
Key Points
-
Pain is the primary reason why people seek medical care; more than 40% of the US population is affected by chronic pain.
-
Opioids, which are the most commonly used and often the most effective class of analgesics, produce tolerance, dependence and constipation, and are associated with major abuse liabilities. The respiratory depression associated with high doses has led to a catastrophic increase in the number of drug overdose deaths in the United States.
-
Several new or previously overlooked targets are gaining significant attention. In the field of G-protein-coupled receptors (GPCRs), these include new ligands targeting opioid receptor heteromers, different opioid receptor subtypes and biased agonists. Non-opioid GPCRs currently being pursued include cannabinoid receptor 2 (CB2), angiotensin type 2 receptor (AT2R) and chemokine receptors.
-
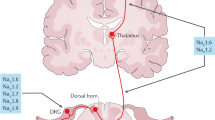
Various academic and industry groups are pursuing ion channel strategies by targeting sodium, potassium and calcium channels — specifically, certain Nav1.7, Nav1.8 and voltage-dependent calcium channel (Cavs) ligands are showing particular promise in early preclinical and clinical trials.
-
Several enzyme targets that modulate pain pathways are also being pursued.
-
Despite considerable efforts, there have been several high-profile failures of novel analgesics in the clinic.
-
Barriers that need to be overcome to develop efficacious analgesics include issues related to the lack of predictability of preclinical models in certain contexts, the translation of pathways from animal models to humans, exaggerated placebo effects and issues with clinical trial design.
Abstract
Acute and chronic pain complaints, although common, are generally poorly served by existing therapies. This unmet clinical need reflects a failure to develop novel classes of analgesics with superior efficacy, diminished adverse effects and a lower abuse liability than those currently available. Reasons for this include the heterogeneity of clinical pain conditions, the complexity and diversity of underlying pathophysiological mechanisms, and the unreliability of some preclinical pain models. However, recent advances in our understanding of the neurobiology of pain are beginning to offer opportunities for developing novel therapeutic strategies and revisiting existing targets, including modulating ion channels, enzymes and G-protein-coupled receptors.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

Change history
23 June 2017
In the original published article, the ligands RB-64 and PZM21 have been shown as attributed to Trevena in table 1 in the 'biased GPCR ligands' row. This error has been corrected in the HTML and PDF versions of the article.
06 October 2017
The compounds APD371,LY2828360, S-777469 and KHK6188 were incorrectly referred to as inhibitors of the cannabinoid receptors CB1 and CB2 in Table 1, when they are cannabinoid receptor agonists. In addition, KHK6188 is not currently in a Phase 2 clinical trial for neuropathic pain as stated in Table 1 and development of this agent has been discontinued. The error has been corrected in the html and pdf versions online.
References
Dubois, M. Y., Gallagher, R. M. & Lippe, P. M. Pain medicine position paper. Pain Med. 10, 972–1000 (2009).
Johannes, C. B., Le, T. K., Zhou, X., Johnston, J. A. & Dworkin, R. H. The prevalence of chronic pain in United States adults: results of an Internet-based survey. J. Pain 11, 1230–1239 (2010).
Volkow, N. D. & McLellan, A. T. Opioid abuse in chronic pain — misconceptions and mitigation strategies. N. Engl. J. Med. 374, 1253–1263 (2016). This review highlights common misconceptions about abuse-related liabilities of prescription opioids and proposes strategies that could help to mitigate these risks.
Decosterd, I. & Woolf, C. J. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87, 149–158 (2000). In this study, the authors describe a technique for modelling peripheral neuropathic pain in laboratory rodents.
Honore, P. et al. Murine models of inflammatory, neuropathic and cancer pain each generates a unique set of neurochemical changes in the spinal cord and sensory neurons. Neuroscience 98, 585–598 (2000).
Costigan, M. et al. Multiple chronic pain states are associated with a common amino acid-changing allele in KCNS1. Brain 133, 2519–2527 (2010). This research article describes a putative human pain gene that could inform the selection of novel drug targets for patients with neuropathic pain. It may help to explain why some, but not all, people with nerve injury progress to chronic pain.
von Hehn, C. A., Baron, R. & Woolf, C. J. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron 73, 638–652 (2012). This work describes how the variable expression of sensory nerve injury symptoms can provide insights into the underlying pathophysiological mechanisms and guidance for the development of personalized pain therapies.
Ji, R. R., Xu, Z. Z. & Gao, Y. J. Emerging targets in neuroinflammation-driven chronic pain. Nat. Rev. Drug Discov. 13, 533–548 (2014). Here, the authors discuss emerging neuroinflammatory pain targets and describe potential therapeutic opportunities to target excessive neuroinflammation.
Latremoliere, A. & Woolf, C. J. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J. Pain 10, 895–926 (2009). This work describes the mechanisms and triggers that underlie the initiation and maintenance of central sensitization, and how they are altered by changes in the properties and expression patterns of glutamate receptors.
Basbaum, A. I., Bautista, D. M., Scherrer, G. & Julius, D. Cellular and molecular mechanisms of pain. Cell 139, 267–284 (2009). In this piece, the authors review the biological underpinnings of somatosensation at the circuit, cellular and subcellular levels, with particular emphasis on pain-related receptors and mechanisms.
Ossipov, M. H., Dussor, G. O. & Porreca, F. Central modulation of pain. J. Clin. Invest. 120, 3779–3787 (2010). This review explores evidence that central modulatory circuits can dramatically change the subjective experience of painful stimuli.
Vardeh, D., Mannion, R. J. & Woolf, C. J. Toward a mechanism-based approach to pain diagnosis. J. Pain 17, T50–T69 (2016). Here, the authors propose that identifying specific mechanisms that underlie chronic pain could provide the basis for a personalized-medicine approach to analgesia.
Woolf, C. J. Overcoming obstacles to developing new analgesics. Nat. Med. 16, 1241–1247 (2010). This article discusses the many complexities that have made the development of new analgesics so challenging.
Andrews, N. A. et al. Ensuring transparency and minimization of methodologic bias in preclinical pain research: PPRECISE considerations. Pain 157, 901–909 (2016). Here, members of the Preclinical Pain Research Consortium for Investigating Safety and Efficacy (PPRECISE) working group propose new voluntary standards of scientific rigour and transparent reporting to promote more-efficient advancement of the search for new pain treatments.
Singla, N. et al. Assay sensitivity of pain intensity versus pain relief in acute pain clinical trials: ACTTION systematic review and meta-analysis. J. Pain 16, 683–691 (2015). This meta-analysis found that for preclinical acute pain trials, readouts of total pain relief may be more sensitive to treatment than summed pain intensity differences.
Finnerup, N. B. et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 14, 162–173 (2015).
Costigan, M., Scholz, J. & Woolf, C. J. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu. Rev. Neurosci. 32, 1–32 (2009).
Patapoutian, A., Tate, S. & Woolf, C. J. Transient receptor potential channels: targeting pain at the source. Nat. Rev. Drug Discov. 8, 55–68 (2009).
Woolf, C. J. Pain: morphine, metabolites, mambas, and mutations. Lancet Neurol. 12, 18–20 (2013).
Woolf, C. J. & Salter, M. W. Neuronal plasticity: increasing the gain in pain. Science 288, 1765–1769 (2000). In this review, the authors conceptualize how plasticity in ascending sensory pathways may elicit pain hypersensitivity by increasing signal gain.
Chiu, I. M., von Hehn, C. A. & Woolf, C. J. Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology. Nat. Neurosci. 15, 1063–1067 (2012).
Woolf, C. J. What is this thing called pain? J. Clin. Invest. 120, 3742–3744 (2010).
Julius, D. & Basbaum, A. I. Molecular mechanisms of nociception. Nature 413, 203–210 (2001). Here, the authors describe molecular mechanisms of primary afferent neurons, their modality sensitivities and various transducers, peptides, lipids and growth factors that signal pain and mediate pain-related signals.
Michaud, K., Bombardier, C. & Emery, P. Quality of life in patients with rheumatoid arthritis: does abatacept make a difference? Clin. Exp. Rheumatol. 25, S35–S45 (2007).
