that may poorly reflect responses of mucosal APCs following vaccination.
Induction of IgA and other mucosal antibodies
In terms of correlates of immunity following mucosal vaccination, induction of antigen-specific IgA is a crucial consideration. IgA is the dominant antibody at many mucosal sites and mediates protection against a range of enteropathogens. Recently, the importance of dimeric IgA in neutralization of SARS-CoV-2 has been highlighted in patients infected with the virus52 and high-avidity IgA can protect against enteropathogens by processes including agglutination and the recently described process of ‘enchained growth’53. For oral vaccination with adjuvanted subunit antigens, multiple doses (at least three) are needed to induce effective secretory IgA responses21. Induction of antigen-specific IgA was observed following two doses of ETVAX, with the addition of the E. coli double-mutant heat-labile toxin (dmLT) adjuvant enhancing the kinetics of induction31. This suggests that suitable adjuvants may exert a dose-sparing effect in mucosal IgA induction. Vaccines utilizing viral vectors and attenuated viruses may induce IgA responses more efficiently — for example, one dose of intranasal live attenuated influenza vaccine can elicit mucosal IgA54 in recipients, and faecal/salivary IgA was observed in recipients of the Vaxart norovirus vaccine candidate, comprising an adjuvanted enterically stable adenovirus type 5 (Ad5) vector55. Although detailed assessment of antigen-specific mucosal immunity is more challenging in humans than in preclinical models, recent advances are facilitating novel means of determining vaccine-induced mucosal IgA responses in clinical trials (Box 2).
Although the importance of mucosal dendritic cell-mediated antigen sampling and trafficking to draining lymph nodes for induction of IgA responses has long been appreciated, Komban et al. recently uncovered a new layer of antigenic crosstalk between microfold (M) cells and B cells in the subepithelial dome region of Peyer’s patches. CC26+CCR1+GL7– B cells were shown to be capable of sampling antigen directly from M cells and trafficking to germinal centres where their activation and population expansion occurs, challenging the idea of dependence on cDC-mediated antigen transfer for optimal antigen-specific IgA induction56. However, whereas transport of secretory antibody (IgA, IgM) via the polymeric immunoglobulin receptor is an essential and highly efficient process in the intestine, this is not the case at all mucosal sites. In the female reproductive tract, IgG rather than IgA can be critical for protective immunity to viral infection57. Likewise, IgG plays an important role in protective immunity in the lower respiratory tract whereas IgA is relatively more important in the nasal compartment58.
Tissue-resident memory T cells in mucosal tissues
Tissue-resident memory T (TRM) cells have been identified at multiple mucosal sites59 and are thought to play decisive roles in rapid responses to infection60 and cancers (Box 3), thus providing another important correlate for mucosal vaccine efficacy. The human duodenal CD4+ T cell compartment was recently shown to be enriched with a population of polyfunctional TH1 cells, which survive for at least 1 year61. This offers significant hope for inducing sustained protective cellular immunity if optimal oral vaccine strategies are designed. There are clearly significant tissue-specific differences in the nature of TRM cell populations so this must be considered in the design of new vaccine approaches. CD8+ TRM cells in the lungs are pivotal for protection against respiratory viral infections but these cells are generally relatively short-lived, and this can compromise responses to subsequent infection62. The latter study found that systemic vaccination (intravenous administration of Listeria monocytogenes expressing influenza virus-associated antigen) could enhance lung TRM cells in mice previously infected with influenza virus by increasing numbers of circulating effector memory T cells. This clearly has implications regarding the potential for systemic booster vaccinations in previously infected or mucosally primed populations to sustain resident memory CD8+ T cells in the lungs. A population of lung-resident helper T cells was recently characterized that was required to support tissue-resident memory B cells and CD8+ cells following influenza virus infection63,64. These cells, which are induced locally in the lung, may be key for promoting long-lived cellular and humoral immunity following vaccination in the respiratory tract, so the optimal strategy for their induction should be addressed. Recent evidence indicates that long-term maintenance of lung TRM cells requires airway vaccination and sustained antigen presence in the lungs, which was facilitated by an adenovirus vector vaccine65. It was recently shown in mouse models that TRM cells migrate to the mediastinal lymph nodes from the lungs during infection in a process termed ‘retrograde migration’. These cells retained a TRM cell phenotype and provided long-term protection66. This may be an important consideration following intranasal vaccination strategies. Further studies from the same group demonstrate that, upon restimulation, TRM cells can undergo retrograde migration and give rise to effector memory T cells and central memory T cells that have a predisposition for homing to their tissue of origin67.
Targeting the genital tract
Although oral, sublingual and nasal routes are more convenient and there are currently no vaccines that specifically target the genital tract in clinical use, vaccination in the genital tract could have significant advantages in targeting STDs, even as a vaccine-boosting approach. In mouse models, vaginal immunization with herpes simplex virus 2 (HSV-2) glycoprotein D antigen and the adjuvant α-galactosylceramide (α-GalCer) induced protective immunity against HSV-2 challenge68. A combined vaccination approach using recombinant influenza virus–HIV vectors administered via intranasal and intravaginal routes (in mice) resulted in HIV-specific CD8+ TRM cells in the vaginal mucosa69. Vaginal immunization of mice with an attenuated HSV-2 strain resulted in the induction of a population of IFNγ+CD4+ TRM cells, which promoted CXCL9-mediated and CXCL10-mediated recruitment of memory B cells upon secondary challenge70. By contrast, primary vaccination did not result in the induction of a tissue-resident population of plasma cells in the female reproductive tract. Thus, vaginal booster vaccination or, possibly, booster vaccination in the large intestine may be an effective strategy following systemic priming to trigger genital tract responses, although these findings must first be confirmed in a human setting. Promising recent data showed that vaginal delivery (by intramucosal vaginal injection or spray) of recombinant glycosylated IL-7 to rhesus macaques acted as an effective mucosal adjuvant, enhancing the induction of antigen-specific IgA/IgG in the vaginal mucosa following subsequent vaginal delivery of diphtheria toxoid71. This could be a broadly applicable strategy that may overcome hypo-responsiveness to vaginal vaccine delivery.
Immune cell populations targeted by mucosal vaccines
Mucosal adjuvants should aim to activate and target local or recruited APCs (Fig. 3) or populations of immune cells enriched in the mucosa (Table 1) in order to mount effective mucosal responses. Innate lymphoid cells (ILCs), mucosal-associated invariant T cells, natural killer T (NKT) cells and γδ T cells are abundant in mucosal tissues and can play crucial roles in mediating and shaping mucosal immunity72,73,74,75,76,77,78,79. Adjuvants can also be exploited in parenteral–mucosal push–pull strategies; for example, dmLT has been shown to imprint mucosal homing markers on T cells when injected80. Similarly, retinoic acid has been identified as a suitable adjuvant in such strategies, imprinting gut-homing markers on T cells and leading to protective intestinal responses following subcutaneous vaccination81. There are currently two ongoing trials investigating parenteral–mucosal push–pull strategies for SARS-CoV-2 vaccination: NCT04732468 and IG/VPIN/CVD19/2001. The former trial involves investigating combinations of oral and subcutaneous immunization with a human adenoviral vector expressing modified SARS-CoV-2 spike and nucleocapsid proteins. By contrast, the latter trial involves combinations of intranasal and intramuscular immunization, with the vaccine composed of the receptor-binding domain of SARS-CoV-2 spike protein adjuvanted with hepatitis B virus nucleocapsid protein when given intranasally and with alum when given intramuscularly.
