
Abstract
Cholangiocarcinoma (CCA) is a heterogeneous group of malignancies with features of biliary tract differentiation. CCA is the second most common primary liver tumour and the incidence is increasing worldwide. CCA has high mortality owing to its aggressiveness, late diagnosis and refractory nature. In May 2015, the “European Network for the Study of Cholangiocarcinoma” (ENS-CCA: www.enscca.org or www.cholangiocarcinoma.eu) was created to promote and boost international research collaboration on the study of CCA at basic, translational and clinical level. In this Consensus Statement, we aim to provide valuable information on classifications, pathological features, risk factors, cells of origin, genetic and epigenetic modifications and current therapies available for this cancer. Moreover, future directions on basic and clinical investigations and plans for the ENS-CCA are highlighted.
Similar content being viewed by others

Main
Cholangiocarcinoma (CCA) is a heterogeneous group of malignancies that can emerge at every point of the biliary tree, from the canals of Hering to the main bile duct1,2. According to their anatomical location, CCAs are classified as intrahepatic (iCCA), perihilar (pCCA) and distal CCA (dCCA), which have particular similarities but also important inter-tumour and intra-tumour differences that can affect the pathogenesis and outcome. CCAs, taken together, represent the second most frequent type of primary liver cancer and ∼3% of all gastrointestinal neoplasias1. However, the epidemiological profile of CCA and its subtypes (Fig. 1) displays enormous geographical variation, reflecting the exposure to different risk factors. Although in most countries CCA is a rare cancer (incidence <6 cases per 100,000 people), its incidence is exceptionally high in some countries and regions, including Chile, Bolivia, South Korea and North Thailand1. In general, a progressive increase in iCCA incidence worldwide was reported up to the end of the past century, reaching a plateau in the past 10 years. By contrast, the incidences of both pCCA and dCCA seem to be decreasing1. CCAs are generally asymptomatic in early stages and are usually diagnosed when the disease has already metastasized, by combining nonspecific biomarkers in serum and/or biopsy samples, as well as imaging methods3,4. Late diagnosis compromises the effective therapeutic options, which are based on surgical resection and/or liver transplantation, whereas chemotherapies are virtually palliative given the marked chemoresistance of this cancer4,5,6. Tumour size and other features such as anatomical location, vascular and lymph node invasion, and metastasis condition the potential surgical and/or radiological options but chances of recurrence are very high4,5. Individual characterization (that is, genomic, epigenetic and molecular) of each tumour might provide valuable information on pathogenesis, prognosis and chemosensitivity, thus indicating the best therapeutic options for each patient as well as new potential targets for therapy.

Worldwide incidence (cases per 100,000) of cholangiocarcinoma (CCA)1,5,30. Data refer to the period 1971–2009. Green colour identifies countries with lower incidence (<6 per 100,000 cases, rare cancer), whereas pink colour indicates countries in which CCA is not a rare cancer (>6 per 100,000 cases). Diagnoses have been classified according to international classification of disease (ICD) codes (ICD-O-1, ICD-O-2, ICD-O-3, ICD-10, ICD-V9, ICD-V10, ICD-O). When available, the more incident form (intrahepatic (IH) versus extrahepatic (EH) CCA) and the temporal trend of incidence (↑increasing trend; ↕stable trend; ↓decreasing trend) have been reported.
In sum, CCA represents a global health problem that warrants considerable attention and thorough investigation (Box 1). In this respect, the European Network for the Study of Cholangiocarcinoma (ENS-CCA) was created in May 2015 (www.enscca.org or www.cholangiocarcinoma.eu) to promote and boost collaborative research projects on CCA at basic, translational and clinical levels. In this Consensus Statement, the ENS-CCA aims to provide knowledge on novel classifications, genetics, epigenetics, pathogenesis, signalling pathways, current emerging therapies and future directions in basic and translational research as well as clinical medicine for CCA.
Methods
The ENS-CCA is constituted by active research groups of nine European countries (Austria, Denmark, France, Germany, Italy, Norway, Spain, Switzerland and the UK) and also involves distinguished International Advisors and/or Collaborators (see Acknowledgements). These research teams have contributed to generate many of the key advancements in the pathophysiology of the biliary tree and development and/or onset of CCA.
To write this Consensus statement, relevant articles were found by searching PubMed with the term “cholangiocarcinoma” in combination with the following terms: “diagnosis”, “progression”, “survival”, “growth factors”, “neuroendocrine peptides”, “estrogen”, “inflammation”, “cancer-associated fibroblast”, “myofibroblasts”, “hepatic stellate cells”, “macrophages”, “tumour-associated macrophage”, “endothelial cells”, “vascular cells”, “inflammation”, “classification”, “histology”, “cell of origin”, “cancer stem cell”, “therapy”, “chemoresistance”. No specific search dates were used.
Classification and pathological features
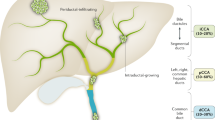
The classification of CCA has become a matter of intense debate. Considering different aspects of these tumours, several classifications have been proposed2,7. On the basis of anatomical location, the latest system classifies CCA into iCCA, pCCA and dCCA (Fig. 2A).

a | Cholangiocarcinomas (CCAs) are classified according to the anatomical location into intrahepatic (iCCA), perihilar (pCCA) and distal (dCCA). b | Concerning the gross appearance, the iCCA can present three different patterns of growth: mass-forming; periductal infiltrating; and intraductal growth.
On the basis of gross appearance, the iCCA can present three different patterns of growth (Fig. 2B): mass-forming (MF-iCCA), periductal infiltrating (PI-iCCA), and intraductal growing (IG-iCCA), in which the MF-iCCA largely represents the most frequent form3,8. For pCCA and dCCA, growth patterns similar to PI-iCCA or IG-iCCA can also be seen; however, pCCA can adopt a nodular plus periductal infiltrating growth pattern that represents the most frequent form (>80%)3,9,10. MF-iCCA usually occurs in chronic non-biliary liver diseases and arises in peripheral small bile ducts, whereas PI-iCCA and IG-iCCA types exclusively involve large (segmental and area) intrahepatic bile ducts11. These differences can be also linked to the bile duct heterogeneity (that is, small versus medium and large bile ducts)12. The PI-iCCA type grows longitudinally along the bile duct, typically determines biliary strictures13 and, in some cases, invades the liver parenchyma adopting combined features of periductal infiltrating and mass-forming types (PI+MF-iCCA). The IG-iCCA type shows papillary growth towards duct lumina11,13 but at present the American Joint Cancer Committee/Union for International Cancer Control (AJCC/UICC) does not recognize this growth pattern13,14. Similar to pCCA and dCCA, PI-iCCA emerging from large intrahepatic bile ducts is often preceded by preinvasive lesions classified as biliary intraepithelial neoplasm, intraductal papillary neoplasm, mucinous cystic neoplasm or intraductal tubular neoplasm11,15,16. By contrast, preinvasive lesions of the MF-CCA are not well known.
Histologically, the vast majority of pCCA and dCCA are mucinous adenocarcinomas. Conversely, iCCAs are highly heterogeneous tumours and several classifications have been proposed11,13,17,18. However, despite differences in nomenclature, iCCAs show two main histological subtypes, reflecting their anatomical origin along the intrahepatic biliary tree: bile ductular type (mixed) (Fig. 3A), arising from small intrahepatic bile ducts, and bile duct type (mucinous) (Fig. 3B), arising from large intrahepatic bile ducts11,13,17,18. Interestingly, this histological subclassification corresponds to different clinicopathological features. The bile ductular type (mixed) iCCAs display an almost exclusively mass-forming growth pattern11,13,17,18, are frequently associated with chronic liver diseases (viral hepatitis or cirrhosis)19 and are not preceded by pre-neoplastic lesions such as biliary intraepithelial neoplasm or intraductal papillary neoplasm11,13,17,18. Notably, bile ductular type (mixed) iCCAs share clinicopathological similarities with cytokeratin (CK) 19-positive hepatocellular carcinoma (HCC)17,20. On the other hand, bile duct type (mucinous) iCCAs might appear grossly as mass-forming, periductal infiltrating or intraductal growing types; they are more frequently associated with primary sclerosing cholangitis (PSC) than bile ductular type (mixed) iCCAs, and can be preceded by preneoplastic lesions such as biliary intraepithelial neoplasm or intraductal papillary neoplasm11,13,17,18. Interestingly, the bile duct type (mucinous) iCCAs share phenotypic traits with pCCA and pancreatic cancers17. In our opinion, this histological subtyping should be taken into serious consideration because it underlines different cell of origin, aetiology, risk factors, molecular profile, clinical outcome and response to treatment.

a | Bile ductular (mixed)-type intrahepatic cholangiocarcinomas (iCCAs) are composed of areas with small tubular cord-like structures (yellow arrows) with irregular lumina (arrowheads) and cordonal areas expressing hepatocyte markers such as Hep-Par1 (hepatocyte-like areas, yellow arrows). The proportion of each area is largely variable. In general, mucin production is low or absent. Scale bars = 200 μm and 100 μm as indicated. b | Bile duct (mucinous)-type iCCAs are well-to-moderately differentiated adenocarcinoma with abundant stroma (asterisks); these tumour subtypes are composed of tubular, acinar or papillary structures; tumour cells are columnar with abundant clear, eosinophilic or mucinous cytoplasm; mucin production is also present in the glandular lumen (arrowheads). CK7, cytokeratin-7; H&E, haematoxylin and eosin; PAS, periodic acid–Schiff.
Risk factors
Although most CCAs are considered de novo without apparent cause, there are also well-established risk factors21,22 (Box 2). Infection with liver flukes (Opisthorchis viverrini and Clonorchis sinensis) is a common risk factor in East Asia where iCCA represents a large proportion (∼85%) of primitive liver cancers23,24,25. The association between CCA (mainly pCCA) and PSC is well established especially in Europe, but PSC is a rare disease23. More relevant from epidemiological and clinical points of view is the association with HBV-related and HCV-related liver diseases that have been identified as definitive risk factors, with a stronger association for iCCA than pCCA26. In general, HCV-related diseases show an increased association with CCA in Europe and other Western countries; the association with HBV is more statistically significant where the prevalence of HBV infection is high, including Asian countries27,28. Occult HBV infection is an emerging risk factor for iCCA29. The increased incidence of iCCA, registered at the end of the past century, has been linked with the burden of HCV infection29,30. Other studies also demonstrate an association between metabolic syndrome and CCA, which could lead to increased incidence in Western countries given the rising prevalence of obesity31,32. Hepatolithiasis, as well as congenital biliary tract malformations such as Caroli disease and bile duct cysts, also predisposes to the development of CCA33,34. All these risk factors share, as a putative pathogenic mechanism, chronic inflammation involving the biliary tract34,35. This process might be favoured by local intrahepatic accumulation of bile acids, even in the absence of net cholestasis36. Several toxic and environmental factors are known or suspected to be related to CCA development, among them nitrosamine-contaminated food, asbestos, dioxins, vinyl chlorides and thorotrast22. Heavy smoking and possibly alcohol consumption represent relevant cofactors.
Cells of origin
The cell of origin, or cancer-initiating cell, is considered to be the normal cell that receives the first cancer-causing mutation37,38. On the other hand, cancer stem cells (CSCs) are the cells that sustain tumour growth and propagation37,38. The phenotype of cell of origin and CSCs might therefore be substantially different37,38.
CCAs of different locations exhibit pronounced heterogeneity, raising the question of potential diverse cellular origins in every type of CCA17. Possible cells of origin are hepatic stem cells, immature neural cell adhesion molecule positive (NCAM+) cholangiocytes, mature (NCAM−) interlobular cholangiocytes and peribiliary gland cells39. According to different observations, pCCAs are thought to originate from mucin-secreting cholangiocytes and/or peribiliary glands40,41 located in hilar bile ducts. First, pCCAs are associated with preneoplastic lesions emerging in surface epithelium3 and peribiliary glands40,41. Second, immunohistochemistry and gene-expression profiling of pCCA have shown a strong similarity to the cylindrical, taller, mucin-producing cholangiocytes (or peribiliary glands) lining hilar bile ducts17,42,43. iCCAs show inter-tumour heterogeneity, leading to the classification into two main different histological subtypes17,44, with both probably having a different cell of origin17. The bile duct (mucinous) type iCCAs arise in large intrahepatic bile ducts that share anatomical (mucin-secreting cholangiocytes and peribiliary glands) and embryological similarities with the extrahepatic biliary tree and pancreatic duct system45,46. This iCCA subtype displays immunohistochemistry, gene expression and a clinicopathological profile that can be superimposed on pCCA17 and, in addition, shows (together with pCCA) large similarities to pancreatic ductal adenocarcinoma47,48,49. Consistently, the pattern of growth and the presence of preneoplastic lesions in cholangiocytes and peribiliary glands lining large intrahepatic bile ducts seem to indicate that these cells are candidate cells of origin40,43. The bile ductular (mixed)-type iCCAs show an immunohistochemical and gene expression profile corresponding to mucin-negative cuboidal cholangiocytes that line the smaller bile ducts (interlobular bile ducts and ductules)17. In addition, the phenotypic and genotypic profiles are similar to cholangiolocellular carcinoma, thought to originate from hepatic progenitor cells17,50,51. A number of observations suggest that bile ductular (mixed)-type iCCAs together with cholangiolocellular carcinoma and CK19+ HCC represent a group of primitive liver cancers originating from hepatic progenitor cells, the different phenotype depending on the step of hepatic progenitor cell differentiation toward cholangiocytes or hepatocytes, in which neoplastic transformation occurs17,20,52,53,54.
Cancer stem cells
CSCs are defined as the cells within a tumour that possess the capacity for self-renewal and generation of heterogeneous lineages. CSCs are highly tumorigenic and responsible for chemoradioresistance and for tumour recurrence38,55.
Several CSC markers are expressed in human CCAs56, including CD133 (Ref. 57), epithelial cell adhesion molecule (EpCAM)58, CD44 (Ref. 59), CD13 (Ref. 60), SOX2 (Ref. 59), CD90 (Ref. 61) and Nanog62, the entity of expression correlating with the worst prognosis. Although in most solid cancers CSCs represent <3% of the total cell population, in CCAs >30% of the tumour mass express CSC markers, pointing to the potential role of CSCs in CCA47. Indeed, all CSC markers characterize normal stem cells located in canals of Hering or bile ductules and/or peribiliary glands, further indicating these structures as the site of origin of most CCAs46. On the other hand, the absence of specific markers limits selective strategies targeting CSCs in CCAs. Consistently, also in liver-fluke-associated CCAs, the vast majority of neoplastic cells co-express CK19 and albumin, a feature characterizing hepatobiliary stem or progenitor cells63. Interestingly, the CSC profile is similar between pCCA and the bile duct (mucinous) type iCCA with high representation of the intestinal CSC marker LGR5, whereas, at variance, CD13+ CSCs characterized bile ductular (mixed)-type iCCA47. Notably, the original human CCA can be reproduced by injecting selected human CSCs into liver of a mouse model of cirrhosis47. Most CCA-associated CSCs co-express epithelial and mesenchymal features and display markers of EMT (epithelial to mesenchymal transition) trait, therefore justifying many typical CCA properties including desmoplastic features, morphopathological heterogeneity, aggressiveness and resistance to chemotherapeutic agents.