Drenth, J. P. & Waxman, S. G. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J. Clin. Invest. 117, 3603–3609 (2007).
Woolf, C. J. Central sensitization: implications for the diagnosis and treatment of pain. Pain 152, S2–S15 (2011).
Hains, B. C. et al. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J. Neurosci. 23, 8881–8892 (2003).
Nassar, M. A. et al. Nerve injury induces robust allodynia and ectopic discharges in Nav1.3 null mutant mice. Mol. Pain 2, 33 (2006).
Dong, X. W. et al. Small interfering RNA-mediated selective knockdown of NaV1.8 tetrodotoxin-resistant sodium channel reverses mechanical allodynia in neuropathic rats. Neuroscience 146, 812–821 (2007).
Jarvis, M. F. et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl Acad. Sci. USA 104, 8520–8525 (2007).
Ekberg, J. et al. muO-conotoxin MrVIB selectively blocks Nav1.8 sensory neuron specific sodium channels and chronic pain behavior without motor deficits. Proc. Natl Acad. Sci. USA 103, 17030–17035 (2006).
Gold, M. S. et al. Redistribution of NaV1.8 in uninjured axons enables neuropathic pain. J. Neurosci. 23, 158–166 (2003).
Joshi, S. K. et al. Involvement of the TTX-resistant sodium channel Nav 1.8 in inflammatory and neuropathic, but not post-operative, pain states. Pain 123, 75–82 (2006).
Roza, C., Laird, J. M., Souslova, V., Wood, J. N. & Cervero, F. The tetrodotoxin-resistant Na+ channel Nav1.8 is essential for the expression of spontaneous activity in damaged sensory axons of mice. J. Physiol. 550, 921–926 (2003).
Fritch, P. C. et al. Novel KCNQ2/Q3 agonists as potential therapeutics for epilepsy and neuropathic pain. J. Med. Chem. 53, 887–896 (2010).
Dost, R., Rostock, A. & Rundfeldt, C. The anti-hyperalgesic activity of retigabine is mediated by KCNQ potassium channel activation. Naunyn Schmiedebergs Arch. Pharmacol. 369, 382–390 (2004).
Lee, S. Pharmacological inhibition of voltage-gated Ca2+ channels for chronic pain relief. Curr. Neuropharmacol. 11, 606–620 (2013).
Stemkowski, P. L., Noh, M. C., Chen, Y. & Smith, P. A. Increased excitability of medium-sized dorsal root ganglion neurons by prolonged interleukin-1β exposure is K+ channel dependent and reversible. J. Physiol. 593, 3739–3755 (2015).
Zogopoulos, P., Vasileiou, I., Patsouris, E. & Theocharis, S. E. The role of endocannabinoids in pain modulation. Fundam. Clin. Pharmacol. 27, 64–80 (2013).
Gutenstein, H. & Akil, H. in Goodman and Gilman's Pharmacological Basis of Therapeutics Ch. 21 (eds Brunton, L., Lazo, J. & Parker, K.) 547–590 (The McGraw Hill companies, 2006). Chapter 21 of this authoritative pharmacology text provides a thorough overview of opioid analgesic pharmacology.
Fries, D. S. in Principles of Medicinal Chemistry Ch. 14 (eds Foye, W. O., Lemke, T. L. & Williams,D. A.) 247–269 (William & Wilkins, 1995). Chapter 14 of this essential medicinal chemistry source describes the medicinal chemistry of common opioid and anti-inflammatory analgesics.
Lesniak, A. & Lipkowski, A. W. Opioid peptides in peripheral pain control. Acta Neurobiol. Exp. (Wars.) 71, 129–138 (2011).
Navratilova, E. et al. Positive emotions and brain reward circuits in chronic pain. J. Comp. Neurol. 524, 1646–1652 (2016).
Navratilova, E. & Porreca, F. Reward and motivation in pain and pain relief. Nat. Neurosci. 17, 1304–1312 (2014).
Kolodny, A. et al. The prescription opioid and heroin crisis: a public health approach to an epidemic of addiction. Annu. Rev. Public Health 36, 559–574 (2015).
Cassidy, T. A., DasMahapatra, P., Black, R. A., Wieman, M. S. & Butler, S. F. Changes in prevalence of prescription opioid abuse after introduction of an abuse-deterrent opioid formulation. Pain Med. 15, 440–451 (2014).
Kunins, H. V. Abuse-deterrent opioid formulations: part of a public health strategy to reverse the opioid epidemic. JAMA Intern. Med. 175, 987–988 (2015).
Chilcoat, H. D., Coplan, P. M., Harikrishnan, V. & Alexander, L. Decreased diversion by doctor-shopping for a reformulated extended release oxycodone product (OxyContin). Drug Alcohol Depend. 165, 221–228 (2016).
Coplan, P. M. et al. The effect of an abuse-deterrent opioid formulation on opioid abuse-related outcomes in the post-marketing setting. Clin. Pharmacol. Ther. 100, 275–286 (2016).
Cicero, T. J., Ellis, M. S. & Surratt, H. L. Effect of abuse-deterrent formulation of OxyContin. N. Engl. J. Med. 367, 187–189 (2012).
Cicero, T. J., Ellis, M. S. & Kasper, Z. A. A tale of 2 ADFs: differences in the effectiveness of abuse-deterrent formulations of oxymorphone and oxycodone extended-release drugs. Pain 157, 1232–1238 (2016).
Walsh, S. L., Strain, E. C., Abreu, M. E. & Bigelow, G. E. Enadoline, a selective kappa opioid agonist: comparison with butorphanol and hydromorphone in humans. Psychopharmacology (Berl.) 157, 151–162 (2001).
Pallasch, T. J. & Gill, C. J. Butorphanol and nalbuphine: a pharmacologic comparison. Oral Surg. Oral Med. Oral Pathol. 59, 15–20 (1985).
Webster, L., Menzaghi, F. & Spencer, R. CR845, a novel peripherally-acting kappa opioid receptor agonist, has low abuse potential compared with pentazocine. J. Pain 16, S81 (2015).
Spahn, V. et al. A nontoxic pain killer designed by modeling of pathological receptor conformations. Science 355, 966–969 (2017).
Milligan, G. The prevalence, maintenance, and relevance of G protein-coupled receptor oligomerization. Mol. Pharmacol. 84, 158–169 (2013).
Rozenfeld, R. & Devi, L. A. Receptor heteromerization and drug discovery. Trends Pharmacol. Sci. 31, 124–130 (2010).
Yekkirala, A. S. Two to tango: GPCR oligomers and GPCR–TRP channel interactions in nociception. Life Sci. 92, 438–445 (2013).
Yekkirala, A. S., Kalyuzhny, A. E. & Portoghese, P. S. An immunocytochemical-derived correlate for evaluating the bridging of heteromeric mu-delta opioid protomers by bivalent ligands. ACS Chem. Biol. 8, 1412–1416 (2013).
Lenard, N. R., Daniels, D. J., Portoghese, P. S. & Roerig, S. C. Absence of conditioned place preference or reinstatement with bivalent ligands containing mu-opioid receptor agonist and delta-opioid receptor antagonist pharmacophores. Eur. J. Pharmacol. 566, 75–82 (2007).
Aceto, M. D. et al. MDAN-21: a bivalent opioid ligand containing mu-agonist and delta-antagonist pharmacophores and its effects in rhesus monkeys. Int. J. Med. Chem. 2012, 327257 (2012).
Daniels, D. J. et al. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc. Natl Acad. Sci. USA 102, 19208–19213 (2005).
Le Naour, M. et al. Bivalent ligands that target mu opioid (MOP) and cannabinoid1 (CB1) receptors are potent analgesics devoid of tolerance. J. Med. Chem. 56, 5505–5513 (2013).
Akgun, E. et al. Ligands that interact with putative MOR-mGluR5 heteromer in mice with inflammatory pain produce potent antinociception. Proc. Natl Acad. Sci. USA 110, 11595–11599 (2013).