Mucosal adjuvant approaches
Enhancing the efficacy of subunit and inactivated antigens
Toxoid adjuvants are the best-characterized class of mucosal adjuvants and the development of safe yet potent derivatives of E. coli heat-labile toxin and cholera toxin (Box 4) has paved the way for their safe incorporation in vaccine formulations. Incorporation of dmLT has been shown to improve clinical responses to several whole-cell antigens, as seen with ETVAX and ACE527 (refs31,82). Excellent overviews of the development and clinical application of dmLT are provided by Clements and Norton80 and Qadri et al.31. Based on a similar approach, the adjuvant multiple mutated cholera toxin (mmCT) has been proposed as an alternative to dmLT83, and in preclinical studies mmCT has been shown to enhance TH1 cell and TH17 cell responses in addition to mucosal and serum antibodies to a whole-cell Helicobacter pylori antigen84. CTA1DD is a cholera toxin-derived adjuvant that was designed to combine the beneficial immunostimulatory effects of the CTA subunit enzyme with the B cell-targeting properties of a D-domain dimer from Staphylococcus aureus, to reduce off-target effects and toxicities85,86. It was recently shown that CTA1DD enhanced the maturation of follicular dendritic cells in lymph nodes following mucosal vaccination in neonatal mice and that oral priming with a construct incorporating the influenza virus M2e antigen (CTA1-3M2e-DD) induced protective immunity in neonates against influenza challenge87. Combination of lipid nanoparticles and CTA1-3M2e-DD generated a highly effective nasal vaccination system that conferred protective immunity against influenza virus infection in mice88. This combination adjuvant was particularly effective in promoting respiratory tract IgA, TH1 cell and TH17 cell responses, holding promise for universal influenza vaccination applications88. However, the efficacy and safety of CTA1DD remains to be determined clinically. The use of toxoid adjuvants intranasally has been somewhat marred by the clinical emergence of Bell’s palsy in some recipients of influenza vaccines adjuvanted with wild-type E. coli heat-labile toxin89 or LTK63, a genetically detoxified E. coli heat-labile toxin derivative90. An alternative is sublingual vaccination, which has shown significant promise as a means of promoting protective immunity in animal models91,92 although immune responses to sublingual dmLT were modest in a clinical trial93. There may be scope to enhance such responses by formulation with agents such as chitosan to enhance antigen and adjuvant residence times (Box 2).
These studies would suggest that nasal delivery of ganglioside-targeting toxoid adjuvants is inadvisable. However, results from a phase II clinical trial on a trivalent influenza vaccine, composed of haemagglutinin and adjuvanted with LThαK, a detoxified E. coli heat-labile toxin derivative, were recently reported94,95 (NCT03784885). An acceptable safety profile was reported following two nasal vaccinations, which induced higher antigen-specific nasal IgA responses than the non-adjuvanted antigen94. LThαK is reported to have no ribosylating activity, correlating with enhanced retention in the nasal passages and the enhanced safety profile. Phase III trials will investigate efficacy in a challenge setting and will further elucidate the safety of LThαK for intranasal incorporation in a larger patient cohort. Importantly, nasal delivery of CTA1DD did not result in trafficking to the olfactory bulb, indicating its safety as a nasal vaccine adjuvant88. In summary, toxoid adjuvants are the most advanced mucosal adjuvants, having demonstrated impressive efficacy in clinical trials for oral whole-cell killed vaccines.
Aside from toxoid adjuvants, there are a small number of mucosal adjuvants with demonstrated safety and efficacy. The invariant NKT cell activator α-GalCer is a promising mucosal adjuvant and potentially an indicator of the potential for targeting innate-like lymphocytes to produce a new generation of mucosal adjuvants. We have demonstrated that oral delivery of a whole-cell killed H. pylori antigen adjuvanted with α-GalCer induced protective immunity from gastric bacterial challenge, characterized by induction of local IgA and TH1 cell immunity, comparable with a cholera toxin adjuvanted vaccine96. The induction of antigen-specific TH1 cell responses was dependent on CD1d, IL-1R1 and IL-17R signalling; therefore, α-GalCer provides a proof of principle for targeting the relatively abundant mucosal invariant NKT cell populations for effective adjuvanticity. We have further characterized α-GalCer as an effective adjuvant with oral whole-cell killed ETEC and cholera antigens including the CFA/I overexpressing JT-49 ETEC vaccine combined into enterically stable smPill mini-spheres. Potent induction of intestinal CFA/I-specific IgA was observed in addition to serum IgG responses97. Oral vaccination with Dukoral and α-GalCer induced stronger intestinal IgA and serum IgG responses than Dukoral alone and was comparable with cholera toxin adjuvanted Dukoral98. Finally, incorporation of α-GalCer in a novel multi-antigen cholera vaccine composed of whole-cell killed double-lipopolysaccharide (LPS) antigen expressing cholera vaccine with CTB promoted robust mucosal immunity with concomitant systemic antibody production, outperforming Dukoral and the whole-cell killed cholera alone98. Our preclinical data provide a rationale for the inclusion of α-GalCer in future whole-cell oral vaccines, which may lead to more durable protection, addressing shortcomings in immunity and response rates.
Chitosan is well established as a mucosal adjuvant/delivery system given its mucoadhesive properties and immunostimulatory effects99. We have demonstrated the effectiveness of chitosan as an intranasal adjuvant in mouse models. Intranasal vaccination with chitosan and pneumococcal surface protein A (PspA) led to the induction of lung PspA-specific IFNγ and IgG1, IgG2c and IgA responses that were dependent on STING signalling100. STING-activating cyclic dinucleotides have been trialled as mucosal adjuvants. An intranasal synthetic cyclic dinucleotide (cyclic diguanylate) adjuvanted subunit vaccine induced protective immunity against M. tuberculosis in mice, correlating with potent induction of TH17 cells101. Other cyclic dinucleotides — including cyclic di-AMP and cyclic di-GMP — have also shown promise as mucosal adjuvants102,103. Mansouri et al. recently highlighted roles for two lung cDC2 populations in intranasal cyclic di-GMP adjuvanticity. Antibody responses were dependent on activation of moDCs by TNFR2– cDC2 populations, with subsequent T follicular helper cell and germinal centre B cell activation, whereas induction of TH1 cell and/or TH17 cell responses was dependent on their TNFR+ cDC2 counterparts104. These studies provide a strong rationale for further development of mucosal adjuvants targeting the STING pathway. In this context, chitosan has specific advantages in its record of clinical use and mucoadhesive properties in addition to STING-dependent adjuvanticity.
Whereas emulsion-based adjuvants have been highly successful in injectable vaccines, such approaches have not reached clinical application mucosally. Bluewillow Biologics currently have a phase I trial underway (NCT04148118) utilizing an intranasally administered nanoemulsion (oil in water emulsion) adjuvanted recombinant protein vaccine against anthrax (BW-1010). Preclinically, this vaccine has previously been shown to be protective in guinea pig models of infection, correlating with induction of systemic and local antibody induction105. The nanoemulsion adjuvant has also been shown to promote TH1 cell immunity and TH17 cell immunity in anthrax and M. tuberculosis vaccine formulations, respectively105,106. Kimoto et al. recently reported a promising mucosal adjuvant with possible applications in oral and intranasal vaccination routes. Two oral doses of HAv (haemagglutinin-based vaccine) adjuvanted with SF10 (a synthetic surfactant adjuvant) led to induction of antigen-specific mucosal IgA and protection from influenza in a preclinical model, outperforming cholera toxin107. These recent studies highlight the promise of adjuvanted mucosal vaccines, with many taking inspiration from bacteria-derived virulence factors and showing promise for inclusion not only in subunit but also whole-cell formulations.