The CSC model is also involved in tumour heterogeneity64,65. Specifically, genetically distinct subclones together with developmental pathways and epigenetic modifications can contribute to functional heterogeneity and chemoresistance65. Furthermore, the tumour microenvironment, composed of cancer-associated macrophages, fibroblasts and vascular cells and functioning as a specialized CSC niche, contributes to the maintenance of stemness and chemoresistance65,66,67.
Genomic and epigenetic alterations
Genomic heterogeneity
The genomic heterogeneity of CCA (Table 1) is not only related to the diverse anatomical location of the tumour (that is, intrahepatic, perihilar or distal) but also to the various risk factors and associated pathologies29,67,68,69,70,71,72,73,74,75. The most prevalent genetic alterations identified in CCA affect key networks such as DNA repair (TP53)72,73,75, the WNT–CTNNB1 pathway67, tyrosine kinase signalling (KRAS, BRAF, SMAD4 and FGFR2)29,68,70,73,74,75, protein tyrosine phosphatase (PTPN3)71, epigenetic (IDH1 and IDH2)69,70,72,74,75 and chromatin-remodelling factors (histone-lysine N-methyltransferase 2C, also known as MLL3)73, including the SWI/SNF complex (ARID1A, PBRM1 and BAP1)69,70,72,75 and deregulated Notch signalling, which is a key component in cholangiocyte differentiation and biliary duct development. Recurrent genetic variants have also been identified in the promoter of the human telomerase reverse transcriptase (TERT)70, which for CCA is found to be associated with chronic hepatitis70. Thus, all these alterations summarize some of the genome defects and pathways involved in CCA development, and represent potential candidates for personalized targeted cancer therapy.
Whole-genome analyses of CCA have provided additional peculiarities. As far as iCCA is concerned, two distinct genomic classes have been characterized: an inflammatory class with predominant activation of inflammatory pathways, and a proliferation class with predominant activation of oncogenes that correlate with worse patient outcome76. Next-generation sequencing of 56 cancer-related genes has been performed in ∼150 CCAs with different localizations. The majority of CCAs showed a driver gene mutation, although tumours from different sites (iCCA versus pCCA and dCCA) had different genetic profiles, with a prevalence of RAS mutations in the dCCA77. Further underlining the complexity of the molecular classification of CCAs, exome-sequencing revealed a unique subtype of CCA without RAS mutations and/or fibroblast growth factor receptor 2 fusion genes78. This apparent multitude of CCA subtypes might very well reflect the diverse underlying risk factors, tumour biology and prognosis, but are not yet ready for clinical application5.
FGFR2 gene fusions
Fusion gene products between the kinase receptor FGFR2 and multiple other genes have been described in CCA. This alteration is not currently identified in other liver cancers, and is targetable and relevant for use in diagnosis of the disease. FGFR2 fusion gene products are FGFR2–BICC1 (Ref. 79), FGFR2–KIAA1598 (Ref. 80), FGFR2–TACC3 (Ref. 80), FGFR2–AHCYL1 (Ref. 78), FGFR2–MGEA5 (Ref. 68), FGFR2–KCTD1 and FGFR2–TXLNA29. FGFR2–BICC1 and other selected FGFR fusions have been shown to facilitate oligomerization and FGFR kinase activation, which result in altered cell morphology and increased cell proliferation79. The in vitro and in vivo oncogenic ability of FGFR2 fusion proteins has effectively been suppressed by treatment with FGFR kinase inhibitors such as BGJ398 and PD173074 (Ref. 78), as well as PD173074 and pazopanib78,79, suggesting that FGFR fusion kinase is a promising candidate for targeted therapy for CCA. Accordingly, the beneficial effect of FGFR2 inhibition in patients with CCA who have FGFR2–MGEA5 and FGFR2–TACC3 fusions after treatment with ponatinib and pazopanib, respectively, has been highlighted68.
Finally, an integrative RNA-sequencing and exome-sequencing analysis revealed the presence of another novel fusion product, FGFR2–PPHLN1 (Ref. 74). This study demonstrated that FGFR2 fusions might represent the most recurrent targetable alteration in CCA. Importantly, FGFR2 fusions could also represent a diagnostic marker as these rearrangements are almost exclusively found in iCCA29,74,78.
Epigenetic modifications
The mechanisms involved in gene regulation controlled by epigenetics include histone modification, DNA methylation and noncoding RNAs. Information related to the effect of altered histone modifications in CCA and to the response of CCA to epigenetic-based therapies is limited81,82,83.
In the epigenetic landscape, frequent mutations have been shown in both IDH1 and IDH2 in CCA84,85. Mutations in IDH are associated with hypermethylation of CpG shores, which suggests global deregulation within the transcriptional programme85. IDH1 was emphasized as an epigenetic rheostat that when mutated was proposed to reshape the genomic landscape with a global consequence on the transcriptional machinery, triggering an altered state in the cellular process of differentiation86. Importantly, mutations in IDH were shown to cause the deregulation of hepatocyte nuclear factor 4α (HNF4α), blocking hepatocytic differentiation and thus promoting bile duct cancer87.
The contribution of chronic liver inflammation alongside an aberrant epigenetic landscape provides survival signals to the tumour (for example, IL-6–STAT3)88,89. Marked reduction in DNA hydroxymethylation characterizes CCA tissue when compared with non-neoplastic tissue and a DNA mCyt content of ≥5.59% in peripheral blood mononuclear cells relates to a favourable outcome in primary liver cancers81. On the other hand, several promoters of genes involved in Wnt signalling are hypermethylated in CCA tissue90. A number of publications strongly suggest that epigenetic changes are early events in the malignant process, linking the tumour epigenetics to the microenvironment82 and opening up opportunities for early detection of CCA83. In particular, overexpression of histone deacetylase 6 (HDAC6) was reported in CCA, which promotes the shortening of the primary cilium and subsequent hyperproliferation91. Molecular and pharmacological targeting of HDAC6 restores the primary cilium and decreases CCA cell growth91. These data suggest that restoration of primary cilia in CCA cells by HDAC6 targeting is a potential therapeutic approach.
Molecular pathways and interactions
Endocrine and neuroendocrine factors
Several hormones and growth factors promote proliferation and exert anti-apoptotic effects on reactive and neoplastic cholangiocytes (Fig. 4)92. CCAs are estrogen-sensitive tumours and the expression of both estrogen receptors (ER-α and ER-β) is generally increased93. Although ER-α activation stimulates proliferation of CCA cells93, the selective stimulation of the ER-β has antineoplastic effects in vitro and in vivo via induction of apoptosis94. Commonly, estrogen-sensitive cancers lose ER-β expression with disease progression; however, the expression of ER-β is maintained in CCA at advanced stages, representing a potential therapeutic target94. Indeed, administration of the ER antagonist, tamoxifen, or a selective ER-β-selective agonist, KB9520, inhibits CCA growth in vivo95. Moreover, estrogens stimulate the expression of IL-6 and vascular endothelial growth factor (VEGF), both crucial mediators of CCA biology96,97.

a | Multiple factors and molecular pathways modulate the proliferative capacity of cholangiocarcinoma (CCA) cells. Reactive and neoplastic cholangiocytes actively secrete a number of neuroendocrine factors that either stimulate or inhibit cellular proliferation in an autocrine or paracrine fashion. Bile acids are able to influence a number of intracellular oncogenic pathways, either by direct binding to bile acid receptors (e.g. S1PR2), transactivation of growth factor receptors or intracellular entry. Inflammatory cytokines can induce DNA damage via induction of inducible nitric oxide synthase (iNOS) and regulate the expression of survival signalling cascades. Lately, pathways involved in biliary embryological development such as Notch and Hedgehog have also been shown to modulate the neoplastic proliferation of CCA cells. Many of these pathways are actively investigated as potential therapeutic targets. b | Escape from apoptosis is equally essential for CCA cell survival. Bile acids, inflammatory cytokines and developmental pathways play crucial roles in apoptosis resistance, mainly via the overexpression of MCL1 and the blockage of caspase activation. A number of neuroendocrine factors have also been shown to induce apoptosis and might prove useful as therapeutic tools. 2-AG, 2-arachidonylglycerol; COX-2, cyclooxygenase 2; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; ER, estrogen receptor; GABA, γ-aminobutyric acid; IL-6R, IL-6 receptor; NO, nitric oxide; OR, opioid receptor; TGF, transforming growth factor.
Several other mediators have also been shown to regulate biliary proliferation in CCA (Fig. 4)98, either by stimulating cell growth or affecting survival. Examples of factors promoting proliferative effects in CCA cells include serotonin, dopamine, leptin, opioids and the endocannabinoid 2-arachidonoylglycerol99,100,101,102,103,104. Interestingly, some of those mediators, such as serotonin or endogenous opioid peptides, limit cholangiocyte hyperplasia in response to damage99,102,105 However, such a function is lost in the course of CCA development, in which they stimulate cell growth and survival99,102,105.
Among neuroendocrine factors that either inhibit proliferation or induce apoptosis secretin, gastrin, γ-aminobutyric acid, endothelin-1 and the endocannabinoid anandamide have been described103,104,106,107,108,109,110,111. Although the activation of histamine H3 and H4 receptors (HRH3 and HRH4) inhibits CCA growth, histamine itself is considered proliferative as it sustains CCA growth by forming an autocrine loop112,113. The proliferative effects of histamine are in part mediated through HRH1, as shown by in vitro blocking experiments using a HRH1 antagonist112,113,114.
Growth factors
Immunohistochemistry studies have shown that the epidermal growth factor receptor (EGFR) is overexpressed in CCA human samples115,116. EGFR activation triggers the oncogenic MAPK–ERK signalling pathway in cholangiocytes, making this receptor a potential target for therapy117. To this extent, mutations and amplifications in the EGFR gene have been found in up to 15% and 5% of CCAs, respectively118,119.
The expression level of hepatocyte growth factor (HGF) receptor (HFGR, also known as c-Met) is also increased in CCA120,121. In iCCA, HGFR overexpression is associated with poor prognosis120. Moreover, the activation of the EGF and HGF pathways has been shown to influence the metastatic potential of CCA. Indeed, EGFR activation contributes to the EMT of CCA cells122, which has been implicated in tumour invasiveness and poor differentiation123,124, and HGF stimulates in vitro cell invasiveness and motility via AKT and ERK pathways125.
Biliary compounds
Cholestatic conditions are well-known risk factors for CCA development. Bile acids are able to activate the EGFR via a transforming growth factor (TGF) α-dependent mechanism, thereby stimulating cholangiocyte proliferation126. Moreover, conjugated bile acids such as glycochenodeoxycholic acid downregulate the expression of the bile acid receptor farnesoid X-activated receptor (FXR, also known as bile acid receptor) and promote CCA growth in vivo; this event is inhibited by the administration of FXR agonists127. Moreover, growth-promoting effects of conjugated bile acids via the activation of sphingosine 1-phosphate receptor 2 have been reported128,129. In sum, in experimental models, bile acid accumulation during cholestatic conditions seems to facilitate carcinogenesis via the induction of biliary proliferation and inflammation rather than by direct mutagenic effects36.
Developmental pathways
Deregulation of developmental pathways is involved in CCA pathophysiology. Cell-fate tracing experiments have demonstrated that combined activation of Notch and AKT can lead to iCCA arising from mature hepatocytes in mice130,131. The expression of the intracellular domain of Notch2 upregulates proliferative genes that sustain the dysplastic proliferation of cholangiocytes in vivo132. Notch2 and the Notch ligand Jagged1 seem to be particularly important, as their inhibition almost eliminates CCA development in a mouse model of liver cancer driven by transfection of activated forms of AKT and Ras oncogenes133,134. Activation of Notch signalling has also been shown to induce EMT and increase the migration of CCA cells135,136. An additional developmental pathway involved in CCA is the Hedgehog (Hh) signalling pathway. The Hh ligand Sonic hedgehog protein is overexpressed in human CCA and the inhibition of its receptor Smoothened by cyclopamine reduces the proliferation and invasion of CCA cells137. CCA cells also exhibit a non-canonical G protein-coupled Hh signalling, which does not require cilia expression and controls chemotaxis and metastasis formation138. Moreover, Hh signalling is a potent survival pathway in CCA. Activation of Hh pathway by myofibroblast-derived PDGF-BB is essential in protecting CCA cells from TRAIL-induced apoptosis139, which acts, at least in part, via modulation of the cell-cycle kinase serine/threonine-protein kinase PLK2 (also known as polo-like kinases 2)140. As such, the fine-tuning of developmental pathways seems to be a promising therapeutic tool for CCA. A number of modulators and inhibitors of Notch and Hh signalling have been described and certainly warrant further investigation141.
Inflammatory mediators
CCA often arises in the context of biliary inflammation. Integrative molecular analysis of iCCA identified two different biological subtypes of the tumour, namely the proliferation and inflammation classes76. The latter is specifically characterized by activation of inflammatory pathways and overexpression of different cytokines. IL-6, which is constitutively secreted by CCA cells, has crucial paracrine stimulatory effects on cholangiocyte growth via the activation of the MAPK pathway or the epigenetic control of gene expression142,143. IL-6 also modulates the survival of CCA cells through the induction of induced myeloid leukaemia cell differentiation protein Mcl-1 (MCL1), an anti-apoptotic member of the Bcl2 family responsible for resistance to TRAIL144,145. MCL1 expression is induced by IL-6 via the modulation of the MAPK, JAK–STAT and AKT pathways144,146,147. Along the same lines, suppressor of cytokine signalling 3, which normally controls IL-6–STAT-3 signalling pathway by a negative feedback loop, is epigenetically silenced in CCA148. Malignant cholangiocytes also overexpress TGFβ and TGFβ receptor II149,150. TGFβ has been shown to induce EMT in CCA cells via modulation of epithelial and mesenchymal markers expression, and thereby promotes invasion and migration of CCA151,152.
The enzyme cyclooxygenase-2 (COX2), responsible for prostaglandin synthesis, also has a role in CCA development. COX2 expression is induced in CCA by both bile acids and oxysterols, oxidation products of cholesterol that are increased in bile during biliary inflammation153,154. The inhibition of COX2 by celecoxib has been shown to reduce proliferation and to increase apoptosis of CCA cells in different studies155,156,157,158,159. The microsomal prostaglandin E synthase-1, which is coupled with COX2 and mediates the synthesis of prostaglandin E2, is also crucial for cholangiocarcinogenesis in vitro and in vivo160.
Inflammatory cytokines might also induce the expression of inducible nitric oxide synthase (iNOS) in CCA161. Nitric oxide (NO) promotes DNA damage directly and also by inhibiting DNA repair mechanisms, thereby promoting carcinogenesis161,162. iNOS activation also stimulates the expression of COX2 (Ref. 163). In this complex scenario, in an integrated mouse model of CCA based on constitutively active AKT and YAP, cholestasis induced by bile duct ligation and IL-33 administration largely recapitulates the molecular pathogenesis of human CCA164,165.
