
Key Points
-
Phospholipase D (PLD) enzymes are one source of receptor-generated phosphatidic acid (PtdOH),which may subsequently be metabolized to diacylglycerol (DAG) and lysophosphatidic acid. There are other pathways that lead to PtdOH generation, but differences in pathways and in the acyl composition of the products seem to provide some specificity.
-
Both direct and indirect inhibitors of PLD activity have been identified despite a long-held suspicion that this pathway was undruggable. The identification of raloxifene and halopemide as direct inhibitors was followed by the systematic development of isoenzyme-preferring compounds that have been used to further differentiate the functions of PLD1 and PLD2.
-
PLD2 in host cells has been associated with viral entry processes and innate immune response pathways such that inhibition blocks efficient infection. This PLD2 pathway has been linked to autophagy via AKT kinases.
-
As a potential target in antiretroviral therapy, PLD1 works through the CAD enzyme (which contains carbamoyl aspartate synthase, aspartate transcarbamylase and dihydro-orotase domains) to modulate pyrimidine biosynthesis.
-
PLD activity and expression have been shown to be upregulated in several types of human cancers, in which PLD enzymes function downstream of a variety of known oncogenes. Inhibition of PtdOH production has a marked effect on tumorigenesis and malignant invasion.
-
PLD1, PLD2 and PLD3 have each been suggested to have a role in Alzheimer disease and other neurodegenerative conditions, but a mechanism has not yet emerged to explain the roles of these proteins in central nervous system pathophysiology.
Abstract
Lipid second messengers have essential roles in cellular function and contribute to the molecular mechanisms that underlie inflammation, malignant transformation, invasiveness, neurodegenerative disorders, and infectious and other pathophysiological processes. The phospholipase D (PLD) isoenzymes PLD1 and PLD2 are one of the major sources of signal-activated phosphatidic acid (PtdOH) generation downstream of a variety of cell-surface receptors, including G protein-coupled receptors (GPCRs), receptor tyrosine kinases (RTKs) and integrins. Recent advances in the development of isoenzyme-selective PLD inhibitors and in molecular genetics have suggested that PLD isoenzymes in mammalian cells and pathogenic organisms may be valuable targets for the treatment of several human diseases. Isoenzyme-selective inhibitors have revealed complex inter-relationships between PtdOH biosynthetic pathways and the role of PtdOH in pathophysiology. PLD enzymes were once thought to be undruggable owing to the ubiquitous nature of PtdOH in cell signalling and concerns that inhibitors would be too toxic for use in humans. However, recent promising discoveries suggest that small-molecule isoenzyme-selective inhibitors may provide novel compounds for a unique approach to the treatment of cancers, neurodegenerative disorders and other afflictions of the central nervous system, and potentially serve as broad-spectrum antiviral and antimicrobial therapeutics.
Similar content being viewed by others
Main
Phospholipase D (PLD; KEGG enzyme commission number 3.1.4.4) enzymes are phosphodiesterases that serve as key components of multiple signalling and metabolic pathways. They are encoded by a superfamily of genes1 and can be defined by several highly conserved motifs. These enzymes catalyse the removal of head groups from glycerophospholipids to generate phosphatidic acid (PtdOH), a reaction that results in the stoichiometric release of the free head group1,2,3,4,5,6,7. One of the four subgroups of PLD enzymes is characterized by a conserved H-X-K-X4-D-X6-G-(G/S) catalytic motif that is commonly known as an HKD motif. Members of this subgroup hydrolyse phosphodiester bonds via the HKD catalytic motif using a generally similar reaction mechanism; however, some family members also exhibit lipid hydrolase activity, whereas others do not. In addition, several PLD enzymes that lack HKD motifs have been described that also generate PtdOH5.
In mammalian cells, the HKD-containing isoenzymes PLD1 and PLD2, which share highly conserved phox and pleckstrin homology (PX–PH) domains, are almost ubiquitous5. These two isoenzymes frequently serve as nodes at points where signalling pathways converge. They are known to participate in cellular functions that require membrane remodelling or biogenesis, such as vesicular transport, endocytosis, degranulation and cell cycle progression. The substrate for PLD1 and PLD2 is typically phosphatidylcholine, but the enzymes are also able to hydrolyse other amine-containing glycerophospholipids, including phosphatidylethanolamine, phosphatidylserine and, to a lesser extent, phosphatidylglycerol.