Mucosal nucleic acid and viral vectored vaccines
Until very recently, there were no licensed nucleic acid vaccines for clinical use. However, mRNA vaccines against SARS-CoV-2 have now been successfully trialled and rolled out for parenteral vaccination, displaying impressive efficacy and paving the way for others to follow108. Mucosal vaccination utilizing nucleic acids poses a greater challenge, as successful candidates must penetrate the mucus layer, translocate into target cells and evade extracellular and intracellular degradation. Vaccination via the oral route poses an added challenge with the low gastric pH and difficulty in ensuring release of the nucleic acid payload at the appropriate location. Innovative protective delivery strategies for nucleic acids have been developed using nanocarriers and biomaterials109,110,111, and in particular the complexing of nucleic acids with polycationic materials including chitosan and polyethylenimine (PEI) and encapsulation of the nucleic acid cargo utilizing liposomes and polymersomes are showing potential. Lipidoid nanoparticles have been shown to effectively deliver small interfering RNA molecules to intestinal epithelial cells in the lower small intestine and colon following oral administration112. Additionally, intranasal delivery of chitosan nanoparticles encapsulating mRNA with a viral protein coating elicited protection from avian influenza in chickens113. The coming years are likely to see great activity in this space, particularly around mobilizing solid lipid nanoparticles for mucosal RNA vaccine development.
Viral vectors are among the most promising strategies for mucosal vaccination, owing to their capacity for intracellular delivery, versatility and intrinsic immunogenicity. Viral vector strategies are applicable to oral vaccination when protection from conditions of the gastrointestinal tract and effective release are addressed. This is exemplified by the technology from Vaxart, whose oral influenza vaccine candidate VXA-A1.1 utilizes an enterically stable tableted delivery system, carrying a cargo of haemagglutinin encoding adenoviral vectors and a double-stranded RNA adjuvant. Data from phase I (NCT01688297) and phase II (NCT02918006) clinical trials demonstrated that VXA-A1.1 is well tolerated114 and, crucially for future adenoviral vector strategies, is not hindered by pre-existing adenoviral immunity when given orally114. Oral vaccination with VXA-A1.1 induced superior protection from influenza A virus challenge compared with the conventional intramuscularly delivered FluZone vaccine115. Whether this platform can be used to develop an effective oral quadrivalent influenza vaccine remains to be demonstrated. Additionally, orally administered RSV vaccines are in preclinical development (VXA-RSV-f)116, and an oral Norovirus vaccine (VXA-G1.1-NN) showed favourable safety and immunogenicity in a phase 1 trial (NCT02868073)55. More recently, an orally administered SARS-CoV-2 vaccine (VXA-CoV2-1) was described that uses the same formulation and is currently in phase I trials (NCT04563702). The efficacy of the double-stranded RNA adjuvant included in this platform is also very promising as an alternative to the canonical toxoid-based adjuvants in various stages of development and broadens the range of PRR targets that can be exploited for oral vaccination. This double-stranded RNA adjuvant will likely effectively target dendritic cell populations for activation owing to their high TLR3 expression. This capacity to successfully adjuvant viral vectors may be critical as they will likely be less effective when given mucosally compared with parenteral routes. Further to this point, it has been recently shown that responses to viral vector (MVA) antigens can be enhanced by the saponin-containing adjuvant matrix M following subcutaneous vaccination117.
Viral vector approaches also hold potential for vaccination in the respiratory tract. Intranasal vaccination of mice with an adenoviral vector encoding influenza virus nucleoprotein induced a population of CD8+ TRM cells in the lungs that was sustained for longer than 1 year65. This was dependent on respiratory vaccination and sustained antigen expression, and contrasted with the situation following parenteral influenza virus infection, where the local CD8+ TRM cell population was rapidly lost. The authors suggested that induction of robust local cellular immunity may address issues surrounding the reliance on systemic antibody responses to haemagglutinin associated with parenteral influenza vaccination65. Nasal delivery of chimpanzee adenoviral (ChAd) vectors may also have potential in SARS-CoV-2 vaccines. Nasal delivery of ChAd-SARS-CoV-2 expressing homotrimeric spike antigen induced promising results in a murine infection model (K18-hACE2 mice). A single dose provided protection from upper and lower respiratory tract infection, correlating with induction of neutralizing antibody titres in the serum and bronchoalveolar lavage alongside the induction of IFNγ+ and granzyme B+ CD8+ T cells118. Whether efficacy would be sufficient clinically with viral vectors alone is unclear but, as with oral delivery, there may be scope to enhance responses with appropriately targeted mucosal adjuvants. With viral (or bacterial) vectored vaccines, the capacity of vaccine-induced secretory antibody responses to compromise responses to booster vaccination must be considered119. However, data from preclinical models have shown that pre-existing intestinal immunity did not compromise efficacy of an oral experimental viral vectored rabies vaccine120. Furthermore, compelling recent clinical data found no detrimental effect of pre-existing influenza-specific nasal IgA responses on the efficacy of nasal live attenuated influenza virus vaccination in children121, and recently Janssen reported no clear impact of pre-existing immunity to their Ad26 vectored vaccine platform on efficacy following priming or boosting vaccinations122. This must be ascertained for each specific vaccine but, moving forwards, the availability of numerous mucosal viral vectors and adjuvant strategies would allow a heterologous prime–boost approach to overcome pre-existing immunity if required.
Concluding remarks
We are currently in the midst of a revolution in vaccine research and development. Cutting-edge research and advances into nucleic acid and viral vector vaccine technologies allowed SARS-CoV-2 vaccines to be developed and produced in an unprecedented short period of time. These advances are yet to impact on clinically used mucosal vaccines, but this will likely change in the near future. Mucosal vaccines offer the significant benefit of triggering immune responses at the principal sites of infection, offering scope for sterilizing immunity achieved by local secretory antibody responses and resident populations of CD4+ and CD8+ T cells. Thus, the outstanding obstacles to mucosal vaccine development are worth the effort as they are far outweighed by the potential immunological and logistical benefits in terms of ease of delivery. One of the major challenges that requires innovative solutions is the ‘tropical barrier’, where responses to oral vaccines in low- and middle-income countries can be lower than those seen throughout clinical trials in high-income countries. Interventions to address this problem are urgently required123 and may include implementation of probiotic supplements prior to or during vaccination124. The potential for adjuvants to overcome suboptimal responses must be addressed as this and increased antigen doses may have a greater impact than other proposed strategies. Indeed, the most advanced mucosal adjuvant, dmLT, has demonstrated efficacy in both high-income and low-income countries31,32. Identifying whether other candidate adjuvants can also increase efficacy of existing oral vaccines as well as facilitating the development of novel vaccines is a priority. Targeting mucosally abundant cellular populations such as ILCs, mucosal-associated invariant T cells and NKT cells has significant promise but clinical validation of these approaches is required. A recent study in mice found that intestinal ILCs can migrate via the lymph to the mesenteric lymph nodes, and in response to infection with S. Typhimurium these migrating ILCs exhibited greater levels of activation and cytokine production. Mobilizing this population using ILC-targeting adjuvants may have significant potential to bolster mucosal immune responses75. In addition to stand-alone mucosal vaccine approaches, parenteral mucosal prime–boost strategies offer promise. These may be enhanced with injectable vaccines that imprint a degree of mucosal homing, for example, with dmLT125 or retinoic acid81, and their relative ability to enhance tissue-resident T cell responses may be key to success. In some cases, antigen alone may be sufficient for mucosal boosting126 although this will depend on the nature and immunogenicity of the antigen and it is likely, in most cases, that an effective adjuvant will be required. In summary, although the leaps forward in injectable vaccine strategies have not yet been seen with mucosal vaccines, this is likely to change in the near future. Advances in our understanding of mucosal protective immunity, developments in measuring human mucosal immunity127 and antigen and adjuvant discovery offer hope that novel mucosal vaccines for infectious diseases and cancer are on the horizon.
Change history
03 August 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41577-021-00599-8
References
WHO. The top 10 causes of death. World Health Organization https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (2020).
Troeger, C. et al. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory infections in 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect. Dis. 18, 1191–1210 (2018).
Shi, T. et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: a systematic review and modelling study. Lancet 390, 946–958 (2017).
Vos, T. et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 396, 1204–1222 (2020).
Hoft, D. F. et al. Comparisons of the humoral and cellular immune responses induced by live attenuated influenza vaccine and inactivated influenza vaccine in adults. Clin. Vaccine Immunol. 24, 1–9 (2017).