Tumour microenvironment
CCA is characterized by a prominent desmoplastic stroma141, which is composed primarily of cancer-associated fibroblasts (CAFs) and a lesser proportion of tumour-associated macrophages (TAMs) and vascular cells. Through reciprocal interactions with malignant cells, stromal cells potentially contribute to the hallmarks of cancer and therapeutic responses166,167. Extracellular vesicles such as microvesicles and exosomes are emerging as important carriers involved in the intercellular communication of cancer cells with tumour microenvironment168. The presence of microRNA-laden extracellular vesicles in human bile has been described in patients with CCA169,170. CCA-cell-derived extracellular vesicles are able to modulate fibroblastic differentiation of mesenchymal stem cells, which in turn can enhance CCA growth in vitro by releasing proinflammatory factors, such as IL-6 (Ref. 171).
The stroma of CCA undergoes profound changes in its composition during cholangiocarcinogenesis with an upregulation of genes related to the cell cycle, extracellular matrix, TGFβ pathway and inflammation172,173. Stromal signature was found to be significantly associated with poor CCA prognosis (enrichment score = 0.52), consistent with a major contribution of the microenvironment to tumour progression173.
Cancer-associated fibroblasts
Although their origin has not been formally proven, CAFs are probably derived from activated hepatic stellate cells and/or portal (or periductal) fibroblasts in the liver174. CAFs, which express α-smooth muscle actin, are able to modulate several processes (that is, proliferation, migration, invasion and EMT) in CCA tumour cells139,175,176,177,178,179,180. Accordingly, patients with high levels of α-smooth muscle actin expression in CCA tissue samples exhibit worse prognosis181,182. Experimental in vitro models of CCA–stromal cells co-cultures have shown that the major signalling axes identified so far involving CAF are: PDGF–PDGFR139,175, SDF-1–CXCR4 (Refs 176,177), HB-EGF–EGFR178, CXCL5–CXCR2–IL-1β179 and Hh180.
Regarding therapeutic implications, a study has demonstrated how the unique properties of CAFs can be used in tumour treatment183. Activation or transdifferentiation of hepatic stellate cells into myofibroblasts enhances their susceptibility to apoptosis, which makes them exceptional targets to impair their reciprocal communication with cancer cells. By using the cytotoxic drug navitoclax (an inhibitor of Bcl-2, Bcl-XL and Bcl-w) apoptosis was induced only in CAFs in a syngeneic rat model of CCA, with the concomitant reduction of desmoplastic extracellular matrix proteins, suppressing tumour growth and improving host survival. Thus, these data strongly support the possibility of targeting CAFs from the tumour stroma as a therapeutic strategy.
Immune cells
TAMs are the most representative infiltrating immune cells of the CCA stromal microenvironment. These cells mainly originate from circulating monocytes, specifically from a minor blood monocyte subpopulation (CD14+CD16+) that is elevated in patients with CCA184,185. A high density of TAMs in patients with CCA has been associated with poor prognosis, reduced overall survival and disease-free survival, and metastasis184,186, which points to the role of these cells in CCA progression. Regarding the molecular crosstalk between TAMs and CCA cells, several molecules with well-known effects on CCA cells have been described as being produced by lipopolysaccharide-activated TAMs (that is, matrix metalloproteinases, interleukins, VEGF-A, TNF and TGFβ)185,186,187, but so far the components of the Wnt pathway are the best characterized in this context. Through the production of Wnt ligands (Wnt3a and Wnt7b)67,188, TAMs activate the canonical Wnt–β-catenin pathway in tumour cells and thereby participate in CCA development. Furthermore, depletion of TAMs or inhibition of Wnt signalling with Wnt inhibitors both in vitro and in mouse and rat CCA models markedly reduced CCA proliferation and increased apoptosis, resulting in tumour regression67.
Vascular cells
Analysis of tumour-associated neovascularization indicates that angiogenesis occurs in CCA189 and is critical for its progression189,190,191,192. iCCAs with high microvessel density more frequently display advanced primary tumours and multiple tumour nodes190. Microvessel density has been identified as an independent prognostic factor for survival after tumour resection190. The 5-year survival of patients with high microvessel density is 2.2% compared with 42.1% in patients with low microvessel density190. However, to date, the molecular interaction between vascular cells and tumour cells has been poorly investigated.
Presentation, diagnosis and staging
Clinical presentation and diagnosis
CCAs are usually asymptomatic in early stages. When symptomatic, the clinical onset of iCCA is heterogeneous with malaise, cachexia, abdominal pain, night sweats, fatigue and/or jaundice, associated or not with systemic manifestations9. In 20–25% of cases, however, diagnosis of iCCA is an incidental finding9. For pCCA, by contrast, jaundice (typically painless) is the most frequent clinical onset9. In patients with PSC, CCA can emerge as a rapid deterioration of clinical conditions, dominant stricture during follow-up, during transplantation work-up or waiting list, or as an incidental finding at transplantation23,193,194,195,196. As aforementioned, iCCA occurs more frequently in patients with chronic liver disease (HBV or HCV infection) or parasitic infestation than pCCA9,197. Nonetheless, the majority of CCA cases occur in the absence of an evident chronic liver disease or other risk factors9,197. In general, the mass-forming type represents the most frequent macroscopic presentation of iCCA (>90%)9,10 appearing, at imaging, as a nodule. If MF-iCCA occurs in context of cirrhotic liver, after exclusion of a metastatic lesion, differential diagnosis with HCC is obligatory4,5. In this context, contrast-enhanced MRI studies on patients with iCCA show a lack of HCC hallmarks (such as contrast medium wash-in in the arterial phase followed by wash-out in the late phase) in all cases; however, by CT, this finding occurs only in large nodules (>3 cm) as smaller nodules frequently show a pattern of contrast medium wash-in and wash-out similar to HCC198,199,200. The most frequent imaging patterns displayed by iCCA in the cirrhotic liver are a progressive homogeneous contrast uptake until the delayed (around 5′) phase (MRI, CT) or an arterial peripheral-rim enhancement (CT)198,199. Currently, identification of rare primitive liver cancers — such as HCC with stem cell features (CK19+ HCC), cholangiolocellular carcinoma and combined HCC–CCA — by imaging procedures is still a challenge17,201. After excluding HCC in cirrhosis, or in the context of a nodule in non-cirrhotic liver, biopsy is necessary4,5. According to most guidelines, biopsy should be avoided in case of surgical resectability because of the risk of tumour seeding4,5; however, this statement lacks supporting evidence.
At histology, differential diagnosis of iCCA versus HCC or metastasis represents an unsolved problem3,4,202, and no specific markers have been validated. A panel of immunohistochemistry markers is required to exclude metastasis, and the cytokeratin profile (CK7+, CK19+, CK20−) in combination with immunohistochemistry for Hep-Par1 is sufficient to exclude HCC203,204. The positivity for N-cadherin205, the study of IDH1 and IDH2 mutations84,85, and the evaluation of albumin expression by in situ hybridization206 have been proposed for iCCA differential diagnosis.
Radiologically, pCCA usually appears as a bile duct stricture207. In this instance, MRI plus magnetic resonance cholangiopancreatography represents the imaging procedure with the highest diagnostic accuracy for localizing and sizing the stricture; thus, the challenge is the definitive demonstration of malignancy208,209,210,211. For a definitive diagnosis, these patients usually undergo endoscopic retrograde cholangiopancreatography and a number of procedures (cytology, brushing, FISH (fluorescence in situ hybridization)-polisomy, biopsy, intraductal ultrasonography, choledochoscopy, cholangioscopy, chromoendoscopy, confocal endoscopy, narrow-band imaging and so on) can be applied for microscopic confirmation, albeit with unsatisfactory sensitivity212,213,214,215. Indeed, at least 40% of patients are sent to surgery without definitive diagnosis and, in 10% of cases after surgery, no evidence of cancer is seen in resected tissues216.
Even more challenging is the diagnosis of CCA in patients with PSC. Biliary strictures, occurring at the time of PSC presentation in 15–20% of patients, might be of malignant nature in 10–15% of the cases207. MRI, CT, endoscopic ultrasonography or 18FDG PET-CT cannot definitively demonstrate the neoplastic nature of the stricture208,209,210,211,217. The only condition (either in patients with or without PSC) that does not require histological confirmation is biliary stricture associated with perihilar mass, hypertrophy–atrophy complex and vascular encasement, but this presentation is very rare. Endoscopic ultrasonography-guided fine-needle aspiration demonstrated a good diagnostic performance for discriminating benign versus malignant biliary strictures and without apparent risk of tumour seeding linked with the procedure218,219,220,221,222. As for iCCA, the risk of tumour seeding after transperitoneal biopsy of pCCA is based on limited evidence223. The role of FISH-polisomy in detecting CCA in patients with PSC has been questioned by a meta-analysis, due to its limited sensitivity215. Better markers are therefore required for early CCA detection215. In this regard, serum CA19-9 levels >130 U/ml in PSC had sensitivity and specificity of 79% and 98% for the detection of CCA, respectively224. However, the CA19-9 serum level is biased by elevation due to cholangitis and cholestasis and, is undetectable in Lewis-antigen-negative patients (average 7%)224. Correction of CA19-9 serum levels for fucosyltransferase (FUT)2 and FUT3 genotype has been proposed to improve sensitivity in patients with CCA and PSC, as individuals lacking FUT3 activity are unable to express the CA19-9 epitope225. A number of biomarkers in serum (trypsinogen-2, serum IL-6, MUC5AC, trypsinogen-2, CYFRA21-1, progranulin), urine (volatile organic compounds, proteomic profiles) and bile (IGF1, microRNA-laden vesicles, proteomic profile, Wisteria floribunda agglutinin-positive mucin 1, molecular profiling on cell-free DNA of bile supernatant) have been proposed, but none have reached clinical application83,226,227,228,229,230. In summary, diagnosis of CCA still requires a combination of clinical, radiological and nonspecific histological and/or biochemical markers.
Staging systems
The goals of CCA management are to determine the surgical resectability and outcomes, and for this purpose correct staging is crucial. Different staging systems have been proposed for iCCA and pCCA. For years, iCCA has been staged by using the same tumour-node metastasis (TNM) system of HCC. In 2010, the 7th edition of the AJCC/UICC staging manual15 proposed a staging system for iCCA based on specific criteria including the number of tumours, vascular invasion, direct invasion of adjacent structures histology and lymph node metastasis. Notably, tumour size was removed as a prognostic factor. This staging system was independently validated for iCCA in France in 2011 (Ref. 14) with the evidence of a better discriminating capacity in predicting survival than with the 5th and 6th editions. The upcoming 8th edition of the AJCC/UICC staging manual (set for publication in 2016) should further improve iCCA staging thanks to new insights on pathology and the relative importance of the different lymph node stations eventually involved. According to European Association for the Study of the Liver guidelines published in 2014 the 7th edition of the AJCC/UICC staging system is preferred for staging iCCA5,15.
As far as pCCA staging is concerned, in 1975, Bismuth and Corlette described their criteria for classifying bile duct involvement by pCCA and this staging system has been used for years to categorize pCCA231. However, the lack of information concerning vascular encasement and distant metastasis makes this classification scarcely helpful for management decisions. By specifically focusing on predicting resectability and outcomes, the Memorial Sloan Kettering Cancer Center group proposed a staging system that classifies pCCA on the basis of the local tumour extension, site of bile duct involvement, portal vein invasion and hepatic lobar atrophy, although the size of the remnant liver is not specified232. In 2011, the Mayo Clinic proposed a staging system comprising the tumour size, the extent of the disease in the biliary system, the involvement of the hepatic artery and portal vein, the involvement of lymph nodes, distant metastasis and the volume of the putative remnant liver after resection233. Although complex, this classification has the merit of finely defining surgical options, standardizing prospective reporting of pCCA and discriminating between prognostic classes. The AJCC/UICC 7th Edition that incorporates the TNM staging system is simple and is the most widely used postoperatively, but it cannot allow evaluation of local resectability of the tumour and, therefore, does not help with the decision about the various surgical options. In comparison with the 6th edition, the 7th edition of the AJCC manual allows improved prediction of survival and stratification of prognostic classes of patients who have undergone resection234.
Therapies and treatment strategies
Surgery
Surgery with complete resection represents the only treatment for CCA with curative intent235. Resection of affected segments or lobe is usually performed in iCCA, pancreatoduodenectomy in dCCA and, depending on the extent of the tumour, resection of the involved intrahepatic and extrahepatic bile ducts, the associated ipsilateral liver, the gallbladder and regional lymph nodes in pCCA4. Survival after resection mainly depends on the presence of tumour-negative margins, absence of vascular invasion and lymph node metastasis, and adequate functional liver remnant236. Overall, 5-year survival after resection has been reported in the range 22–44% for iCCA, 11–41% for pCCA and 27–37% for dCCA4. Traditionally, less than one-third of the patients have been classified as having a resectable tumour at the time of diagnosis owing to advanced local tumour infiltration or peritoneal or distant metastasis, lack of biliary reconstruction options and inadequate future liver remnant4. However, differing resectability criteria and surgical strategies have been applied between regions, in particular between Western (USA and Europe) and Eastern centres, and a more aggressive surgical approach (including extended hepatic resection and combined vascular resection in early-stage pCCA) has led to increased rates of actual resection and improved outcomes in the East Asia217,237,238.
Liver transplantation has been associated with rapid tumour recurrence and low survival (10–25%), and has historically not been recommended as treatment for unresectable CCA239. However, in selected patients with early stage (I–II) pCCA, the rate of recurrence-free survival after 5 years has been reported in the range 65–68% after liver transplantation following protocols using neoadjuvant therapy (including external beam radiotherapy combined with radiosensitizing chemotherapy, endoluminal brachytherapy and maintenance chemotherapy)239,240,241,242. Notably, the patient selection criteria used in these transplantation series have been rigorous, therefore survival outcomes after transplantation are not directly comparable to that observed in historical resection series, including a less-selective patient group243. Indeed, in similar-staged patients, performance of extended hepatic surgery in pCCA has demonstrated survival comparable to that observed after liver transplantation (the disease-specific survival: 67.1% at year 5)237.
Chemotherapy
For patients presenting with unresectable or metastatic CCA, systemic chemotherapy remains the mainstay palliative treatment modality. A meta-analysis combining the results from two randomized trials (ABC-02, phase III and BT22, phase II; see Box 3 and Supplementary information S1 (table)), provides supportive evidence for use of gemcitabine combined with cisplatin as first-line treatment in this patient group244,245,246. Gemcitabine combined with cisplatin also represents a cost-effective alternative compared with gemcitabine alone247. Although the combination of these drugs improves the progression-free and overall survival compared with gemcitabine alone, the median overall survival is still modestly approaching 1 year in metastatic CCA244. If cisplatin is contraindicated (for example, in renal insufficiency), safety and efficacy using gemcitabine in combination with oxaliplatin have been demonstrated in several phase II studies248,249. When gemcitabine and cisplatin fail, no established standard regimens in the second-line setting are available250. A systematic review published in 2014 of second-line trials concluded that insufficient evidence is available to recommend second-line chemotherapy250. In medical practice, a fluoropyrimidine-based regimen is often used when gemcitabine-based treatment fails. The role of adjuvant chemotherapy is not clearly defined in CCA, but for patients with local recurrence after resection of pCCA, chemotherapy has been recommended217. Several ongoing phase III clinical trials might potentially provide practice-changing results (Supplementary information S1 (table)). The antidiabetic drug metformin has been reported to inhibit tumour growth in CCA, and might also represent a promising option for prevention and treatment of CCA in the future251.
