
Abstract
The definition of acute-on-chronic liver failure (ACLF) remains contested. In Europe and North America, the term is generally applied according to the European Association for the Study of the Liver-Chronic Liver Failure (EASL-CLIF) Consortium guidelines, which defines this condition as a syndrome that develops in patients with cirrhosis and is characterized by acute decompensation, organ failure and high short-term mortality. One-third of patients who are hospitalized for acute decompensation present with ACLF at admission or develop the syndrome during hospitalization. ACLF frequently occurs in a closed temporal relationship to a precipitating event, such as bacterial infection or acute alcoholic, drug-induced or viral hepatitis. However, no precipitating event can be identified in approximately 40% of patients. The mechanisms of ACLF involve systemic inflammation due to infections, acute liver damage and, in cases without precipitating events, probably intestinal translocation of bacteria or bacterial products. ACLF is graded into three stages (ACLF grades 1–3) on the basis of the number of organ failures, with higher grades associated with increased mortality. Liver and renal failures are the most common organ failures, followed by coagulation, brain, circulatory and respiratory failure. The 28-day mortality rate associated with ACLF is 30%. Depending on the grade, ACLF can be reversed using standard therapy in only 16–51% of patients, leaving a considerable proportion of patients with ACLF that remains steady or progresses. Liver transplantation in selected patients with ACLF grade 2 and ACLF grade 3 increases the 6-month survival from 10% to 80%.
Similar content being viewed by others

Introduction
Cirrhosis is a progressive chronic liver disease characterized by diffuse fibrosis, severe disruption of the intrahepatic venous flow, portal hypertension and liver failure. The course of cirrhosis is divided into two stages1 (Fig. 1). Compensated cirrhosis defines the period between the onset of cirrhosis and the first major complication. During this period, which is relatively long in most patients (>10 years), symptoms are absent or minor, but liver lesions and portal pressure steadily progress. The term decompensated cirrhosis defines the period following the development of ascites (that is, the accumulation of large amounts of fluid within the peritoneal cavity), variceal haemorrhage and/or hepatic encephalopathy2–4. This period is associated with short-term survival (3–5 years).

Acute-on-chronic liver failure (ACLF) can develop at any stage from compensated to decompensated cirrhosis, and can involve hepatic or extrahepatic precipitating events. A considerable proportion of patients have no identifiable triggering event. In this figure, paracentesis means ‘large volume paracentesis’ (>5 litres). Acute decompensation of cirrhosis defines the acute development of clinically evident ascites, hepatic encephalopathy, gastrointestinal haemorrhage or any combination of these in patients with or without prior history of these complications. Although bacterial infections are not specific complications of cirrhosis, they are considered as such in patients with prior history of ascites, haemorrhage or encephalopathy because of their high prevalence and their association with abnormalities related to cirrhosis, including bacterial translocation and impaired leukocyte function1–8. DILI, drug-induced liver injury; TIPS, transjugular intrahepatic portosystemic shunt. Figure is adapted from an image provided courtesy of Jordi Bozzo, Avinguda de la Generalitat, Barcelona, Spain.
Concepts about cirrhosis are rapidly changing. First, cirrhosis is no longer considered to be an irreversible progressive disease. Indeed, decompensated cirrhosis may return to compensated cirrhosis or even to precirrhotic phases if the cause of the disease is removed5. Second, the list of organ or system dysfunctions in cirrhosis (hepatic, renal, brain and circulatory) has been expanded to include the immune system, intestines, heart, lungs, adrenal glands, muscles and thyroid glands. Third, new mechanisms involved in the pathogenesis of cirrhotic complications, such as dysbiosis of the microbiota6 and systemic inflammation7 have been recognized. Last, it is increasingly evident that patients rarely die as a consequence of an end-stage irreversible destruction of the liver. Rather, in most patients, the cause of death is an acute deterioration in their clinical condition promoted by a precipitating event — a syndrome termed acute-on-chronic liver failure (ACLF)8.
More than 13 distinct definitions of ACLF have been proposed. These definitions are generally based on personal experience or consensus agreements9–15 (Box 1).
The Asian Pacific Association for the Study of the Liver (APASL) definition has received major attention11,12 (Box 1). This definition is based on positive and negative criteria. The main positive criteria are: prior diagnosis of chronic liver disease (cirrhotic or non-cirrhotic, excluding isolated steatosis); a precipitating event that has a direct effect on the liver; and acute hepatic insult that causes acute liver failure. The main negative criteria in the APASL definition are: no prior history of acute decompensation in patients with cirrhosis (decompensated cirrhosis would represent the presence of end-stage progressive liver disease); and no extrahepatic precipitating event, such as bacterial infection. The APASL definition was based on a consensus conference.
The APASL proposal did not reach wide diffusion in Europe and North America for several reasons. First, the most common form of ACLF in these areas occurs in patients with decompensated cirrhosis in a closed temporal relationship with bacterial infections or active alcoholism8,13 (Fig. 1), and these patients are not included in the APASL definition. Second, the concept that decompensated cirrhosis represents a terminal phase of the disease is not the experience of European centres. Third, extrahepatic organ failure is the most characteristic differential feature between patients with ACLF and those with acute decompensation in European patients. Last, ACLF in patients with non-cirrhotic chronic liver disease is exceptional in Europe and North America owing to the low prevalence of hepatitis A virus, hepatitis B virus (HBV) and hepatitis E virus infections.
For these reasons, in 2009, the European Association for the Study of the Liver-Chronic Liver Failure (EASL-CLIF) Consortium started a prospective, multicentre European observational study in 1,343 patients who were hospitalized for acute decompensation of cirrhosis (the CANONIC study). This study aimed to define ACLF in cirrhosis, to assess the prevalence and clinical course of the syndrome and to improve the accuracy of the prognostic scores currently available8,16–20 through an evidence-based pragmatic approach. The analysis of this study lead to a new definition with three major characteristics (Box 1): acute decompensation of cirrhosis; the presence of organ failure (or failures, which can be either hepatic or extrahepatic); and a high probability of short-term (28-day) mortality. Following the publication of the main articles derived from the CANONIC study, the definition, grading of severity of ACLF and prognostic scores proposed are widely used in Europe, Asia and North America for the assessment and treatment of patients with decompensated cirrhosis21–32.
It has been suggested that differences between APASL and EASL-CLIF Consortium definitions are logical consequences of the distinct epidemiology of liver diseases in the east and in the west28,33. However, the differences are more likely to be related to the distinct objectives by which both definitions were designed. The ACLF definition by the APASL consensus group was designed to identify a condition that anticipates the development of extrahepatic or multiple organ failure and death11,12. By contrast, the goal of the CANONIC study was to characterize a syndrome in which organ failure (or failures) and high short-term mortality are central features8.
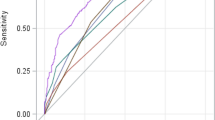
Attempts to unify both definitions have not been successful34. Recent investigations from Asia have compared the two definitions in a large series of patients with cirrhosis23,26,35. Figure 2 compares the results of the largest Asian series35 with those in the CANONIC study8. A coincident diagnosis by the APASL and the EASL-CLIF Consortium definitions was observed in only a minority of patients in both series, indicating that the two ACLF definitions selected different patient populations (Fig. 2a). The EASL-CLIF Consortium definition was significantly more accurate in predicting prognosis than the APASL definition in both the eastern and the western populations (Fig. 2b). Significant differences in mortality depending on the diagnostic criteria were also observed in two other cohorts of patients from China23 and India26. This Primer on ACLF in cirrhosis uses the EASL-CLIF Consortium definition. The reader is referred to a review by Sarin and Choudhury36 for a discussion of ACLF that is based on the APASL criteria.

a | The proportion of patients diagnosed with acute-on-chronic liver failure (ACLF) according to the Asian Pacific Association for the Study of the Liver (APASL) definition (green), the European Association for the Study of the Liver-Chronic Liver Failure (EASL-CLIF) Consortium definition (red) and both definitions (blue). Data are from a large series of patients from Korea35 (1,470 patients, of which 1,352 patients had cirrhosis with or without prior history of decompensation) and from the CANONIC study8 carried out in Europe. b | The 90-day probability of survival in both series of patients depending on the ACLF diagnosis. Figures with European data derive from unpublished results of the CANONIC study (R.M. and the CANONIC trialists, unpublished observations). Part b (left panel) adapted from Ref. 35.
Epidemiology
Worldwide prevalence and mortality
ACLF is a major worldwide medical problem, with prevalence rates in at-risk populations in the region of 20–35% (Table 1). The worldwide reported mortality of ACLF according to the EASL-CLIF Consortium definition ranges between 30% and 50% and correlates closely with the number of organ failures. In Europe, the average 28-day mortality rate without liver transplantation reported by the CANONIC study was 1.9% in patients with decompensated cirrhosis without ACLF and 32.8% in patients with ACLF (23% in patients with ACLF grade 1, 31% in patients with ACLF grade 2 and 74% in patients with ACLF grade 3; see Box 2 for details)8.
In the United States, a study using the North-American Consortium for the Study of End Stage Liver Disease (NACSELD) criteria (Box 1) reported that the 30-day mortality rate associated with infected decompensated cirrhosis without ACLF was 8% and this rate increased to 27% in patients with one, 49% in patients with two, 64% in patients with three and 77% in patients with four organ failures13. In addition, in Asia, no significant reduction in mortality in patients with ACLF has been observed over the past two decades, with mortality in the nationwide sample approaching 50%36.
In China, the average 28-day transplant-free mortality reported by Li et al.24 in patients with decompensated cirrhosis due to chronic HBV infection was 2.6% in patients without ACLF and 44% in patients with ACLF. This study used the EASL-CLIF Consortium definition of ACLF and found mortality rates of 23.6% in patients with ACLF grade 1, 40.8% in patients with ACLF grade 2 and 60.2% in patients with ACLF grade 3 (Ref. 24). Zhang et al.23 reported similar findings in Chinese patients with decompensated cirrhosis of different aetiologies. The 90-day mortality rate in patients without ACLF was 2.1% and in patients with ACLF grade 1 was 39.9%, ACLF grade 2 was 54.1% and ACLF grade 3 was 84.7% (using the EASL-CLIF Consortium definition)23.
Precipitating events
Precipitating events of ACLF vary according to geographical areas and can be classified as hepatic or extrahepatic depending on their site of origin14,34,37–39 (Fig. 1). Reactivation of chronic HBV, acute hepatitis A virus or hepatitis E virus infection38, acute alcoholic hepatitis and acute bacterial infection are the most frequent precipitating events of ACLF in Asia23. In the west, the most common precipitating events are active alcoholism and bacterial infections, although in a considerable proportion of patients there is no recognizable precipitating event8. The potential role of drug-induced liver injury (DILI) as a precipitating event in ACLF has been insufficiently explored in both the east and the west.
Organ failures
In the CANONIC study8, among the different organ and system failures in ACLF, the most frequently affected organs or systems were the kidneys (55.8% of patients), followed by the liver (43.6% of patients), coagulation (27.7% of patients), the brain (24.1% of patients), circulation (16.8% of patients) and the lungs (9.2% of patients). At first glance, it might be surprising that not all patients with ACLF had liver failure, but there are two important issues that should be taken into account. First, the level of bilirubin used to define liver failure was very high (≥12 mg per dl) and most (if not all) patients without liver failure also had abnormal bilirubin values, which implies a variable degree of impairment of liver function in these patients. Second, it is important to note that the definition of ACLF goes beyond the classic concept of decompensation of cirrhosis and includes the consequences of cirrhosis on the function of other organs7.