Lartey, S. et al. Live-attenuated influenza vaccine induces tonsillar follicular T helper cell responses that correlate with antibody induction. J. Infect. Dis. 221, 21–32 (2020).
Jahnmatz, M. et al. Safety and immunogenicity of the live attenuated intranasal pertussis vaccine BPZE1: a phase 1b, double-blind, randomised, placebo-controlled dose-escalation study. Lancet Infect. Dis. 20, 1290–1301 (2020).
Lin, A. et al. Live attenuated pertussis vaccine BPZE1 induces a broad antibody response in humans. J. Clin. Invest. 130, 2332–2346 (2020). This study provides evidence supporting the safety and immunogenicity of a novel live attenuated nasal B. pertussis vaccine in humans.
Bull, N. C. et al. Enhanced protection conferred by mucosal BCG vaccination associates with presence of antigen-specific lung tissue-resident PD-1+KLRG1−CD4+ T cells. Mucosal Immunol. 12, 555–564 (2019).
WHO. WHO coronavirus disease (COVID-19) dashboard. World Health Organization https://covid19.who.int/ (2021)
International Monetary Fund. World Economic Outlook Update June 2020 — A Crisis Like No Other, An Uncertain Recovery (International Monetary Fund, 2020).
Lamers, M. M. et al. SARS-CoV-2 productively infects human gut enterocytes. Science 369, 50–54 (2020).
Giurgea, L. T., Han, A. & Memoli, M. J. Universal coronavirus vaccines: the time to start is now. NPJ Vaccines 5, 43 (2020).
Troeger, C. et al. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of diarrhoea in 195 countries: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect. Dis. 18, 1211–1228 (2018).
WHO. Global task force on cholera control: overview of ending cholera — a global roadmap to 2030 (WHO, 2018).
Sharma, T. et al. Development of Hillchol®, a low-cost inactivated single strain Hikojima oral cholera vaccine. Vaccine 38, 7998–8009 (2020).
WHO. Cervical cancer. World Health Organization https://www.who.int/health-topics/cervical-cancer#tab=tab_1 (2021).
Cavarelli, M. & Scarlatti, G. HIV-1 infection: the role of the gastrointestinal tract. Am. J. Reprod. Immunol. 71, 537–542 (2014).
Jones, A. T. et al. HIV-1 vaccination by needle-free oral injection induces strong mucosal immunity and protects against SHIV challenge. Nat. Commun. 10, 798 (2019).
Russell, M. W., Moldoveanu, Z., Ogra, P. L. & Mestecky, J. Mucosal immunity in COVID-19: a neglected but critical aspect of SARS-CoV-2 infection. Front. Immunol. 11, 1–5 (2020).
Lycke, N. Recent progress in mucosal vaccine development: potential and limitations. Nat. Rev. Immunol. 12, 592–605 (2012).
Strugnell, R. A. & Wijburg, O. L. C. The role of secretory antibodies in infection immunity. Nat. Rev. Microbiol. 8, 656–667 (2010).
Perez-Lopez, A., Behnsen, J., Nuccio, S.-P. & Raffatellu, M. Mucosal immunity to pathogenic intestinal bacteria. Nat. Rev. Immunol. 16, 135–148 (2016).
Ahmad, R., Sorrell, M. F., Batra, S. K., Dhawan, P. & Singh, A. B. Gut permeability and mucosal inflammation: bad, good or context dependent. Mucosal Immunol. 10, 307–317 (2017).
LaMere, M. W. et al. Regulation of antinucleoprotein IgG by systemic vaccination and its effect on influenza virus clearance. J. Virol. 85, 5027–5035 (2011).
Wang, N., Shang, J., Jiang, S. & Du, L. Subunit vaccines against emerging pathogenic human coronaviruses. Front. Microbiol. 11, 298 (2020).
Pardi, N., Hogan, M. J., Porter, F. W. & Weissman, D. mRNA vaccines — a new era in vaccinology. Nat. Rev. Drug Discov. 17, 261–279 (2018).
Heyningen, S. V. Cholera toxin: interaction of subunits with ganglioside GM1. Science 183, 656–657 (1974).
Antonio-Herrera, L. et al. The nontoxic cholera B subunit is a potent adjuvant for intradermal DC-targeted vaccination. Front. Immunol. 9, 2212 (2018).
Eriksson, K., Fredriksson, M., Nordström, I. & Holmgren, J. Cholera toxin and its B subunit promote dendritic cell vaccination with different influences on TH1 and TH2 development. Infect. Immun. 71, 1740–1747 (2003).
Qadri, F. et al. Safety and immunogenicity of the oral, inactivated, enterotoxigenic Escherichia coli vaccine ETVAX in Bangladeshi children and infants: a double-blind, randomised, placebo-controlled phase 1/2 trial. Lancet Infect. Dis. 20, 208–219 (2020). This study shows that the inclusion of dmLT adjuvant in an oral ETEC vaccine enhances intestinal IgA responses in infants.
Akhtar, M. et al. Evaluation of the safety and immunogenicity of the oral inactivated multivalent enterotoxigenic Escherichia coli vaccine ETVAX in Bangladeshi adults in a double-blind, randomized, placebo-controlled phase I trial using electrochemiluminescence and ELISA assays for immunogenicity analyses. Vaccine 37, 5645–5656 (2019).
Engeroff, P. & Bachmann, M. F. The 5th Virus-Like Particle and Nano-Particle Vaccines (VLPNPV) conference. Expert Rev. Vaccines 18, 1–3 (2019).
Mohsen, M. O., Zha, L., Cabral-Miranda, G. & Bachmann, M. F. Major findings and recent advances in virus-like particle (VLP)-based vaccines. Semin. Immunol. 34, 123–132 (2017).
Mörbe, U. M. et al. Human gut-associated lymphoid tissues (GALT); diversity, structure, and function. Mucosal Immunol. 14, 793–802 (2021).
Macpherson, A. J., McCoy, K. D., Johansen, F.-E. & Brandtzaeg, P. The immune geography of IgA induction and function. Mucosal Immunol. 1, 11–22 (2008).
Agace, W. Generation of gut-homing T cells and their localization to the small intestinal mucosa. Immunol. Lett. 128, 21–23 (2010).
van Splunter, M. et al. Oral cholera vaccination promotes homing of IgA+ memory B cells to the large intestine and the respiratory tract. Mucosal Immunol. 11, 1254–1264 (2018). This study shows that oral cholera vaccination (Dukoral) in humans leads to the induction of IgA+ memory B cells expressing homing markers for the airways and colon.
Helander, H. F. & Fändriks, L. Surface area of the digestive tract — revisited. Scand. J. Gastroenterol. 49, 681–689 (2014).
Caruso, R., Lo, B. C. & Núñez, G. Host–microbiota interactions in inflammatory bowel disease. Nat. Rev. Immunol. 20, 411–426 (2020).
Bain, C. C. et al. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 6, 498–510 (2013).
Esterházy, D. et al. Compartmentalized gut lymph node drainage dictates adaptive immune responses. Nature 569, 126–130 (2019).
Zhu, Q. et al. Large intestine-targeted, nanoparticle-releasing oral vaccine to control genitorectal viral infection. Nat. Med. 18, 1291–1296 (2012).
Mowat, A. M. I., Scott, C. L. & Bain, C. C. Barrier-tissue macrophages: functional adaptation to environmental challenges. Nat. Med. 23, 1258–1270 (2017).
Jakubzick, C. V., Randolph, G. J. & Henson, P. M. Monocyte differentiation and antigen-presenting functions. Nat. Rev. Immunol. 17, 349–362 (2017).
Mazzini, E., Massimiliano, L., Penna, G. & Rescigno, M. Oral tolerance can be established via gap junction transfer of fed antigens from CX3CR1+ macrophages to CD103+ dendritic cells. Immunity 40, 248–261 (2014).