Locoregional therapy
The role of locoregional therapies, such as transarterial chemoembolization (TACE) and transarterial radioembolization (TARE), has increasingly been investigated for patients with CCA. In retrospective studies, TACE with cisplatin has been shown to improve survival in unresectable iCCA (12.2 versus 3.3 months)252. A retrospective multicentre study published in 2013, including close to 200 patients with iCCA, concluded that intra-arterial therapy was safe and this approach resulted in stable disease (62%) or partial to complete response (26%)253. The majority of patients were treated with conventional TACE or 90Y-TARE, and there was no statistically significant difference in overall survival (median 13.2 months) between these two groups. In addition, 90Y-TARE has provided partial response (27%) or stable disease (68%) in patients with iCCA254. Hepatic arterial infusion includes a catheter-based intra-arterial infusion of chemotherapeutic agents, without embolization253. This method has also been reported to be of benefit in cases of iCCA, but involves implantation of an infusion port or pump and might predispose to more complications than TACE and TARE255.
Radiofrequency ablation seems to prolong survival in inoperable iCCA256. Intraductal radiofrequency ablation has been shown to be safe and feasible in the treatment of extrahepatic CCA257 and seems to be a safe way of improving stent patency258. On the other hand, photodynamic therapy can improve survival in patients with unresectable CCA259,260,261. The role of radiation therapy in CCA is still not clearly outlined, but radiation with concurrent chemotherapy has been recommended in margin-positive and node-positive iCCA and pCCA, and in unresectable pCCA ineligible for liver transplantation201,217. Thus, although results have been promising from the various locoregional types of therapy, conclusive evidence for efficacy in patients with CCA is still lacking and larger prospective randomized studies are warranted.
Biliary stenting
Although debated, guidelines recommend that routine biliary drainage should be avoided before staging and assessment of resectability of CCA and preoperatively4. Preoperative drainage is indicated in cases of cholangitis, jaundice in conjunction with preoperative antineoplastic therapy, severe malnutrition, hepatic or renal insufficiency, and in patients undergoing portal vein embolization4,217,262. A multidisciplinary approach is important to select the best approach in each case. Biliary drainage might be beneficial as a palliative treatment in patients with unresectable CCA, with longer survival (19 months for the endoscopic group versus 16.5 months for the surgical group) and less cost than surgical treatment263. Both plastic stents and self-expandable metallic stents can be utilized, but self-expandable metallic stents seem to offer several advantages, including higher patency duration than plastic stents264.
Targeted therapy
Several clinical trials are evaluating the effect of specific molecular agents targeting various signalling pathways in CCAs, for example tyrosine kinase inhibitors (for example, erlotinib, bevacizumab, cetuximab, panitimumab and lapatinib) (Box 3 and Supplementary information S2 and S3 (tables)). Existing data demonstrate no or only very modest survival benefits of the agents tested. Larger clinical CCA studies, and also improved patient selection based on localization as well as molecular alterations, are needed29.
Novel molecular alterations have been identified in CCA68,72,74, and a large comprehensive study published in 2015 demonstrates that nearly 40% of the patients harbour genetic alterations that are potentially targetable29. Several preclinical (Supplementary information S4 (table)) and clinical phase I studies have been initiated265, evaluating some of these novel targets for therapy, including IDH266, microRNAs267,268 and fusion genes68,74,79,269. Targeted anti-fibrotic therapy is also under investigation183. In addition, the expression in tumour cells of the specific uptake transporter for bile acids, the apical sodium-dependent bile acid transporter (ASBT), has suggested the possibility of using cytostatic bile acid derivatives, such as the ursodeoxycholic acid–cisplatin conjugate BAMET-UD2, in targeted chemotherapy for CCA270.
In 2015, the rationale for immunotherapy with checkpoint-molecule-specific monoclonal antibodies in patients bearing iCCA without defects in HLA class I antigen expression has been provided271 but, so far, no clinical trial has been performed. Crucially, it must be underscored that the management of CCAs should only be undertaken in dedicated tertiary units with access to multidisciplinary approaches.
Mechanisms of chemoresistance
CCAs are highly chemoresistant tumours, which means that pharmacological therapies are generally unsuccessful. One of the goals of modern pharmacology is the identification and overcoming of the mechanisms of chemoresistance (MOC) by addressing the marked multidrug resistance phenotype of different tumours, including CCA, which usually becomes exacerbated in response to chemotherapy (Table 2). Most MOC are already present in healthy cholangiocytes, where they are involved in defense against toxic compounds from blood and/or bile6.
In lowering the intracellular amount of drug (MOC-1)6, an important part is played by reduced uptake (MOC-1a) through solute carrier (SLC) transporters, such as organic anion-transporting polypeptides (OATPs). Substrates of OATP1A2, which is highly expressed in cholangiocytes, include methotrexate, taxanes and imatinib, whose uptake by CCA cells in vitro can be impaired by OATP1A2 downregulation272 or the expression of less active genetic variants273. Uptake of cationic drugs (for example, platinum derivatives and tyrosine kinase inhibitors) is mediated in part by organic cationic transporters (OCT), which are downregulated (OCT3)274 or very poorly expressed (OCT1)275 in CCA. Gemcitabine and 5-fluorouracil are taken up through nucleoside transporters, equilibrative nucleoside transporters (ENT) and concentrative nucleoside transporters (CNT). In CCA cells, low expression of ENT1 is associated with poor response to these drugs276,277,278, and CNT1 expression is also impaired279. Lack of sensitivity to cisplatin is due, in part, to reduced uptake through the copper transporter CTR1, whose expression in CCA is decreased279.
Export pumps of the ATP-binding cassette (ABC) superfamily of proteins are key elements determining the intracellular concentration of drugs (MOC-1b). Multidrug resistance protein 1, highly expressed in healthy cholangiocytes280, is able to export a large variety of anticancer drugs (for example, doxorubicin, etoposide, paclitaxel and vinblastine)281,282, hence reducing their efficacy in CCA283,284. Members of the ABCC family, MRP1, MRP3, MRP4 and MRP5, might also be involved in CCA chemoresistance279,285,286.
Intracellular mechanisms account for decreased prodrug activation or enhanced inactivation of active agents (MOC-2). Downregulation and/or impaired activity of enzymes involved in gemcitabine and 5-fluorouracil activation, such as thymidine phosphorylase, uridine phosphorylase 1 and uridine monophosphate synthetase, reduce the sensitivity of CCA to these drugs287,288. The phase II enzyme glutathione S-transferase P is highly expressed in CCA, which might have an important role in inactivating drugs by conjugation with glutathione289,290.
Changes in molecular targets could also affect the response to chemotherapy (MOC-3). Thymidylate synthase has a key role in the sensitivity of CCA to 5- fluorouracil291. Although some controversy exists278,288,290, simvastatin has been shown to boost the 5-fluorouracil effect in CCA cells by suppressing thymidylate synthase expression292. Expression of ERs in CCA93 justifies its sensitivity to tamoxifen293 and KB9520, which activates apoptosis in experimental CCA94. Decreased ER expression might account for a weaker response to this type of drug than CCAs with a high ER expression. Upregulation of EGFR decreases the sensitivity of CCA cells to erlotinib294. Insulin-like growth factor type 1 receptor (IGF1R) contributes to tumour angiogenesis through upregulation of VEGF. Given the upregulation of IGF1R in CCA93,295, antibodies against the IGF1R or against its ligands might have limited therapeutic applications296,297.
Different repair strategies permit cancer cells to tackle different types of drug-induced DNA lesions (MOC-4). In 5-fluorouracil-resistant CCA cells, uracil-DNA glycosylase is upregulated, which activates base-excision repair, the major route to repair 5-fluoruracil-induced misincorporation of fluoronucleotides278. The endonuclease involved in nucleotide-excision repair, DNA excision repair protein ERCC-1(ERCC1), removes a wide variety of bulky DNA adducts. ERCC1 has been associated with the response to cisplatin of several tumours, including CCA298. RAD51, upregulated in most CCA299, is a recombinase involved in repairing DNA double-strand breaks and is associated with poor sensitivity to cyclophosphamide, epirubicin and docetaxel300. The DNA mismatch repair system recognizes and repairs erroneous insertion of nucleotides as well as short insertions and deletions. Downregulation of MutS (MSH2, MSH3 and MSH6) and MutLa (hMLH1 and PMS2) protein complexes involved in DNA mismatch repair results in genetic instability, poorer prognosis and higher chemoresistance in CCAs in comparison with tumours without MutS and MutLa downregulation301,302,303. Under the direct control of Tp53, upregulation of ribonucleotide reductase p53R2 increases the supply of nucleotides for repairing DNA damage. p53R2 expression is suggested as a predictive marker for resistance to gemcitabine in CCA304. Serine/threonine-protein kinase tousled-like kinase 1 is highly expressed in CCA, regulating chromatin assembly and the repair of cisplatin-induced DNA damage305.
As the final goal of most anticancer drugs is to induce apoptosis, downregulation and/or inactivation of pro-apoptotic mediators (MOC-5a) or enhanced expression and/or activity of anti-apoptotic factors (MOC-5b) resulted in decreased efficacy of chemotherapy. NK4 is a fragment of hepatocyte growth factor (HGF) that blocks its binding to HGF-receptor (HGFR), which enhances 5-fluorouracil-induced activation of caspases 3 and 9 (Ref. 306). Downregulation of NK4 in response to 5-fluorouracil treatment constitutes an intrinsic mechanism of CCA resistance to this drug306. In addition, CCAs are frequently resistant to TRAIL-mediated apoptosis307,308. Interaction of Fas cell surface death receptor with calmodulin leads to the inhibition of Fas-induced apoptosis and might be involved in CCA chemoresistance309. Moreover, mutations in the pro-apoptotic tumour suppressor TP53 have been suggested as predictors of CCA outcome209. Among anti-apoptotic factors, several members of the Bcl-2 protein family are upregulated in CCA cell lines310, causing chemoresistance to cisplatin and 5-fluorouracil311. The E3 ubiquitin protein ligase (XIAP, IAP3 or BIRC4), which inhibits caspases 3 and 9 and TRAIL pathway, shows increased activity in CCA-enhancing chemoresistance in vitro312. Likewise, the anti-apoptotic protein survivin is also highly expressed in CCA279. Finally, inhibition of the AKT signalling pathway in CCA cells leads to apoptosis via Bcl-2 downregulation and Bax upregulation, and sensitizes cells to cisplatin311.
The demonstration that high numbers of CSCs are present in iCCA and pCCA is an additional explanation for the marked chemoradioresistance and for the high rate of recurrence of this cancer47.
Future perspectives
CCAs display pronounced inter-tumoural and intra-tumoural heterogeneity caused, among others, by the inter-relationships between cancer cells, CSCs and the tumour microenvironment, clonal evolution and molecular (genetic and epigenetic) abnormalities. Indeed, studies on different cohorts described an extreme heterogeneity of molecular profiling with subclassification of CCAs in different molecular subtypes, associated with potential therapeutic targets and prognostic indicators. Unfortunately, most clinical trials have been performed without accurate molecular profile analyses and, therefore, the evaluation of outcomes for specific subgroups of patients with CCA is absolutely inadequate. For the future, it should be desiderable that clinical studies on CCA will take into consideration the clinical–pathological subtyping (mixed versus mucin, CCAs associated with HCV, HBV, PSC or liver fluke) and the relative genetic background. With these considerations in mind, a major mission of the ENS-CCA is to support and coordinate a roadmap of future translational works to fill the gap between basic science and clinical studies exploring biomarkers for screening and surveillance of populations at risk, early diagnosis, prognosis and targeted therapies.
From a clinical point of view, the regulatory authorities should consider that CCA must be managed by dedicated centres provided with multidisciplinary expertise where personalized diagnostic work-up and management can be performed. This approach is fundamental given the heterogeneity and complexity of the disease. From a scientific point of view, a number of key issues need to be addressed in the near future. CCA is surgically curable if diagnosed at early stages and, therefore, every effort must be made to identify populations at risk for strict follow-up and early diagnosis. As CCA emerges in the context of bile duct inflammation, biomarkers or radiological tools finely evidencing bile duct inflammation and/or activation of reactive cholangiocytes and peribiliary glands are required. Molecular, biochemical or biological tumour markers are needed not only for diagnostic but also for screening and prognostic purposes. Moreover, the fine interplay between neoplastic cells and the microenvironment needs more investigation. In this regard, signalling pathways driving EMT and emergence of CSC traits need to be defined together with a better clarification of the part played by CAFs, TAMs and vascular cells in tumour growth and spread; exploring these issues will help not only to understand CCA pathophysiology but, principally, to develop effective targeted therapies. Finally, it is evident that CCA cells display complex mechanisms of chemoradioresistance and, therefore, elucidating these mechanisms seems crucial for CCA treatment.
Conclusions
CCAs comprise a group of cancers with different locations and pronounced inter-tumoural and intra-tumoural heterogeneity. Apart from the anatomical location, CCA heterogeneity is caused by different variables including the inter-relationships between cancer cells, CSCs and the tumour microenvironment, clonal evolution and molecular (genetic and epigenetic) abnormalities. Specific efforts have been undertaken in the past to classify CCAs on the basis of anatomical location, genetic background, pathology, risk factors and molecular profile. However, a lot of work still remains to update clinical-pathological classification and to investigate biomarkers and/or imaging hallmarks specific for each CCA subtype. Studies, focused on molecular profiling, described different CCA subtypes and this should represent the background for clinical trials addressing targeted therapies against specific CCA subgroups. Waiting for these future acquisitions, surgery with complete resection, including liver transplantation in highly selected cases, is still the only curative therapy for CCA. Unfortunately, curative surgical resection is applicable in a minority of cases and therefore a main challenge is to increase the number of resectable cases by expanding early diagnosis. As a worldwide accepted statement, a personalized CCA diagnostic work-up and therapeutic approach must be managed by dedicated centres with multidisciplinary expertise and where translation from basic science to clinic can rapidly take place.
References
Global Burden of Disease Cancer Collaboration. The global burden of cancer 2013. JAMA Oncol. 1, 505–527 (2015).
Nakeeb, A. et al. Cholangiocarcinoma: a spectrum of intrahepatic, perihilar, and distal tumors. Ann. Surg. 224, 463–473; discussion 473–475 (1996).
Blechacz, B., Komuta, M., Roskams, T. & Gores, G. J. Clinical diagnosis and staging of cholangiocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 8, 512–522 (2011).
Khan, S. A. et al. Guidelines for the diagnosis and treatment of cholangiocarcinoma: an update. Gut 61, 1657–1669 (2012).