Mechanisms/pathophysiology
ACLF during the course of cirrhosis
As indicated, cirrhosis is a progressive disease that inevitably leads to death unless the aetiological mechanism is suppressed by appropriate treatment or a liver transplantation is performed. Indeed, there is good evidence that discontinuation of alcohol ingestion in alcoholic cirrhosis, antiviral treatment in chronic HBV-related and hepatitis C virus-related cirrhosis and immunosuppressive therapy in autoimmune cirrhosis may transform decompensated cirrhosis to compensated cirrhosis or even to precirrhotic phases5. By contrast, if the aetiological mechanisms persist in patients with compensated cirrhosis, hepatic fibrosis increases progressively as a consequence of continuous liver cell necrosis and inflammation, giving rise to progressive distortion of the liver architecture, reduction in liver parenchyma cells, increase in the intrahepatic resistance to the portal venous flow, portal hypertension, liver insufficiency and acute decompensation of the disease (Fig. 1).
The development of complications, mainly ascites and, less frequently, variceal haemorrhage or hepatic encephalopathy, marks the onset of decompensated cirrhosis. Decompensated cirrhosis is characterized by impairment in the function of the liver and extrahepatic organs and systems, including: the brain (disturbances affecting cognitive, psychiatric and motor functions ranging from subclinical alterations to severe stupor and coma); the kidneys (impairment in renal sodium and free water excretion, intrarenal haemodynamics, renal perfusion and glomerular filtration rate); circulation (splanchnic arterial vasodilation leading to reduction in systemic vascular resistances and high cardiac output); the lungs (impairment in the ventilation/perfusion ratio leading to hypoxia and hypocapnia); the heart (impairment in chronotropic and left ventricular systolic and diastolic functions); coagulation (as a result of impairment in the hepatic synthesis of coagulant and anticoagulant factors and increased fibrinolysis); the adrenal glands (impaired ability to provide adequate cortisol release in response to stress); the intestines (reduced motility, bacterial overgrowth and increased permeability of the mucosal barrier leading to increased translocation of bacteria and/or bacterial products from the intestinal lumen to the systemic circulation); the immune system (systemic inflammation and impaired function of polymorphonuclear leukocytes and monocytes); the thyroid glands (impaired hormonal secretion); and muscles (sarcopaenia) (Fig. 1).
ACLF may develop at any phase of the disease from compensated to early or late decompensated cirrhosis (Fig. 1). Thus, it is not a terminal event of a long-standing decompensated cirrhosis. As indicated above (Box 2), organ failure is defined by an intense impairment in the function of six specific organs or systems that are important in determining prognosis (the liver, the kidneys and the brain and the coagulation, circulatory and respiratory systems)8. Organ failure is the feature that differentiates ACLF from decompensated cirrhosis without ACLF. By contrast, organ dysfunction, which defines a less severe impairment in the function of these (and other) organs and systems, is the differential feature of decompensated cirrhosis versus compensated cirrhosis. For instance, according to the CANONIC study8, brain failure is defined by a hepatic encephalopathy grade 3 or grade 4 of the West Haven classification, whereas brain dysfunction is defined by a hepatic encephalopathy grade 1 or grade 2. Similarly, renal dysfunction is defined by a serum creatinine level of 1.5–1.9 mg per dl, whereas renal failure is defined be a serum creatinine level of ≥2 mg per dl.
Inflammation in ACLF
ACLF is associated with features of systemic inflammation. For example, white blood cell count and plasma levels of C-reactive protein and pro-inflammatory cytokines and chemokines, such as IL-6, IL-1β and IL-8, are higher in patients with ACLF than in patients with cirrhosis without ACLF8,22. Moreover, among patients with ACLF, the higher the ACLF severity, as estimated by the number of organ failures, the higher the plasma pro-inflammatory cytokine or chemokine levels (R.M. and the CANONIC trialists, unpublished observations). The excessive systemic production of pro-inflammatory cytokines and chemokines — or the ‘cytokine storm’ — by the patient's immune system might cause collateral tissue damage40, a process termed immunopathology41. As such, a cytokine storm might also be a prominent contributor to the development of organ failures in patients with cirrhosis. Of note, in patients with ACLF, a subset of CD14+ monocytes show overexpression of the tyrosine-protein kinase MER (encoded by MERTK), which results in the inhibition of the production of inflammatory cytokines by these cells22, suggesting that a form of compensatory immunosuppression develops in parallel to the systemic inflammatory response.
There are two categories of ACLF: those in which the inducer (or inducers) of inflammation (for example, bacterial infection or excessive alcohol intake) are identified and those in which there is no clinically identifiable trigger (or triggers)8. In this Primer, the latter category is called ‘ACLF with no clinically identifiable trigger’. Inducers of inflammation are either exogenous or endogenous42. Among exogenous inducers, we discuss only bacterial inducers because the others are beyond the scope of this Primer and have been described elsewhere42. Although much of the molecular detail of how inflammation triggers ACLF remains to be elucidated, it is likely that the following general processes play a key part. ‘Bacterial inducers of inflammation’ and ‘endogenous inducers of inflammation’ are potential mechanisms of inflammation in ACLF.
Bacterial inducers of inflammation. Bacterial pathogens can induce inflammation through two distinct classes of molecules: pathogen-associated molecular patterns (PAMPs)42–44 and virulence factors42,45. PAMPs are recognized by the host via dedicated receptors called pattern recognition receptors (PRRs) and examples of PRRs for bacterial ligands are detailed in Fig. 3a42–44. The engagement of PRRs results in the stimulation of signalling cascades that activate transcription factors43. PRR-activated transcription factors can induce an array of genes that encode molecules involved in inflammation, including pro-inflammatory cytokines43,45,46 (Fig. 3b).

a | Examples of pattern recognition receptors (PRRs) that detect unique molecular structures of bacterial pathogen-associated molecular patterns (PAMPs) and their subcellular localizations. PRRs for bacterial ligands include Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene 1 (RIG-I; a member of the RIG-I-like receptor family) and cytosolic DNA sensors (including interferon-γ (IFNγ)-inducible protein 16 (IFI16), absent in melanoma 2 (AIM2) and cyclic GMP-AMP synthase (cGAS))43–48,167. b | An example of PRR-mediated inflammation is the activation of inflammatory signalling pathways by extracellular and intracellular lipopolysaccharide (LPS). Detection of extracellular LPS by TLR4 engages two intracellular signalling conduits: the myeloid differentiation primary response protein 88 (MYD88) pathway (grey), involving the adaptor Toll–IL-1 receptor (TIR) domain-containing adaptor protein (TIRAP); and the TIR domain-containing adaptor protein-inducing IFNβ (TRIF) pathway (light green), involving the adaptor TIR domain-containing adaptor molecule 2 (TICAM2)43. The MYD88 pathway via tumour necrosis factor (TNF) receptor-associated factor 6 (TRAF6) stimulates different kinases (dark green), including inhibitor of nuclear factor-κB kinase (IKK) and mitogen-activated protein kinases (MAPKs), that activate master transcription factors (orange), NF-κB and activator protein 1 (AP-1), respectively. The MYD88 pathway also activates the transcription factor IFN regulatory factor 5 (IRF5). These activated transcription factors contribute to the induction of inflammatory genes (red), such as pro-inflammatory genes including TNF and IL6 and anti-inflammatory genes such as IL10 and IL1RN. The TRIF pathway involves TRAF3 activation of the transcription factor IRF3, which then contributes with other transcription factors to the induction of type I IFNs43. Intracellular LPS is recognized by the inflammatory caspases (caspase 4 and caspase 5 in humans and caspase 11 in mice) that stimulate the non-canonical NOD-, LRR- and pyrin domain-containing 3 (NLRP3) inflammasome167,168. This results in the activation of caspase 1 (not shown), which promotes cleavage of IL-1β and IL-18 (Refs 167,168). Activation of caspase 4 and caspase 5 by intracellular LPS can trigger a programmed cell death called pyroptosis169. dsDNA, double-stranded DNA; NAIP5, neuronal apoptosis inhibitory protein 5; ssRNA, single-stranded RNA.
The second class of bacterial inducers of inflammation includes a large number of virulence factors42,44. Unlike PAMPs, most of these factors are generally not recognized by dedicated receptors but can be sensed by the effects of their activity (a process called functional feature recognition)38,46–48.
Endogenous inducers of inflammation. Endogenous inducers are released by necrotic cells or produced by extracellular matrix (ECM) breakdown in an injured tissue (such as the diseased liver in the case of ACLF)42,43 and are called damage-associated molecular patterns (DAMPs)49. DAMPs can be recognized by certain receptors of the host, with this recognition resulting in ‘sterile’ inflammation. For example, high mobility group box 1 protein (HMGB1) engages the advanced glycosylation end product-specific receptor (RAGE), which cooperates with Toll-like receptors (TLRs; a class of PRRs) to induce an inflammatory response42,43,49. Additional factors that might also be involved in ACLF include necrotic cells, which may release members of the IL-1 family such as IL-1α and IL-33 that trigger inflammation through their respective myeloid differentiation primary response protein 88 (MYD88)-coupled cognate receptors50.
Outcomes of the inflammatory response. The purpose of the inflammatory response to bacterial infection is to promote host resistance by reducing bacterial burden, whereas that of sterile inflammation is to promote tissue repair51–54. However, when these two categories of inflammatory responses are excessive, they may induce tissue damage52. During bacterial infection, the acute phase of the inflammatory response can be excessive and can cause immunopathology. For example, effectors of the immune response, such as recruited neutrophils and inflammatory monocytes, activated T helper 1 (TH1) and TH17 cells, and cytotoxic T cells, are known to be associated with a high risk of immunopathology44. There are also some examples of DAMP-induced excessive inflammatory response causing major tissue damage. Mice deficient for receptor-interacting serine/threonine kinase 1 (Ripk1) develop RIPK3–mixed lineage kinase domain-like protein (MLKL)-mediated necroptosis resulting in systemic inflammation, multiple organ injury and death within 3 days of birth50. In this model, IL-33 (a DAMP) drives systemic inflammation and severity. Therefore, the initial tissue injury caused by necroptosis may result in further tissue damage. In the context of severe bacterial infection, cell necrosis can occur (as a feature of immunopathology) and can result in DAMP release. In this case, released DAMPs can perpetuate or accentuate inflammation originally triggered by bacterial inducers (PAMPs and virulence factors)51.
ACLF with identified inducers of inflammation
The relative contribution of these inflammatory processes to ACLF probably differs depending on the trigger, and considerable research is still needed to fully elucidate the aetiological pathways of this syndrome. Of all the recognized precipitating events in ACLF, the mechanisms underlying two — sepsis and severe alcoholic hepatitis — are the best characterized and detailed below.
Sepsis-induced ACLF. Organ dysfunction caused by a dysfunctional host immune response to bacterial infection defines sepsis-induced ACLF. 30% of patients with cirrhosis and ACLF have bacterial sepsis as an identifiable trigger of the syndrome8. However, ACLF can also predispose to bacterial infection; indeed, a proportion of patients with ACLF develop bacterial infection during the course of the syndrome8. Among bacterial infections, spontaneous bacterial peritonitis (SBP), sepsis and pneumonia were more frequently associated with ACLF than other infections in the CANONIC study8. In patients with cirrhosis and ascites, viable intestinal bacteria can cross the intestinal barrier and migrate to the general circulation and colonize the ascitic fluid55,56.
During the first hours of bacterial infection, patients with cirrhosis have higher plasma levels of pro-inflammatory cytokines than patients without cirrhosis. This finding suggests the existence of excessive inflammation in cirrhosis57,58. The mechanisms that underlie this excessive inflammatory response to bacterial infection are incompletely understood59. In fact, most of our knowledge is based on experiments investigating the innate immune response to lipopolysaccharide (LPS), a PAMP recognized by TLR4 (Refs 59–61) (Fig. 3). The response to LPS has been studied in ex vivo studies carried out in freshly isolated monocytes or peripheral blood mononuclear cells (PBMCs) from patients with and without cirrhosis. LPS-stimulated production of pro-inflammatory cytokines and chemokines is higher in cells from patients with cirrhosis than in control cells62–66. The mechanisms of the LPS-induced cytokine storm associated with cirrhosis are poorly understood. Ex vivo experiments have shown that PBMCs or monocytes from patients with cirrhosis show defects in the following negative-feedback mechanisms of TLR4 signalling: the activation of the phosphoinositide 3-kinase (PI3K)–AKT pathway61,65; inhibition of glycogen synthase kinase 3 activity66; and the induction of IL-1 receptor-associated kinase M (IRAKM; also known as IRKA3)62 and of the anti-inflammatory cytokine IL-10 (Refs 61,65). Nevertheless, several other crucial mechanisms known to downregulate the TLR-mediated inflammatory response under non-cirrhotic conditions (in particular, the induction of tumour necrosis factor-α-induced protein 3 (A20; also known as TNFAIP3)) have not yet been investigated in the context of cirrhosis.