Leonardi, I. et al. CX3CR1+, mononuclear phagocytes control immunity to intestinal fungi. Science 359, 232–236 (2018).
Esterházy, D. et al. Classical dendritic cells are required for dietary antigen-mediated induction of peripheral Treg cells and tolerance. Nat. Immunol. 17, 545–555 (2016).
Koscsó, B. et al. Gut-resident CX3CR1hi macrophages induce tertiary lymphoid structures and IgA response in situ. Sci. Immunol. 5, eaax0062 (2020).
Granot, T. et al. Dendritic cells display subset and tissue-specific maturation dynamics over human life. Immunity 46, 504–515 (2017).
Bedford, J. G. et al. Unresponsiveness to inhaled antigen is governed by conventional dendritic cells and overridden during infection by monocytes. Sci. Immunol 5, eabb5439 (2020).
Wang, Z. et al. Enhanced SARS-CoV-2 neutralization by dimeric IgA. Sci. Transl Med. 13, eabf1555 (2021).
Moor, K. et al. High-avidity IgA protects the intestine by enchaining growing bacteria. Nature 544, 498–502 (2017). This study reports the mechanism by which vaccine-induced intestinal IgA can mediate protection against enteric bacterial infection.
Barría, M. I. et al. Localized mucosal response to intranasal live attenuated influenza vaccine in adults. J. Infect. Dis. 207, 115–124 (2013).
Kim, L. et al. Safety and immunogenicity of an oral tablet norovirus vaccine, a phase I randomized, placebo-controlled trial. JCI insight 3, 1–12 (2018).
Komban, R. J. et al. Activated Peyer′s patch B cells sample antigen directly from M cells in the subepithelial dome. Nat. Commun. 10, 1–15 (2019).
Parr, E. L. & Parr, M. B. Immunoglobulin G, plasma cells, and lymphocytes in the murine vagina after vaginal or parenteral immunization with attenuated herpes simplex virus type 2. J. Virol. 72, 5137–5145 (1998).
Renegar, K. B., Small, P. A., Boykins, L. G. & Wright, P. F. Role of IgA versus IgG in the control of influenza viral infection in the murine respiratory tract. J. Immunol. 173, 1978–1986 (2004).
Mueller, S. N. & Mackay, L. K. Tissue-resident memory T cells: local specialists in immune defence. Nat. Rev. Immunol. 16, 79–89 (2016).
Schenkel, J. M. & Masopust, D. Tissue-resident memory T cells. Immunity 41, 886–897 (2014).
Bartolomé-Casado, R. et al. CD4+ T cells persist for years in the human small intestine and display a TH1 cytokine profile. Mucosal Immunol. 14, 402–410 (2021). This study shows that CD4+ TRM cells in the human intestine are long lived.
Slütter, B. et al. Dynamics of influenza-induced lung-resident memory T cells underlie waning heterosubtypic immunity. Sci. Immunol. 2, eaag2031 (2017).
Son, Y. M. et al. Tissue-resident CD4+ T helper cells assist the development of protective respiratory B and CD8+ T cell memory responses. Sci. Immunol. 6, eabb6852 (2021).
Swarnalekha, N. et al. T resident helper cells promote humoral responses in the lung. Sci. Immunol 6, eabb6808 (2021). This study provides evidence that lung-resident T helper cells should be induced by vaccination to support local antibody responses to influenza virus.
Uddbäck, I. et al. Long-term maintenance of lung resident memory T cells is mediated by persistent antigen. Mucosal Immunol. 14, 92–99 (2021). This study shows that long-lived CD8+ TRM cell responses require airway vaccination and antigen persistence in the lungs.
Stolley, J. M. et al. Retrograde migration supplies resident memory T cells to lung-draining LN after influenza infection. J. Exp. Med. 217, e20192197 (2020).
Fonseca, R. et al. Developmental plasticity allows outside-in immune responses by resident memory T cells. Nat. Immunol. 21, 412–421 (2020).
Lindqvist, M., Persson, J., Thörn, K. & Harandi, A. M. The mucosal adjuvant effect of α-galactosylceramide for induction of protective immunity to sexually transmitted viral infection. J. Immunol. 182, 6435–6443 (2009).
Tan, H. X. et al. Induction of vaginal-resident HIV-specific CD8 T cells with mucosal prime-boost immunization. Mucosal Immunol. 11, 994–1007 (2018).
Oh, J. E. et al. Migrant memory B cells secrete luminal antibody in the vagina. Nature 571, 122–126 (2019).
Logerot, S. et al. IL-7-adjuvanted vaginal vaccine elicits strong mucosal immune responses in non-human primates. Front. Immunol. 12, 1–17 (2021).
Panda, S. K. & Colonna, M. Innate lymphoid cells in mucosal immunity. Front. Immunol. 10, 1–13 (2019).
Sonnenberg, G. F. & Hepworth, M. R. Functional interactions between innate lymphoid cells and adaptive immunity. Nat. Rev. Immunol. 19, 599–613 (2019).
Klose, C. S. N. & Artis, D. Innate lymphoid cells control signaling circuits to regulate tissue-specific immunity. Cell Res. 30, 475–491 (2020).
Kästele, V. et al. Intestinal-derived ILCs migrating in lymph increase IFNγ production in response to Salmonella typhimurium infection. Mucosal Immunol. 14, 717–727 (2021).
Meierovics, A., Yankelevich, W. J. C. & Cowley, S. C. MAIT cells are critical for optimal mucosal immune responses during in vivo pulmonary bacterial infection. Proc. Natl Acad. Sci. USA 110, 3119–3128 (2013).
Toubal, A., Nel, I., Lotersztajn, S. & Lehuen, A. Mucosal-associated invariant T cells and disease. Nat. Rev. Immunol. 19, 643–657 (2019).
Trottein, F. & Paget, C. Natural killer T cells and mucosal-associated invariant T cells in lung infections. Front. Immunol. 9, 1–17 (2018).
McCarthy, N. E. & Eberl, M. Human γδ T-cell control of mucosal immunity and inflammation. Front. Immunol. 9, 985 (2018).
Clements, J. D. & Norton, E. B. The mucosal vaccine adjuvant LT(R192G/L211A) or dmLT. mSphere 3, 1–17 (2018).
Hammerschmidt, S. I. et al. Retinoic acid induces homing of protective T and B cells to the gut after subcutaneous immunization in mice. J. Clin. Invest. 121, 3051–3061 (2011).
Harro, C. et al. Live attenuated enterotoxigenic Escherichia coli (ETEC) vaccine with dmLT adjuvant protects human volunteers against virulent experimental ETEC challenge. Vaccine 37, 1978–1986 (2019).
Lebens, M. et al. Construction and preclinical evaluation of mmCT, a novel mutant cholera toxin adjuvant that can be efficiently produced in genetically manipulated Vibrio cholerae. Vaccine 34, 2121–2128 (2016).
Holmgren, J. et al. Preclinical immunogenicity and protective efficacy of an oral Helicobacter pylori inactivated whole cell vaccine and multiple mutant cholera toxin: a novel and non-toxic mucosal adjuvant. Vaccine 36, 6223–6230 (2018).
Ågren, L., Löwenadler, B. & Lycke, N. A novel concept in mucosal adjuvanticity: the CTA1-DD adjuvant is a B cell-targeted fusion protein that incorporates the enzymatically active cholera toxin A1 subunit. Immunol. Cell Biol. 76, 280–287 (1998).
Eriksson, A. M., Schön, K. M. & Lycke, N. Y. The cholera toxin-derived CTA1-DD vaccine adjuvant administered intranasally does not cause inflammation or accumulate in the nervous tissues. J. Immunol. 173, 3310–3319 (2004).
Schussek, S. et al. The CTA1-DD adjuvant strongly potentiates follicular dendritic cell function and germinal center formation, which results in improved neonatal immunization. Mucosal Immunol. 13, 545–557 (2020).