Bridgewater, J. et al. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J. Hepatol. 60, 1268–1289 (2014).
Marin, J. J. et al. Molecular bases of chemoresistance in cholangiocarcinoma. Curr. Drug Targets http://dx.doi.org/10.2174/1389450116666150223121508 (2015).
Ebata, T. et al. Proposal to modify the International Union Against Cancer staging system for perihilar cholangiocarcinomas. Br. J. Surg. 101, 79–88 (2014).
Yamasaki, S. Intrahepatic cholangiocarcinoma: macroscopic type and stage classification. J. Hepatobiliary Pancreat. Surg. 10, 288–291 (2003).
Alvaro, D. et al. Cholangiocarcinoma in Italy: a national survey on clinical characteristics, diagnostic modalities and treatment. Results from the 'cholangiocarcinoma' committee of the Italian Association for the Study of Liver disease. Dig. Liver Dis. 43, 60–65 (2011).
De Rose, A. M. et al. Prognostic significance of tumor doubling time in mass-forming type cholangiocarcinoma. J. Gastrointest. Surg. 17, 739–747 (2013).
Nakanuma, Y. et al. Pathological classification of intrahepatic cholangiocarcinoma based on a new concept. World J. Hepatol. 2, 419–427 (2010).
Han, Y. et al. Recent advances in the morphological and functional heterogeneity of the biliary epithelium. Exp. Biol. Med. (Maywood) 238, 549–565 (2013).
Aishima, S. & Oda, Y. Pathogenesis and classification of intrahepatic cholangiocarcinoma: different characters of perihilar large duct type versus peripheral small duct type. J. Hepatobiliary Pancreat. Sci. 22, 94–100 (2015).
Farges, O. et al. AJCC 7th edition of TNM staging accurately discriminates outcomes of patients with resectable intrahepatic cholangiocarcinoma: by the AFC-IHCC-2009 study group. Cancer 117, 2170–2177 (2011).
Edge, S. B. & Compton, C. C. The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann. Surg. Oncol. 17, 1471–1474 (2010).
Sato, Y. et al. Pathological diagnosis of flat epithelial lesions of the biliary tract with emphasis on biliary intraepithelial neoplasia. J. Gastroenterol. 49, 64–72 (2014).
Komuta, M. et al. Histological diversity in cholangiocellular carcinoma reflects the different cholangiocyte phenotypes. Hepatology 55, 1876–1888 (2012).
Liau, J. Y. et al. Morphological subclassification of intrahepatic cholangiocarcinoma: etiological, clinicopathological, and molecular features. Mod. Pathol. 27, 1163–1173 (2014).
Nakanuma, Y. et al. Pathological spectrum of intrahepatic cholangiocarcinoma arising in non-biliary chronic advanced liver diseases. Pathol. Int. 61, 298–305 (2011).
Komuta, M. et al. Clinicopathological study on cholangiolocellular carcinoma suggesting hepatic progenitor cell origin. Hepatology 47, 1544–1556 (2008).
Palmer, W. C. & Patel, T. Are common factors involved in the pathogenesis of primary liver cancers? A meta-analysis of risk factors for intrahepatic cholangiocarcinoma. J. Hepatol. 57, 69–76 (2012).
Patel, T. Cholangiocarcinoma — controversies and challenges. Nat. Rev. Gastroenterol. Hepatol. 8, 189–200 (2011).
Burak, K. et al. Incidence and risk factors for cholangiocarcinoma in primary sclerosing cholangitis. Am. J. Gastroenterol. 99, 523–526 (2004).
Kobayashi, M. et al. Incidence of primary cholangiocellular carcinoma of the liver in japanese patients with hepatitis C virus-related cirrhosis. Cancer 88, 2471–2477 (2000).
Lee, T. Y. et al. Hepatitis B virus infection and intrahepatic cholangiocarcinoma in Korea: a case–control study. Am. J. Gastroenterol. 103, 1716–1720 (2008).
Shin, H. R. et al. Hepatitis B and C virus, Clonorchis sinensis for the risk of liver cancer: a case-control study in Pusan, Korea. Int. J. Epidemiol. 25, 933–940 (1996).
Plentz, R. R. & Malek, N. P. Clinical presentation, risk factors and staging systems of cholangiocarcinoma. Best Pract. Res. Clin. Gastroenterol. 29, 245–252 (2015).
Zhou, H. B., Hu, J. Y. & Hu, H. P. Hepatitis B virus infection and intrahepatic cholangiocarcinoma. World J. Gastroenterol. 20, 5721–5729 (2014).
Nakamura, H. et al. Genomic spectra of biliary tract cancer. Nat. Genet. 47, 1003–1010 (2015).
Cardinale, V. et al. Intra-hepatic and extra-hepatic cholangiocarcinoma: new insight into epidemiology and risk factors. World J. Gastrointest. Oncol. 2, 407–416 (2010).
Grainge, M. J., West, J., Solaymani-Dodaran, M., Aithal, G. P. & Card, T. R. The antecedents of biliary cancer: a primary care case-control study in the United Kingdom. Br. J. Cancer 100, 178–180 (2009).
Welzel, T. M. et al. Metabolic syndrome increases the risk of primary liver cancer in the United States: a study in the SEER-Medicare database. Hepatology 54, 463–471 (2011).
Nishihara, K., Koga, A., Sumiyoshi, K., Kayashima, K. & Koso, E. Intrahepatic calculi associated with cholangiocarcinoma. Jpn J. Surg. 16, 367–370 (1986).
Soreide, K., Korner, H., Havnen, J. & Soreide, J. A. Bile duct cysts in adults. Br. J. Surg. 91, 1538–1548 (2004).
Khan, S. A., Thomas, H. C., Davidson, B. R. & Taylor-Robinson, S. D. Cholangiocarcinoma. Lancet 366, 1303–1314 (2005).
Lozano, E. et al. Cocarcinogenic effects of intrahepatic bile acid accumulation in cholangiocarcinoma development. Mol. Cancer Res. 12, 91–100 (2014).
Rycaj, K. & Tang, D. G. Cell-of-origin of cancer versus cancer stem cells: assays and interpretations. Cancer Res. 75, 1–9 (2015).
Visvader, J. E. Cells of origin in cancer. Nature 469, 314–322 (2011).
Cardinale, V., Carpino, G., Reid, L., Gaudio, E. & Alvaro, D. Multiple cells of origin in cholangiocarcinoma underlie biological, epidemiological and clinical heterogeneity. World J. Gastrointest. Oncol. 4, 94–102 (2012).
Carpino, G. et al. Activation of biliary tree stem cells within peribiliary glands in primary sclerosing cholangitis. J. Hepatol. 63, 1220–1228 (2015).
Sato, Y., Harada, K., Sasaki, M. & Nakanuma, Y. Cystic and micropapillary epithelial changes of peribiliary glands might represent a precursor lesion of biliary epithelial neoplasms. Virchows Arch. 464, 157–163 (2014).
Nakanuma, Y. & Sato, Y. Cystic and papillary neoplasm involving peribiliary glands: a biliary counterpart of branch-type intraductal papillary mucinous [corrected] neoplasm? Hepatology 55, 2040–2041 (2012).
Cardinale, V. et al. Mucin-producing cholangiocarcinoma might derive from biliary tree stem/progenitor cells located in peribiliary glands. Hepatology 55, 2041–2042 (2012).
Cardinale, V., Carpino, G., Reid, L. M., Gaudio, E. & Alvaro, D. Cholangiocarcinoma: a cancer in search of the right classification. Hepatology 56, 1585–1586; author reply 1586 (2012).
Carpino, G. et al. Biliary tree stem/progenitor cells in glands of extrahepatic and intraheptic bile ducts: an anatomical in situ study yielding evidence of maturational lineages. J. Anat. 220, 186–199 (2012).
Cardinale, V. et al. The biliary tree — a reservoir of multipotent stem cells. Nat. Rev. Gastroenterol. Hepatol. 9, 231–240 (2012).
Cardinale, V. et al. Profiles of cancer stem cell subpopulations in cholangiocarcinomas. Am. J. Pathol. 185, 1724–1739 (2015).
Gandou, C. et al. Hilar cholangiocarcinoma and pancreatic ductal adenocarcinoma share similar histopathologies, immunophenotypes, and development-related molecules. Hum. Pathol. 44, 811–821 (2013).
Nakanuma, Y., Harada, K., Sasaki, M. & Sato, Y. Proposal of a new disease concept 'biliary diseases with pancreatic counterparts'. Anatomical and pathological bases. Histol. Histopathol. 29, 1–10 (2014).
Rizvi, S. & Gores, G. J. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology 145, 1215–1229 (2013).
Raggi, C., Invernizzi, P. & Andersen, J. B. Impact of microenvironment and stem-like plasticity in cholangiocarcinoma: molecular networks and biological concepts. J. Hepatol. 62, 198–207 (2015).
Chen, W. T. et al. Liver-specific knockout of GRP94 in mice disrupts cell adhesion, activates liver progenitor cells, and accelerates liver tumorigenesis. Hepatology 59, 947–957 (2014).
Vander Borght, S. et al. Expression of multidrug resistance-associated protein 1 in hepatocellular carcinoma is associated with a more aggressive tumour phenotype and may reflect a progenitor cell origin. Liver Int. 28, 1370–1380 (2008).
Govaere, O. et al. Keratin 19: a key role player in the invasion of human hepatocellular carcinomas. Gut 63, 674–685 (2014).
Magee, J. A., Piskounova, E. & Morrison, S. J. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell 21, 283–296 (2012).
Yamashita, T. & Wang, X. W. Cancer stem cells in the development of liver cancer. J. Clin. Invest. 123, 1911–1918 (2013).
Leelawat, K., Thongtawee, T., Narong, S., Subwongcharoen, S. & Treepongkaruna, S. A. Strong expression of CD133 is associated with increased cholangiocarcinoma progression. World J. Gastroenterol. 17, 1192–1198 (2011).
Sulpice, L. et al. Epithelial cell adhesion molecule is a prognosis marker for intrahepatic cholangiocarcinoma. J. Surg. Res. 192, 117–123 (2014).
Gu, M. J. & Jang, B. I. Clinicopathologic significance of Sox2, CD44 and CD44v6 expression in intrahepatic cholangiocarcinoma. Pathol. Oncol. Res. 20, 655–660 (2014).
Haraguchi, N. et al. CD13 is a therapeutic target in human liver cancer stem cells. J. Clin. Invest. 120, 3326–3339 (2010).
Sukowati, C. H. et al. The expression of CD90/Thy-1 in hepatocellular carcinoma: an in vivo and in vitro study. PLoS ONE 8, e76830 (2013).
Kemmerling, R. et al. Association of stem cell marker expression pattern and survival in human biliary tract cancer. Int. J. Oncol. 41, 511–522 (2012).
Lederer, A. et al. Metastasis-associated in colon cancer 1 is an independent prognostic biomarker for survival in klatskin tumor patients. Hepatology 62, 841–850 (2015).
O'Connor, M. L. et al. Cancer stem cells: a contentious hypothesis now moving forward. Cancer Lett. 344, 180–187 (2014).
Kreso, A. & Dick, J. E. Evolution of the cancer stem cell model. Cell Stem Cell 14, 275–291 (2014).
Govaere, O. et al. Laminin-332 sustains chemoresistance and quiescence as part of the human hepatic cancer stem cell niche. J. Hepatol. 64, 609–617 (2015).
Boulter, L. et al. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J. Clin. Invest. 125, 1269–1285 (2015).
Borad, M. J. et al. Integrated genomic characterization reveals novel, therapeutically relevant drug targets in FGFR and EGFR pathways in sporadic intrahepatic cholangiocarcinoma. PLoS Genet. 10, e1004135 (2014).
Chan-On, W. et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat. Genet. 45, 1474–1478 (2013).
Fujimoto, A. et al. Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat. Commun. 6, 6120 (2015).
Gao, Q. et al. Activating mutations in PTPN3 promote cholangiocarcinoma cell proliferation and migration and are associated with tumor recurrence in patients. Gastroenterology 146, 1397–1407 (2014).
Jiao, Y. et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat. Genet. 45, 1470–1473 (2013).
Ong, C. K. et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat. Genet. 44, 690–693 (2012).
Sia, D. et al. Massive parallel sequencing uncovers actionable FGFR2–PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat. Commun. 6, 6087 (2015).
Zou, S. et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat. Commun. 5, 5696 (2014).
Sia, D. et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 144, 829–840 (2013).
Simbolo, M. et al. Multigene mutational profiling of cholangiocarcinomas identifies actionable molecular subgroups. Oncotarget 5, 2839–2852 (2014).
Arai, Y. et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 59, 1427–1434 (2014).
Wu, Y. M. et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 3, 636–647 (2013).
Ross, J. S. et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist 19, 235–242 (2014).
Udali, S. et al. Global DNA methylation and hydroxymethylation differ in hepatocellular carcinoma and cholangiocarcinoma and relate to survival rate. Hepatology 62, 496–504 (2015).
Chiang, N. J., Shan, Y. S., Hung, W. C. & Chen, L. T. Epigenetic regulation in the carcinogenesis of cholangiocarcinoma. Int. J. Biochem. Cell Biol. 67, 110–114 (2015).
Andresen, K. et al. Four DNA methylation biomarkers in biliary brush samples accurately identify the presence of cholangiocarcinoma. Hepatology 61, 1651–1659 (2015).
Borger, D. R. et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 17, 72–79 (2012).
Wang, P. et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 32, 3091–3100 (2013).
Turcan, S. et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483 (2012).
Saha, S. K. et al. Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 513, 110–114 (2014).
Grivennikov, S. I. & Karin, M. Inflammation and oncogenesis: a vicious connection. Curr. Opin. Genet. Dev. 20, 65–71 (2010).
Sica, A., Invernizzi, P. & Mantovani, A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 59, 2034–2042 (2014).
Goeppert, B. et al. Global alterations of DNA methylation in cholangiocarcinoma target the Wnt signaling pathway. Hepatology 59, 544–554 (2014).
Gradilone, S. A. et al. HDAC6 inhibition restores ciliary expression and decreases tumor growth. Cancer Res. 73, 2259–2270 (2013).
Franchitto, A. et al. Recent advances on the mechanisms regulating cholangiocyte proliferation and the significance of the neuroendocrine regulation of cholangiocyte pathophysiology. Ann. Transl. Med. 1, 27 (2013).
Alvaro, D. et al. Estrogens and insulin-like growth factor 1 modulate neoplastic cell growth in human cholangiocarcinoma. Am. J. Pathol. 169, 877–888 (2006).
Marzioni, M. et al. An oestrogen receptor β-selective agonist exerts anti-neoplastic effects in experimental intrahepatic cholangiocarcinoma. Dig. Liver Dis. 44, 134–142 (2012).
Pawar, P. et al. Molecular mechanisms of tamoxifen therapy for cholangiocarcinoma: role of calmodulin. Clin. Cancer Res. 15, 1288–1296 (2009).
Mancino, A. et al. Estrogens stimulate the proliferation of human cholangiocarcinoma by inducing the expression and secretion of vascular endothelial growth factor. Dig. Liver Dis. 41, 156–163 (2009).