Akgun, E. et al. Inhibition of inflammatory and neuropathic pain by targeting a mu opioid receptor/chemokine receptor5 heteromer (MOR-CCR5). J. Med. Chem. 58, 8647–8657 (2015).
Yekkirala, A. S. et al. N-Naphthoyl-beta-naltrexamine (NNTA), a highly selective and potent activator of mu/kappa-opioid heteromers. Proc. Natl Acad. Sci. USA 108, 5098–5103 (2011).
Chakrabarti, S., Liu, N. J. & Gintzler, A. R. Formation of mu-/kappa-opioid receptor heterodimer is sex-dependent and mediates female-specific opioid analgesia. Proc. Natl Acad. Sci. USA 107, 20115–20119 (2010).
Ding, H. et al. A novel orvinol analog, BU08028, as a safe opioid analgesic without abuse liability in primates. Proc. Natl Acad. Sci. USA 113, E5511–E5518 (2016).
Pasternak, G. W. & Pan, Y. X. Mu opioids and their receptors: evolution of a concept. Pharmacol. Rev. 65, 1257–1317 (2013).
Marrone, G. F. et al. Truncated mu opioid GPCR variant involvement in opioid-dependent and opioid-independent pain modulatory systems within the CNS. Proc. Natl Acad. Sci. USA 113, 3663–3668 (2016).
Majumdar, S. et al. Truncated G protein-coupled mu opioid receptor MOR-1 splice variants are targets for highly potent opioid analgesics lacking side effects. Proc. Natl Acad. Sci. USA 108, 19778–19783 (2011).
Wieskopf, J. S. et al. Broad-spectrum analgesic efficacy of IBNtxA is mediated by exon 11-associated splice variants of the mu-opioid receptor gene. Pain 155, 2063–2070 (2014).
Liu, X. Y. et al. Unidirectional cross-activation of GRPR by MOR1D uncouples itch and analgesia induced by opioids. Cell 147, 447–458 (2011).
White, K. L. et al. The G protein-biased kappa-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J. Pharmacol. Exp. Ther. 352, 98–109 (2015).
Tang, W., Strachan, R. T., Lefkowitz, R. J. & Rockman, H. A. Allosteric modulation of beta-arrestin-biased angiotensin II type 1 receptor signaling by membrane stretch. J. Biol. Chem. 289, 28271–28283 (2014).
Drake, M. T. et al. β-Arrestin-biased agonism at the β2-adrenergic receptor. J. Biol. Chem. 283, 5669–5676 (2008).
Wisler, J. W., Xiao, K., Thomsen, A. R. & Lefkowitz, R. J. Recent developments in biased agonism. Curr. Opin. Cell Biol. 27, 18–24 (2014). In this review, the authors describe how different ligands can induce distinct receptor conformations at the same GPCR to elicit unique downstream signalling profiles. They provide support for how these properties could be exploited to develop analgesics with different or reduced side-effect profiles.
Shukla, A. K. et al. Distinct conformational changes in beta-arrestin report biased agonism at seven-transmembrane receptors. Proc. Natl Acad. Sci. USA 105, 9988–9993 (2008).
Whalen, E. J., Rajagopal, S. & Lefkowitz, R. J. Therapeutic potential of beta-arrestin- and G protein-biased agonists. Trends Mol. Med. 17, 126–139 (2011).
Zidar, D. A., Violin, J. D., Whalen, E. J. & Lefkowitz, R. J. Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc. Natl Acad. Sci. USA 106, 9649–9654 (2009).
Gesty-Palmer, D. et al. A beta-arrestin-biased agonist of the parathyroid hormone receptor (PTH1R) promotes bone formation independent of G protein activation. Sci. Transl Med. 1, 1ra1 (2009).
Strachan, R. T. et al. Divergent transducer-specific molecular efficacies generate biased agonism at a G protein-coupled receptor (GPCR). J. Biol. Chem. 289, 14211–14224 (2014).
Violin, J. D., Crombie, A. L., Soergel, D. G. & Lark, M. W. Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol. Sci. 35, 308–316 (2014).
Soergel, D. G. et al. Biased agonism of the mu-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: a randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain 155, 1829–1835 (2014).
Soergel, D. G. et al. First clinical experience with TRV130: pharmacokinetics and pharmacodynamics in healthy volunteers. J. Clin. Pharmacol. 54, 351–357 (2014).
Manglik, A. et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 537, 185–190 (2016).
Tabrizi, M. A., Baraldi, P. G., Borea, P. A. & Varani, K. Medicinal chemistry, pharmacology, and potential therapeutic benefits of cannabinoid CB2 receptor agonists. Chem. Rev. 116, 519–560 (2016).
Han, S., Thatte, J., Buzard, D. J. & Jones, R. M. Therapeutic utility of cannabinoid receptor type 2 (CB2) selective agonists. J. Med. Chem. 56, 8224–8256 (2013).
Nevalainen, T. Recent development of CB2 selective and peripheral CB1/CB2 cannabinoid receptor ligands. Curr. Med. Chem. 21, 187–203 (2014).
Anand, U. et al. Mechanisms underlying clinical efficacy of angiotensin II type 2 receptor (AT2R) antagonist EMA401 in neuropathic pain: clinical tissue and in vitro studies. Mol. Pain 11, 38 (2015).
Danser, A. H. & Anand, P. The angiotensin II type 2 receptor for pain control. Cell 157, 1504–1506 (2014).
Rice, A. S. et al. EMA401, an orally administered highly selective angiotensin II type 2 receptor antagonist, as a novel treatment for postherpetic neuralgia: a randomised, double-blind, placebo-controlled phase 2 clinical trial. Lancet 383, 1637–1647 (2014).
Anand, U. et al. Angiotensin II type 2 receptor (AT2 R) localization and antagonist-mediated inhibition of capsaicin responses and neurite outgrowth in human and rat sensory neurons. Eur. J. Pain 17, 1012–1026 (2013).
Smith, M. T., Woodruff, T. M., Wyse, B. D., Muralidharan, A. & Walther, T. A small molecule angiotensin II type 2 receptor (AT2R) antagonist produces analgesia in a rat model of neuropathic pain by inhibition of p38 mitogen-activated protein kinase (MAPK) and p44/p42 MAPK activation in the dorsal root ganglia. Pain Med. 14, 1557–1568 (2013).
Marion, E. et al. Mycobacterial toxin induces analgesia in buruli ulcer by targeting the angiotensin pathways. Cell 157, 1565–1576 (2014).
Lemmens, S., Brone, B., Dooley, D., Hendrix, S. & Geurts, N. Alpha-adrenoceptor modulation in central nervous system trauma: pain, spasms, and paralysis — an unlucky triad. Med. Res. Rev. 35, 653–677 (2015).
Giovannitti, J. A. Jr, Thoms, S. M. & Crawford, J. J. Alpha-2 adrenergic receptor agonists: a review of current clinical applications. Anesth. Prog. 62, 31–39 (2015).
Mori, K. et al. Effects of norepinephrine on rat cultured microglial cells that express alpha1, alpha2, beta1 and beta2 adrenergic receptors. Neuropharmacology 43, 1026–1034 (2002).
Lavand'homme, P. M. & Eisenach, J. C. Perioperative administration of the alpha2-adrenoceptor agonist clonidine at the site of nerve injury reduces the development of mechanical hypersensitivity and modulates local cytokine expression. Pain 105, 247–254 (2003).
Feng, X. et al. Intrathecal administration of clonidine attenuates spinal neuroimmune activation in a rat model of neuropathic pain with existing hyperalgesia. Eur. J. Pharmacol. 614, 38–43 (2009).
Wei, H. & Pertovaara, A. Spinal and pontine alpha2-adrenoceptors have opposite effects on pain-related behavior in the neuropathic rat. Eur. J. Pharmacol. 551, 41–49 (2006).
Schnabel, A., Meyer-Friessem, C. H., Reichl, S. U., Zahn, P. K. & Pogatzki-Zahn, E. M. Is intraoperative dexmedetomidine a new option for postoperative pain treatment? A meta-analysis of randomized controlled trials. Pain 154, 1140–1149 (2013).