Bernasconi, V. et al. A vaccine combination of lipid nanoparticles and a cholera toxin adjuvant derivative greatly improves lung protection against influenza virus infection. Mucosal Immunol. 14, 523–536 (2021).
Mutsch, M. et al. Use of the inactivated intranasal influenza vaccine and the risk of Bell’s palsy in Switzerland. N. Engl. J. Med. 350, 896–903 (2004).
Lewis, D. J. M. et al. Transient facial nerve paralysis (Bell’s palsy) following intranasal delivery of a genetically detoxified mutant of Escherichia coli heat labile toxin. PLoS ONE 4, 1–5 (2009).
Eickhoff, C. S., Blazevic, A., Killoran, E. A., Morris, M. S. & Hoft, D. F. Induction of mycobacterial protective immunity by sublingual BCG vaccination. Vaccine 37, 5364–5370 (2019).
Ottsjö, L. S., Flach, C. F., Raghavan, S., Holmgren, J. & Clements, J. A double mutant heat-labile toxin from Escherichia coli, LT(R192G/L211A), is an effective mucosal adjuvant for vaccination against Helicobacter pylori infection. Infect. Immun. 81, 1532–1540 (2013).
Bernstein, D. I. et al. A phase 1 dose escalating study of double mutant heat-labile toxin LTR192G/L211A (dmLT) from enterotoxigenic Escherichia coli (ETEC) by sublingual or oral immunization. Vaccine 37, 602–611 (2019).
Pan, S. C. et al. A double-blind, randomized controlled trial to evaluate the safety and immunogenicity of an intranasally administered trivalent inactivated influenza vaccine with the adjuvant LTh(αK): a phase II study. Vaccine 38, 1048–1056 (2020).
Pan, S. C. et al. A randomized, double-blind, controlled clinical trial to evaluate the safety and immunogenicity of an intranasally administered trivalent inactivated influenza vaccine with adjuvant LTh(αK): a phase I study. Vaccine 37, 1994–2003 (2019).
Longet, S. et al. An oral α-galactosylceramide adjuvanted Helicobacter pylori vaccine induces protective IL-1R- and IL-17R-dependent TH1 responses. NPJ Vaccines 4, 45 (2019).
Davitt, C. J. H. et al. A novel adjuvanted capsule based strategy for oral vaccination against infectious diarrhoeal pathogens. J. Control. Rel. 233, 162–173 (2016).
Davitt, C. J. H. et al. α-Galactosylceramide enhances mucosal immunity to oral whole-cell cholera vaccines. Mucosal Immunol. 12, 1055–1064 (2019).
Moran, H. B. T., Turley, J. L., Andersson, M. & Lavelle, E. C. Immunomodulatory properties of chitosan polymers. Biomaterials 184, 1–9 (2018).
Carroll, E. C. et al. The vaccine adjuvant chitosan promotes cellular immunity via DNA sensor cGAS-STING-dependent induction of type I interferons. Immunity 44, 597–608 (2016).
Van Dis, E. et al. STING-activating adjuvants elicit a TH17 immune response and protect against Mycobacterium tuberculosis infection. Cell Rep. 23, 1435–1447 (2018).
Ebensen, T. et al. Mucosal administration of cycle-di-nucleotide-adjuvanted virosomes efficiently induces protection against influenza H5N1 in mice. Front. Immunol. 8, 1–15 (2017).
Madhun, A. S. et al. Intranasal c-di-GMP-adjuvanted plant-derived H5 influenza vaccine induces multifunctional TH1 CD4+ cells and strong mucosal and systemic antibody responses in mice. Vaccine 29, 4973–4982 (2011).
Mansouri, S. et al. Immature lung TNFR2− conventional DC 2 subpopulation activates moDCs to promote cyclic di-GMP mucosal adjuvant responses in vivo. Mucosal Immunol. 12, 277–289 (2019).
Bielinska, A. U. et al. Mucosal immunization with a novel nanoemulsion-based recombinant anthrax protective antigen vaccine protects against Bacillus anthracis spore challenge. Infect. Immun. 75, 4020–4029 (2007).
Ahmed, M. et al. A novel nanoemulsion vaccine induces mucosal interleukin-17 responses and confers protection upon Mycobacterium tuberculosis challenge in mice. Vaccine 35, 4983–4989 (2017).
Kimoto, T., Kim, H., Sakai, S., Takahashi, E. & Kido, H. Oral vaccination with influenza hemagglutinin combined with human pulmonary surfactant-mimicking synthetic adjuvant SF-10 induces efficient local and systemic immunity compared with nasal and subcutaneous vaccination and provides protective immunity in mice. Vaccine 37, 612–622 (2019).
Dolgin, E. How COVID unlocked the power of RNA vaccines. Nature 589, 189–191 (2021).
Ferber, S., Gonzalez, R. J., Cryer, A. M., von Andrian, U. H. & Artzi, N. Immunology-guided biomaterial design for mucosal cancer vaccines. Adv. Mater. 32, e1903847 (2020).
Lim, M. et al. Engineered nanodelivery systems to improve DNA vaccine technologies. Pharmaceutics 12, 1–29 (2020).
O’Driscoll, C. M., Bernkop-Schnürch, A., Friedl, J. D., Préat, V. & Jannin, V. Oral delivery of non-viral nucleic acid-based therapeutics—do we have the guts for this? Eur. J. Pharm. Sci. 133, 190–204 (2019).
Ball, R. L., Bajaj, P. & Whitehead, K. A. Oral delivery of siRNA lipid nanoparticles: fate in the GI tract. Sci. Rep. 8, 1–12 (2018).
Hajam, I. A., Senevirathne, A., Hewawaduge, C., Kim, J. & Lee, J. H. Intranasally administered protein coated chitosan nanoparticles encapsulating influenza H9N2 HA2 and M2e mRNA molecules elicit protective immunity against avian influenza viruses in chickens. Vet. Res. 51, 37 (2020).
Liebowitz, D., Lindbloom, J. D., Brandl, J. R., Garg, S. J. & Tucker, S. N. High titre neutralising antibodies to influenza after oral tablet immunisation: a phase 1, randomised, placebo-controlled trial. Lancet Infect. Dis. 15, 1041–1048 (2015).
Liebowitz, D. et al. Efficacy, immunogenicity, and safety of an oral influenza vaccine: a placebo-controlled and active-controlled phase 2 human challenge study. Lancet Infect. Dis. 20, 435–444 (2020). This paper reports that an oral tablet-based influenza vaccine induces protective immunity against viral shedding in a human influenza virus challenge model.
Joyce, C. et al. Orally administered adenoviral-based vaccine induces respiratory mucosal memory and protection against RSV infection in cotton rats. Vaccine 36, 4265–4277 (2018).
Magnusson, S. E. et al. Matrix-MTM adjuvant enhances immunogenicity of both protein- and modified vaccinia virus Ankara-based influenza vaccines in mice. Immunol. Res. 66, 224–233 (2018).
Hassan, A. O. et al. A single-dose intranasal ChAd vaccine protects upper and lower respiratory tracts against SARS-CoV-2. Cell 183, 1–16 (2020).
Frey, S. E. et al. A phase I, dose-escalation trial in adults of three recombinant attenuated Salmonella typhi vaccine vectors producing Streptococcus pneumoniae surface protein antigen PspA. Vaccine 31, 4874–4880 (2013).
Xiang, Z. Q. et al. Oral vaccination of mice with adenoviral vectors is not impaired by preexisting immunity to the vaccine carrier. J. Virol. 79, 3888–3888 (2005).
Cole, M. E. et al. Pre-existing influenza-specific nasal IgA or nasal viral infection does not affect live attenuated influenza vaccine immunogenicity in children. Clin. Exp. Immunol. 204, 125–133 (2021). This report shows that pre-existing antigen-specific nasal IgA does not compromise live attenuated influenza vaccine immunogenicity in children.