Isse, K. et al. Estrogen stimulates female biliary epithelial cell interleukin-6 expression in mice and humans. Hepatology 51, 869–880 (2010).
Francis, H., Alpini, G. & DeMorrow, S. Recent advances in the regulation of cholangiocarcinoma growth. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G1–G9 (2010).
Alpini, G. et al. Serotonin metabolism is dysregulated in cholangiocarcinoma, which has implications for tumor growth. Cancer Res. 68, 9184–9193 (2008).
Coufal, M. et al. Increased local dopamine secretion has growth-promoting effects in cholangiocarcinoma. Int. J. Cancer 126, 2112–2122 (2010).
Fava, G. et al. Leptin enhances cholangiocarcinoma cell growth. Cancer Res. 68, 6752–6761 (2008).
Marzioni, M. et al. Human cholangiocarcinoma development is associated with dysregulation of opioidergic modulation of cholangiocyte growth. Dig. Liver Dis. 41, 523–533 (2009).
DeMorrow, S. et al. Opposing actions of endocannabinoids on cholangiocarcinoma growth: recruitment of Fas and Fas ligand to lipid rafts. J. Biol. Chem. 282, 13098–13113 (2007).
Frampton, G., Coufal, M., Li, H., Ramirez, J. & DeMorrow, S. Opposing actions of endocannabinoids on cholangiocarcinoma growth is via the differential activation of Notch signaling. Exp. Cell Res. 316, 1465–1478 (2010).
Marzioni, M. et al. Endogenous opioids modulate the growth of the biliary tree in the course of cholestasis. Gastroenterology 130, 1831–1847 (2006).
Onori, P. et al. Secretin inhibits cholangiocarcinoma growth via dysregulation of the cAMP-dependent signaling mechanisms of secretin receptor. Int. J. Cancer 127, 43–54 (2010).
Kanno, N. et al. Gastrin inhibits cholangiocarcinoma growth through increased apoptosis by activation of Ca2+-dependent protein kinase C-α. J. Hepatol. 34, 284–291 (2001).
Fava, G. et al. γ-aminobutyric acid inhibits cholangiocarcinoma growth by cyclic AMP-dependent regulation of the protein kinase A/extracellular signal-regulated kinase 1/2 pathway. Cancer Res. 65, 11437–11446 (2005).
Fava, G. et al. Endothelin inhibits cholangiocarcinoma growth by a decrease in the vascular endothelial growth factor expression. Liver Int. 29, 1031–1042 (2009).
DeMorrow, S. et al. The endocannabinoid anandamide inhibits cholangiocarcinoma growth via activation of the noncanonical Wnt signaling pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 295, G1150–G1158 (2008).
Huang, L. et al. Anandamide exerts its antiproliferative actions on cholangiocarcinoma by activation of the GPR55 receptor. Lab. Invest. 91, 1007–1017 (2011).
Francis, H. et al. H3 histamine receptor-mediated activation of protein kinase Cα inhibits the growth of cholangiocarcinoma in vitro and in vivo. Mol. Cancer Res. 7, 1704–1713 (2009).
Meng, F. et al. The H4 histamine receptor agonist, clobenpropit, suppresses human cholangiocarcinoma progression by disruption of epithelial mesenchymal transition and tumor metastasis. Hepatology 54, 1718–1728 (2011).
Francis, H. et al. Inhibition of histidine decarboxylase ablates the autocrine tumorigenic effects of histamine in human cholangiocarcinoma. Gut 61, 753–764 (2012).
Harder, J. et al. EGFR and HER2 expression in advanced biliary tract cancer. World J. Gastroenterol. 15, 4511–4517 (2009).
Yoshikawa, D. et al. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br. J. Cancer 98, 418–425 (2008).
Yoon, J. H. et al. Enhanced epidermal growth factor receptor activation in human cholangiocarcinoma cells. J. Hepatol. 41, 808–814 (2004).
Gwak, G. Y. et al. Detection of response-predicting mutations in the kinase domain of the epidermal growth factor receptor gene in cholangiocarcinomas. J. Cancer Res. Clin. Oncol. 131, 649–652 (2005).
Nakazawa, K. et al. Amplification and overexpression of c-erbB-2, epidermal growth factor receptor, and c-met in biliary tract cancers. J. Pathol. 206, 356–365 (2005).
Miyamoto, M. et al. Prognostic significance of overexpression of c-Met oncoprotein in cholangiocarcinoma. Br. J. Cancer 105, 131–138 (2011).
Terada, T., Nakanuma, Y. & Sirica, A. E. Immunohistochemical demonstration of MET overexpression in human intrahepatic cholangiocarcinoma and in hepatolithiasis. Hum. Pathol. 29, 175–180 (1998).
Claperon, A. et al. EGF/EGFR axis contributes to the progression of cholangiocarcinoma through the induction of an epithelial–mesenchymal transition. J. Hepatol. 61, 325–332 (2014).
Ryu, H. S. et al. Overexpression of epithelial–mesenchymal transition-related markers according to cell dedifferentiation: clinical implications as an independent predictor of poor prognosis in cholangiocarcinoma. Hum. Pathol. 43, 2360–2370 (2012).
Fabris, L. et al. Nuclear expression of S100A4 calcium-binding protein increases cholangiocarcinoma invasiveness and metastasization. Hepatology 54, 890–899 (2011).
Menakongka, A. & Suthiphongchai, T. Involvement of PI3K and ERK1/2 pathways in hepatocyte growth factor-induced cholangiocarcinoma cell invasion. World J. Gastroenterol. 16, 713–722 (2010).
Werneburg, N. W., Yoon, J. H., Higuchi, H. & Gores, G. J. Bile acids activate EGF receptor via a TGF-α-dependent mechanism in human cholangiocyte cell lines. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G31–G36 (2003).
Dai, J. et al. Impact of bile acids on the growth of human cholangiocarcinoma via FXR. J. Hematol. Oncol. 4, 41 (2011).
Liu, R. et al. Conjugated bile acids promote cholangiocarcinoma cell invasive growth through activation of sphingosine 1-phosphate receptor 2. Hepatology 60, 908–918 (2014).
Maroni, L., Alpini, G. & Marzioni, M. Cholangiocarcinoma development: the resurgence of bile acids. Hepatology 60, 795–797 (2014).
Fan, B. et al. Cholangiocarcinomas can originate from hepatocytes in mice. J. Clin. Invest. 122, 2911–2915 (2012).
Sekiya, S. & Suzuki, A. Intrahepatic cholangiocarcinoma can arise from Notch-mediated conversion of hepatocytes. J. Clin. Invest. 122, 3914–3918 (2012).
Dill, M. T. et al. Constitutive Notch2 signaling induces hepatic tumors in mice. Hepatology 57, 1607–1619 (2013).
Huntzicker, E. G. et al. Differential effects of targeting Notch receptors in a mouse model of liver cancer. Hepatology 61, 942–952 (2015).
Greenhill, C. Liver cancer: different effects of the Notch receptors in liver cancer revealed. Nat. Rev. Gastroenterol. Hepatol. 11, 703 (2014).
Zhou, Q., Wang, Y., Peng, B., Liang, L. & Li, J. The roles of Notch1 expression in the migration of intrahepatic cholangiocarcinoma. BMC Cancer 13, 244 (2013).
Wu, W. R. et al. Notch1 is overexpressed in human intrahepatic cholangiocarcinoma and is associated with its proliferation, invasiveness and sensitivity to 5-fluorouracil in vitro. Oncol. Rep. 31, 2515–2524 (2014).
El Khatib, M. et al. Inhibition of hedgehog signaling attenuates carcinogenesis in vitro and increases necrosis of cholangiocellular carcinoma. Hepatology 57, 1035–1045 (2013).
Razumilava, N. et al. Non-canonical Hedgehog signaling contributes to chemotaxis in cholangiocarcinoma. J. Hepatol. 60, 599–605 (2014).
Fingas, C. D. et al. Myofibroblast-derived PDGF-BB promotes Hedgehog survival signaling in cholangiocarcinoma cells. Hepatology 54, 2076–2088 (2011).
Fingas, C. D. et al. Polo-like kinase 2 is a mediator of hedgehog survival signaling in cholangiocarcinoma. Hepatology 58, 1362–1374 (2013).
Sirica, A. E. & Gores, G. J. Desmoplastic stroma and cholangiocarcinoma: clinical implications and therapeutic targeting. Hepatology 59, 2397–2402 (2014).
Park, J., Tadlock, L., Gores, G. J. & Patel, T. Inhibition of interleukin 6-mediated mitogen-activated protein kinase activation attenuates growth of a cholangiocarcinoma cell line. Hepatology 30, 1128–1133 (1999).
Wehbe, H., Henson, R., Meng, F., Mize-Berge, J. & Patel, T. Interleukin-6 contributes to growth in cholangiocarcinoma cells by aberrant promoter methylation and gene expression. Cancer Res. 66, 10517–10524 (2006).
Kobayashi, S., Werneburg, N. W., Bronk, S. F., Kaufmann, S. H. & Gores, G. J. Interleukin-6 contributes to Mcl-1 up-regulation and TRAIL resistance via an Akt-signaling pathway in cholangiocarcinoma cells. Gastroenterology 128, 2054–2065 (2005).
Taniai, M. et al. Mcl-1 mediates tumor necrosis factor-related apoptosis-inducing ligand resistance in human cholangiocarcinoma cells. Cancer Res. 64, 3517–3524 (2004).
Isomoto, H. et al. Interleukin 6 upregulates myeloid cell leukemia-1 expression through a STAT3 pathway in cholangiocarcinoma cells. Hepatology 42, 1329–1338 (2005).
Meng, F., Yamagiwa, Y., Ueno, Y. & Patel, T. Over-expression of interleukin-6 enhances cell survival and transformed cell growth in human malignant cholangiocytes. J. Hepatol. 44, 1055–1065 (2006).
Isomoto, H. et al. Sustained IL-6/STAT-3 signaling in cholangiocarcinoma cells due to SOCS-3 epigenetic silencing. Gastroenterology 132, 384–396 (2007).
Lu, J. P. et al. In situ detection of TGF betas, TGF beta receptor II mRNA and telomerase activity in rat cholangiocarcinogenesis. World J. Gastroenterol. 9, 590–594 (2003).
Benckert, C. et al. Transforming growth factor β1 stimulates vascular endothelial growth factor gene transcription in human cholangiocellular carcinoma cells. Cancer Res. 63, 1083–1092 (2003).
Araki, K. et al. E/N-cadherin switch mediates cancer progression via TGF-β-induced epithelial-to-mesenchymal transition in extrahepatic cholangiocarcinoma. Br. J. Cancer 105, 1885–1893 (2011).
Sato, Y. et al. Epithelial–mesenchymal transition induced by transforming growth factor-β1/Snail activation aggravates invasive growth of cholangiocarcinoma. Am. J. Pathol. 177, 141–152 (2010).
Yoon, J. H., Higuchi, H., Werneburg, N. W., Kaufmann, S. H. & Gores, G. J. Bile acids induce cyclooxygenase-2 expression via the epidermal growth factor receptor in a human cholangiocarcinoma cell line. Gastroenterology 122, 985–993 (2002).
Yoon, J. H., Canbay, A. E., Werneburg, N. W., Lee, S. P. & Gores, G. J. Oxysterols induce cyclooxygenase-2 expression in cholangiocytes: implications for biliary tract carcinogenesis. Hepatology 39, 732–738 (2004).
Sirica, A. E., Lai, G. H., Endo, K., Zhang, Z. & Yoon, B. I. Cyclooxygenase-2 and ERBB-2 in cholangiocarcinoma: potential therapeutic targets. Semin. Liver Dis. 22, 303–313 (2002).
Zhang, Z., Lai, G. H. & Sirica, A. E. Celecoxib-induced apoptosis in rat cholangiocarcinoma cells mediated by Akt inactivation and Bax translocation. Hepatology 39, 1028–1037 (2004).
Wu, T., Leng, J., Han, C. & Demetris, A. J. The cyclooxygenase-2 inhibitor celecoxib blocks phosphorylation of Akt and induces apoptosis in human cholangiocarcinoma cells. Mol. Cancer Ther. 3, 299–307 (2004).
Han, C., Leng, J., Demetris, A. J. & Wu, T. Cyclooxygenase-2 promotes human cholangiocarcinoma growth: evidence for cyclooxygenase-2-independent mechanism in celecoxib-mediated induction of p21waf1/cip1 and p27kip1 and cell cycle arrest. Cancer Res. 64, 1369–1376 (2004).
Yeh, C. N. et al. Reappraisal of the therapeutic role of celecoxib in cholangiocarcinoma. PLoS ONE 8, e69928 (2013).
Lu, D., Han, C. & Wu, T. Microsomal prostaglandin E synthase-1 inhibits PTEN and promotes experimental cholangiocarcinogenesis and tumor progression. Gastroenterology 140, 2084–2094 (2011).
Jaiswal, M., LaRusso, N. F., Burgart, L. J. & Gores, G. J. Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res. 60, 184–190 (2000).
Jaiswal, M., LaRusso, N. F., Shapiro, R. A., Billiar, T. R. & Gores, G. J. Nitric oxide-mediated inhibition of DNA repair potentiates oxidative DNA damage in cholangiocytes. Gastroenterology 120, 190–199 (2001).
Ishimura, N., Bronk, S. F. & Gores, G. J. Inducible nitric oxide synthase upregulates cyclooxygenase-2 in mouse cholangiocytes promoting cell growth. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G88–G95 (2004).
Yamada, D. et al. IL-33 facilitates oncogene-induced cholangiocarcinoma in mice by an interleukin-6-sensitive mechanism. Hepatology 61, 1627–1642 (2015).
Leake, I. Biliary tract. A new mouse model that closely resembles human cholangiocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 12, 122 (2015).
Hanahan, D. & Coussens, L. M. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21, 309–322 (2012).
Junttila, M. R. & de Sauvage, F. J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 501, 346–354 (2013).
Lemoinne, S. et al. The emerging roles of microvesicles in liver diseases. Nat. Rev. Gastroenterol. Hepatol. 11, 350–361 (2014).
Li, L. et al. Human bile contains microRNA-laden extracellular vesicles that can be used for cholangiocarcinoma diagnosis. Hepatology 60, 896–907 (2014).
Patel, T. Extracellular vesicle noncoding RNA: new players in the diagnosis and pathogenesis of cholangiocarcinoma. Hepatology 60, 782–784 (2014).
Haga, H. et al. Tumour cell-derived extracellular vesicles interact with mesenchymal stem cells to modulate the microenvironment and enhance cholangiocarcinoma growth. J. Extracell. Vesicles 4, 24900 (2015).
Sulpice, L. et al. Molecular profiling of stroma identifies osteopontin as an independent predictor of poor prognosis in intrahepatic cholangiocarcinoma. Hepatology 58, 1992–2000 (2013).
Andersen, J. B. et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology 142, 1021–1031.e15 (2012).
Sirica, A. E. The role of cancer-associated myofibroblasts in intrahepatic cholangiocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 9, 44–54 (2012).