Many HKD motif-containing PLD enzymes also catalyse an alternative reaction to hydrolysis (that is, transphosphatidylation), in which short-chain primary alcohols compete with water as a nucleophile, generating a phosphatidyl alcohol product, such as phosphatidylbutanol (PtdBuOH) or phosphatidylethanol (PtdEtOH). This alcohol-mediated transphosphatidylation reaction (Fig. 1) uses physiological substrates and has catalysis rates comparable to those of hydrolysis. In some cases, the phosphatidyl alcohol products mimic PtdOH binding to downstream targets, thereby activating some signalling pathways downstream of PLD enzymes, while blocking others. Erroneously, primary alcohols have widely been referred to as PLD 'inhibitors' in publications, and it is likely that some roles previously ascribed to PLD enzymes in studies that used alcohols as 'inhibitors' are really attributable to nonspecific effects and should be re-examined2. Details of the sequence homology among members of the PLD superfamily, and the enzymology, signalling and functions of respective PLD proteins, have been reviewed previously3,4,5,6.

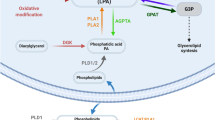
a | Recent findings have implicated phospholipase D (PLD) enzymes as therapeutic targets in a variety of human diseases. b | Most PLD enzymes mediate both a hydrolysis reaction that generates phosphatidic acid (PtdOH) directly and a transphosphatidylation reaction in which primary alcohols serve as alternative substrates for the generation of a phosphatidyl alcohol lipid product. Allosteric small-molecule inhibitors block both reactions. PtdOH is metabolized to diacylglycerol (DAG) by lipid phosphate phosphatase (LPP) enzymes. PtdOH species are also generated downstream of PLC enzymes, which directly produce DAG; subsequent phosphorylation of DAG by DAG kinases (DGKs) generates PtdOH. The mechanism of transphosphatidylation has been reviewed in detail elsewhere5. BuOH, butanol; PtdBuOH, phosphatidylbutanol. *denotes long-chain fatty acid residues.
Recently, theoretical work was presented that describes the possible mechanisms underlying the catalytic activity of HKD motif-containing PLD enzymes using computational methods and models that are based on reaction kinetics, thermodynamics and quantitative insights from studies of the Streptomyces spp. strain PMF PLD enzyme (PLDPMF)7. The mechanism of catalytic activity includes the following steps: first, the formation of a five-coordinate phosphohistidine intermediate and initial phosphoryl transfer during which the head group is cleaved; second, the hydrolysis of the phosphohistidine intermediate and bond dissociation of the hydrolysed substrate; and third, the formation of a thermodynamically stable four-coordinate phosphohistidine intermediate7. These individual steps are highly conserved among enzymes that contain the HKD motif, which supports speculation that the large number of highly diverse PLD enzymes evolved as a consequence of differences in the mechanism of regulation by constituents of distinct cell signalling and metabolic pathways to fulfil a variety of cellular functions for which PtdOH generation is vital.
Despite their essential roles in cellular function, it is curious that one or more PLD isoenzymes can be genetically ablated in mice without causing lethality or even overt severe morbidity phenotypes. However, some isoform-specific functions have been identified in these models8,9,10. It can be postulated that PtdOH production is so indispensable to cellular function and organism survival that multiple redundant pathways have evolved to prevent the global loss of PtdOH. Both metabolic and signalling pathways depend on PtdOH generation, from the initial steps of the de novo glycerophospholipid biosynthetic pathway (that is, the Kennedy pathway) to signalling pathways that are initiated by specific classes of GPCRs, RTKs and integrins, which lead to the rapid generation of signalling pools of PtdOH5 (Box 1; Fig. 2). In mammalian cells, GPCR signal-activated PtdOH generation occurs predominantly via the PLD1 and PLD2 isoenzymes, and phospholipase C (PLC)–diacylglycerol kinase (DGK) signalling. From this perspective, one or both of the isoenzymes may be involved both temporally and spatially in a specific cellular function, but PtdOH depletion can be compensated for if an isoenzyme is inactivated. If the causal misregulation step is due to a signalling pathway that modulates the specific PLD isoenzyme in a physiological circuit, then one of the parallel PtdOH-generating pathways may not be similarly affected. As an example, purinergic receptors can induce PtdOH production via both PLD and PLC–DGK signalling in astrocytomas11. These pathways are interconnected and seem to constitutively cross-regulate one another in the production of primary PtdOH lipid products as well as that of downstream products such as diacylglycerol (DAG). When one pathway is blocked the other compensates, thereby ensuring the continued production of essential lipid signalling products. However, the resulting molecular species differ in acyl composition depending on whether they were generated via the PLD pathway or the PLC–DGK pathway. In some cases, acyl composition may be important for the activation of a specific target. For example, a recent report showed that only unsaturated fatty acids containing PtdOH dissociate the dishevelled, EGL-10 and pleckstrin (DEP) domain-containing mTOR-interacting protein (DEPTOR) from mechanistic target of rapamycin complex 1 (mTORC1)12. This finding extends previous suggestions that PLD enzymes may have a primary role in the regulation of mTORC1 and that other enzymatic sources of PtdOH have a secondary role. By extension, if an individual isoform of PLD is dysfunctional and thus contributes to a disease process, then it is likely that isoform-specific small-molecule inhibitors would have less adverse consequences than would pan-reactive inhibitors.