JanssenMD. Janssen COVID-19 vaccine immunity to the Ad26 vector. JanssenMD https://www.janssenmd.com/janssen-covid19-vaccine/clinical-use/janssen-covid19-vaccine-preexisting-immunity-to-the-ad26-vector (2021).
Church, J. A., Parker, E. P., Kirkpatrick, B. D., Grassly, N. C. & Prendergast, A. J. Interventions to improve oral vaccine performance: a systematic review and meta-analysis. Lancet Infect. Dis. 19, 203–214 (2019).
Zimmermann, P. & Curtis, N. The influence of probiotics on vaccine responses — a systematic review. Vaccine 36, 207–213 (2018).
Frederick, D. R. et al. Adjuvant selection regulates gut migration and phenotypic diversity of antigen-specific CD4+ T cells following parenteral immunization. Mucosal Immunol. 11, 549–561 (2018). This study shows that intradermal injection of the adjuvant dmLT promoted intestinal homing of antigen-specific CD4+ T cells.
McEntee, C. P. et al. Type I IFN signalling is required for cationic adjuvant formulation (CAF)01-induced cellular immunity and mucosal priming. Vaccine 38, 635–643 (2020).
Wagar, L. E. et al. Modeling human adaptive immune responses with tonsil organoids. Nat. Med. 27, 125–135 (2021). This paper shows human tonsil organoids can be used to assess responses to vaccines and adjuvants.
Krämer, B. et al. Compartment-specific distribution of human intestinal innate lymphoid cells is altered in HIV patients under effective therapy. PLoS Pathog. 13, 1–24 (2017).
Marquardt, N. et al. Human lung natural killer cells are predominantly comprised of highly differentiated hypofunctional CD69−CD56dim cells. J. Allergy Clin. Immunol. 139, 1321–1330.e4 (2017).
De Grove, K. C. et al. Characterization and quantification of innate lymphoid cell subsets in human lung. PLoS ONE 11, 1–12 (2016).
Golebski, K. et al. IL-1β, IL-23, and TGF-β drive plasticity of human ILC2s towards IL-17-producing ILCs in nasal inflammation. Nat. Commun. 10, 1–15 (2019).
Deusch, K. et al. A major fraction of human intraepithelial lymphocytes simultaneously expresses the γ/δ T cell receptor, the CD8 accessory molecule and preferentially uses the Vδ1 gene segment. Eur. J. Immunol. 21, 1053–1059 (1991).
Trejdosiewicz, L. K. et al. γδ T cell receptor-positive cells of the human gastrointestinal mucosa: occurrence and V region gene expression in Heliobacter pylori-associated gastritis, coeliac disease and inflammatory bowel disease. Clin. Exp. Immunol. 84, 440–444 (1991).
Fukushima, K., Masuda, T., Ohtani, H. & Nagura, H. Immunohistochemical characterization, distribution, and ultrastructure of lymphocytes bearing T-cell receptor γδ in inflammatory bowel disease. Gastroenterology 101, 670–678 (1991).
Ullrich, R., Schieferdecker, H. L., Ziegler, K., Riecken, E. O. & Zeitz, M. γδ T cells in the human intestine express surface markers of activation and are preferentially located in the epithelium. Cell. Immunol. 128, 619–627 (1990).
Spencer, J., Isaacson, P. G., Diss, T. C. & Macdonald, T. T. Expression of disulfide-linked and non-disulfide-linked forms of the T cell receptor γ/δ heterodimer in human intestinal intraepithelial lymphocytes. Eur. J. Immunol. 19, 1335–1338 (1989).
Jarry, A., Cerf-bensussan, N., Brousse, N., Selz, F. & Guy-grand, D. Subsets of CD3+ (T cell receptor α/β or γ/δ) and CD3− lymphocytes isolated from normal human gut epithelium display phenotypical features different from their counterparts in peripheral blood. Eur. J. Immunol. 20, 1097–1103 (1990).
Zeissig, S., Kaser, A., Dougan, S. K., Nieuwenhuis, E. E. S. & Blumberg, R. S. Role of NKT cells in the digestive system. III. Role of NKT cells in intestinal immunity. Am. J. Physiol. Gastrointest. Liver Physiol. 293, 1101–1105 (2007).
Hama, I. et al. Different distribution of mucosal-associated invariant T cells within the human cecum and colon. Cent. Eur. J. Immunol. 44, 75–83 (2019).
Dusseaux, M. et al. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood 117, 1250–1259 (2011).
Tominaga, K. et al. Possible involvement of mucosal-associated invariant T cells in the progression of inflammatory bowel diseases. Biomed. Res. 38, 111–121 (2017).
Gold, M. C. et al. Human mucosal associated invariant T cells detect bacterially infected cells. PLoS Biol. 8, 1–14 (2010).
Holmgren, J., Lonnroth, I. & Svennerholm, L. Tissue receptor for cholera exotoxin: postulated structure from studies with G(M1) ganglioside and related glycolipids. Infect. Immun. 8, 208–214 (1973).
Sethi, A. et al. Cell type and receptor identity regulate cholera toxin subunit B (CTB) internalization. Interface Focus 9, 20180076 (2019).
Wands, A. M. et al. Fucosylation and protein glycosylation create functional receptors for cholera toxin. eLife 4, 1–38 (2015).
Gustafsson, T. et al. Direct interaction between cholera toxin and dendritic cells is required for oral adjuvant activity. Eur. J. Immunol. 43, 1779–1788 (2013).
Lycke, N. & Lebrero-Fernández, C. ADP-ribosylating enterotoxins as vaccine adjuvants. Curr. Opin. Pharmacol. 41, 42–51 (2018).
Smits, H. H. et al. Cholera toxin B suppresses allergic inflammation through induction of secretory IgA. Mucosal Immunol. 2, 331–339 (2009).
Sun, J.-B., Raghavan, S., Sjöling, Å., Lundin, S. & Holmgren, J. Oral tolerance induction with antigen conjugated to cholera toxin B subunit generates both Foxp3+CD25+ and Foxp3−CD25−CD4+ regulatory T cells. J. Immunol. 177, 7634–7644 (2006).
Baldauf, K. J. et al. Oral administration of a recombinant cholera toxin B subunit promotes mucosal healing in the colon. Mucosal Immunol. 10, 887–900 (2017).
Jertborn, M., Nordström, I., Kilander, A., Czerkinsky, C. & Holmgren, J. Local and systemic immune responses to rectal administration of recombinant cholera toxin B subunit in humans. Infect. Immun. 69, 4125–4128 (2001).
Bergquist, C., Johansson, E. L., Lagergård, T., Holmgren, J. & Rudin, A. Intranasal vaccination of humans with recombinant cholera toxin B subunit induces systemic and local antibody responses in the upper respiratory tract and the vagina. Infect. Immun. 65, 2676–2684 (1997).
Harris, J. B. Cholera: immunity and prospects in vaccine development. J. Infect. Dis. 218, S141–S146 (2018).
Clemens, J. D. et al. Cross-protection by B subunit-whole cell cholera vaccine against diarrhea associated with heat-labile toxin-producing enterotoxigenic Escherichia coli: results of a large-scale field trial. J. Infect. Dis. 158, 372–377 (1988).
Scerpella, E. G. et al. Safety, immunogenicity, and protective efficacy of the whole-cell/recombinant B subunit (WC/rBS) oral cholera vaccine against travelers’ diarrhea. J. Travel. Med. 2, 22–27 (1995).
Holmgren, J. et al. Correlates of protection for enteric vaccines. Vaccine 35, 3355–3363 (2017).
Plotkin, S. A. Updates on immunologic correlates of vaccine-induced protection. Vaccine 38, 2250–2257 (2020).
Tomic, A., Tomic, I., Dekker, C. L., Maecker, H. T. & Davis, M. M. The FluPRINT dataset, a multidimensional analysis of the influenza vaccine imprint on the immune system. Sci. Data 6, 214 (2019).