Cadamuro, M. et al. Platelet-derived growth factor-D and Rho GTPases regulate recruitment of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology 58, 1042–1053 (2013).
Ohira, S. et al. Possible regulation of migration of intrahepatic cholangiocarcinoma cells by interaction of CXCR4 expressed in carcinoma cells with tumor necrosis factor-α and stromal-derived factor-1 released in stroma. Am. J. Pathol. 168, 1155–1168 (2006).
Gentilini, A. et al. Role of the stromal-derived factor-1 (SDF-1)–CXCR4 axis in the interaction between hepatic stellate cells and cholangiocarcinoma. J. Hepatol. 57, 813–820 (2012).
Claperon, A. et al. Hepatic myofibroblasts promote the progression of human cholangiocarcinoma through activation of epidermal growth factor receptor. Hepatology 58, 2001–2011 (2013).
Okabe, H. et al. Identification of CXCL5/ENA-78 as a factor involved in the interaction between cholangiocarcinoma cells and cancer-associated fibroblasts. Int. J. Cancer 131, 2234–2241 (2012).
Kim, Y. et al. Hedgehog signaling between cancer cells and hepatic stellate cells in promoting cholangiocarcinoma. Ann. Surg. Oncol. 21, 2684–2698 (2014).
Okabe, H. et al. Hepatic stellate cells may relate to progression of intrahepatic cholangiocarcinoma. Ann. Surg. Oncol. 16, 2555–2564 (2009).
Chuaysri, C. et al. Alpha-smooth muscle actin-positive fibroblasts promote biliary cell proliferation and correlate with poor survival in cholangiocarcinoma. Oncol. Rep. 21, 957–969 (2009).
Mertens, J. C. et al. Therapeutic effects of deleting cancer-associated fibroblasts in cholangiocarcinoma. Cancer Res. 73, 897–907 (2013).
Subimerb, C. et al. Tissue invasive macrophage density is correlated with prognosis in cholangiocarcinoma. Mol. Med. Rep. 3, 597–605 (2010).
Subimerb, C. et al. Circulating CD14+ CD16+ monocyte levels predict tissue invasive character of cholangiocarcinoma. Clin. Exp. Immunol. 161, 471–479 (2010).
Hasita, H. et al. Significance of alternatively activated macrophages in patients with intrahepatic cholangiocarcinoma. Cancer Sci. 101, 1913–1919 (2010).
Techasen, A. et al. Cytokines released from activated human macrophages induce epithelial mesenchymal transition markers of cholangiocarcinoma cells. Asian Pac. J. Cancer Prev. 13 (Suppl.), 115–118 (2012).
Loilome, W. et al. Activated macrophages promote Wnt/β-catenin signaling in cholangiocarcinoma cells. Tumour Biol. 35, 5357–5367 (2014).
Tang, D. et al. Angiogenesis in cholangiocellular carcinoma: expression of vascular endothelial growth factor, angiopoietin-1/2, thrombospondin-1 and clinicopathological significance. Oncol. Rep. 15, 525–532 (2006).
Thelen, A. et al. Tumor-associated angiogenesis and lymphangiogenesis correlate with progression of intrahepatic cholangiocarcinoma. Am. J. Gastroenterol. 105, 1123–1132 (2010).
Thelen, A. et al. Microvessel density correlates with lymph node metastases and prognosis in hilar cholangiocarcinoma. J. Gastroenterol. 43, 959–966 (2008).
Shirabe, K. et al. Prognostic factors in node-negative intrahepatic cholangiocarcinoma with special reference to angiogenesis. Am. J. Surg. 187, 538–542 (2004).
Angulo, P. & Lindor, K. D. Primary sclerosing cholangitis. Hepatology 30, 325–332 (1999).
Boberg, K. M. et al. Cholangiocarcinoma in primary sclerosing cholangitis: risk factors and clinical presentation. Scand. J. Gastroenterol. 37, 1205–1211 (2002).
Morris-Stiff, G. et al. Cholangiocarcinoma complicating primary sclerosing cholangitis: a 24-year experience. Dig. Surg. 25, 126–132 (2008).
Bjornsson, E. & Angulo, P. Cholangiocarcinoma in young individuals with and without primary sclerosing cholangitis. Am. J. Gastroenterol. 102, 1677–1682 (2007).
Singal, A. G. et al. The clinical presentation and prognostic factors for intrahepatic and extrahepatic cholangiocarcinoma in a tertiary care centre. Aliment. Pharmacol. Ther. 31, 625–633 (2010).
Rimola, J. et al. Cholangiocarcinoma in cirrhosis: absence of contrast washout in delayed phases by magnetic resonance imaging avoids misdiagnosis of hepatocellular carcinoma. Hepatology 50, 791–798 (2009).
Iavarone, M. et al. Contrast enhanced CT-scan to diagnose intrahepatic cholangiocarcinoma in patients with cirrhosis. J. Hepatol. 58, 1188–1193 (2013).
Kim, S. J. et al. Peripheral mass-forming cholangiocarcinoma in cirrhotic liver. AJR Am. J. Roentgenol 189, 1428–1434 (2007).
Weber, S. M. et al. Intrahepatic cholangiocarcinoma: expert consensus statement. HPB (Oxford) 17, 669–680 (2015).
Bledsoe, J. R., Shinagare, S. A. & Deshpande, V. Difficult diagnostic problems in pancreatobiliary neoplasia. Arch. Pathol. Lab Med. 139, 848–857 (2015).
Rullier, A. et al. Cytokeratin 7 and 20 expression in cholangiocarcinomas varies along the biliary tract but still differs from that in colorectal carcinoma metastasis. Am. J. Surg. Pathol. 24, 870–876 (2000).
Lau, S. K., Prakash, S., Geller, S. A. & Alsabeh, R. Comparative immunohistochemical profile of hepatocellular carcinoma, cholangiocarcinoma, and metastatic adenocarcinoma. Hum. Pathol. 33, 1175–1181 (2002).
Mosnier, J. F. et al. N-cadherin serves as diagnostic biomarker in intrahepatic and perihilar cholangiocarcinomas. Mod. Pathol. 22, 182–190 (2009).
Ferrone, C. R. et al. The ability to diagnose intrahepatic cholangiocarcinoma definitively using novel branched DNA-enhanced albumin RNA in situ hybridization technology. Ann. Surg. Oncol. 23, 290–296 (2014).
Abu-Wasel, B., Keough, V., Renfrew, P. D. & Molinari, M. Biliary stent therapy for dominant strictures in patients affected by primary sclerosing cholangitis. Pathobiology 80, 182–193 (2013).
Romagnuolo, J. et al. Magnetic resonance cholangiopancreatography: a meta-analysis of test performance in suspected biliary disease. Ann. Intern. Med. 139, 547–557 (2003).
Fevery, J. & Verslype, C. An update on cholangiocarcinoma associated with primary sclerosing cholangitis. Curr. Opin. Gastroenterol. 26, 236–245 (2010).
Campbell, W. L. et al. Using CT and cholangiography to diagnose biliary tract carcinoma complicating primary sclerosing cholangitis. AJR Am. J. Roentgenol. 177, 1095–1100 (2001).
Rosch, T. et al. A prospective comparison of the diagnostic accuracy of ERCP, MRCP, CT, and EUS in biliary strictures. Gastrointest. Endosc. 55, 870–876 (2002).
Chapman, R. et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology 51, 660–678 (2010).
European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of cholestatic liver diseases. J. Hepatol. 51, 237–267 (2009).
Lindor, K. D., Kowdley, K. V., Harrison, M. E. & American College of Gastroenterology. ACG clinical guideline: primary sclerosing cholangitis. Am. J. Gastroenterol. 110, 646–659 (2015).
Navaneethan, U. et al. Comparative effectiveness of biliary brush cytology and intraductal biopsy for detection of malignant biliary strictures: a systematic review and meta-analysis. Gastrointest. Endosc. 81, 168–176 (2015).
Nuzzo, G. et al. Improvement in perioperative and long-term outcome after surgical treatment of hilar cholangiocarcinoma: results of an Italian multicenter analysis of 440 patients. Arch. Surg. 147, 26–34 (2012).
Mansour, J. C. et al. Hilar cholangiocarcinoma: expert consensus statement. HPB (Oxford) 17, 691–699 (2015).
Fritscher-Ravens, A. et al. EUS-guided fine-needle aspiration cytodiagnosis of hilar cholangiocarcinoma: a case series. Gastrointest. Endosc. 52, 534–540 (2000).
Fritscher-Ravens, A. et al. EUS-guided fine-needle aspiration of suspected hilar cholangiocarcinoma in potentially operable patients with negative brush cytology. Am. J. Gastroenterol. 99, 45–51 (2004).
Rosch, T. et al. ERCP or EUS for tissue diagnosis of biliary strictures? A prospective comparative study. Gastrointest. Endosc. 60, 390–396 (2004).
Lee, J. H., Salem, R., Aslanian, H., Chacho, M. & Topazian, M. Endoscopic ultrasound and fine-needle aspiration of unexplained bile duct strictures. Am. J. Gastroenterol. 99, 1069–1073 (2004).
Byrne, M. F. et al. Yield of endoscopic ultrasound-guided fine-needle aspiration of bile duct lesions. Endoscopy 36, 715–719 (2004).
Heimbach, J. K., Sanchez, W., Rosen, C. B. & Gores, G. J. Trans-peritoneal fine needle aspiration biopsy of hilar cholangiocarcinoma is associated with disease dissemination. HPB (Oxford) 13, 356–360 (2011).
Levy, C. et al. The value of serum CA 19–9 in predicting cholangiocarcinomas in patients with primary sclerosing cholangitis. Dig. Dis. Sci. 50, 1734–1740 (2005).
Wannhoff, A. et al. FUT2 and FUT3 genotype determines CA19-9 cut-off values for detection of cholangiocarcinoma in patients with primary sclerosing cholangitis. J. Hepatol. 59, 1278–1284 (2013).
Alvaro, D. et al. Serum and biliary insulin-like growth factor I and vascular endothelial growth factor in determining the cause of obstructive cholestasis. Ann. Intern. Med. 147, 451–459 (2007).
Navaneethan, U. et al. Volatile organic compounds in bile for early diagnosis of cholangiocarcinoma in patients with primary sclerosing cholangitis: a pilot study. Gastrointest. Endosc. 81, 943–949.e1 (2015).
Navaneethan, U. et al. Bile proteomics for differentiation of malignant from benign biliary strictures: a pilot study. Gastroenterol. Rep. (Oxf.) 3, 136–143 (2015).
Alvaro, D. Progranulin and cholangiocarcinoma: another bad boy on the block! Gut 61, 170–171 (2012).
Alvaro, D. Serum and bile biomarkers for cholangiocarcinoma. Curr. Opin. Gastroenterol. 25, 279–284 (2009).
Bismuth, H. & Corlette, M. B. Intrahepatic cholangioenteric anastomosis in carcinoma of the hilus of the liver. Surg. Gynecol. Obstet. 140, 170–178 (1975).
Jarnagin, W. R. et al. Staging, resectability, and outcome in 225 patients with hilar cholangiocarcinoma. Ann. Surg. 234, 507–517; discussion 517–519 (2001).
Deoliveira, M. L. et al. New staging system and a registry for perihilar cholangiocarcinoma. Hepatology 53, 1363–1371 (2011).
Juntermanns, B. et al. Comparison of the sixth and the seventh editions of the UICC classification for perihilar cholangiocarcinoma. Ann. Surg. Oncol. 20, 277–284 (2013).
Nathan, H. et al. Trends in survival after surgery for cholangiocarcinoma: a 30-year population-based SEER database analysis. J. Gastrointest Surg. 11, 1488–1496; discussion 1496–1497 (2007).
Zaydfudim, V. M., Rosen, C. B. & Nagorney, D. M. Hilar cholangiocarcinoma. Surg. Oncol. Clin. N. Am. 23, 247–263 (2014).
Nagino, M. et al. Evolution of surgical treatment for perihilar cholangiocarcinoma: a single-center 34-year review of 574 consecutive resections. Ann. Surg. 258, 129–140 (2013).
Song, S. C. et al. Surgical outcomes of 230 resected hilar cholangiocarcinoma in a single centre. ANZ J. Surg. 83, 268–274 (2013).
Rosen, C. B., Heimbach, J. K. & Gores, G. J. Liver transplantation for cholangiocarcinoma. Transpl. Int. 23, 692–697 (2010).
Darwish Murad, S. et al. Efficacy of neoadjuvant chemoradiation, followed by liver transplantation, for perihilar cholangiocarcinoma at 12 US centers. Gastroenterology 143, 88–98.e3 (2012).
Darwish Murad, S. et al. Predictors of pretransplant dropout and posttransplant recurrence in patients with perihilar cholangiocarcinoma. Hepatology 56, 972–981 (2012).
Friman, S. et al. Liver transplantation for cholangiocarcinoma: selection is essential for acceptable results. Scand. J. Gastroenterol. 46, 370–375 (2011).
Kelley, R. K., Hirose, R. & Venook, A. P. Can we cure cholangiocarcinoma with neoadjuvant chemoradiation and liver transplantation? Time for a multicenter trial. Liver Transpl. 18, 509–513 (2012).
Valle, J. W. et al. Cisplatin and gemcitabine for advanced biliary tract cancer: a meta-analysis of two randomised trials. Ann. Oncol. 25, 391–398 (2014).
Valle, J. et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 362, 1273–1281 (2010).
Okusaka, T. et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: a comparative multicentre study in Japan. Br. J. Cancer 103, 469–474 (2010).
Roth, J. A. & Carlson, J. J. Cost-effectiveness of gemcitabine + cisplatin versus gemcitabine monotherapy in advanced biliary tract cancer. J. Gastrointest. Cancer 43, 215–223 (2012).
Andre, T. et al. Gemcitabine and oxaliplatin in advanced biliary tract carcinoma: a Phase II study. Br. J. Cancer 99, 862–867 (2008).
Jang, J. S. et al. Gemcitabine and oxaliplatin in patients with unresectable biliary cancer including gall bladder cancer: a Korean Cancer Study Group Phase II trial. Cancer Chemother. Pharmacol. 65, 641–647 (2010).
Lamarca, A., Hubner, R. A., David Ryder, W. & Valle, J. W. Second-line chemotherapy in advanced biliary cancer: a systematic review. Ann. Oncol. 25, 2328–2338 (2014).
Jiang, X. et al. Metformin inhibits tumor growth by regulating multiple miRNAs in human cholangiocarcinoma. Oncotarget 6, 3178–3194 (2015).
Park, S. Y. et al. Transarterial chemoembolization versus supportive therapy in the palliative treatment of unresectable intrahepatic cholangiocarcinoma. Clin. Radiol. 66, 322–328 (2011).
Hyder, O. et al. Intra-arterial therapy for advanced intrahepatic cholangiocarcinoma: a multi-institutional analysis. Ann. Surg. Oncol. 20, 3779–3786 (2013).
Ibrahim, S. M. et al. Treatment of unresectable cholangiocarcinoma using yttrium-90 microspheres: results from a pilot study. Cancer 113, 2119–2128 (2008).