Metabolic pathways that lead to the generation of phosphatidic acid (PtdOH) include de novo biosynthesis as well as the phospholipase D (PLD) and PLC–diacylglycerol kinase (DGK)-mediated signalling pathways. The generation of PtdOH seems to be broadly exploited by pathogens as part of the infection process and thus represents a novel therapeutic opportunity; the design of novel small-molecule inhibitors may lead to new treatments for infection. The figure illustrates three routes of cellular PtdOH generation via the indicated enzyme-mediated pathways. ATX, autotaxin; FA, fatty acid; G3P, glycerol-3-phosphate; GK, glycerol kinase; GPAT, glycerol-3-phosphate acyltransferase; LPP, lipid phosphate phosphatase; LPAAT, lyso-PtdOH acyltransferase; LPCAT, lysophosphatidylcholine acyltransferase; MGAT, monoacylglycerol acyltransferase; MGK, monoacylglycerol kinase; MGL, monoacylglycerol lipase; PI, phosphatidylinositol; PIP, PI phosphate; PI(4,5)P2, PI 4,5-bisphosphate; R1, R2, fatty acyl moieties.
Interest in targeting PLD isoenzymes with small-molecule inhibitors has steadily grown since PLD family members were implicated in a variety of human diseases, ranging from cancer to neurodegeneration and viral infections. Moreover, viruses such as human cytomegalovirus (HCMV)13, influenza virus14 and HIV-1 (Ref. 15) have been shown to use PtdOH in various aspects of cellular entry, intracellular trafficking and the disruption of innate immune responses.
Understanding the mechanism of action of the HKD motif-containing superfamily, as well as the roles of specific PLD isoforms in cellular signalling metabolism and cellular function, is paramount to the development of optimal therapeutic compounds. Here, we provide an overview of the development of isoenzyme-preferring small-molecule inhibitors of PLD enzymes, discuss the role of PLD family members in infectious diseases, cancers and central nervous system (CNS) disorders, and provide an outlook on possible applications of isoformspecific PLD inhibitors.
PLD isoenzyme inhibitors
Until 2009, few chemical tools existed to directly study and thereby elucidate the function of PLD enzymes, and no small molecules were available to dissect the individual roles of the two main mammalian PLD isoenzymes (that is, PLD1 and PLD2) or other therapeutically relevant PLD enzymes, such as the endocannabinoid regulator N-acyl phosphatidylethanolamine-specific phospholipase D (NAPE-PLD) or bacterial PldA3,5,16,17,18,19,20. Historically, the field relied on the overexpression of catalytically active or inactive forms of the enzymes in question, or on blocking their expression using RNA-mediated interference (RNAi); however, these biochemical and genetic approaches did little to garner attention from the pharmaceutical industry3,5,16,17,18.
For PLD inhibition to emerge as a viable therapeutic approach, target validation with selective small molecules was required. Although early efforts throughout the 2000s did identify small molecules that were capable of modulating the function of some PLD enzymes— both indirectly (for example, polyphenolic natural products such as resveratrol (Fig. 3a, compound 1) or steroids such as triptolide (Fig. 3a, compound 2)) and directly (phosphate mimetics such as tungstate (Fig. 3a, compound 3) and vanadate (Fig. 3a, compound 4), as well as natural products such as SCH420789 (Fig. 3a, compound 5) and calphostin C (Fig. 3a, compound 6)) — these ligands failed to accelerate development in the field in part owing to their obvious lack of specificity. Thus, over the past 30 years, the most commonly used class of molecules to study the function of PLD enzymes was primary alcohols (for example, n-butanol (Fig. 3a, compound 7)). As emphasized above, alcohols such as n-butanol block PLD-catalysed PtdOH production by competing with water as a nucleophile. However, the blockade of PtdOH production is frequently incomplete under experimental conditions, and thus caution must be exercised in interpreting data from studies that have used primary alcohols such as n-butanol1,3,5,16.

a | Representative early indirect and direct inhibitors of phospholipase D (PLD). b | Second-generation PLD inhibitors that are based on halopemide (providing the first isoenzyme-selective PLD1 inhibitors). IC50, concentration that inhibits PLD activity by 50%; FIPI, 5-fluoro-2 indolyl des-chlorohalopemide.