Carlsson, B., Zaman, S., Mellander, L., Jalil, F. & Hanson, L. A. Secretory and serum immunoglobulin class-specific antibodies to poliovirus after vaccination. J. Infect. Dis. 152, 1238–1244 (1985).
Ainai, A. et al. Intranasal vaccination with an inactivated whole influenza virus vaccine induces strong antibody responses in serum and nasal mucus of healthy adults. Hum. Vaccines Immunother. 9, 1962–1970 (2013).
Akhtar, M. et al. Kinetics of antibody-secreting cell and fecal IgA responses after oral cholera vaccination in different age groups in a cholera endemic country. Vaccine 35, 321–328 (2017).
Aase, A. et al. Salivary IgA from the sublingual compartment as a novel noninvasive proxy for intestinal immune induction. Mucosal Immunol. 9, 884–893 (2016).
Saletti, G., Çuburu, N., Yang, J. S., Dey, A. & Czerkinsky, C. Enzyme-linked immunospot assays for direct ex vivo measurement of vaccine-induced human humoral immune responses in blood. Nat. Protoc. 8, 1073–1087 (2013).
Chang, H. S. & Sack, D. A. Development of a novel in vitro assay (ALS assay) for evaluation of vaccine-induced antibody secretion from circulating mucosal lymphocytes. Clin. Diagn. Lab. Immunol. 8, 482–488 (2001).
Chakraborty, S. et al. Characterization of mucosal immune responses to enterotoxigenic Escherichia coli vaccine antigens in a human challenge model: response profiles after primary infection and homologous rechallenge with strain H10407. Clin. Vaccine Immunol. 23, 55–64 (2016).
Uddin, T. et al. Mucosal immunologic responses in cholera patients in Bangladesh. Clin. Vaccine Immunol. 18, 506–512 (2011).
Quiding, M. et al. Intestinal immune responses in humans. Oral cholera vaccination induces strong intestinal antibody responses and interferon-γ production and evokes local immunological memory. J. Clin. Invest. 88, 143–148 (1991).
Åhrén, C., Jertborn, M. & Svennerholm, A. M. Intestinal immune responses to an inactivated oral enterotoxigenic Escherichia coli vaccine and associated immunoglobulin a responses in blood. Infect. Immun. 66, 3311–3316 (1998).
Mottram, L., Lundgren, A., Svennerholm, A. M. & Leach, S. A systems biology approach identifies B cell maturation antigen (BCMA) as a biomarker reflecting oral vaccine induced IgA antibody responses in humans. Front. Immunol. 12, 647873 (2021).
WHO. Cancer Tomorrow (International Agency for Research on Cancer, 2020).
Nizard, M. et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat. Commun. 8, 15221 (2017). This study shows that mucosal vaccine-induced TRM cells enhance antitumour immunity in mice.
Blanc, C. et al. Targeting resident memory T cells for cancer immunotherapy. Front. Immunol. 9, 1722 (2018).
Sandoval, F. et al. Mucosal imprinting of vaccine-induced CD8+ T cells is crucial to inhibit the growth of mucosal tumors. Sci. Transl Med. 5, 172ra20 (2013).
Sun, Y. Y. et al. Local HPV recombinant vaccinia boost following priming with an HPV DNA vaccine enhances local HPV-specific CD8+ T-cell-mediated tumor control in the genital tract. Clin. Cancer Res. 22, 657–669 (2016).
Hildner, K. et al. Batf3 deficiency reveals a critical role for CD8+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100 (2008).
Salmon, H. et al. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity 44, 924–938 (2016).
Fuertes, M. B. et al. Host type I IFN signals are required for antitumor CD8 + T cell responses through CD8α + dendritic cells. J. Exp. Med. 208, 2005–2016 (2011).
Roberts, E. W. et al. Critical role for CD103+/CD141+ dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell 30, 324–336 (2016).
Williford, J. M. et al. Recruitment of CD103+ dendritic cells via tumor-targeted chemokine delivery enhances efficacy of checkpoint inhibitor immunotherapy. Sci. Adv. 5, 1–16 (2019).
Parkhurst, M. R. et al. Unique neoantigens arise from somatic mutations in patients with gastrointestinal cancers. Cancer Discov. 9, 1022–1035 (2019).
Jiang, T. et al. Heterogeneity of neoantigen landscape between primary lesions and their matched metastases in lung cancer. Transl Lung Cancer Res. 9, 246–256 (2020).
Sahin, U. et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 (2017).
Ott, P. A. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017).
Sagiv-Barfi, I. et al. Eradication of spontaneous malignancy by local immunotherapy. Sci. Transl Med. 10, eaan4488 (2018).
Norton, E. B., Lawson, L. B., Freytag, L. C. & Clements, J. D. Characterization of a mutant Escherichia coli heat-labile toxin, LT(R192G/L211A), as a safe and effective oral adjuvant. Clin. Vaccine Immunol. 18, 546–551 (2011).
Terrinoni, M., Holmgren, J., Lebens, M. & Larena, M. Requirement for cyclic AMP/protein kinase A-dependent canonical NFκB signaling in the adjusvant action of cholera toxin and its non-toxic derivative mmCT. Front. Immunol. 10, 269 (2019).
Valli, E., Baudier, R. L., Harriett, A. J. & Norton, E. B. LTA1 and dmLT enterotoxin-based proteins activate antigen-presenting cells independent of PKA and despite distinct cell entry mechanisms. PLoS ONE 15, 1–21 (2020).
Larena, M., Holmgren, J., Lebens, M., Terrinoni, M. & Lundgren, A. Cholera toxin, and the related nontoxic adjuvants mmCT and dmLT, promote human TH17 responses via cyclic AMP–protein kinase A and inflammasome-dependent IL-1 signaling. J. Immunol. 194, 3829–3839 (2015).
Acknowledgements
The authors thank N. Muñoz-Wolf for critical reading of the manuscript. The mucosal vaccine research from the Lavelle laboratory has been funded by Science Foundation Ireland (SFI) (Grant numbers 12/IA/1421, 19/FFP/6484, SFI/12/RC/2278_2, 20/SPP/3685), the Irish Research Council and the European Union FP7 programme (FP7-SME-2012-1).
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information
Nature Reviews Immunology thanks J. Holmgren and J. Mestecky for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Glossary
- Chitosan
-
A cationic polymer derived from chitin with mucosal adjuvant properties. In addition to its mucoadhesive attributes, chitosan promotes adaptive immunity through activation of the cGAS–STING and NLRP3 inflammasome pathways.
- Enchained growth
-
A process where high-affinity IgA specific for surface antigens cross-links bacteria, preventing daughter cell separation after division and contributing to clearance of mucosal pathogens.
- Microfold (M) cells
-
Specialized antigen sampling epithelial cells generally found in the follicle-associated epithelium overlying organized mucosal lymphoid tissue.
- Polymersomes
-
Hollow vesicles generally comprising block copolymers that can incorporate vaccine antigens and adjuvants in the aqueous phase.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lavelle, E.C., Ward, R.W. Mucosal vaccines — fortifying the frontiers. Nat Rev Immunol 22, 236–250 (2022). https://doi.org/10.1038/s41577-021-00583-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-021-00583-2
This article is cited by
-
Oral administration of DNA alginate nanovaccine induced immune-protection against Helicobacter pylori in Balb/C mice
BMC Immunology (2024)
-
Immunity to Cryptosporidium: insights into principles of enteric responses to infection
Nature Reviews Immunology (2024)
-
An intranasal live-attenuated SARS-CoV-2 vaccine limits virus transmission
Nature Communications (2024)
-
Intranasal immunization of mice with chimera of Salmonella Typhi protein elicits protective intestinal immunity
npj Vaccines (2024)
-
Nanotechnology of inhalable vaccines for enhancing mucosal immunity
Drug Delivery and Translational Research (2024)