Boehm, L. M. et al. Comparative effectiveness of hepatic artery based therapies for unresectable intrahepatic cholangiocarcinoma. J. Surg. Oncol. 111, 213–220 (2015).
Han, K. et al. Radiofrequency ablation in the treatment of unresectable intrahepatic cholangiocarcinoma: systematic review and meta-analysis. J. Vasc. Interv. Radiol. 26, 943–948 (2015).
Laquiere, A. et al. Safety and feasibility of endoscopic biliary radiofrequency ablation treatment of extrahepatic cholangiocarcinoma. Surg. Endosc. 30, 1242–1248 (2015).
Dolak, W. et al. Endoscopic radiofrequency ablation for malignant biliary obstruction: a nationwide retrospective study of 84 consecutive applications. Surg. Endosc. 28, 854–860 (2014).
Ortner, M. E. et al. Successful photodynamic therapy for nonresectable cholangiocarcinoma: a randomized prospective study. Gastroenterology 125, 1355–1363 (2003).
Leggett, C. L. et al. Photodynamic therapy for unresectable cholangiocarcinoma: a comparative effectiveness systematic review and meta-analyses. Photodiagnosis Photodyn. Ther. 9, 189–195 (2012).
Lu, Y., Liu, L., Wu, J. C., Bie, L. K. & Gong, B. Efficacy and safety of photodynamic therapy for unresectable cholangiocarcinoma: a meta-analysis. Clin. Res. Hepatol. Gastroenterol. 39, 718–724 (2015).
Uppal, D. S. & Wang, A. Y. Advances in endoscopic retrograde cholangiopancreatography for the treatment of cholangiocarcinoma. World J. Gastrointest. Endosc. 7, 675–687 (2015).
Martin, R. C. et al. Cost comparison of endoscopic stenting versus surgical treatment for unresectable cholangiocarcinoma. Surg. Endosc. 16, 667–670 (2002).
Liberato, M. J. & Canena, J. M. Endoscopic stenting for hilar cholangiocarcinoma: efficacy of unilateral and bilateral placement of plastic and metal stents in a retrospective review of 480 patients. BMC Gastroenterol. 12, 103 (2012).
Rizvi, S. & Gores, G. J. Molecular pathogenesis of cholangiocarcinoma. Dig. Dis. 32, 564–569 (2014).
Rizvi, S., Borad, M. J., Patel, T. & Gores, G. J. Cholangiocarcinoma: molecular pathways and therapeutic opportunities. Semin. Liver Dis. 34, 456–464 (2014).
Yao, L. et al. Omega-3 polyunsaturated fatty acids upregulate 15-PGDH expression in cholangiocarcinoma cells by inhibiting miR-26a/b expression. Cancer Res. 75, 1388–1398 (2015).
Zhang, J. et al. miR-21 inhibition reduces liver fibrosis and prevents tumor development by inducing apoptosis of CD24+ progenitor cells. Cancer Res. 75, 1859–1867 (2015).
Saborowski, A. et al. Mouse model of intrahepatic cholangiocarcinoma validates FIG-ROS as a potent fusion oncogene and therapeutic target. Proc. Natl Acad. Sci. USA 110, 19513–19518 (2013).
Lozano, E. et al. Enhanced antitumour drug delivery to cholangiocarcinoma through the apical sodium-dependent bile acid transporter (ASBT). J. Control Release 216, 93–102 (2015).
Sabbatino, F. et al. PD-L1 and HLA class I antigen expression and clinical course of the disease in intrahepatic cholangiocarcinoma. Clin. Cancer Res. 22, 470–478 (2015).
Wlcek, K. et al. The analysis of organic anion transporting polypeptide (OATP) mRNA and protein patterns in primary and metastatic liver cancer. Cancer Biol. Ther. 11, 801–811 (2011).
Franke, R. M., Scherkenbach, L. A. & Sparreboom, A. Pharmacogenetics of the organic anion transporting polypeptide 1A2. Pharmacogenomics 10, 339–344 (2009).
Lautem, A. et al. Downregulation of organic cation transporter 1 (SLC22A1) is associated with tumor progression and reduced patient survival in human cholangiocellular carcinoma. Int. J. Oncol. 42, 1297–1304 (2013).
Herraez, E. et al. Expression of SLC22A1 variants may affect the response of hepatocellular carcinoma and cholangiocarcinoma to sorafenib. Hepatology 58, 1065–1073 (2013).
Sasaki, H. et al. Concurrent analysis of human equilibrative nucleoside transporter 1 and ribonucleotide reductase subunit 1 expression increases predictive value for prognosis in cholangiocarcinoma patients treated with adjuvant gemcitabine-based chemotherapy. Br. J. Cancer 111, 1275–1284 (2014).
Borbath, I. et al. Human equilibrative nucleoside transporter 1 (hENT1) expression is a potential predictive tool for response to gemcitabine in patients with advanced cholangiocarcinoma. Eur. J. Cancer 48, 990–996 (2012).
Namwat, N. et al. Characterization of 5-fluorouracil-resistant cholangiocarcinoma cell lines. Chemotherapy 54, 343–351 (2008).
Martinez-Becerra, P. et al. No correlation between the expression of FXR and genes involved in multidrug resistance phenotype of primary liver tumors. Mol. Pharm. 9, 1693–1704 (2012).
Gigliozzi, A. et al. Molecular identification and functional characterization of Mdr1a in rat cholangiocytes. Gastroenterology 119, 1113–1122 (2000).
Hunter, J., Hirst, B. H. & Simmons, N. L. Epithelial secretion of vinblastine by human intestinal adenocarcinoma cell (HCT-8 and T84) layers expressing P-glycoprotein. Br. J. Cancer 64, 437–444 (1991).
Ambudkar, S. V. et al. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 39, 361–398 (1999).
Cao, L. et al. Expression of MDR1 mRNA and encoding P-glycoprotein in archival formalin-fixed paraffin-embedded gall bladder cancer tissues. Eur. J. Cancer 34, 1612–1617 (1998).
Wang, B. L. et al. Clinical relationship between MDR1 gene and gallbladder cancer. Hepatobiliary Pancreat. Dis. Int. 3, 296–299 (2004).
Srimunta, U. et al. High expression of ABCC1 indicates poor prognosis in intrahepatic cholangiocarcinoma. Asian Pac. J. Cancer Prev. 13 (Suppl.), 125–130 (2012).
Rau, S. et al. Expression of the multidrug resistance proteins MRP2 and MRP3 in human cholangiocellular carcinomas. Eur. J. Clin. Invest. 38, 134–142 (2008).
Evrard, A., Cuq, P., Ciccolini, J., Vian, L. & Cano, J. P. Increased cytotoxicity and bystander effect of 5-fluorouracil and 5-deoxy-5-fluorouridine in human colorectal cancer cells transfected with thymidine phosphorylase. Br. J. Cancer 80, 1726–1733 (1999).
Hahnvajanawong, C. et al. Orotate phosphoribosyl transferase mRNA expression and the response of cholangiocarcinoma to 5-fluorouracil. World J. Gastroenterol. 18, 3955–3961 (2012).
Nakajima, T. et al. Reversal of multiple drug resistance in cholangiocarcinoma by the glutathione S-transferase-π-specific inhibitor O1-hexadecyl-γ-glutamyl-S-benzylcysteinyl-d-phenylglycine ethylester. J. Pharmacol. Exp. Ther. 306, 861–869 (2003).
Tepsiri, N. et al. Drug sensitivity and drug resistance profiles of human intrahepatic cholangiocarcinoma cell lines. World J. Gastroenterol. 11, 2748–2753 (2005).
Habara, K., Ajiki, T., Kamigaki, T., Nakamura, T. & Kuroda, Y. High expression of thymidylate synthase leads to resistance to 5-fluorouracil in biliary tract carcinoma in vitro. Jpn J. Cancer Res. 92, 1127–1132 (2001).
Cai, J. P. et al. Simvastatin enhances the chemotherapeutic efficacy of S-1 against bile duct cancer: E2F-1/TS downregulation might be the mechanism. Anticancer Drugs 24, 1020–1029 (2013).
Sampson, L. K., Vickers, S. M., Ying, W. & Phillips, J. O. Tamoxifen-mediated growth inhibition of human cholangiocarcinoma. Cancer Res. 57, 1743–1749 (1997).
Jimeno, A. et al. Epidermal growth factor receptor dynamics influences response to epidermal growth factor receptor targeted agents. Cancer Res. 65, 3003–3010 (2005).
Kornprat, P., Rehak, P., Ruschoff, J. & Langner, C. Expression of IGF-I, IGF-II, and IGF-IR in gallbladder carcinoma. A systematic analysis including primary and corresponding metastatic tumours. J. Clin. Pathol. 59, 202–206 (2006).
Zhang, H. & Yee, D. The therapeutic potential of agents targeting the type I insulin-like growth factor receptor. Expert Opin. Investig. Drugs 13, 1569–1577 (2004).
Sachdev, D. & Yee, D. Disrupting insulin-like growth factor signaling as a potential cancer therapy. Mol. Cancer Ther. 6, 1–12 (2007).
Hwang, I. G. et al. Different relation between ERCC1 overexpression and treatment outcomes of two platinum agents in advanced biliary tract adenocarcinoma patients. Cancer Chemother. Pharmacol. 68, 935–944 (2011).
Obama, K. et al. Enhanced expression of RAD51 associating protein-1 is involved in the growth of intrahepatic cholangiocarcinoma cells. Clin. Cancer Res. 14, 1333–1339 (2008).
Asakawa, H. et al. Prediction of breast cancer sensitivity to neoadjuvant chemotherapy based on status of DNA damage repair proteins. Breast Cancer Res. 12, R17 (2010).
Momoi, H. et al. Microsatellite instability and alternative genetic pathway in intrahepatic cholangiocarcinoma. J. Hepatol. 35, 235–244 (2001).
Liu, D., Momoi, H., Li, L., Ishikawa, Y. & Fukumoto, M. Microsatellite instability in thorotrast-induced human intrahepatic cholangiocarcinoma. Int. J. Cancer 102, 366–371 (2002).
Limpaiboon, T. et al. Promoter hypermethylation is a major event of hMLH1 gene inactivation in liver fluke related cholangiocarcinoma. Cancer Lett. 217, 213–219 (2005).
Sato, J. et al. Gene expression analysis for predicting gemcitabine resistance in human cholangiocarcinoma. J. Hepatobiliary Pancreat. Sci. 18, 700–711 (2011).
Takayama, Y. et al. Silencing of Tousled-like kinase 1 sensitizes cholangiocarcinoma cells to cisplatin-induced apoptosis. Cancer Lett. 296, 27–34 (2010).
Ge, X. et al. NK4 regulates 5-fluorouracil sensitivity in cholangiocarcinoma cells by modulating the intrinsic apoptosis pathway. Oncol. Rep. 30, 448–454 (2013).
Tanaka, S., Sugimachi, K., Shirabe, K., Shimada, M. & Wands, J. R. Expression and antitumor effects of TRAIL in human cholangiocarcinoma. Hepatology 32, 523–527 (2000).
Panichakul, T., Intachote, P., Wongkajorsilp, A., Sripa, B. & Sirisinha, S. Triptolide sensitizes resistant cholangiocarcinoma cells to TRAIL-induced apoptosis. Anticancer Res. 26, 259–265 (2006).
Fernandez, T. F., Samal, A. B., Bedwell, G. J., Chen, Y. & Saad, J. S. Structural and biophysical characterization of the interactions between the death domain of Fas receptor and calmodulin. J. Biol. Chem. 288, 21898–21908 (2013).
Harnois, D. M., Que, F. G., Celli, A., LaRusso, N. F. & Gores, G. J. Bcl-2 is overexpressed and alters the threshold for apoptosis in a cholangiocarcinoma cell line. Hepatology 26, 884–890 (1997).
Yoon, H., Min, J. K., Lee, J. W., Kim, D. G. & Hong, H. J. Acquisition of chemoresistance in intrahepatic cholangiocarcinoma cells by activation of AKT and extracellular signal-regulated kinase (ERK)1/2. Biochem. Biophys. Res. Commun. 405, 333–337 (2011).
Wehrkamp, C. J., Gutwein, A. R., Natarajan, S. K., Phillippi, M. A. & Mott, J. L. XIAP antagonist embelin inhibited proliferation of cholangiocarcinoma cells. PLoS ONE 9, e90238 (2014).
Acknowledgements
Authors thank ENS-CCA International Advisors or Collaborators for their continuous support and help: G. Gores, N. F. LaRusso and L. Roberts (Mayo Clinic, Rochester, Minnesota, USA); T. Patel (Mayo Clinic, Jacksonville, Florida, USA); S. Gradilone (Hormel Institute, University of Minnesota, Austin, Minnesota, USA); G. Alpini (Texas A&M University, Temple, Texas, USA); L. Reid (University of North Carolina, Chapel Hill, North Carolina, USA). ENS-CCA recognizes O. Echaniz (University of the Basque Country, San Sebastian, Spain) and A. Santos (Biodonostia Research Health Institute, San Sebastian, Spain) for their constant technical assistance. Authors also thank M. Consiglia Bragazzi for her help in preparing data for Fig. 1 and A. Cardinale for the graphic design. Finally, authors thank R. Breeze (University of Navarra, Spain) for the English revision of the manuscript.
Author information
Authors and Affiliations
Contributions
All listed authors contributed equally to all aspects of this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
FURTHER INFORMATION
Supplementary information
Supplementary information S1 (table)
Ongoing and completed phase III trials including chemotherapeutic agents in CCA (PDF 77 kb)
Supplementary information S2 (table)
Clinical trials of targeted therapies in CCA (PDF 124 kb)
Supplementary information S3 (table)
Ongoing trials (phase II or III and selected phase 1/2) (based on ClinicalTrials.gov database, August 2015) (PDF 94 kb)
Supplementary information S4 (table)
Examples of pre-clinical studies analyzing novel targets or target combinations for therapy in CCA not yet included in clinical trials (PDF 130 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Banales, J., Cardinale, V., Carpino, G. et al. Cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat Rev Gastroenterol Hepatol 13, 261–280 (2016). https://doi.org/10.1038/nrgastro.2016.51
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrgastro.2016.51
This article is cited by
-
Human Vγ9Vδ2 T cell expansion and their cytotoxic responses against cholangiocarcinoma
Scientific Reports (2024)
-
Granzyme B+ B cells detected by single-cell sequencing are associated with prognosis in patients with intrahepatic cholangiocarcinoma following liver transplantation
Cancer Immunology, Immunotherapy (2024)
-
Systemic chemotherapy plus transarterial chemoembolization versus systemic chemotherapy alone for unresectable intrahepatic cholangiocarcinoma: a multicenter retrospective cohort study
La radiologia medica (2024)
-
Factors Impacting Survival After Transarterial Radioembolization in Patients with Unresectable Intrahepatic Cholangiocarcinoma: A Combined Analysis of the Prospective CIRT Studies
CardioVascular and Interventional Radiology (2024)
-
Cancer-specific survival in patients with cholangiocarcinoma after radical surgery: a Novel, dynamic nomogram based on clinicopathological features and serum markers
BMC Cancer (2023)