Following in vivo LPS challenge, plasma tumour necrosis factor (TNF) levels are significantly higher in cirrhotic than in non-cirrhotic animals67–71. Moreover, in this setting, animals with, but not without, cirrhosis develop hepatocyte apoptosis and necrosis70. In addition, compared with normal livers, in cirrhotic livers, LPS elicits prolonged endoplasmic reticulum stress and a subsequent unfolded protein response that is responsible for sustained phosphorylation of eukaryotic translation initiation factor 2 subunit-α (eIF2α)70. eIF2α phosphorylation is known to attenuate the translation of most RNAs72. In this context, hepatocyte TNF-mediated cell death might occur in cirrhotic livers because of the lack of translation of nuclear factor-κB (NF-κB)-dependent survival mRNAs into proteins. In support of this hypothesis, normal hepatocytes exposed to high levels of TNF are protected against cell death because of the induction of NF-κB-dependent prosurvival proteins73. Together, these findings led to the theory that, in cirrhosis, LPS recognition might result in severe liver damage that is due not only to an excessive innate immune response but also to the impairment of mechanisms involved in hepatocyte endoplasmic reticulum homeostasis.
Future studies should investigate the inflammatory response and tissue damage induced by the recognition of PAMPs other than LPS. It should also be noted that the role of inducers of inflammation, other than PAMPs, such as virulence factors and DAMPs, have not yet been studied in the context of sepsis-induced ACLF.
Severe alcoholic hepatitis. Results of the CANONIC study8 suggest that 20% of cases of ACLF are caused by severe alcoholic hepatitis. In alcoholic hepatitis, the liver shows features of cell death and inflammation74,75. However, the underlying mechanisms that explain these features are still poorly understood75 and most of the following mechanisms require confirmation.
Excessive alcohol consumption alters the gut microbiota and increases intestinal permeability75. In addition, chronic and excessive systemic inflammation causes damage to the intestinal barrier. These alterations might favour the translocation of bacteria into the bloodstream76–78 (Fig. 4). Regardless of whether these bacteria cause infection, they release PAMPs (such as LPS) that can reach the liver where they are recognized by TLRs expressed in resident macrophages (called Kupffer cells). This recognition stimulates the production of pro-inflammatory CXC chemokines, such as IL-8 (Ref. 79), that attract and activate neutrophils80. Neutrophil infiltration is a hallmark of alcoholic hepatitis75. Hepatocyte necrosis, which has been documented in severe alcoholic hepatitis81, might result in the release of DAMPs that would be recognized by different receptors mediating an inflammatory response, as described above.

Cirrhosis is associated with quantitative differences (bacterial overgrowth) and compositional changes of the gut microbiota, so called dysbiosis. Several factors might contribute to dysbiosis of the gut microbiota during cirrhosis including diet, use of antibiotics, decreased bile flow and intestinal motility, changes in gastric pH and impaired mucosal immunity. A second important feature of patients with cirrhosis is the translocation of bacteria. Disruption of tight junctions allows pathogen-associated molecular patterns (PAMPs) and possibly other microbial metabolites to use the paracellular route between adjacent intestinal epithelial cells for translocation. Intestinal permeability is already increased in precirrhotic stages, whereas translocation of viable bacteria is a characteristic of cirrhosis, particularly during decompensation. Bacteria probably use the transcellular route (transcytosis) through epithelial cells. PAMPs might also activate immune cells, including monocytes, macrophages and T cells, in the lamina propria of the intestines, leading to secretion of inflammatory mediators. Cytokines, such as tumour necrosis factor (TNF), IL-6, IL-17, nitric oxide (NO) and interferon-γ (IFNγ), are increased in the intestines of patients or animal models with cirrhosis76–78. Several of these mediators are known to contribute to a dysfunction of tight junctions. Conversely, the intestinal immune surveillance response might be impaired to remove translocated bacteria in the lamina propria.
Mitochondrial DNA (mtDNA) is a type of DAMP, and mtDNA stress might also contribute to inflammation in the context of alcoholic hepatitis. Acetaldehyde metabolism results in hepatocyte reactive oxygen species (ROS) production68. ROS production is also stimulated by TNF65. In the context of chronic alcohol consumption82 or after LPS challenge83, ROS overproduction induces mtDNA stress. In a mouse model of moderate mtDNA stress, mtDNA was shown to escape to the cytosol where it engaged a cell-intrinsic response involving the innate cytosolic DNA sensor cyclic GMP-AMP synthase (cGAS) (Fig. 3a). cGAS engagement with mtDNA, in turn, mediates type I interferon (IFN) production and subsequent autocrine and paracrine induction of IFN target genes84. Thus, a cell-intrinsic response to mtDNA stress might become an inflammatory response at the tissue level and thereby might contribute to liver failure.
Recent results suggest that the inhibition of liver regeneration might be involved in liver failure associated with severe alcoholic hepatitis85. Although hepatic progenitor cells are activated in livers with severe alcoholic hepatitis, these cells are committed to differentiate into cholangiocytes (epithelial cells lining the bile duct) instead of hepatocytes85. Thus, it is possible that no replacement of hepatocytes that die as a result of alcoholic hepatitis occurs. Together, these findings suggest that severe alcoholic hepatitis might be caused by both immunopathology and impaired hepatocyte regeneration.
ACLF with no identifiable trigger
The trigger of ACLF is unknown in approximately 40% of cases8. Although these patients show features of systemic inflammation8, one cannot clearly explain how the systemic inflammation is stimulated. Three hypotheses might explain the mechanisms that underlie inflammation in ACLF with no clinically identifiable trigger.
The first hypothesis is based on the existence of dysbiosis of the gut microbiota in patients with cirrhosis (Fig. 4). Dysbiosis associated with cirrhosis is typically characterized by a decrease in diversity, a decrease in Lachnospiraceae, Ruminococcaceae, Bacteroidaceae and Family XIV incertae sedis at the family level and a decrease in Bacteroides spp. at the genus level6,86–89. In addition, dysbiosis associated with cirrhosis involves an increase in Gram-negative Enterobacteriaceae, Fusobacteriaceae and Porphyromonadaceae and in Gram-positive Streptococcaceae at the family level6,86–89. Decompensation of cirrhosis results in additional distinct compositional changes of the microbiota compared with compensated stages of liver disease87. There is a robust positive correlation between the abundance of certain bacterial family members and plasma levels of inflammatory cytokines (including IL-6 and TNF in patients with ACLF)90. These findings suggest that metabolites produced by gut microbiota might contribute to systemic inflammation (Fig. 4).
The second hypothesis is that some patients might have intestinal translocation of PAMPs, such as LPS or bacterial CpG DNA91 (Fig. 3a). These ligands might reach the liver and systemic circulation and then be recognized by TLRs. Thus, TLR recognition is generally not dependent on microbial viability or invasiveness. During the peak phase of ACLF, systemic levels of LPS are higher than before the onset of ACLF and during remission of survivors87,91, suggesting that higher systemic LPS levels correlate with disease severity. Interestingly, small intestinal bacterial overgrowth is a risk factor for increased systemic LPS levels in patients with cirrhosis92. Increased systemic levels of CpG DNA are found in patients with decompensated cirrhosis and without overt bacterial infection, and correlate with mortality93, suggesting that increased levels of CpG DNA might be involved in the development of ACLF in patients with acutely decompensated cirrhosis.
The third mechanism explaining inflammation in ACLF with no clinically identifiable trigger might be the release of DAMPs, for example, by necrotic hepatocytes. In patients and animals with acute liver failure, various DAMPs, such as HMGB1, that might contribute to inflammation are released83, but nothing is known about DAMPs in patients with ACLF.
Finally, another possible explanation for a failure to identify a precipitating event in ACLF could relate to a failure of current diagnostic tests or the testing protocol to identify infection or DILI.
Diagnosis, screening and prevention
Defining organ failure and ACLF
Diagnostic criteria of organ failure. One of the assumptions made to define the EASL-CLIF criteria is that extrahepatic organ failure (or failures) is a major differential feature of ACLF. The CLIF-Sequential Organ Failure Assessment (CLIF-SOFA) score was the original scale used to define organ failure in the CANONIC study8. It was derived from the SOFA score, a scale widely used in intensive care16–19, which was then adapted to patients with chronic liver disease on the basis of published studies. Cut-off values were established after assessing the risk increase of 28-day mortality rates in each of the last four CLIF-SOFA score categories compared with that of the previous one in the patients enrolled in the CANONIC study. A simplified version of the CLIF-SOFA score, the CLIF Consortium Organ Failure (CLIF-C OF) score (Table 2) with identical criteria to diagnose organ failure and similar prognostic accuracy, has been developed20.
Mortality criteria. Another predefined criterion for the diagnosis of ACLF in the development of the EASL-CLIF criteria was an expected 28-day mortality rate of ≥15%. In the CANONIC series8, this criterion was present in patients with two or more organ failures, but not in patients with one organ failure (a 28-day mortality rate of 14.6%). Additional risk factors were used to further categorize patients in this low-risk subgroup. This analysis produced subgroups of patients that fulfilled the three predefined criteria for ACLF (Fig. 5): patients with two or more organ failures; patients with one organ failure (specifically kidney failure); and patients with non-renal single organ failure if these failures are associated with renal and/or brain dysfunction.

28-day mortality rates of patients with decompensated cirrhosis with (red bars) and without (green bars) acute-on-chronic liver failure (ACLF) according to the diagnostic criteria proposed in the CANONIC study8. Patients are divided into the following categories: patients with no organ failure (OF); patients with a single non-kidney organ failure without kidney dysfunction (KD; a serum creatinine level of 1.5–1.9 mg per dl) or brain dysfunction (BD; grade 1–2 hepatic encephalopathy); patients with a single kidney failure; patients with a single non-kidney organ failure with kidney dysfunction and/or brain dysfunction; patients with two organ failures; and patients with three or more organ failures. Data from Ref. 8.
Grades of severity of ACLF
Patients with decompensated cirrhosis can be stratified into four groups of severity — no ACLF or ACLF grades 1–3 — on the basis of the type and the number of organ failures they have (Box 2). Kidney failure is the most prevalent organ failure in ACLF grade 1. For ACLF grade 2, liver failure is the most prevalent organ failure followed by kidney, brain and coagulation failure. For ACLF grade 3, the prevalence of all organ failures is high.
In the CANONIC study8, 23% of patients admitted to hospital had ACLF at admission. Furthermore, 11% of patients with no ACLF at admission developed the syndrome during hospitalization, which gives a total prevalence of ACLF of 31%. Among patients with ACLF, 51% had ACLF grade 1, 35% had ACLF grade 2 and 13% had ACLF grade 3. Besides providing the diagnosis of the syndrome, these criteria also provide data for rapid prognostic information, with the ACLF grade associated with different rates of mortality (Table 3). The usefulness of these classification criteria as well as that of CLIF-SOFA and CLIF-C OF scores in assessing prognosis have been validated in independent series of patients26,29–31,94,95.