After decades of 'false starts', a brief report was published in 2007 in which a group at Novartis described halopemide (Fig. 3b, compound 8; concentration that inhibits PLD2 activity by 50% (IC50) = 1.5 μM) and a set of 14 analogues as PLD2 inhibitors. These analogues included an indole amide congener that is now referred to as 5-fluoro-2-indolyl des-chlorohalopemide (FIPI; Fig. 3b, compound 9; PLD2 IC50 = 200 nM)21. This manuscript only reported PLD2 activity, with no mention of PLD1 inhibition, which prompted further pharmacological evaluation of these compounds in both cell-based and biochemical assays with the purified PLD1 and PLD2 enzymes16. Interestingly, halopemide potently inhibited both PLD1 (cellular IC50 = 21 nM; biochemical IC50 = 220 nM) and PLD2 (cellular IC50 = 300 nM; biochemical IC50 = 310 nM), as does FIPI (PLD1 cellular IC50 = 1 nM; PLD1 biochemical IC50 = 9.5 nM; PLD2 cellular IC50 = 44 nM; PLD2 biochemical IC50 = 17 nM); therefore, halopemide and the reported analogues21 are more accurately described as dual PLD1–PLD2 inhibitors. Although isoenzyme selectivity remained elusive, this discovery was an informative lead. Halopemide was originally developed by Janssen in the 1980s as an atypical antipsychotic that has potent antagonistic activity at D2 dopamine receptors (as well as at more than 30 additional biogenic amine receptors) and was called a “psychic energizer” (Refs 22,23). In clinical trials, halopemide was shown to have effects on the negative as well as the positive symptoms of schizophrenia, without the extrapyramidal side effects that are common to standard atypical antipsychotic agents22,23. Importantly, halopemide achieved high plasma exposure and displayed no adverse events or abnormal biochemistry. At these exposures, halopemide fully inhibited both PLD1 and PLD2, which suggests that the mechanism of inhibition of PLD1 and PLD2 by this chemotype would be safe in humans; this represented a major step towards de-risking this therapeutic approach. However, halopemide was not a panacea. Many laboratories began to use both halopemide and FIPI as key proof-of-concept tools for PLD1 and PLD2 target validation, which led to potentially flawed conclusions, as both compounds display a considerable amount of ancillary pharmacology at a multitude of biogenic amine receptors, as expected for atypical antipsychotics that have GPCR-privileged structures as a core motif22,23,24,25,26,27,28,29,30.
Thus, we initiated an optimization programme that used halopemide as one starting point to develop isoenzyme-selective PLD inhibitors that had improved ancillary pharmacology and drug metabolism and pharmacokinetics (DMPK) profiles, and were suitable for in vivo proof-of-concept studies16. The design and optimization strategy was based on results from two types of in vitro catalytic assays that were conducted in parallel: a cell-based assay and a biochemical assay that presented defined substrates to human or rat recombinant PLD enzymes16. Through a combination of diversity-oriented synthesis, matrix libraries and subsequent iterative parallel synthesis, more than 1,000 analogues were prepared and assayed, leading to a series of the first highly PLD1-selective inhibitors, which are exemplified by VU0359595 (Fig. 3b, compound 10; PLD1 IC50 = 3.7 nM); VU0359595 has more than 1,700-fold selectivity for PLD1 versus PLD2 (Refs 16,25) (Fig. 3). In this instance, the addition of a (S)-methyl group to the ethyl linker increased selectivity for PLD1 versus PLD2 by more than 150-fold across multiple examples in this sub-series by both diminishing activity at PLD2 and enhancing inhibitory activity at PLD1 (Fig. 4). By virtue of the (S)-methyl group, a multitude of the resulting analogues displayed more than 200-fold selectivity for PLD1 over PLD2 within a traditionally PLD2-preferring scaffold16,25. This unanticipated finding is a striking example of the 'magic methyl' effect, and this modification to the linker promotes a U-shaped ligand topology. Importantly, VU0359595 had markedly improved ancillary pharmacology relative to halopemide and FIPI, and displayed an attractive DMPK profile, which enabled in vivo proof-of-concept studies16,25.

The phospholipase D 1 (PLD1) IC50 values (concentration that inhibits PLD activity by 50%) were determined in cellular PLD1 assay with Calu-1 cells. The PLD2 IC50 values were determined in a cellular assay with HEK293-GFP PLD2 cells. Each IC50 was determined in triplicate measurements. X denotes H or halogen, and R1 denotes the aryl- or cyclopropylaryl moieties (as listed in the table).