Schnabel, A. et al. Efficacy and safety of intraoperative dexmedetomidine for acute postoperative pain in children: a meta-analysis of randomized controlled trials. Paediatr. Anaesth. 23, 170–179 (2013).
Melik Parsadaniantz, S., Rivat, C., Rostene, W. & Reaux-Le Goazigo, A. Opioid and chemokine receptor crosstalk: a promising target for pain therapy? Nat. Rev. Neurosci. 16, 69–78 (2015).
Abbadie, C. et al. Chemokines and pain mechanisms. Brain Res. Rev. 60, 125–134 (2009).
Reaux-Le Goazigo, A., Van Steenwinckel, J., Rostene, W. & Melik Parsadaniantz, S. Current status of chemokines in the adult CNS. Prog. Neurobiol. 104, 67–92 (2013).
Van Steenwinckel, J. et al. CCL2 released from neuronal synaptic vesicles in the spinal cord is a major mediator of local inflammation and pain after peripheral nerve injury. J. Neurosci. 31, 5865–5875 (2011).
Xie, F., Wang, Y., Li, X., Chao, Y. C. & Yue, Y. Early repeated administration of CXCR4 antagonist AMD3100 dose-dependently improves neuropathic pain in rats after L5 spinal nerve ligation. Neurochem. Res. 41, 2289–2299 (2016).
Talbot, S., Foster, S. L. & Woolf, C. J. Neuroimmunity: physiology and pathology. Annu. Rev. Immunol. 34, 421–447 (2016).
Szabo, I. et al. Heterologous desensitization of opioid receptors by chemokines inhibits chemotaxis and enhances the perception of pain. Proc. Natl Acad. Sci. USA 99, 10276–10281 (2002).
Szabo, I. et al. Selective inactivation of CCR5 and decreased infectivity of R5 HIV-1 strains mediated by opioid-induced heterologous desensitization. J. Leukoc. Biol. 74, 1074–1082 (2003).
Grimm, M. C. et al. Opiates transdeactivate chemokine receptors: delta and mu opiate receptor-mediated heterologous desensitization. J. Exp. Med. 188, 317–325 (1998).
Pello, O. M. et al. Ligand stabilization of CXCR4/delta-opioid receptor heterodimers reveals a mechanism for immune response regulation. Eur. J. Immunol. 38, 537–549 (2008).
Rivat, C. et al. Src family kinases involved in CXCL12-induced loss of acute morphine analgesia. Brain Behav. Immun. 38, 38–52 (2014).
Zhao, C. M. et al. Spinal MCP-1 contributes to the development of morphine antinociceptive tolerance in rats. Am. J. Med. Sci. 344, 473–479 (2012).
Zhang, N., Rogers, T. J., Caterina, M. & Oppenheim, J. J. Proinflammatory chemokines, such as C-C chemokine ligand 3, desensitize mu-opioid receptors on dorsal root ganglia neurons. J. Immunol. 173, 594–599 (2004).
Ye, D. et al. Activation of CXCL10/CXCR3 signaling attenuates morphine analgesia: involvement of Gi protein. J. Mol. Neurosci. 53, 571–579 (2014).
Rittner, H. L. et al. Pain control by CXCR2 ligands through Ca2+-regulated release of opioid peptides from polymorphonuclear cells. FASEB J. 20, 2627–2629 (2006).
Wilson, N. M., Jung, H., Ripsch, M. S., Miller, R. J. & White, F. A. CXCR4 signaling mediates morphine-induced tactile hyperalgesia. Brain Behav. Immun. 25, 565–573 (2011).
Kalliomaki, J. et al. A randomized, double-blind, placebo-controlled trial of a chemokine receptor 2 (CCR2) antagonist in posttraumatic neuralgia. Pain 154, 761–767 (2013).
Padi, S. S. et al. Attenuation of rodent neuropathic pain by an orally active peptide, RAP-103, which potently blocks CCR2- and CCR5-mediated monocyte chemotaxis and inflammation. Pain 153, 95–106 (2012).
Szallasi, A., Cortright, D. N., Blum, C. A. & Eid, S. R. The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof-of-concept. Nat. Rev. Drug Discov. 6, 357–372 (2007).
Habib, A. M., Wood, J. N. & Cox, J. J. Sodium channels and pain. Handb. Exp. Pharmacol. 227, 39–56 (2015).
Waxman, S. G. et al. Sodium channel genes in pain-related disorders: phenotype-genotype associations and recommendations for clinical use. Lancet Neurol. 13, 1152–1160 (2014).
Caterina, M. J. et al. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389, 816–824 (1997).
Nilius, B. & Szallasi, A. Transient receptor potential channels as drug targets: from the science of basic research to the art of medicine. Pharmacol. Rev. 66, 676–814 (2014).
Moran, M. M., McAlexander, M. A., Biro, T. & Szallasi, A. Transient receptor potential channels as therapeutic targets. Nat. Rev. Drug Discov. 10, 601–620 (2011).
Carnevale, V. & Rohacs, T. TRPV1: a target for rational drug design. Pharmaceuticals (Basel) 9, E52 (2016).
Lehto, S. G. et al. Antihyperalgesic effects of (R,E)-N-(2-hydroxy-2,3-dihydro-1H-inden-4-yl)-3-(2-(piperidin-1-yl)-4-(trifluorom ethyl)phenyl)-acrylamide (AMG8562), a novel transient receptor potential vanilloid type 1 modulator that does not cause hyperthermia in rats. J. Pharmacol. Exp. Ther. 326, 218–229 (2008).
Watabiki, T. et al. Amelioration of neuropathic pain by novel transient receptor potential vanilloid 1 antagonist AS1928370 in rats without hyperthermic effect. J. Pharmacol. Exp. Ther. 336, 743–750 (2011).
Chiche, D., Brown, W. & Walker, P. NEO6860, a novel modality selective TRPV1 antagonist: results from a phase I, double-blind, placebo-controlled study in healthy subjects. J. Pain 17, S79 (2016).
Gao, Y., Cao, E., Julius, D. & Cheng, Y. TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 534, 347–351 (2016).
Cao, L. et al. Pharmacological reversal of a pain phenotype in iPSC-derived sensory neurons and patients with inherited erythromelalgia. Sci. Transl Med. 8, 335ra56 (2016).
Dib-Hajj, S. D., Yang, Y., Black, J. A. & Waxman, S. G. The NaV1.7 sodium channel: from molecule to man. Nat. Rev. Neurosci. 14, 49–62 (2013).
Cox, J. J. et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature 444, 894–898 (2006).
Cox, J. J. et al. Congenital insensitivity to pain: novel SCN9A missense and in-frame deletion mutations. Hum. Mut. 31, E1670–E1686 (2010).
Nassar, M. A. et al. Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc. Natl Acad. Sci. USA 101, 12706–12711 (2004).
Minett, M. S. et al. Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat. Commun. 3, 791 (2012).
Gingras, J. et al. Global Nav1.7 knockout mice recapitulate the phenotype of human congenital indifference to pain. PLoS ONE 9, e105895 (2014).
Bagal, S. K., Marron, B. E., Owen, R. M., Storer, R. I. & Swain, N. A. Voltage gated sodium channels as drug discovery targets. Channels (Austin) 9, 360–366 (2015).
Sun, S., Cohen, C. J. & Dehnhardt, C. M. Inhibitors of voltage-gated sodium channel Nav1.7: patent applications since 2010. Pharm. Pat. Anal. 3, 509–521 (2014).
Focken, T. et al. Discovery of aryl sulfonamides as isoform-selective inhibitors of NaV1.7 with efficacy in rodent pain models. ACS Med. Chem. Lett. 7, 277–282 (2016).
McCormack, K. et al. Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proc. Natl Acad. Sci. USA 110, E2724–E2732 (2013).
Butt, M. et al. Morphologic, stereologic, and morphometric evaluation of the nervous system in young cynomolgus monkeys (Macaca fascicularis) following maternal administration of tanezumab, a monoclonal antibody to nerve growth factor. Toxicol. Sci. 142, 463–476 (2014).