Natural history of ACLF
ACLF is a syndrome that has potential for reversibility96. However, data from the CANONIC study clearly show that, despite this feature, mortality of patients with ACLF increases cumulatively even after these patients are discharged from the hospital. Specifically, mortality increases from approximately 20% at 28 days to >35% at 90 days in patients with ACLF grade 1 and from 30% to approximately 50% at 90 days in patients with ACLF grade 2 (Ref. 8). Although there is considerable variability between patients20,21, some broad principles regarding the course of the condition can be put forward. In general, at days 3–7 from presentation, approximately 50% patients with ACLF grade 1 will improve to having no ACLF, with a consequent 28-day mortality rate of approximately 7%. In addition, 25% of patients with ACLF grade 1 will remain unchanged (that is, their ACLF grade will not change) with a 28-day mortality rate of 24%. By contrast, approximately 25% of patients with ACLF grade 1 progress to ACLF grade 2 or ACLF grade 3; their 28-day mortality rate is 53% and 88%, respectively. In patients presenting with ACLF grade 2, only 35% improve to having no ACLF or ACLF grade 1 at days 3–7 post presentation. Those patients who do improve have low 28-day mortality rates of approximately 5%. In addition, approximately 50% of patients with ACLF grade 2 deteriorate to ACLF grade 3 (a 28-day mortality rate of 90%) or remain the same (a 28-day mortality rate of 26%). In patients presenting with ACLF grade 3, mortality rates remain very high with only approximately 13% improving to no ACLF or ACLF grade 1. The factors that were independently related to progression to more-advanced grades were the CLIF-C ACLF score (discussed below) and the presence of liver failure. These data indicate that the syndrome is indeed very dynamic and that early intervention is crucial to minimize the risk of death.
Clinical risk factors
There is evidence that the clinical course of ACLF outlined above is true for all aetiologies of ACLF. In a retrospective study from China in patients who predominantly had HBV-related cirrhosis, short-term outcomes, which were the most closely associated with ACLF, were not affected by the nature of the precipitating illness97. Patients with hepatic precipitants, such as reactivation of HBV, had a short-term (28-day and 90-day) mortality similar to patients with an extrahepatic precipitant, such as infection. This pattern was also observed in a second Chinese study24 in patients with cirrhosis due to chronic HBV infection who developed ACLF. The 28-day and 90-day mortality rates for any given grade of ACLF in this study24 were similar to those reported by the CANONIC study8 and were unrelated to the presence and type of precipitating events (Table 4). These studies, therefore, suggest that it is the number of organ failures and not the aetiology of cirrhosis or precipitating events that is the main risk factor of mortality.
A previous history of episodes of acute decompensation was absent in 23% of patients with ACLF in the CANONIC study, indicating that the development of ACLF as the initial manifestation of decompensated cirrhosis is a relatively common feature. These patients without prior history of decompensation were younger, more-frequently alcoholics, had more-severe systemic inflammation and ACLF grade, and had higher short-term mortality (42% versus 30%) than patients with ACLF with prior history of acute decompensation.
Prediction of prognosis
As ACLF is a dynamic syndrome, prognostic scores need to be dynamic so that they can be updated sequentially on a daily basis, which would allow assessment of response to intervention, escalation for the need for urgent liver transplantation and determination of futility of ongoing treatment. The CANONIC study indicates that follow-up data within the first 3–7 days following diagnosis of ACLF are extremely important to predict clinical course since resolution; improvement or worsening of ACLF following standard medical therapy occur within this early time period in most patients96.
A prognostic model was developed and validated for patients with ACLF, referred to as the CLIF-C ACLF score, and for patients with acute decompensation who did not fulfil criteria for the diagnosis of ACLF, which is called the CLIF-C Acute Decompensation (CLIF-C AD; www.clifconsortium.com) score. These two scores were designed because a single score was insufficient to satisfactorily delineate the prognosis associated with acute decompensation and ACLF8.
The CLIF-C ACLF score comprises the CLIF-C OF score, age and white blood cell count20,98. The score is a number from 0 to 100; the higher the number, the greater the risk of death. The score was validated using prospectively collected data from a series of patients not included in the CANONIC study. The CLIF-C ACLF score provided a significantly better estimate of the risk of death at 28 days, 90 days, 6 months and 12 months post-presentation compared with the Model for End-Stage Liver Disease (MELD) score, the MELD-Sodium score and the Child-Pugh score24,32,95,99–101. Compared with the CLIF-C ACLF score, the MELD score underestimated the risk of death of patients by 20–30%, implying that organ allocation for transplantations using the MELD score seriously disadvantages the patient with ACLF20. The performance of the CLIF-C ACLF score improved over the period of follow-up, suggesting that it should be updated daily20.
The CLIF-C AD score was developed in patients with acute decompensation without ACLF98. Variables that were found to be independently associated with survival were age, serum sodium level, serum creatinine level, white blood cell count and international normalized ratio. These generated a score between 0 and 100, which was also significantly more accurate in predicting prognosis than the MELD, MELD-Sodium and Child-Pugh scores24,98. Patients with a CLIF-C AD score of <45 had a 28-day mortality rate of <3% and this category might identify a group of patients who could be discharged early from the hospital. Conversely, patients with a CLIF-C AD score of >60 were at high risk of progression to full-blown ACLF and had a 28-day mortality rate of approximately 20%, indicating that this is probably a ‘pre-ACLF’ group. The CLIF-C AD score was also validated for sequential use.
Prevention
Early diagnosis and treatment of potential precipitating events are essential in the prevention of ACLF, and several preventive measures have been shown to be effective5,55,102–105. These all involve treating infections before they can go on to trigger ACLF and include: prompt administration of antibiotics tailored to the local epidemiological pattern of resistance in patients with suspected infections; long-term suppression of HBV infection or sustained eradication of hepatitis C virus infection in patients with compensated or decompensated cirrhosis; and intravenous administration of albumin at infection diagnosis in patients with SBP. In patients with SBP, albumin is highly effective in preventing the development of type 1 hepatorenal syndrome (HRS), which is a special form of ACLF characterized by rapidly progressive renal failure. This effect is probably as a consequence of plasma volume expansion and the modulatory effect of albumin on the systemic inflammation associated with PAMPs (such as LPS)104,105. There is no evidence that intravenous albumin is effective in other bacterial infections106,107.
There is also indirect evidence in support of other potential preventive measures for ACLF. For example, long-term oral norfloxacin administration reduces the rate of SBP (and of other bacterial infections) and type 1 HRS in patients with decompensated cirrhosis55,102,108–110. Norfloxacin acts by selectively reducing the Gram-negative microbiota, decreasing the permeability of the gut barrier via stimulation of IL-10 release and modulating the immune response to bacterial translocation111–113. In addition, treatment of patients with severe acute alcoholic hepatitis with pentoxifylline, an inhibitor of macrophage production of TNF, or with the combination of prednisolone and intravenous N-acetylcysteine has been shown to reduce the incidence of type 1 HRS in some studies114,115, presumably by modulating hepatic inflammation, but this has not been confirmed in a recent investigation116. Finally, short-term administration of the combination of granulocyte colony-stimulating factor (G-CSF) plus darbepoetin (a synthetic analogue of erythropoietin) has been shown to improve liver function, to reduce the incidence of severe sepsis and to increase 1-year survival in comparison to placebo in patients with decompensated cirrhosis117.
Management
Medical management
Medical management of ACLF consists of early recognition, treatment of the precipitating event and supportive care8,14,118,119. Early treatment of the trigger is proven to reduce mortality, for example, in treatment of reactivated HBV infection with tenofovir or alcoholic hepatitis with steroids75,117,119–121. However, most of ACLF management is focused on supportive care118.
Antibacterial therapy. As discussed above, bacterial infections are the precipitating event of ACLF in approximately 35% of patients14 (Table 4). As such, there should be a low threshold for early initiation of antibiotics in patients with cirrhosis who have a bacterial infection. In patients with septic shock, every hour delay beyond presentation is associated with an adjusted odds ratio of overall death of 1.1 (Refs 122,123). Broad spectrum antibiotics should be used, particularly in patients with nosocomial or health care-associated infections or in those with septic shock, as inappropriate initial antimicrobial therapy increases the adjusted odds ratio of death by tenfold122,123. Equally important to early initiation of antibiotics is prompt de-escalation of antibiotics once an organism is identified and/or the patient shows clinical improvement. If no organism is identified and there is persistent clinical deterioration in the setting of broad spectrum antibiotics, antifungals should be considered123. Measures to prevent superinfections (secondary infections that occur on top of a primary infection) should be implemented in patients with ACLF, including bundles of prevention and control of ventilator-associated pneumonia, catheter-related bacteraemia and urinary tract infections, hand hygiene, barrier precautions and avoiding unnecessary instrumentation124.
Infected and non-infected patients admitted with ACLF are highly predisposed to developing new bacterial infections during hospitalization (R.M. and the CANONIC trialists, unpublished observations). These infections act as a ‘second hit’ of the syndrome. Thus, prevention, early diagnosis and treatment of these secondary infections are major issues in ACLF.
HBV-specific therapy. Reactivation of HBV is a frequent precipitating event of ACLF in patients with cirrhosis in Asia. Antiviral treatment in patients with hepatitis B-related ACLF improves liver function and increases short-term and long-term survival119–121,125,126. Thus, early treatment with antiviral agents (such as lamivudine, tenofovir, entecavir or telbuvidine) should be started as soon as possible121,127.
Immunomodulation. Patients with ACLF might benefit from treatments that aim to restore immune function, such as albumin, N-acetylcysteine and G-CSF117,128. Indeed, results from a recent randomized controlled trial suggest that the administration of G-CSF prevents the development of sepsis and improves short-term survival in patients without severe forms of ACLF, who did not have sepsis, brain failure or multiple organ failure128. G-CSF is thought to act by mobilizing stem cells from the bone marrow to the periphery, including the liver, thus improving liver regeneration.
Renal dysfunction and failure. Acute kidney injury (AKI) is the most frequent organ failure in ACLF8,129. Common causes of AKI include pre-renal, intrinsic causes and HRS129. Management of AKI differs depending on the underlying aetiology, and urinary biomarkers are helpful in identifying the cause of AKI129,130. Volume resuscitation with crystalloids and/or albumin should be used in patients with pre-renal AKI (that is, impairment in renal function related to hypovolaemia caused by excessive diuretic treatment). Terlipressin or noradrenaline are the first-choice treatment for HRS combined with volume expansion with albumin129. Terlipressin or noradrenaline are given to reduce the splanchnic arterial vasodilation causing systemic circulatory dysfunction and renal vasoconstriction in HRS. The effect of albumin was initially thought to be due to plasma volume expansion. However, a potential effect of albumin in modulating the systemic inflammation of patients with ACLF has recently been proposed7,105. Renal replacement therapy is used as a bridge to liver transplantation or liver–kidney transplantation in patients with severe AKI, although the dose and timing of dialysis has not been fully studied129.
Cardiovascular failure. As in the management of sepsis, aggressive volume resuscitation and the initiation of vasoconstrictor agents (that is, noradrenaline) to maintain an adequate blood pressure for organ perfusion is crucial to counter the vasodilatory state that occurs with ACLF14,131. The goal mean arterial pressure is >60 mmHg and careful attention should be made to volume administration with crystalloids given the predisposition of volume overload in patients with cirrhosis118. Colloids, including albumin, may also trigger volume overload. Terlipressin or vasopressin can be used as an adjunctive agent. There is also growing evidence that adrenal insufficiency in ACLF can further compromise haemodynamics132. Although still controversial, evaluation for adrenal insufficiency can be done by measuring random cortisol levels in the morning. If these levels are indeterminate, adrenal insufficiency can be confirmed with an adrenocorticotropic hormone (ACTH) test (also called the cosyntropin, tetracosactide or Synacthen test), which estimates the response of the adrenal glands to stress.
Brain dysfunction and failure. Treatment of encephalopathy with tap water enemas, lactulose and oral non-absorbable antibiotics (such as rifaximin and neomycin), by improving the level of consciousness, can prevent bronchial aspiration, aspiration pneumonias and respiratory failure14. It is important to titrate lactulose dose and enemas appropriately to prevent hypovolaemia that results from diarrhoea and to prevent hypernatraemia (a rise in serum sodium levels) that results from lactulose. Goal stool output in a day should be 3–4 bowel movements133. Lower stool output is insufficient to reduce the intestinal production of ammonia and to increase ammonia clearance from blood. Higher stool output may induce hypernatraemic dehydration. Patients with grade III–IV encephalopathy should be intubated, as they have a high risk of experiencing bronchial aspiration (of saliva or gastric fluid). Intracranial pressure monitoring and the use of mannitol is not recommended in these patients as cerebral oedema and intracranial hypertension are exceptional in patients with hepatic encephalopathy associated with ACLF12.