Within the piperidine benzimidazolone core analogues, preference for PLD2 was elusive; thus, a number of bioisosteres of the piperidine benzimidazolone, as well as alternative GPCR-privileged structures, were surveyed26,27,28,29,30. The structure–activity relationship (SARs) of these compounds were shallow, and more than 99% of the variants studied were devoid of PLD-inhibitory activity; however, an N-phenyl triazaspirone congener (Fig. 5, compound 11) displayed a modest preference for PLD2 and represented a considerable departure from the halopemide-based PLD inhibitors26. A subsequent lead optimization campaign focusing on the N-phenyl triazaspirone congener encountered a shallow SAR, yet the addition of a fluorine atom in the three-position of the N-phenyl moiety provided VU0364739 (Fig. 5, compound 12), the first highly selective PLD2 inhibitor (PLD2 IC50 = 20 nM; 75-fold selective for PLD2 versus PLD1)27. Moreover, similarly to VU0359595, VU0364739 was a direct PLD inhibitor, and it had potent activity in biochemical assays with purified PLD proteins. In addition, VU0364739 had improved ancillary pharmacology relative to halopemide and FIPI, and a DMPK profile that was suitable for in vivo proof-of-concept studies27. Interestingly, PLD1-preferring VU0359595 was peripherally restricted, whereas PLD2-preferring VU0364739 was CNS penetrant (brain-to-plasma ratio (Kp) = 0.7)25,26,27. An intriguing observation within this novel PLD2-preferring series relates to the effect of the (S)-methyl group, which had a marked impact on PLD1 selectivity in the benzimidazolone series. In the benzimidazolone series, the addition of a (S)-methyl group into N-phenyl triazaspirone congener converted an approximately tenfold PLD2-selective inhibitor into the dual PLD1–PLD2 inhibitor compound 13 (Fig. 5), which was approximately sixfold selective for PLD1 versus PLD2. These subtle structural features that promote substantial changes in PLD isoform selectivity are reminiscent of the 'molecular switches' that are prevalent in allosteric GPCR ligands25,26,27,28.

VU0364739 (also known as JWJ; compound 12) emerged as an important phospholipase D2 (PLD2)-selective compound. IC50, concentration that inhibits PLD activity by 50%.
The unprecedented level of PLD isoenzyme selectivity and the presence of apparent 'molecular switches' led us to question the mechanism by which these compounds inhibit PLD enzymes. These features, along with the structural similarity of these compounds to previously reported piperidine benzimidazolone-based allosteric inhibitors of AKT31 — that is, they have a two-site binding mode within both the catalytic domain and the pleckstrin homology domain — led us to postulate that VU0359595 and VU0364739 were allosteric inhibitors31,32. Initial studies with halopemide and FIPI in assays using a PLD1.d311 truncation construct (which lacked a portion of the pleckstrin homology domain) showed a slight reduction of PLD activity16; however, both VU0359595 and VU0364739 significantly reduced PLD activity (by more than tenfold), which suggests that the pleckstrin homology domain (a domain that is far away from the HKD catalytic active site) is key for the binding of these molecules. Additional enzyme kinetic studies further increased our confidence that these compounds were allosteric16,25,26,27,28,29,30. Using a backscattering interferometry technique33, it was found that VU0359595 binds to two sites on the PLD1 protein (one with high affinity and the other with low affinity)34, which further supports an allosteric mode of inhibition akin to that of allosteric AKT inhibitors31,32. Of course, in the absence of high-resolution structural determinations, the sites of interaction are somewhat speculative, but some suggestions have been reported. In 2015, evidence was published for two sites of inhibitor action that involved both the enzyme catalytic centre and an allosteric phosphoinositide-binding pocket35. In this study, FIPI was shown to interact with S757 of PLD2 (and also other PLD isoenzymes); by contrast, VU0364739 interacted with two different PLD2 sites, with one in the HKD (at S757 or S648) and the other an allosteric site (R210 or R212) that is natively occupied by phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2). Binding of VU0364739 to the allosteric site blocked PI(4,5)P2 binding. In combination with F244-L245-L246, the PI(4,5)P2-binding site forms a hydrophobic pocket in the pleckstrin homology domain that is identical to that of allosteric AKT inhibitors31, and thus promotes a high degree of PLD2 selectivity35. Not only have these second-generation, halopemide-based and triazaspirone-based PLD inhibitors achieved high selectivity for the individual PLD1 and PLD2 isoenzymes, they have also provided a novel allosteric mechanism for PLD inhibition16,25,26,27,28,29,30.