Wainger, B. J. et al. Modeling pain in vitro using nociceptor neurons reprogrammed from fibroblasts. Nat. Neurosci. 18, 17–24 (2015).
Talbot, S. et al. Silencing nociceptor neurons reduces allergic airway inflammation. Neuron 87, 341–354 (2015).
Bauer, C. S. et al. The increased trafficking of the calcium channel subunit alpha2delta-1 to presynaptic terminals in neuropathic pain is inhibited by the alpha2delta ligand pregabalin. J. Neurosci. 29, 4076–4088 (2009).
Lawrence, J. Nav1.7: a new channel for pain treatment. Pharm. J. http://dx.doi.org/10.1211/PJ.2016.20200841 (2016).
Bowman, C. J. et al. Developmental toxicity assessment of tanezumab, an anti-nerve growth factor monoclonal antibody, in cynomolgus monkeys (Macaca fascicularis). Reprod. Toxicol. 53, 105–118 (2015).
Murray, J. K. et al. Single residue substitutions that confer voltage-gated sodium ion channel subtype selectivity in the NaV1.7 inhibitory peptide GpTx-1. J. Med. Chem. 59, 2704–2717 (2016).
Weiss, J. et al. Loss-of-function mutations in sodium channel Nav1.7 cause anosmia. Nature 472, 186–190 (2011).
Minett, M. S. et al. Endogenous opioids contribute to insensitivity to pain in humans and mice lacking sodium channel Nav1.7. Nat. Commun. 6, 8967 (2015).
Kort, M. E. et al. Subtype-selective NaV1.8 sodium channel blockers: identification of potent, orally active nicotinamide derivatives. Bioorg. Med. Chem. Lett. 20, 6812–6815 (2010).
Bagal, S. K. et al. Recent progress in sodium channel modulators for pain. Bioorg. Med. Chem. Lett. 24, 3690–3699 (2014).
Wilson, M. J. et al. μ-Conotoxins that differentially block sodium channels NaV1.1 through 1.8 identify those responsible for action potentials in sciatic nerve. Proc. Natl Acad. Sci. USA 108, 10302–10307 (2011).
Rush, A. M., Cummins, T. R. & Waxman, S. G. Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J. Physiol. 579, 1–14 (2007).
Dib-Hajj, S. D., Black, J. A. & Waxman, S. G. NaV1.9: a sodium channel linked to human pain. Nat. Rev. Neurosci. 16, 511–519 (2015).
Goral, R. O., Leipold, E., Nematian-Ardestani, E. & Heinemann, S. H. Heterologous expression of NaV1.9 chimeras in various cell systems. Pflugers Arch. 467, 2423–2435 (2015).
Owsianik, G., Talavera, K., Voets, T. & Nilius, B. Permeation and selectivity of TRP channels. Annu. Rev. Physiol. 68, 685–717 (2006).
Hellwig, N. et al. TRPV1 acts as proton channel to induce acidification in nociceptive neurons. J. Biol. Chem. 279, 34553–34561 (2004).
Meyers, J. R. et al. Lighting up the senses: FM1-43 loading of sensory cells through nonselective ion channels. J. Neurosci. 23, 4054–4065 (2003).
Binshtok, A. M., Bean, B. P. & Woolf, C. J. Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature 449, 607–610 (2007).
Puopolo, M. et al. Permeation and block of TRPV1 channels by the cationic lidocaine derivative QX-314. J. Neurophysiol. 109, 1704–1712 (2013).
Brenneis, C. et al. Bupivacaine-induced cellular entry of QX-314 and its contribution to differential nerve block. Br. J. Pharmacol. 171, 438–451 (2014).
Virginio, C., MacKenzie, A., Rassendren, F. A., North, R. A. & Surprenant, A. Pore dilation of neuronal P2X receptor channels. Nat. Neurosci. 2, 315–321 (1999).
Khakh, B. S., Bao, X. R., Labarca, C. & Lester, H. A. Neuronal P2X transmitter-gated cation channels change their ion selectivity in seconds. Nat. Neurosci. 2, 322–330 (1999).
Yan, Z., Li, S., Liang, Z., Tomic, M. & Stojilkovic, S. S. The P2X7 receptor channel pore dilates under physiological ion conditions. J. Gen. Physiol. 132, 563–573 (2008).
Binshtok, A. M. et al. Coapplication of lidocaine and the permanently charged sodium channel blocker QX-314 produces a long-lasting nociceptive blockade in rodents. Anesthesiology 111, 127–137 (2009).
Roberson, D. P., Binshtok, A. M., Blasl, F., Bean, B. P. & Woolf, C. J. Targeting of sodium channel blockers into nociceptors to produce long-duration analgesia: a systematic study and review. Br. J. Pharmacol. 164, 48–58 (2011).
Leffler, A. et al. The vanilloid receptor TRPV1 is activated and sensitized by local anesthetics in rodent sensory neurons. J. Clin. Invest. 118, 763–776 (2008).
Leffler, A., Lattrell, A., Kronewald, S., Niedermirtl, F. & Nau, C. Activation of TRPA1 by membrane permeable local anesthetics. Mol. Pain 7, 62 (2011).
Roberson, D. P. et al. Activity-dependent silencing reveals functionally distinct itch-generating sensory neurons. Nat. Neurosci. 16, 910–918 (2013).
Zamponi, G. W., Striessnig, J., Koschak, A. & Dolphin, A. C. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol. Rev. 67, 821–870 (2015).
Zamponi, G. W. Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nat. Rev. Drug Discov. 15, 19–34 (2016). In this Review, Zamponi discusses various challenges and opportunities for using calcium channels as drug targets for neurological disorders.
Abbadie, C. et al. Analgesic effects of a substituted N-triazole oxindole (TROX-1), a state-dependent, voltage-gated calcium channel 2 blocker. J. Pharmacol. Exp. Ther. 334, 545–555 (2010).
Patel, R. et al. Electrophysiological characterization of activation state-dependent CaV2 channel antagonist TROX-1 in spinal nerve injured rats. Neuroscience 297, 47–57 (2015).
Shao, P. P. et al. Aminopiperidine sulfonamide Cav2.2 channel inhibitors for the treatment of chronic pain. J. Med. Chem. 55, 9847–9855 (2012).
Lipscombe, D. & Andrade, A. Calcium channel CaVα1 splice isoforms — tissue specificity and drug action. Curr. Mol. Pharmacol. 8, 22–31 (2015).
Bourinet, E. et al. Silencing of the Cav3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J. 24, 315–324 (2005).
Choi, S. et al. Attenuated pain responses in mice lacking CaV3.2 T-type channels. Genes Brain Behav. 6, 425–431 (2007).
Jarvis, M. F. et al. A peripherally acting, selective T-type calcium channel blocker, ABT-639, effectively reduces nociceptive and neuropathic pain in rats. Biochem. Pharmacol. 89, 536–544 (2014).
Wallace, M., Duan, R., Liu, W., Locke, C. & Nothaft, W. A. Randomized, double-blind, placebo-controlled, crossover study of the T-type calcium channel blocker ABT-639 in an intradermal capsaicin experimental pain model in healthy adults. Pain Med. 17, 551–560 (2015).
Ziegler, D., Duan, W. R., An, G., Thomas, J. W. & Nothaft, W. A randomized double-blind, placebo-, and active-controlled study of T-type calcium channel blocker ABT-639 in patients with diabetic peripheral neuropathic pain. Pain 156, 2013–2020 (2015).
Francois, A. et al. State-dependent properties of a new T-type calcium channel blocker enhance CaV3.2 selectivity and support analgesic effects. Pain 154, 283–293 (2013).
Xu, J. et al. A mixed Ca2+ channel blocker, A-1264087, utilizes peripheral and spinal mechanisms to inhibit spinal nociceptive transmission in a rat model of neuropathic pain. J. Neurophysiol. 111, 394–404 (2014).
Zhu, C. Z. et al. Mechanistic insights into the analgesic efficacy of A-1264087, a novel neuronal Ca2+ channel blocker that reduces nociception in rat preclinical pain models. J. Pain 387, e1–e14 (2014).