Coagulopathy. Coagulopathy in patients with ACLF is often difficult to manage in the setting of fluctuations between a prothrombotic and an ineffective haemostatic state134,135. In the setting of active bleeding and severe coagulopathy, transfusion of platelets, cryoprecipitate (a frozen blood product prepared from plasma used to increase fibrinogen levels) and blood should be considered. However, patients should not be prophylactically transfused with plasma for an increased international normalized ratio. Patients with portal vein thrombosis may require anticoagulant therapy to prevent recurrent variceal bleeding.
Intensive care and liver support devices
Admission to critical care units is mandatory in cases of vascular, respiratory or brain failure and is recommended in those with renal failure. Patients with liver and coagulation failure can still be treated in regular wards, but require strict clinical monitoring. In the CANONIC study8, 50% of the patients with ACLF were admitted to the intensive care unit (86% of those with ACLF grade 3). Owing to the high mortality rate in patients with ACLF, treatments that are able to bridge the time between admission of patients with severe disease (ACLF grade 2 or grade 3 at 3–7 days following admission) to liver transplantation are clearly needed.
Extracorporeal liver support systems are potential treatments for ACLF136,137. Bioartificial liver support systems use hollow-fibre bioreactors containing hepatic cells to support the metabolic and synthetic function of the diseased liver. Currently, only tumour hepatocyte or porcine hepatocyte lines (the Vital Therapies ELAD and the Alliqua HepatAssist 2000 systems) are available. A recent randomized trial comparing ELAD versus standard medical treatment did not find any significant effect on survival136. Non-biological systems consist of albumin dialysis techniques and are based on the capacity of this molecule to remove water-insoluble substances and pro-inflammatory molecules (such as PAMPs and ROS) retained in plasma as a consequence of liver failure and systemic inflammation105. Moreover, the physiology of albumin function is markedly impaired in patients with decompensated cirrhosis because of severe oxidation of the molecule by endogenous ROS, alterations in its molecular structure and saturation of other binding sites by water-insoluble substances produced and/or retained as a consequence of liver failure (such as bilirubin, bile salts and drugs). Three different albumin dialysis systems are currently available: the Gambro molecular adsorbent and recirculating system (MARS), the Prometheus fractionated plasma separation and absorption (FPSA) system and the Fresenius Medical Care single-pass albumin dialysis (SPAD). MARS, the system that is most extensively evaluated in ACLF, improves systemic haemodynamics and severe hepatic encephalopathy136,137. However, two large randomized, multicentre studies failed to show an improvement in survival using MARS and FPSA138,139. Finally, an artificial liver device (the University College London-Liver Dialysis Device) that aims to remove and replace the dysfunctional albumin of patients with cirrhosis and to reduce circulating endotoxaemia is currently under evaluation140. Plasma exchange, a detoxification system that increases survival in patients with acute liver failure141, improves hepatic encephalopathy and liver function in non-randomized studies in patients with ACLF.
Liver transplantation
Liver transplantation represents the only definitive therapeutic option for patients with ACLF. However, very few studies have assessed its feasibility, selection criteria (indications and contraindications), timing and efficacy96,142–148. In contrast to patients with acute liver failure, patients with ACLF cannot currently be included in the high-urgency transplantation list. Moreover, as the clinical course of ACLF evolves rapidly, the time frame for evaluation and listing is frequently very short. Advanced age, active alcoholism, uncontrolled infections and multiple organ failure are the main reasons for contraindication to transplantation or delisting. There is general agreement in considering that transplantation must be avoided in patients with severe circulatory or respiratory failure and ongoing sepsis. By contrast, for the majority of experienced centres, organ support (renal replacement therapy and mechanical ventilation) does not contraindicate transplantation in ACLF. Current data indicate that less than half of patients with ACLF are listed and that the procedure is feasible in only 10–25% of patients, as >50–70% of the listed patients die on the waiting list147. A recent study in the United States showed that patients with cirrhosis, ACLF and a high MELD score (>40) have higher waiting-list mortality (almost twofold higher) than status 1A candidates (that is, patients with acute liver failure)145. Defining criteria to select and prioritize patients with ACLF on the waiting list will help to improve outcomes by providing timely liver transplantation. Several studies show that both living donor liver transplantation and deceased donor transplantation offer similar results in this setting146–148. The reported outcome of patients transplanted for ACLF is good (Fig. 6), ranging between 74% and 90% at 5 years, a number similar to that observed in patients transplanted for other indications96,144–148.

Survival probability curves of patients with grade 2 or grade 3 acute-on-chronic liver failure (ACLF) at days 3–7 following diagnosis who were submitted to early (<28 days) liver transplantation (red curve; n = 21) and patients with the same ACLF grades who did not receive a liver transplant (green curve; n = 120)96. Percentages over the curves show the 28-day, 90-day and 6-month probability of survival. Adapted with permission from Ref. 96, John Wiley and Sons.
Management algorithm
The three new scoring systems derived from the CANONIC study — the CLIF-C OF score (or CLIF-SOFA score), the CLIF-C ACLF score and the CLIF-C AD score — can be used to risk-stratify patients with cirrhosis and acute decompensation, to indicate early liver transplantation and to assess intensive care unit treatment futility20,98 (Fig. 7). The prognosis in ACLF depends not only on the number of organ failures or the CLIF-C ACLF score at diagnosis but also on the early response to treatment96. As nearly 20% of patients with ACLF grade 3 improve after treatment, patients with three or more organ failures should be admitted to the intensive care unit and should receive unrestricted organ support for a short period of time (3–7 days). The persistence of three or more organ failures after this intervention may lead to the need to consider a limitation in life-sustaining treatments as a fatal outcome is almost invariable in the absence of ‘salvage’ liver transplantation19. A high CLIF-C ACLF score (>64 points) after initial intervention (at 3–7 days) has also been suggested as a potential futility rule in patients without possibilities of early liver transplantation96. However, these criteria require further validation. Patients who are potential candidates for early liver transplantation, including living donor liver transplantation, must not be limited in their treatment. In those without options for transplantation, scores and a pragmatic case-by-case evaluation should be used for the decision.

A proposed management strategy for patients with acute-on-chronic liver failure (ACLF) based on mortality rate data from the CANONIC study8. The first step is the assessment of ACLF grade at days 3–7 after initiation of medical management, including organ support. Liver transplantation should be assessed in all patients with ACLF because of high 90-day mortality rates (>20%). Liver transplantation should be performed as early as possible in patients with ACLF grade 2 and grade 3 as they are at considerable risk of short-term (28-day) mortality. In the case of contraindication of liver transplantation, the presence of four or more organ failures (OFs) or a Chronic Liver Failure Consortium (CLIF-C) ACLF score of >64 at days 3–7 after diagnosis could indicate the futility of care. ICU, intensive care unit. Adapted with permission from Ref. 96, John Wiley and Sons.
Regenerative therapy
A few studies have evaluated the effect of G-CSF therapy in small groups of patients with ACLF128,149,150. This cytokine mobilizes bone marrow-derived stem cells, restores neutrophil function and promotes hepatic regeneration. G-CSF administration in non-severe forms of ACLF reduces the risk of developing organ failure (or failures) and sepsis and improves survival. G-CSF therapy seems to be ineffective in patients with sepsis and in those who have more-severe forms of ACLF. Hepatocyte and stem cell transplantation have also been proposed as potential treatments in ACLF151.
Quality of life
Following dismissal from hospital, patients recovering from ACLF may return to functioning in their community, receive a liver transplantation, be sent to intermediate care facilities such as a nursing home or be re-hospitalized. The 30-day hospital readmission rate is approximately 25%152. In the long-term follow-up of 6 months in the NACSELD study153, 27% of patients died, 14% were transplanted and 59% were alive without liver transplantation. After discharge, 45% of patients had subsequent infections. Patients who had repeat infections were older and were more likely to use proton pump inhibitors, rifaximin or prophylactic therapy for SBP with norfloxacin154,155. In these last three circumstances, the predisposition to bacterial infections is probably related to gut dysbiosis or colonization by drug multiresistant bacteria. Of crucial importance, patients with infection-related ACLF were more likely to be delisted for liver transplantation.
Outlook
ACLF definition
The challenge of obtaining a universal definition of ACLF is an important issue. However, differences between the APASL and western definitions are too important to be solved by consensus agreements alone. These definitions differ not only in terms of the characteristics of the patients, diagnostic criteria and clinical course but also, and most importantly, in the conceptual view of the disease. The APASL conference definition postulates that the sequence of events in ACLF starts with a hepatic insult that causes acute liver failure and, as a consequence, extrahepatic organ failure (or failures). By contrast, the western definition relies on the concept that the acute impairment in liver function (which, if intense, is defined as liver failure) develops simultaneously to an impairment in the function of other organs (which, if intense, are also defined as organ failures) as a consequence of intrahepatic or extrahepatic mechanisms. These mechanisms could include intense systemic inflammation related to a massive release of DAMPs from the diseased liver (in the case of acute alcoholic hepatitis, viral hepatitis or DILI) or of PAMPs (by invading bacteria in the case of sepsis or from the intestinal microbiota in patients without clear precipitating events). Such controversy can only be solved by promoting research in this compelling new syndrome.
Clinical challenges
Investigations of ACLF have generally been carried out after the diagnosis of the syndrome. As such, there are few data within the critical period before ACLF development. Prospective observational studies within this period are, therefore, essential, particularly those assessing biomarkers or panels of biomarkers of systemic inflammation that could be of value as predictors of treatment response and survival. Liver pathology in ACLF has also been insufficiently investigated. In patients with cirrhosis due to HBV infection, ACLF occurs in the setting of submassive hepatic necrosis27. In patients with alcoholic cirrhosis and active alcoholism, severe alcoholic hepatitis superimposed on cirrhosis is probably the predominant liver histology. Finally, two recent studies have reported severe ductular bilirubinostasis and cholestasis, a lesion that is also seen in patients with sepsis who do not have cirrhosis, as a specific lesion in ACLF156,157.
Insights into pathophysiology
Sequential studies of the innate and adaptive immune system function before and after ACLF are lacking. Such studies are essential to understand the mechanism of ACLF. Moreover, the immune system function might change during the clinical course of the syndrome. As it occurs in sepsis158, an initial activation of the immune system in ACLF might be followed by a period of immunosuppression, which would favour further bacterial translocation and progression of organ failure (or failures).
The mechanism of organ or system failure in ACLF is of major interest. Renal failure in cirrhosis is considered to be secondary to systemic circulatory dysfunction and impaired renal perfusion. However, recent evidence from studies in sepsis suggest that renal failure might also be a consequence of a direct effect of renal inflammation, which impairs renal microcirculation and cell function159. In fact, there is evidence that inflammation might be involved in the pathogenesis of cardiac dysfunction, encephalopathy, relative adrenal insufficiency and pulmonary dysfunction in cirrhosis7.
A major difficulty for research in ACLF is the lack of appropriate animal models. Carbon tetrachloride-induced cirrhosis in rats is an excellent model of cirrhosis, but animals die prior to the development of extrahepatic organ failure160. Bile duct-ligated rats represent an acute model of liver failure and ascites and, in combination with the acute intraperitoneal administration of LPS, have been used as a model of ACLF161. However, this model differs markedly from cirrhosis in humans and no extrahepatic organ failure has been documented.
Treatment
The recognition of systemic inflammation as the main mechanism of ACLF opens up new fields in the design of new therapeutic procedures. This knowledge will promote the development of new artificial liver support systems capable of removing not only potentially harmful molecules retained as a consequence of organ failure but also pro-inflammatory molecules that cause ACLF. Total plasma exchange141 is clearly an alternative method to remove PAMPs, DAMPs and free radicals.