PLD2 has recently emerged as a target in oncology and infectious diseases14,16,28,29,36, and VU0364739 has served as a proof-of-concept compound28; however, a better tool was required. Although VU0364739 is 75-fold selective for PLD2 over PLD1, the selectivity is driven by potency at PLD2 (IC50 = 20 nM), but the compound still inhibits PLD1 at modest concentrations (IC50 = 1500 nM); therefore, a third-generation optimization campaign was launched27. This effort led to the synthesis of ML298 (Fig. 6, compound 14), which is more than 53-fold selective for PLD2 (IC50 = 355 nM) and has negligible activity against PLD1 (IC50 ≈20 μM). Incorporation of the (S)-methyl group used in the piperidine benzimidazolone series had a substantial impact on enhancing PLD1 inhibitory activity (increasing it by more than 250-fold) and provided ML299 (Fig. 6, compound 15), which is a potent, dual PLD1–PLD2 inhibitor (PLD1 IC50 = 6 nM; PLD2 IC50 = 20 nM)28. Once again, ancillary pharmacology and physiochemical properties were improved, and interestingly, ML298 was found to be peripherally restricted, whereas ML299 was CNS penetrant (Kp = 0.44)28.

Based on the ability of the triazaspirone core to provide a range of PLD pharmacological profiles, a variety of non-N-Ar (aromatic) moieties that are not bound to nitrogen were evaluated in an attempt to develop a PLD2 inhibitor that was comparable to ML298 but was CNS penetrant28. This effort identified ML395 (Fig. 6, compound 16), which is a pyridylmethyl congener with good PLD2 potency (IC50 = 380 nM), no measurable activity at PLD1 (IC50 > 30 μM), exceptional physiochemical properties and an excellent DMPK profile, including high CNS penetration (Kp = 1.48)29. As a result, a series of potent and isoenzyme-selective, as well as dual, PLD inhibitors were developed; these compounds had DMPK and ancillary pharmacology profiles that were suitable for in vivo proof-of-concept experiments aimed at dissecting the roles of the individual mammalian PLD enzymes (described below)16,25,26,27,28,29,30.
Although a major advance in the PLD field, these halopemide-derived and novel triazaspirone-based series of mammalian PLD inhibitors were devoid of activity against both the bacterial Pseudomonas aeruginosa PldA and the endocannabinoid regulator NAPE-PLD18. Importantly, no ligands existed that could modulate these structurally divergent PLD enzymes, thus hindering our understanding of their potential as therapeutic agents. The search for inhibitors of these PLD enzymes led to the re-examination of certain selective oestrogen receptor modulators (SERMs), which have been used in the successful treatment of oestrogen-receptor-positive breast cancer37,38. A previous report suggested a potential ER-independent mechanism for the efficacy of SERMs such as raloxifene (Fig. 7, compound 17) and 4-hydroxy-tamoxifen (Fig. 7, compound 19; the active metabolite of tamoxifen (compound 18)) that may be due to off-target inhibition of mammalian PLD enzymes39.

17–20 selective oestrogen receptor modulator (SERM) chemotypes have inhibitory activity against mammalian phospholipase D1 (PLD1), PLD2 and Pseudomonas aeruginosa PLD (PldA). IC50, concentration that inhibits PLD activity by 50%.
The potential of SERM chemotypes as PLD inhibitors was not fully explored until recently, in part because the PLD-inhibitory activity required mid-micromolar concentrations, and SERM activity might complicate definitive target validation. However, these novel chemotypes might provide a novel scaffold that could achieve universal PLD inhibition across diverse PLD enzymes, and they have a binding mode that is distinct from the halopemide-derived and triazaspirone-based series of mammalian PLD inhibitors16,25,26,27,28,29,30. Intriguingly, raloxifene, 4-hydroxytamoxifen and tamoxifen all showed modest inhibitory activity against the bacterial PldA enzyme, which prompted the evaluation of desketoraloxifene (Figs 7,8, compound 20), which is a SERM that has a unique planar topology and is a stronger activator of the activator protein 1 (AP-1)-binding site of ESR1 (which encodes oestrogen receptor-α)) compared with that of ESR2 (which encodes oestrogen receptor-β)18. Desketoraloxifene was a breakthrough, and had comparable inhibitory activity against mammalian PLD1, mammalian PLD2 and bacterial PldA (Fig. 8). This unexpected finding led to a new multidimensional optimization campaign that focused on desketoraloxifene and produced hundreds of novel analogues. Taking into account the well-established SERM pharmacophore model, these analogues were designed to abolish SERM activity (Fig. 8), and they were then measured using in vitro enzymatic assays against PLD1, PLD2, PldA and NAPE-PLD18. This effort provided compound 21 (Fig. 8), which was devoid of the 6-OH moiety that is crucial for oestrogen receptor binding and antiproliferative action. It also contains an N,N-dimethylamino moiety, which is known to greatly reduce SERM activity in this core40, but retained good activity against PLD1 (IC50 = 4.7 μM), PLD2 (IC50 = 7.1 μM), PldA (IC50 = 7.5 μM) and even NAPE-PLD (IC50 = 67 μM)18. This finding also led to the profiling of desketoraloxifene against NAPE-PLD, and it was found to be slightly more potent than was compound 21 (NAPE-PLD IC50 = 58 μM). Thus, both desketoraloxifene and compound 21 represented the first entry into potentially universal inhibition of structurally and phylogenetically distinct PLD enzymes18. Although an advance, compound 21 is a lead compound (that is, the 'halopemide of the mammalian chemotypes') from which to develop a more optimized compound for target validation of PldA and NAPE-PLD.