Scott, V. E. et al. A-1048400 is a novel, orally active, state-dependent neuronal calcium channel blocker that produces dose-dependent antinociception without altering hemodynamic function in rats. Biochem. Pharmacol. 83, 406–418 (2012).
Marsh, B., Acosta, C., Djouhri, L. & Lawson, S. N. Leak K+ channel mRNAs in dorsal root ganglia: relation to inflammation and spontaneous pain behaviour. Mol. Cell. Neurosci. 49, 375–386 (2012).
Pollema-Mays, S. L., Centeno, M. V., Ashford, C. J., Apkarian, A. V. & Martina, M. Expression of background potassium channels in rat DRG is cell-specific and down-regulated in a neuropathic pain model. Mol. Cell. Neurosci. 57, 1–9 (2013).
Zheng, Q. et al. Suppression of KCNQ/M (Kv7) potassium channels in dorsal root ganglion neurons contributes to the development of bone cancer pain in a rat model. Pain 154, 434–448 (2013).
Laumet, G. et al. G9a is essential for epigenetic silencing of K+ channel genes in acute-to-chronic pain transition. Nat. Neurosci. 18, 1746–1755 (2015).
Lu, R. et al. Slack channels expressed in sensory neurons control neuropathic pain in mice. J. Neurosci. 35, 1125–1135 (2015).
Lyu, C. et al. G protein-gated inwardly rectifying potassium channel subunits 1 and 2 are down-regulated in rat dorsal root ganglion neurons and spinal cord after peripheral axotomy. Mol. Pain 11, 44 (2015).
Maljevic, S. & Lerche, H. Potassium channels: a review of broadening therapeutic possibilities for neurological diseases. J. Neurol. 260, 2201–2211 (2013).
Tsantoulas, C. & McMahon, S. B. Opening paths to novel analgesics: the role of potassium channels in chronic pain. Trends Neurosci. 37, 146–158 (2014).
Tsantoulas, C. Emerging potassium channel targets for the treatment of pain. Curr. Opin. Support. Palliat. Care 9, 147–154 (2015).
Wickenden, A. D. & McNaughton-Smith, G. Kv7 channels as targets for the treatment of pain. Curr. Pharm. Des. 15, 1773–1798 (2009).
Wu, Y. J. et al. Discovery of (S,E)-3-(2-fluorophenyl)-N-(1-(3-(pyridin-3-yloxy)phenyl)ethyl)-acrylamide as a potent and efficacious KCNQ2 (Kv7.2) opener for the treatment of neuropathic pain. Bioorg. Med. Chem. Lett. 23, 6188–6191 (2013).
Zheng, Y. et al. Activation of peripheral KCNQ channels relieves gout pain. Pain 156, 1025–1035 (2015).
Mathie, A. & Veale, E. L. Two-pore domain potassium channels: potential therapeutic targets for the treatment of pain. Pflugers Arch. 467, 931–943 (2015).
Wang, H. S. et al. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282, 1890–1893 (1998).
Pan, Z. et al. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J. Neurosci. 26, 2599–2613 (2006).
Cooper, E. C. Made for “anchorin”: Kv7.2/7.3 (KCNQ2/KCNQ3) channels and the modulation of neuronal excitability in vertebrate axons. Semin. Cell Dev. Biol. 22, 185–192 (2011).
Blackburn-Munro, G. & Jensen, B. S. The anticonvulsant retigabine attenuates nociceptive behaviours in rat models of persistent and neuropathic pain. Eur. J. Pharmacol. 460, 109–116 (2003).
Li, H. et al. Antinociceptive efficacy of retigabine in the monosodium lodoacetate rat model for osteoarthritis pain. Pharmacology 95, 251–257 (2015).
Rose, K. et al. Transcriptional repression of the M channel subunit Kv7.2 in chronic nerve injury. Pain 152, 742–754 (2011).
King, C. H., Lancaster, E., Salomon, D., Peles, E. & Scherer, S. S. Kv7.2 regulates the function of peripheral sensory neurons. J. Comp. Neurol. 522, 3262–3280 (2014).
King, C. H. & Scherer, S. S. Kv7.5 is the primary Kv7 subunit expressed in C-fibers. J. Comp. Neurol. 520, 1940–1950 (2012).
Feliciangeli, S., Chatelain, F. C., Bichet, D. & Lesage, F. The family of K2P channels: salient structural and functional properties. J. Physiol. 593, 2587–2603 (2015).
Busserolles, J., Tsantoulas, C., Eschalier, A. & Lopez Garcia, J. A. Potassium channels in neuropathic pain: advances, challenges, and emerging ideas. Pain 157 (Suppl. 1), S7–S14 (2016).
Devilliers, M. et al. Activation of TREK-1 by morphine results in analgesia without adverse side effects. Nat. Commun. 4, 2941 (2013).
Bagriantsev, S. N. et al. A high-throughput functional screen identifies small molecule regulators of temperature- and mechano-sensitive K2P channels. ACS Chem. Biol. 8, 1841–1851 (2013).
Rodrigues, N. et al. Synthesis and structure-activity relationship study of substituted caffeate esters as antinociceptive agents modulating the TREK-1 channel. Eur. J. Med. Chem. 75, 391–402 (2014).
Bocksteins, E. & Snyders, D. J. Electrically silent Kv subunits: their molecular and functional characteristics. Physiology (Bethesda) 27, 73–84 (2012).
Bocksteins, E. Kv5, Kv6, Kv8, and Kv9 subunits: no simple silent bystanders. J. Gen. Physiol. 147, 105–125 (2016).
Tsantoulas, C. et al. Sensory neuron downregulation of the Kv9.1 potassium channel subunit mediates neuropathic pain following nerve injury. J. Neurosci. 32, 17502–17513 (2012).
Tsantoulas, C. et al. Kv2 dysfunction after peripheral axotomy enhances sensory neuron responsiveness to sustained input. Exp. Neurol. 251, 115–126 (2014).
Bocksteins, E. et al. Kv2.1 and silent Kv subunits underlie the delayed rectifier K+ current in cultured small mouse DRG neurons. Am. J. Physiol. Cell Physiol. 296, C1271–C1278 (2009).
Stas, J. I., Bocksteins, E., Labro, A. J. & Snyders, D. J. Modulation of closed-state inactivation in Kv2.1/Kv6.4 heterotetramers as mechanism for 4-AP induced potentiation. PLoS ONE 10, e0141349 (2015).
Knabl, J. et al. Reversal of pathological pain through specific spinal GABAA receptor subtypes. Nature 451, 330–334 (2008).
Bonin, R. P. & De Koninck, Y. Restoring ionotropic inhibition as an analgesic strategy. Neurosci Lett. 557, 43–51 (2013).
Klinger, F. et al. δ subunit-containing GABAA receptors are preferred targets for the centrally acting analgesic flupirtine. Br. J. Pharmacol. 172, 4946–4958 (2015).
Zeilhofer, H. U., Ralvenius, W. T. & Acuna, M. A. Restoring the spinal pain gate: GABAA receptors as targets for novel analgesics. Adv. Pharmacol. 73, 71–96 (2015).
Tegeder, I. et al. GTP cyclohydrolase and tetrahydrobiopterin regulate pain sensitivity and persistence. Nat. Med. 12, 1269–1277 (2006).
Latremoliere, A. et al. Reduction of neuropathic and inflammatory pain through inhibition of the tetrahydrobiopterin pathway. Neuron 86, 1393–1406 (2015).
Chidley, C., Haruki, H., Pedersen, M. G., Muller, E. & Johnsson, K. A yeast-based screen reveals that sulfasalazine inhibits tetrahydrobiopterin biosynthesis. Nat. Chem. Biol. 7, 375–383 (2011).
Chandrasekhar, S. et al. Identification and characterization of novel microsomal prostaglandin E synthase-1 inhibitors for analgesia. J. Pharmacol. Exp. Ther. 356, 635–644 (2016).
Jin, Y. et al. Pharmacodynamic comparison of LY3023703, a novel microsomal prostaglandin E synthase 1 inhibitor, with celecoxib. Clin. Pharmacol. Ther. 99, 274–284 (2016).