However, a major issue in the management of ACLF is prevention. There are three potential effective treatments that should be explored. The first consists of the prevention of bacterial translocation by long-term oral administration of poorly absorbable antibiotics107,110,162. Long-term weekly administration of intravenous albumin is the second approach. Preliminary data indicate that this technique prevented bacterial infections, AKI and hepatic encephalopathy and improved survival in a large Italian randomized controlled trial in patients with decompensated cirrhosis163. Last, recent investigations have suggested a central role for defective bile acid receptor (also known as farnesoid X-activated receptor) signalling in hepatic inflammation and intestinal bacterial translocation, factors that are known to shape ACLF164–166. Obeticholic acid is a potent bile acid receptor agonist. Recent studies in animals have demonstrated that obeticholic acid lowers portal hypertension and improves bacterial translocation165,166, suggesting that it might be of potential benefit in patients with ACLF.
References
Ginés, P. et al. Compensated cirrhosis: natural history and prognostic factors. Hepatology 7, 122–128 (1987).
Schrier, R. W. et al. Peripheral arterial vasodilation hypothesis: a proposal for the initiation of renal sodium and water retention in cirrhosis. Hepatology 8, 1151–1157 (1998).
Fraser, C. L. & Arieff, A. I. Hepatic encephalopathy. N. Engl. J. Med. 313, 865–873 (1985).
Polio, J. & Groszmann, R. J. Hemodynamic factors involved in the development and rupture of esophageal varices: a pathophysiologic approach to treatment. Semin. Liver Dis. 6, 318–331 (1986).
Ramachandran, P., Iredale, J. P. & Fallowfield, J. A. Resolution of liver fibrosis: basic mechanisms and clinical relevance. Semin. Liver Dis. 35, 119–131 (2015).
Qin, N. et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 513, 59–64 (2014).
Bernardi, M., Moreau, R., Angeli, P., Schnabl, B. & Arroyo, V. Mechanisms of decompensation and organ failure in cirrhosis: from peripheral arterial vasodilation to systemic inflammation hypothesis. J. Hepatol. 63, 1272–1284 (2015).
Moreau, R. et al. Acute-on-chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology 144, 1426–1437.e9 (2013). This article describes the first prospective investigation assessing the diagnostic criteria (EASL-CLIF Consortium definition of ACLF), prevalence, precipitating events, grading of severity and prognosis of ACLF in a large series of European patients hospitalized with decompensated cirrhosis using a pragmatic approach.
Wlodzimirow, K. A., Eslami, S., Abu-Hanna, A., Nieuwoudt, M. & Chamuleau, R. A. F. M. A systematic review on prognostic indicators of acute on chronic liver failure and their predictive value for mortality. Liver Int. 33, 40–52 (2013).
Jalan, R. & Williams, R. Acute-on-chronic liver failure: pathophysiological basis of therapeutic options. Blood Purif. 20, 252–261 (2002).
Sarin, S. K. et al. Acute-on-chronic liver failure: consensus recommendations of the Asian Pacific Association for the Study of the Liver (APASL). Hepatol. Int. 3, 269–282 (2009). This article describes the results of a consensus conference promoted by the APASL aiming to assess the diagnostic definition of ACLF.
Sarin, S. K. et al. Acute-on-chronic liver failure: consensus recommendations of the Asian Pacific Association for the Study of the Liver (APASL) 2014. Hepatol. Int. 8, 453–471 (2014).
Bajaj, J. S. et al. Survival in infection-related acute-on-chronic liver failure is defined by extra-hepatic organ failures. Hepatology 60, 250–256(2014). This study shows the prevalence of ACLF and associated mortality in a large series of patients with cirrhosis and bacterial infection from the United States.
Jalan, R. et al. Acute-on chronic liver failure. J. Hepatol. 57, 1336–1348 (2012).
Liver Failure and Artificial Liver Group, Chinese Society of Infectious Diseases, Chinese Medical Association; Severe Liver Diseases and Artificial Liver Group, Chinese Society of Hepatology, Chinese Medical Association. [Diagnostic and treatment guidelines for liver failure (2012 version)]. Zhonghua Gan Zang Bing Za Zhi 21, 177–183 (in Chinese) (2013).
Vincent, J. L. et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. 22, 707–710 (1996).
Wehler, M., Kokoska, J., Reulbach, U., Hahn, E. G. & Strauss, R. Short-term prognosis in critically ill patients with cirrhosis assessed by prognostic scoring systems. Hepatology 34, 255–261 (2001).
Das, V. et al. Cirrhotic patients in the medical intensive care unit: early prognosis and long-term survival. Crit. Care Med. 38, 2108–2116 (2010).
Levesque, E. et al. Prospective evaluation of the prognostic scores for cirrhotic patients admitted to an intensive care unit. J. Hepatol. 56, 95–102 (2012).
Jalan, R. et al. Development and validation of a prognostic score to predict mortality in patients with acute on chronic liver failure. J. Hepatol. 61, 1038–1047 (2014). This study describes a prognostic score specifically designed for patients with ACLF. The accuracy of this score (CLIF-C ACLF score) is significantly higher than that of all scores currently used in clinical practice.
Bernal, W. et al. Acute-on-chronic liver failure. Lancet 386, 1576–1587 (2015).
Bernsmeier, C. et al. Patients with acute-on-chronic liver failure have increased numbers of regulatory immune cells expressing the receptor tyrosine kinase MERTK. Gastroenterology 148, 603–615.e14 (2015).
Zhang, Q. et al. Comparison of current diagnostic criteria for acute-on-chronic liver failure. PLoS ONE 10, e0122158 (2015).
Li, H. et al. Characteristics, diagnosis and prognosis of acute-on-chronic liver failure in cirrhosis associated to hepatitis B. Sci. Rep. 6, 25487 (2016). This study indicates that the EASL-CLIF Consortium definition of ACLF designed for European patients can also be used in Chinese patients with cirrhosis due to HBV infection with no major differences in prevalence, severity and prognosis.
Dirchwolf, M. et al. Immune dysfunction in cirrhosis: distinct cytokines phenotypes according to cirrhosis severity. Cytokine 77, 14–25 (2015).
Amarapurkar, D. et al. Acute-on-chronic liver failure: a prospective study to determine the clinical profile, outcome, and factors predicting mortality. Indian J. Gastroenterol. 34, 216–224 (2015).
Li, H. et al. Submassive hepatic necrosis distinguishes HBV-associated acute on chronic liver failure from cirrhotic patients with acute decompensation. J. Hepatol. 63, 50–59 (2015).
Singh, H. & Pai, C. G. Defining acute-on-chronic liver failure: east, west or middle ground? World J. Hepatol. 7, 2571–2577 (2015).
Agrawal, S., Duseja, A., Gupta, T., Dhiman, R. K. & Chawla, Y. Simple organ failure count versus CANONIC grading system for predicting mortality in acute-on-chronic liver failure. J. Gastroenterol. Hepatol. 30, 575–581 (2015).
Lee, M. et al. CLIF-SOFA scoring system accurately predicts short-term mortality in acutely decompensated patients with alcoholic cirrhosis: a retrospective analysis. Liver Int. 35, 46–57 (2015).
Dhiman, R. K., Agrawal, S., Gupta, T., Duseja, A. & Chawla, Y. Chronic Liver Failure-Sequential Organ Failure Assessment is better than the Asia-Pacific Association for the Study of Liver criteria for defining acute-on-chronic liver failure and predicting outcome. World J. Gastroenterol. 20, 14934–14941 (2014).
Kim, H. Y. et al. Characterization of acute-on-chronic liver failure and prediction of mortality in Asian patients with active alcoholism. J. Gastroenterol. Hepatol. 31, 427–433 (2016).
Kim, T. Y. & Kim, D. J. Acute-on-chronic liver failure. Clin. Mol. Hepatol. 19, 349–359 (2013).
Jalan, R. et al. Toward an improved definition of acute-on-chronic liver failure. Gastroenterology 147, 4–10 (2014).
Kim, T. Y. et al. Characteristics and discrepancies in acute-on-chronic liver failure: need for a unified definition. PLoS ONE 11, e0146745 (2016). This study is the first to show that the EASL-CLIF Consortium definition and the APASL definition of ACLF include different populations of patients.
Sarin, S. K. & Choudhury, A. Acute-on-chronic liver failure: terminology, mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 13, 131–149 (2016).
Olson, J. C. & Kamath, P. S. Acute-on-chronic liver failure: concept, natural history, and prognosis. Curr. Opin. Crit. Care 17, 165–169 (2011).
Singh, K. K., Panda, S. K., & Shalimar Acharya, S. K. Patients with diabetes mellitus are prone to develop severe hepatitis and liver failure due to hepatitis virus infection. J. Clin. Exp. Hepatol. 3, 275–280 (2013).
Verbeke, L., Nevens, F. & Laleman, W. Bench-to-beside review: acute-on-chronic liver failure — linking the gut, liver and systemic circulation. Crit. Care 15, 233 (2011).
Suntharalingam, G. et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 355, 1018–1028 (2006).
Medzhitov, R., Schneider, D. S. & Soares, M. P. Disease tolerance as a defense strategy. Science 335, 936–941 (2012).
Medzhitov, R. Origin and physiological roles of inflammation. Nature 454, 428–435 (2008).
Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010).
Iwasaki, A. & Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 16, 343–353 (2015).
Wu, J. & Chen, Z. J. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 32, 461–488 (2014).
Xu, H. et al. Innate immune sensing of bacterial modifications of Rho GTPases by the pyrin inflammasome. Nature 513, 237–241 (2014).
Zhao, Y. et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477, 596–600 (2011).
Martinon, F., Mayor, A. & Tschopp, J. The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27, 229–265 (2009).
Kono, H. & Rock, K. L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 8, 279–289 (2008).
Rickard, J. A. et al. RIPK1 regulates RIPK3–MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 157, 1175–1188 (2014).
Angus, D. C. & van der Poll, T. Severe sepsis and septic shock. N. Engl. J. Med. 369, 840–851 (2013).
Medzhitov, R. Inflammation 2010: new adventures of an old flame. Cell 140, 771–776 (2010).
Iwasaki, A. & Medzhitov, R. Regulation of adaptive immunity by the innate immune system. Science 327, 291–295 (2010).
Chovatiya, R. & Medzhitov, R. Stress, inflammation, and defense of homeostasis. Mol. Cell 54, 281–288 (2014).
Jalan, R. et al. Bacterial Infections in cirrhosis. A position statement based on the EASL Special Conference 2013. J. Hepatol. 60, 1310–1324(2014).
Wiest, R., Lawson, M. & Geuking, M. Pathological bacterial translocation in liver cirrhosis. J. Hepatol. 60, 197–209 (2014).
Byl, B., Roucloux, I., Crusiaux, A., Dupont, E. & Devière, J. Tumor necrosis factor alpha and interleukin 6 plasma levels in infected cirrhotic patients. Gastroenterology 104, 1492–1497 (1993).
Navasa, M. et al. Tumor necrosis factor and interleukin-6 in spontaneous bacterial peritonitis in cirrhosis: relationship with the development of renal impairment and mortality. Hepatology 27, 1227–1232 (1998). References 57 and 58 show for the first time an in vivo overproduction of pro-inflammatory cytokines in patients with cirrhosis.
Gustot, T., Durand, F., Lebrec, D., Vincent, J.-L. & Moreau, R. Severe sepsis in cirrhosis. Hepatology 50, 2022–2033 (2009).
Devière, J. et al. Excessive in vitro bacterial lipopolysaccharide-induced production of monokines in cirrhosis. Hepatology 11, 628–634 (1990). This paper shows for the first time that the ex vivo innate immune response to LPS is deregulated in monocytes from patients with alcoholic cirrhosis.
Le Moine, O. et al. Role of defective monocyte interleukin-10 release in tumor necrosis factor-alpha overproduction in alcoholics cirrhosis. Hepatology 22, 1436–1439 (1995).
Tazi, K. A. et al. Upregulation of TNF-alpha production signaling pathways in monocytes from patients with advanced cirrhosis: possible role of Akt and IRAK-M. J. Hepatol. 45, 280–289 (2006). This paper describes the intracellular mechanisms involved in the excessive innate immune response to LPS in monocytes from patients with decompensated cirrhosis.