Chemical optimization of desketoraloxifene (compound 20) delivered compound 21, which is the first universal inhibitor of phylogenetically and structurally diverse PLD enzymes that is devoid of selective oestrogen receptor modulator (SERM) activity. IC50, concentration that inhibits PLD activity by 50%; NAPE-PLD, N-acyl phosphatidylethanolamine-specific phospholipase D.
The strategic use of diverse, interdisciplinary experimental approaches — including lipidomics, metabolomics, phosphoproteomics, medicinal chemistry, molecular pharmacology, biochemistry and DMPK optimization — has led to the development of selective compounds that have allowed the roles of the individual mammalian PLD1 and PLD2 isoenzymes to be dissected; have uncovered an allosteric mechanism of inhibition; and have provided entry into a chemotype that provides potentially universal PLD inhibition. These compounds provide initial starting points for the development of inhibitors of PldA and NAPE-PLD.
The development of these compounds offers a new set of tools for analysing the role of PLD enzymes generally and that of specific PLD isoenzymes in diverse biological conditions. Although early work (described above) implicated PLD enzymes in a number of normal and pathological processes, the selectivity provided by these compounds allowed the direct interrogation of PLD1 and PLD2 in the host response to infectious diseases, including influenza virus and HIV infections, and in cancer. Although genetic approaches, including inducible knockout systems, have the advantages of achieving complete deletion of an enzyme and its activity, highly selective compounds can be used in diverse cell types and animal models, including those in which genetic ablation can be difficult. In addition, the deletion of PLD enzymes is likely to have effects distinct from those of short-term inhibition, the latter of which provides a better model of therapeutic intervention.
PLD enzymes in infectious diseases
All viruses co-opt the resources of their target host to complete their replication cycle. This parasitism includes the use of cellular processes and metabolite resource pools. Every stage of the virus life cycle requires host cell cooperation, from virus entry (which often exploits host-cell trafficking pathways and endosome formation) to nucleic acid transport and replication, protein production, protein trafficking and modification, and finally culminating in viral budding. Host cell energy, nucleic acids, amino acids and lipids are all ingredients for the production of new virus particles.
PLD enzymes can influence each of these pathways. Most directly, the role of PLD enzymes in generating PtdOH makes them key components of the nascent small endosomes that are used for entry by many viruses, including influenza virus, respiratory syncytial virus, hepatitis C virus and coronaviruses41,42,43,44. PtdOH is enriched in endosomal membranes, as it is required for membrane curvature5. Although not yet directly demonstrated, the inhibition of PLD enzymes could potentially limit the efficiency of endosome formation and provide an early block in the virus replication cycle. Furthermore, the activation of PLD enzymes and the production of PtdOH can support multiple signal transduction cascades, including those mediated by mitogen-activated protein kinases (MAPKs; for example, p38), which have been shown to be necessary for the activation of early endosomal trafficking molecules such as RAB5 and early endosome antigen 1 (EEA1)45.
Making use of the extensive library of available compounds that inhibit diverse forms of PLD, studies have been undertaken that dissect the roles of PLD enzymes in the host response to infectious diseases, including influenza virus and HIV, and the role of pathogen PLD enzymes in specific bacterial infections.
Influenza virus. The potential role of PLD enzymes in crucial events in the virus life cycle led us to investigate the function of these enzymes during influenza virus infection14,29. Indeed, following challenge of host cells with influenza virus in vitro, PLD catalytic activity was markedly increased, as measured by increases in PtdBuOH mass. Pretreatment of respiratory epithelial cells with a primary alcohol or RNAi targeting PLD1 and PLD2 resulted in a significant decrease in the number of cells that were infected with influenza virus and in the infectious titre produced by the culture.
A panel of small-molecule PLD inhibitors were tested in similar assays, and isoform-specific inhibitors of PLD2 — including VU0364739, ML298 and ML395 —showed potent antiviral activity. In in vitro experiments, these compounds inhibited cellular infection and viral replication of a range of clinically relevant influenza viruses, such as the 2009 pandemic H1N1 influenza virus strain, the recently emerged avian H7N9 influenza virus strain and a representative of the highly pathogenic H5N1 influenza virus subtype14,29. The addition of VU0364739 to the culture medium reversed the accumulation of PtdBuOH in influenza virus-infected cells, which indicates that the catalytic activity of PLD2 has a role in influenza virus infection14.