Wagner, K., Yang, J., Inceoglu, B. & Hammock, B. D. Soluble epoxide hydrolase inhibition is antinociceptive in a mouse model of diabetic neuropathy. J. Pain 15, 907–914 (2014).
Inceoglu, B. et al. Endoplasmic reticulum stress in the peripheral nervous system is a significant driver of neuropathic pain. Proc. Natl Acad. Sci. USA 112, 9082–9087 (2015).
Sisignano, M. et al. 5,6-EET is released upon neuronal activity and induces mechanical pain hypersensitivity via TRPA1 on central afferent terminals. J. Neurosci. 32, 6364–6372 (2012).
Brenneis, C. et al. Soluble epoxide hydrolase limits mechanical hyperalgesia during inflammation. Mol. Pain 7, 78 (2011).
Lam, D. K., Dang, D., Zhang, J., Dolan, J. C. & Schmidt, B. L. Novel animal models of acute and chronic cancer pain: a pivotal role for PAR2. J. Neurosci. 32, 14178–14183 (2012).
Christianson, C. A. et al. Spinal matrix metalloproteinase 3 mediates inflammatory hyperalgesia via a tumor necrosis factor-dependent mechanism. Neuroscience 200, 199–210 (2012).
Berta, T. et al. Extracellular caspase-6 drives murine inflammatory pain via microglial TNF-alpha secretion. J. Clin. Invest. 124, 1173–1186 (2014).
Clark, A. K., Grist, J., Al-Kashi, A., Perretti, M. & Malcangio, M. Spinal cathepsin S and fractalkine contribute to chronic pain in the collagen-induced arthritis model. Arthritis Rheum. 64, 2038–2047 (2012).
Vicuna, L. et al. The serine protease inhibitor SerpinA3N attenuates neuropathic pain by inhibiting T cell-derived leukocyte elastase. Nat. Med. 21, 518–523 (2015).
Landry, R. P., Jacobs, V. L., Romero-Sandoval, E. A. & DeLeo, J. A. Propentofylline, a CNS glial modulator does not decrease pain in post-herpetic neuralgia patients: in vitro evidence for differential responses in human and rodent microglia and macrophages. Exp. Neurol. 234, 340–350 (2012).
Ji, R. R. Mitogen-activated protein kinases as potential targets for pain killers. Curr. Opin. Investig. Drugs 5, 71–75 (2004).
Ostenfeld, T. et al. A randomized, placebo-controlled trial of the analgesic efficacy and safety of the p38 MAP kinase inhibitor, losmapimod, in patients with neuropathic pain from lumbosacral radiculopathy. Clin. J. Pain 31, 283–293 (2015).
Smith, M. T., Wyse, B. D. & Edwards, S. R. Small molecule angiotensin II type 2 receptor (AT2R) antagonists as novel analgesics for neuropathic pain: comparative pharmacokinetics, radioligand binding, and efficacy in rats. Pain Med. 14, 692–705 (2013).
Bevan, S., Quallo, T. & Andersson, D. A. TRPV1. Handb. Exp. Pharmacol. 222, 207–245 (2014).
Girardin, F. Membrane transporter proteins: a challenge for CNS drug development. Dialogues Clin. Neurosci. 8, 311–321 (2006).
Huggins, J. P., Smart, T. S., Langman, S., Taylor, L. & Young, T. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain 153, 1837–1846 (2012).
Keith, J. M. et al. Preclinical characterization of the FAAH inhibitor JNJ-42165279. ACS Med. Chem. Lett. 6, 1204–1208 (2015).
Pawsey, S. et al. Safety, tolerability and pharmacokinetics of FAAH inhibitor V158866: a double-blind, randomised, placebo-controlled phase I study in healthy volunteers. Drugs R. D 16, 181–191 (2016).
Cajanus, K. et al. Effect of endocannabinoid degradation on pain: role of FAAH polymorphisms in experimental and postoperative pain in women treated for breast cancer. Pain 157, 361–369 (2016).
Woolf, C. J., Safieh-Garabedian, B., Ma, Q. P., Crilly, P. & Winter, J. Nerve growth factor contributes to the generation of inflammatory sensory hypersensitivity. Neuroscience 62, 327–331 (1994).
Gimbel, J. S. et al. Long-term safety and effectiveness of tanezumab as treatment for chronic low back pain. Pain 155, 1793–1801 (2014).
Bramson, C. et al. Exploring the role of tanezumab as a novel treatment for the relief of neuropathic pain. Pain Med. 16, 1163–1176 (2015).
Schnitzer, T. J. & Marks, J. A. A systematic review of the efficacy and general safety of antibodies to NGF in the treatment of OA of the hip or knee. Osteoarthritis Cartilage 23 (Suppl. 1), S8–S17 (2015).
Hochberg, M. C. et al. When is osteonecrosis not osteonecrosis? Adjudication of reported serious adverse joint events in the tanezumab clinical development program. Arthritis Rheumatol. 68, 382–391 (2016).
Sorge, R. E. et al. Olfactory exposure to males, including men, causes stress and related analgesia in rodents. Nat. Methods 11, 629–632 (2014).
Baron, R. et al. Peripheral neuropathic pain: a mechanism-related organizing principle based on sensory profiles. Pain 158, 261–272 (2016).
Mogil, J. S., Davis, K. D. & Derbyshire, S. W. The necessity of animal models in pain research. Pain 151, 12–17 (2010). In this review, Mogil and colleagues describe the crucial role of animal models in the development of clinical analgesics and propose advancements in animal models of pain that could improve the relevance and translatability of preclinical studies.
Whiteside, G. T., Pomonis, J. D. & Kennedy, J. D. An industry perspective on the role and utility of animal models of pain in drug discovery. Neurosci. Lett. 557, 65–72 (2013).
Hill, R. NK1 (substance P) receptor antagonists — why are they not analgesic in humans? Trends Pharmacol. Sci. 21, 244–246 (2000).
Sorge, R. E. et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat. Neurosci. 18, 1081–1083 (2015).
Edwards, R. R., Dworkin, R. H., Sullivan, M. D., Turk, D. C. & Wasan, A. D. The role of psychosocial processes in the development and maintenance of chronic pain. J. Pain 17, T70–T92 (2016).
Smith, S. M. et al. Pain intensity rating training: results from an exploratory study of the ACTTION PROTECCT system. Pain 157, 1056–1064 (2016).
Dodd, S., Dean, O. M., Vian, J. & Berk, M. A. Review of the theoretical and biological understanding of the nocebo and placebo phenomena. Clin. Ther. 39, 469–476 (2017).
Paice, J. A. et al. AAPT diagnostic criteria for chronic cancer pain conditions. J. Pain 18, 233–246 (2017).
Smith, S. M. et al. The potential role of sensory testing, skin biopsy, and functional brain imaging as biomarkers in chronic pain clinical trials: IMMPACT considerations. J. Pain http://dx.doi.org/10.1016/j.jpain.2017.02.429 (2017).
Edwards, R. R. et al. Patient phenotyping in clinical trials of chronic pain treatments: IMMPACT recommendations. Pain 157, 1851–1871 (2016).
Viscusi, E. R. et al. A randomized, phase 2 study investigating TRV130, a biased ligand of the mu-opioid receptor, for the intravenous treatment of acute pain. Pain 157, 264–272 (2016).
Lu, Z. et al. Mediation of opioid analgesia by a truncated 6-transmembrane GPCR. J. Clin. Invest. 125, 2626–2630 (2015).
Moriconi, A. et al. Targeting the minor pocket of C5aR for the rational design of an oral allosteric inhibitor for inflammatory and neuropathic pain relief. Proc. Natl Acad. Sci. USA 111, 16937–16942 (2014).
Altun, A. et al. Attenuation of morphine antinociceptive tolerance by cannabinoid CB1 and CB2 receptor antagonists. J. Physiol. Sci. 65, 407–415 (2015).