Tazi, K. A. et al. Protein array technology to investigate cytokine production by monocytes from patients with advanced alcoholic cirrhosis: an ex vivo pilot study. Hepatol. Res. 39, 706–715 (2009).
Galbois, A. et al. Ex vivo effects of high-density lipoprotein exposure on the lipopolysaccharide-induced inflammatory response in patients with severe cirrhosis. Hepatology 49, 175–184 (2009).
Coant, N. et al. Glycogen synthase kinase 3 involvement in the excessive proinflammatory response to LPS in patients with decompensated cirrhosis. J. Hepatol. 55, 784–793 (2011).
Gandoura, S. et al. Gene- and exon-expression profiling reveals an extensive LPS-induced response in immune cells in patients with cirrhosis. J. Hepatol. 58, 936–948 (2013).
Heller, J. et al. Effects of lipopolysaccharide on TNFα production, hepatic NOS2 activity, and hepatic toxicity in rats with cirrhosis. J. Hepatol. 33, 376–381 (2000).
Moreau, R. et al. Terlipressin inhibits in vivo aortic iNOS expression induced by lipopolysaccharide in rats with biliary cirrhosis. Hepatology 36, 1070–1078 (2002).
Urbanowicz, W. et al. Tezosentan, an endothelin receptor antagonist, limits liver injury in endotoxin challenged cirrhotic rats. Gut 53, 1844–1849 (2004).
Tazi, K. A. et al. In vivo altered unfolded protein response and apoptosis in livers from lipopolysaccharide-challenged cirrhotic rats. J. Hepatol. 46, 1075–1088 (2007). This paper shows for the first time that LPS-induced hepatic endoplasmic reticulum stress inhibits the accumulation of NF-κB-dependent anti-apoptotic proteins in livers from rats with cirrhosis.
Thabut, D. et al. High-density lipoprotein administration attenuates liver proinflammatory response, restores liver endothelial nitric oxide synthase activity, and lowers portal pressure in cirrhotic rats. Hepatology 46, 1893–1906 (2007).
Malhi, H. & Kaufman, R. J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 54, 795–809 (2011).
Chaisson, M. L., Brooling, J. T., Ladiges, W., Tsai, S. & Fausto, N. Hepatocyte-specific inhibition of NF-κB leads to apoptosis after TNF treatment, but not after partial hepatectomy. J. Clin. Invest. 110, 193–202 (2002).
Louvet, A. et al. Infection in patients with severe alcoholic hepatitis treated with steroids: early response to therapy is the key factor. Gastroenterology 137, 541–548 (2009).
Lucey, M. R., Mathurin, P. & Morgan, T. R. Alcoholic hepatitis. N. Engl. J. Med. 360, 2758–2769 (2009).
Úbeda, M. et al. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J. Hepatol. 64, 1049–1057 (2015).
Du Plessis, J. et al. Activated intestinal macrophages in patients with cirrhosis release NO and IL-6 that may disrupt intestinal barrier function. J. Hepatol. 58, 1125–1132 (2013).
Francés, R. et al. Bacterial translocation is downregulated by anti-TNFα monoclonal antibody administration in rats with cirrhosis and ascites. J. Hepatol. 46, 797–803 (2007).
Dominguez, M. et al. Hepatic expression of CXC chemokines predicts portal hypertension and survival in patients with alcoholic hepatitis. Gastroenterology 136, 1639–1650 (2009). This paper shows that neutrophil-attracting chemokines are overexpressed in livers from patients with severe alcoholic cirrhosis.
Charo, I. F. & Ransohoff, R. M. The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 354, 610–621 (2006).
Kubes, P. & Mehal, W. Z. Sterile inflammation in the liver. Gastroenterology 143, 1158–1172 (2012).
Larosche, I. et al. Prolonged ethanol administration depletes mitochondrial DNA in MnSOD-overexpressing transgenic mice, but not in their wild type littermates. Toxicol. Appl. Pharmacol. 234, 326–338 (2009).
Choumar, A. et al. Lipopolysaccharide-induced mitochondrial DNA depletion. Antioxid. Redox Signal. 15, 2837–2854 (2011).
West, A. P. et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557 (2015).
Dubuquoy, L. et al. Progenitor cell expansion and impaired hepatocyte regeneration in explanted livers from alcoholic hepatitis. Gut 64, 1949–1960 (2015).
Brenner, D. A., Paik, Y.-H. & Schnabl, B. Role of gut microbiota in liver disease. J. Clin. Gastroenterol. 49, S25–S27 (2015).
Bajaj, J. S. et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J. Hepatol. 60, 940–947 (2014).
Bajaj, J. S. et al. Linkage of gut microbiome with cognition in hepatic encephalopathy. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G168–G175 (2012).
Chen, Y. et al. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 54, 562–572 (2011).
Chen, Y. et al. Gut dysbiosis in acute-on-chronic liver failure and its predictive value for mortality. J. Gastroenterol. Hepatol. 30, 1429–1437 (2015).
Pan, C. et al. Dynamic changes of lipopolysaccharide levels in different phases of acute on chronic hepatitis B liver failure. PLoS ONE 7, e49460 (2012).
Bauer, T. M. et al. Small intestinal bacterial overgrowth in human cirrhosis is associated with systemic endotoxemia. Am. J. Gastroenterol. 97, 2364–2370 (2002). This paper shows an association between microbiota alterations and systemic endotoxaemia in patients with cirrhosis.
Zapater, P. et al. Serum and ascitic fluid bacterial DNA: a new independent prognostic factor in noninfected patients with cirrhosis. Hepatology 48, 1924–1931 (2008).
McPhail, M. J. W. et al. Increased survival for patients with cirrhosis and organ failure in liver intensive care and validation of the Chronic Liver Failure-Sequential Organ Failure Scoring System. Clin. Gastroenterol. Hepatol. 13, 1353–1360.e8 (2015).
Silva, P. E. S. E. et al. Single-centre validation of the EASL-CLIF Consortium definition of acute-on-chronic liver failure and CLIF-SOFA for prediction of mortality in cirrhosis. Liver Int. 35, 1516–1523 (2015).
Gustot, T. et al. Clinical course of acute-on-chronic liver failure syndrome and effects on prognosis. Hepatology 62, 243–252 (2015). This study is the first investigation defining the clinical course of ACLF within 28 days following diagnosis. It shows that ACLF is an extremely dynamic syndrome that may improve, worsen or follow a steady course. Prognosis is highly dependent on the clinical course within the first week after diagnosis.
Shi, Y. et al. Acute-on-chronic liver failure precipitated by hepatic injury is distinct from that precipitated by extrahepatic insults. Hepatology 62, 232–242 (2015). This study confirms the results of the CANONIC study in European patients, showing that short-term (28-day and 90-day) mortality in Asian patients with ACLF depends on the number of organ failures and not on the aetiology (the type of precipitating events) of the syndrome.
Jalan, R. et al. The CLIF Consortium Acute Decompensation score (CLIF-C ADs) for prognosis of hospitalised cirrhotic patients without acute-on-chronic liver failure. J. Hepatol. 62, 831–840 (2015).
Kamath, P. S. & Kim, W. R. The model for end-stage liver disease (MELD). Hepatology 45, 797–805 (2007).
Durand, F. & Valla, D. Assessment of the prognosis of cirrhosis: Child-Pugh versus MELD. J. Hepatol. 42, S100–S107 (2005).
Biggins, S. W. Use of serum sodium for liver transplant graft allocation: a decade in the making, now is it ready for primetime? Liver Transpl. 21, 279–281 (2015).
Fernandez, J. & Arroyo, V. Bacterial infections in cirrhosis: a growing problem with significant implications. Clin. Liver Dis. 2, 102–105 (2013).
Sarin, S. K. et al. Asian-Pacific clinical practice guidelines on the management of hepatitis B: a 2015 update. Hepatol. Int. 10, 1–98 (2016).
Sort, P. et al. Effect of intravenous albumin on renal impairment and mortality in patients with cirrhosis and spontaneous bacterial peritonitis. N. Engl. J. Med. 341, 403–409 (1999). This randomized controlled trial was the first to show that intravenous albumin administration (1.5 g per kg of body weight at infection diagnosis and 1 g per kg of body weight at the third day) is highly effective in preventing type 1 HRS and mortality in patients with cirrhosis and spontaneous bacterial peritonitis.
Arroyo, V., García-Martinez, R. & Salvatella, X. Human serum albumin, systemic inflammation, and cirrhosis. J. Hepatol. 61, 396–407 (2014).
Guevara, M. et al. Albumin for bacterial infections other than spontaneous bacterial peritonitis in cirrhosis. A randomized, controlled study. J. Hepatol. 57, 759–765 (2012).
Thévenot, T. et al. Effect of albumin in cirrhotic patients with infection other than spontaneous bacterial peritonitis. A randomized trial. J. Hepatol. 62, 822–830 (2015).
Ginés, P. et al. Norfloxacin prevents spontaneous bacterial peritonitis recurrence in cirrhosis: results of a double-blind, placebo-controlled trial. Hepatology 12, 716–724 (1990).
Soriano, G. et al. Norfloxacin prevents bacterial infection in cirrhotics with gastrointestinal hemorrhage. Gastroenterology 103, 1267–1272 (1992).
Fernández, J. et al. Primary prophylaxis of spontaneous bacterial peritonitis delays hepatorenal syndrome and improves survival in cirrhosis. Gastroenterology 133, 818–824 (2007). This randomized controlled trial was the first to show that long-term oral administration of norfloxacin prevents the development of spontaneous bacterial peritonitis and type 1 HRS and improves survival in patients with cirrhosis and severe liver and renal dysfunction.
Zapater, P. et al. Norfloxacin modulates the inflammatory response and directly affects neutrophils in patients with decompensated cirrhosis. Gastroenterology 137, 1669–1679.e1 (2009).
Gómez-Hurtado, I. et al. Interleukin-10-mediated heme oxygenase 1-induced underlying mechanism in inflammatory down-regulation by norfloxacin in cirrhosis. Hepatology 53, 935–944 (2011).
Gómez-Hurtado, I. et al. Role of interleukin 10 in norfloxacin prevention of luminal free endotoxin translocation in mice with cirrhosis. J. Hepatol. 61, 799–808 (2014).
Akriviadis, E. et al. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: a double-blind, placebo-controlled trial. Gastroenterology 119, 1637–1648 (2000).
Nguyen-Khac, E. et al. Glucocorticoids plus N-acetylcysteine in severe alcoholic hepatitis. N. Engl. J. Med. 365, 1781–1789 (2011).
Thursz, M. R. et al. Prednisolone or pentoxifylline for alcoholic hepatitis. N. Engl. J. Med. 372, 1619–1628 (2015).
Kedarisetty, C. K. et al. Combination of granulocyte colony-stimulating factor and erythropoietin improves outcomes of patients with decompensated cirrhosis. Gastroenterology 148, 1362–1370.e7 (2015).
Arroyo, V., Moreau, R., Jalan, R. & Ginès, P. Acute-on-chronic liver failure: a new syndrome that will re-classify cirrhosis. J. Hepatol. 62, S131–S143 (2015).
Seto, W.-K., Lai, C.-L. & Yuen, M.-F. Acute-on-chronic liver failure in chronic hepatitis B. J. Gastroenterol. Hepatol. 27, 662–669 (2012).
Garg, H. et al. Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology 53, 774–780 (2011).
Philips, C. A. & Sarin, S. K. Potent antiviral therapy improves survival in acute on chronic liver failure due to hepatitis B virus reactivation. World J. Gastroenterol. 20, 16037–16052 (2014). The results of this study are a clear indication that potent antiviral therapy significantly improves the clinical course and survival of patients with ACLF due to reactivation of HBV.
Arabi, Y. M. et al. Antimicrobial therapeutic determinants of outcomes from septic shock among patients with cirrhosis. Hepatology 56, 2305–2315 (2012).