Haugh, O., Penman, J., Irving, A. J. & Campbell, V. A. The emerging role of the cannabinoid receptor family in peripheral and neuro-immune interactions. Curr. Drug Targets 17, 1834–1840 (2016).
Deliu, E. et al. The lysophosphatidylinositol receptor GPR55 modulates pain perception in the periaqueductal gray. Mol. Pharmacol. 88, 265–272 (2015).
Kort, M. E. & Kym, P. R. TRPV1 antagonists: clinical setbacks and prospects for future development. Prog. Med. Chem. 51, 57–70 (2012).
Manitpisitkul, P. et al. Safety, tolerability and pharmacokinetic and pharmacodynamic learnings from a double-blind, randomized, placebo-controlled, sequential group first-in-human study of the TRPV1 antagonist, JNJ-38893777, in healthy men. Clin. Drug Investig. 35, 353–363 (2015).
Quiding, H. et al. TRPV1 antagonistic analgesic effect: a randomized study of AZD1386 in pain after third molar extraction. Pain 154, 808–812 (2013).
De Petrocellis, L. & Moriello, A. S. Modulation of the TRPV1 channel: current clinical trials and recent patents with focus on neurological conditions. Recent Pat. CNS Drug Discov. 8, 180–204 (2013).
Ghosh, S. et al. Full fatty acid amide hydrolase inhibition combined with partial monoacylglycerol lipase inhibition: augmented and sustained antinociceptive effects with reduced cannabimimetic side effects in mice. J. Pharmacol. Exp. Ther. 354, 111–120 (2015).
Grim, T. W. et al. Combined inhibition of FAAH and COX produces enhanced anti-allodynic effects in mouse neuropathic and inflammatory pain models. Pharmacol. Biochem. Behav. 124, 405–411 (2014).
Starowicz, K. et al. Full inhibition of spinal FAAH leads to TRPV1-mediated analgesic effects in neuropathic rats and possible lipoxygenase-mediated remodeling of anandamide metabolism. PLoS ONE 8, e60040 (2013).
Schlosburg, J. E. et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat. Neurosci. 13, 1113–1119 (2010).
Luz, J. G. et al. Crystal structures of mPGES-1 inhibitor complexes form a basis for the rational design of potent analgesic and anti-inflammatory therapeutics. J. Med. Chem. 58, 4727–4737 (2015).
Leclerc, P. et al. Characterization of a human and murine mPGES-1 inhibitor and comparison to mPGES-1 genetic deletion in mouse models of inflammation. Prostaglandins Other Lipid Mediat. 107, 26–34 (2013).
Sjogren, T. et al. Crystal structure of microsomal prostaglandin E2 synthase provides insight into diversity in the MAPEG superfamily. Proc. Natl Acad. Sci. USA 110, 3806–3811 (2013).
Hellio le Graverand, M. P. et al. A 2-year randomised, double-blind, placebo-controlled, multicentre study of oral selective iNOS inhibitor, cindunistat (SD-6010), in patients with symptomatic osteoarthritis of the knee. Ann. Rheum. Dis. 72, 187–195 (2013).
Wozniak, K. M. et al. The orally active glutamate carboxypeptidase II inhibitor E2072 exhibits sustained nerve exposure and attenuates peripheral neuropathy. J. Pharmacol. Exp. Ther. 343, 746–754 (2012).
Vornov, J. J. et al. Pharmacokinetics and pharmacodynamics of the glutamate carboxypeptidase II inhibitor 2-MPPA show prolonged alleviation of neuropathic pain through an indirect mechanism. J. Pharmacol. Exp. Ther. 346, 406–413 (2013).
Huang, J. L., Chen, X. L., Guo, C. & Wang, Y. X. Contributions of spinal D-amino acid oxidase to bone cancer pain. Amino Acids 43, 1905–1918 (2012).
Acknowledgements
The authors acknowledge generous funding support from the following US National Institutes of Health grants: NS039518 and NS038253 (US National Institute of Neurological Disorders and Stroke (NINDS) to C.J.W); NS072040 and NS036855 (NINDS to B.P.B.); and DA041912 (US National Institute on Drug Abuse to A.S.Y.).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
A.S.Y and D.P.R. are co-founders of Blue Therapeutics in Allston, Massachusetts, USA, which is focused on developing non-addictive painkillers targeting G-protein-coupled receptor heteromers. A.S.Y. holds a patent on an analgesic agent. B.P.B. is a co-founder of Flex Pharma in Boston, Massachusetts, USA, and co-holds patents on using charged sodium channel blockers for pain relief and other indications. C.J.W is co-founder and scientific advisor to Quartet Medicine in Cambridge, Massachusetts, USA, which is focused on developing treatments for chronic pain and inflammation targeting the tetrahydrobiopterin pathway; he is also a consultant and stock holder for Abide Therapeutics in San Diego, California, USA, and holds several patents related to methods and approaches for studying and treating pain.
Related links
DATABASES
FURTHER INFORMATION
Glossary
- Analgesics
-
Pharmacological agents or ligands that produce analgesia.
- Tolerance
-
A state in which the drug no longer produces the same effect and a higher dose is therefore needed.
- Dependence
-
An adaptive state that develops when a pharmacological agent is used repeatedly and leads to withdrawal on cessation of the drug regimen.
- Hyperalgesia
-
Enhanced nociceptive response to a noxious stimulus, leading to greater discomfort than before.
- Allodynia
-
Nociceptive response elicited even to previously non-noxious stimuli.
- Phenocopy
-
When an organism shows phenotypic characteristics that reflect a different genotype from its own.
- Neuropathic pain
-
A condition leading to pain due to damage or disease of nervous system tissues.
- Central sensitization
-
A condition of the nervous system in which neurons in the central nervous system are in a state of prolonged increase in excitability and synaptic efficacy, coupled with the loss of inhibitory activity.
- Analgesia
-
A lack of, or insensibility to, pain.
- Nociception
-
Sensory neuronal responses to noxious or damaging stimuli that attribute the sensation of pain.
- Depersonalization
-
A state in which an individual's thoughts, feelings and emotions seem to not belong to them.
- Antinociception
-
Inhibition of sensory neuronal response to noxious stimuli that leads to reduction of pain sensation.
- Ligand bias
-
Occurs when a ligand shows selectivity or preference to a particular signal transduction mechanism for a target receptor. Also called 'functional selectivity'.
- Psychotomimetic effects
-
A state of psychosis of the mind leading to delusions, hallucinations, and so on that are precipitated by a pharmacological agent or ligand.
- Post-herpetic neuralgia
-
Pain caused by nerve damage due to infection with varicella zoster virus.
- Withdrawal
-
Symptoms such as anxiety and shaking that develop on cessation of a drug that has been used repeatedly.
- Trigeminal neuralgia
-
A painful condition caused by disease affecting, dysfunction of or damage to the trigeminal nerve.
- Anosmia
-
Loss of the sense of smell.
- Allosteric modulators
-
Ligands that alter the activity of an agonist, antagonist or inverse agonist of a target by binding to a site distinct from the active site.
- Phenotypic screens
-
Unbiased screening strategies in which the functional output is a pre-determined alteration of phenotype.
Rights and permissions
About this article

Cite this article
Yekkirala, A., Roberson, D., Bean, B. et al. Breaking barriers to novel analgesic drug development. Nat Rev Drug Discov 16, 545–564 (2017). https://doi.org/10.1038/nrd.2017.87
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrd.2017.87
This article is cited by
-
Single-nucleus transcriptomic atlas of glial cells in human dorsal root ganglia
Anesthesiology and Perioperative Science (2023)
-
Network pharmacological analysis to reveal the mechanism governing the effect of Qin Xi Tong on osteoarthritis and rheumatoid arthritis
Clinical Rheumatology (2023)
-
Nociceptin Receptor-Related Agonists as Safe and Non-addictive Analgesics
Drugs (2023)
-
Characterization of the Dahl salt-sensitive rat as a rodent model of inherited, widespread, persistent pain
Scientific Reports (2022)
-
Drug Repurposing to Target Neuroinflammation and Sensory Neuron-Dependent Pain
Drugs (2022)