Tandon, P. & Garcia-Tsao, G. Bacterial infections, sepsis, and multiorgan failure in cirrhosis. Semin. Liver Dis. 28, 26–42 (2008).
Fernández, J., Tandon, P., Mensa, J. & Garcia-Tsao, G. Antibiotic prophylaxis in cirrhosis: good and bad. Hepatology 63, 2019–2031(2016).
Chen, T. et al. Nucleoside analogues improve the short-term and long-term prognosis of patients with hepatitis B virus-related acute-on-chronic liver failure. Clin. Exp. Med. 12, 159–164 (2012).
Xie, F. et al. Effects of nucleoside analogue on patients with chronic hepatitis B-associated liver failure: meta-analysis. PLoS ONE 8, e54773 (2013).
Yang, J. et al. Initial combination anti-viral therapy with lamivudine and adefovir dipivoxil decreases short-term fatality rate of hepatitis-B-virus-related acute-on-chronic liver failure. Virol. J. 12, 97 (2015).
Garg, V. et al. Granulocyte colony-stimulating factor mobilizes CD34+ cells and improves survival of patients with acute-on-chronic liver failure. Gastroenterology 142, 505–512.e1 (2012).
Angeli, P. et al. Diagnosis and management of acute kidney injury in patients with cirrhosis: revised consensus recommendations of the International Club of Ascites. J. Hepatol. 62, 968–974 (2015). This article provides the most modern clinical guidelines on the diagnosis and treatment of AKI in cirrhosis.
Belcher, J. M. et al. Urinary biomarkers and progression of AKI in patients with cirrhosis. Clin. J. Am. Soc. Nephrol. 9, 1857–1867 (2014).
Rivers, E. et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N. Engl. J. Med. 345, 1368–1377 (2001).
Fede, G. et al. Adrenocortical dysfunction in liver disease: a systematic review. Hepatology 55, 1282–1291 (2012).
Vilstrup, H. et al. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver. Hepatology 60, 715–735 (2014).
Bianchini, M., De Pietri, L. & Villa, E. Coagulopathy in liver diseases: complication or therapy? Dig. Dis. 32, 609–614 (2014).
de Franchis, R. & Baveno VI Faculty. Expanding consensus in portal hypertension: report of the Baveno VI Consensus Workshop: stratifying risk and individualizing care for portal hypertension. J. Hepatol. 63, 743–752 (2015). This article provides the most modern clinical guidelines on the diagnosis management of portal hypertension and gastrointestinal haemorrhage in cirrhosis. There is a detailed description on the management of coagulopathy in patients with portal vein thrombosis.
Lee, S. Y., Kim, H. J. & Choi, D. Cell sources, liver support systems and liver tissue engineering: alternatives to liver transplantation. Int. J. Stem Cells 8, 36–47 (2015).
Struecker, B., Raschzok, N. & Sauer, I. M. Liver support strategies: cutting-edge technologies. Nat. Rev. Gastroenterol. Hepatol. 11, 166–176 (2014). This article contains a comprehensive review of the artificial liver support strategies in patients with acute liver failure and ACLF.
Bañares, R. et al. Extracorporeal albumin dialysis with the molecular adsorbent recirculating system in acute-on-chronic liver failure: the RELIEF trial. Hepatology 57, 1153–1162 (2013).
Kribben, A. et al. Effects of fractionated plasma separation and adsorption on survival in patients with acute-on-chronic liver failure. Gastroenterology 142, 782–789.e3 (2012).
Lee, K. C. L. et al. Extracorporeal liver assist device to exchange albumin and remove endotoxin in acute liver failure: results of a pivotal pre-clinical study. J. Hepatol. 63, 634–642 (2015).
Larsen, F. S. et al. High-volume plasma exchange in patients with acute liver failure: an open randomised controlled trial. J. Hepatol. 64, 69–78 (2016).
Bahirwani, R., Shaked, O., Bewtra, M., Forde, K. & Reddy, K. R. Acute-on-chronic liver failure before liver transplantation: impact on posttransplant outcomes. Transplantation 92, 952–957 (2011).
Duan, B.-W. et al. Liver transplantation in acute-on-chronic liver failure patients with high model for end-stage liver disease (MELD) scores: a single center experience of 100 consecutive cases. J. Surg. Res. 183, 936–943 (2013).
Finkenstedt, A. et al. Acute-on-chronic liver failure: excellent outcomes after liver transplantation but high mortality on the wait list. Liver Transpl. 19, 879–886 (2013).
Sharma, P., Schaubel, D. E., Gong, Q., Guidinger, M. & Merion, R. M. End-stage liver disease candidates at the highest model for end-stage liver disease scores have higher wait-list mortality than status-1A candidates. Hepatology 55, 192–198 (2012).
Chan, A. C. et al. Liver transplantation for acute-on-chronic liver failure. Hepatol. Int. 3, 571–581 (2009).
Chan, A. C. Y. & Fan, S. T. Criteria for liver transplantation in ACLF and outcome. Hepatol. Int. 9, 355–359 (2015).
Reddy, M. S., Rajalingam, R. & Rela, M. Liver transplantation in acute-on-chronic liver failure: lessons learnt from acute liver failure setting. Hepatol. Int. 9, 508–513 (2015).
Khanam, A. et al. Altered frequencies of dendritic cells and IFN-γ-secreting T cells with granulocyte colony-stimulating factor (G-CSF) therapy in acute-on- chronic liver failure. Liver Int. 34, 505–513 (2014).
Duan, X.-Z. et al. Granulocyte-colony stimulating factor therapy improves survival in patients with hepatitis B virus-associated acute-on-chronic liver failure. World J. Gastroenterol. 19, 1104–1110 (2013).
Ma, X.-R. et al. Transplantation of autologous mesenchymal stem cells for end-stage liver cirrhosis: a meta-analysis based on seven controlled trials. Gastroenterol. Res. Pract. 2015, 908275 (2015).
Volk, M. L., Tocco, R. S., Bazick, J., Rakoski, M. O. & Lok, A. S. Hospital readmissions among patients with decompensated cirrhosis. Am. J. Gastroenterol. 107, 247–252 (2012).
Reddy, K. R. et al. High risk of delisting or death in liver transplant candidates following infections: results from the North American Consortium for the Study of End-Stage Liver Disease. Liver Transpl. 21, 881–888 (2015).
O'Leary, J. G. et al. Long-term use of antibiotics and proton pump inhibitors predict development of infections in patients with cirrhosis. Clin. Gastroenterol. Hepatol. 13, 753–759.e2 (2015).
Battle, C. E., Davies, G. & Evans, P. A. Long term health-related quality of life in survivors of sepsis in south west Wales: an epidemiological study. PLoS ONE 9, e116304 (2014).
Vanwijngaerden, Y.-M. et al. Critical illness evokes elevated circulating bile acids related to altered hepatic transporter and nuclear receptor expression. Hepatology 54, 1741–1752 (2011).
Katoonizadeh, A. et al. Early features of acute-on-chronic alcoholic liver failure: a prospective cohort study. Gut 59, 1561–1569 (2010).
Hotchkiss, R. S., Monneret, G. & Payen, D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 13, 862–874 (2013).
Gomez, H. et al. A unified theory of sepsis-induced acute kidney injury: inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock 41, 3–11 (2014). This article contains a comprehensive review of the mechanisms of AKI associated with systemic inflammation in sepsis. Concepts might be extended to AKI and other types of organ failure in ACLF.
Jiménez, W., Clária, J., Arroyo, V. & Rodés, J. Carbon tetrachloride induced cirrhosis in rats: a useful tool for investigating the pathogenesis of ascites in chronic liver disease. J. Gastroenterol. Hepatol. 7, 90–97 (1992).
Harry, D. et al. Increased sensitivity to endotoxemia in the bile duct-ligated cirrhotic rat. Hepatology 30, 1198–1205 (1999).
Bass, N. M. et al. Rifaximin treatment in hepatic encephalopathy. N. Engl. J. Med. 362, 1071–1081 (2010). This article reports a randomized controlled trial showing that oral rifaximin is highly effective in preventing the recurrence of hepatic encephalopathy in cirrhosis.
Bernardi, M. et al. Long-term use of human albumin for the treatment of ascites in patients with hepatic cirrhosis: the interim analysis of the ANSWER study. Dig. Liv. Dis. 47 (Suppl. 1), e6 (2015).
Neuschwander-Tetri, B. A. et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 385, 956–965 (2015).
Verbeke, L. et al. Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology 59, 2286–2298 (2014).
Verbeke, L. et al. The FXR agonist obeticholic acid prevents gut barrier dysfunction and bacterial translocation in cholestatic rats. Am. J. Pathol. 185, 409–419 (2015). This article reports a study in experimental cirrhosis showing that oral obeticholic acid improves gut permeability, intestinal inflammation and bacterial translocation.
Kayagaki, N. et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249 (2013).
Shi, J. et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192 (2014).
Broz, P. Immunology: caspase target drives pyroptosis. Nature 526, 642–643 (2015).
Sargenti, K., Prytz, H., Nilsson, E. & Kalaitzakis, E. Predictors of mortality among patients with compensated and decompensated liver cirrhosis: the role of bacterial infections and infection-related acute-on-chronic liver failure. Scand. J. Gastroenterol. 50, 875–883 (2015).
Blei, A. T. & Córdoba, J. Hepatic encephalopathy. Am. J. Gastroenterol. 96, 1968–1976 (2001).
Acknowledgements
The European Association for the Study of the Liver-Chronic Liver Failure (EASL-CLIF) Consortium is endorsed by the European Association for the Study of the Liver and supported by an unrestricted grant from Grifols. The authors thank D. J. Kim for the supply of the published Korean data and the Data Management Centre of the EASL-CLIF Consortium for providing the unpublished European data used in Figure 2.
Author information
Authors and Affiliations
Contributions
Introduction (V.A.); Epidemiology (P.S.K.); Mechanisms/pathophysiology (R.M. and B.S.); Diagnosis, screening and prevention (P.G., V.A. and J.F.); Management (R.J., G.G.-T., U.T. and J.F.); Quality of life (P.S.K.); Outlook (F.N. and V.A.); Overview of Primer (V.A.). V.A. and R.M. contributed equally to this work.
Corresponding author
Ethics declarations
Competing interests
V.A. has received research funding from Grifols and has served on the scientific advisory board for Takeda. P.G. has received research funding from Grifols, served on the scientific advisory board for Ferring and Squana Medical and received research funding from Sequana Medical. R.J. has received research funding from Vital Therapies, has served on the scientific advisory board for Conatus Pharma and Takeda, has ongoing research collaborations with Gambro and Grifiols and is the principal investigator of an industry sponsored study (Sequana Medical). F.N. has served on the scientific advisory board of Center Fract, Croix Rouge Belgium, Intercept, Gore, Bristol-Myers Squibb, AbbVie, Novartis, MSD, Janssen-Cilag, Promethera Biosciences and Gilead, and has received grants from Roche, Astellas, Ferring, Novartis, Janssen-Cilag and AbbVie. All other authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Arroyo, V., Moreau, R., Kamath, P. et al. Acute-on-chronic liver failure in cirrhosis. Nat Rev Dis Primers 2, 16041 (2016). https://doi.org/10.1038/nrdp.2016.41
Published:
DOI: https://doi.org/10.1038/nrdp.2016.41
This article is cited by
-
A novel prognostic model to predict mortality in patients with acute-on-chronic liver failure in intensive care unit
Internal and Emergency Medicine (2024)
-
Cell therapy in end-stage liver disease: replace and remodel
Stem Cell Research & Therapy (2023)
-
Partial splenic embolization as a rescue and emergency treatment for portal hypertension and gastroesophageal variceal hemorrhage
BMC Gastroenterology (2023)
-
Risk factors for the mortality of hepatitis B virus-associated acute-on-chronic liver failure: a systematic review and meta-analysis
BMC Gastroenterology (2023)
-
T2-weighted imaging and dynamic contrast‑enhanced imaging in predicting the prognosis in patients with acute-on-chronic liver failure
BMC Gastroenterology (2023)