Prophylactic treatment with VU0364739 provided significant protection in a mouse model of lethal challenge with several influenza virus strains, including the pathogenic H7N9 avian influenza virus A/Anhui/1/2013 (Ref. 14). The low solubility of VU0364739 limited the ability to deliver it at high doses, and the vehicle had some toxicity on its own, which suggests that more robust protection from mouse lethality might be obtained with the newer generation of inhibitors that have improved DMPK and higher solubility in non-toxic delivery systems. Importantly, the compound itself showed no added toxicity beyond the vehicle, as measured by extensive blood chemistry profiles, liver toxicity panels and a modified Irwin Neurological Battery14, which measures a wide variety of parameters in the autonomic nervous system and the somatomotor system.
These promising results led to the investigation of the mechanisms by which PLD inhibition promoted survival after influenza virus infection in vivo and prevented productive infection in vitro. As noted, there are multiple stages of the virus life cycle that could be affected by PLD inhibition. Using a combination of microscopy and RNAi, it was demonstrated that viral entry was significantly delayed after prophylactic treatment of human A549 adenocarcinoma alveolar cells with VU0364739 in vitro14. This was true for infectious viruses and for non-infectious transferrin uptake, which is indicative of a general defect in endocytosis. However, the block was not complete, and some virus was eventually able to enter the cell. The maturation of the endosome was also affected, and the expression of RAB5, EEA1 and CD63 was significantly delayed when comparing VU0364739-treated cells with cells treated with the vehicle control14.
Importantly, this 'mechanical block' was not sufficient to confer protection. Rather, the host-cell innate immune response — which was mediated by retinoic acid-inducible gene I (RIG-I), interferon-regulatory factor 3 (IRF3) and MxA (also known as Mx1), as determined by RNAi — was required for the protective effect of the PLD2 inhibitor14 (Fig. 9). The delay in virus endocytosis permitted a more efficient and effective antiviral response, which limited subsequent productive virus replication. This novel mechanism demonstrated the potential for broad-spectrum antiviral activity of PLD2 inhibition and gave some indication of why the treatment showed remarkably low toxicity (that is, although endocytosis was slowed, it was not blocked, thus permitting normal cell metabolism and cellular processes to continue, albeit with altered kinetics). These data also suggested that virus exocytosis, as measured by RAB10-mediated accumulation of surface-trafficking endosomes, was delayed, probably through mechanisms similar to those that that limited virus entry. This effect seemed to be less striking than the entry phenotype, but continued exploration of the potential for multiple pathways of action of PLD2 inhibition on influenza virus replication is ongoing.

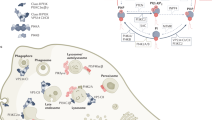
The phospholipase D2 (PLD2) signalling axis is involved in both the entry and egress of influenza virus, as well as the process of autophagy, which can have antiviral function. PLD activity facilitates viral entry, whereas PLD inhibition delays the kinetics of viral endocytosis14, thus widening the window of time in which the host cell can effectively prevent virus replication by producing antiviral effector molecules, such as interferon-induced transmembrane protein 3 (IFITM3) and MxA. Similarly, viral egress appears to be impaired by PLD inhibition14. The regulation of AKT activity by PLD2 has also been shown to activate the autophagy pathway, which can limit virus production. PLD-generated phosphatidic acid (PtdOH) recruits AKT to cellular membranes, and AKT subsequently phosphorylates beclin 1 at serine 295. This leads to dissociation of the beclin 1–rubicon complex, a process that promotes autophagy. PLD inhibitors reduce PtdOH concentrations, reduce AKT-mediated phosphorylation of beclin 1 and stabilize the beclin 1–rubicon complex to stall autophagy. The changes depicted in membrane lipid composition are based on previous work that investigated the contributions of PtdOH to membrane curvature115,116.
In summary, PLD2 inhibition can protect against influenza virus infection by delaying, but not blocking, endocytosis, which allows the host-cell innate immune system to mount an effective response against influenza virus. This 'extended window' of antiviral activity can significantly reduce productive virus replication (the steps in which PtdOH contributes to infection are summarized in Fig. 9).
HIV. The generalized pathway by which PLD2 inhibition protected against influenza virus infection suggested that similar efficacy might be seen with other enveloped viruses. Supporting this hypothesis, the PLD1 inhibitor VU0359595 greatly reduced the ability of HIV to replicate in CD4+ T cells in vitro15. Surprisingly, there were distinctive elements in the mechanism of suppression of HIV and influenza virus by PLD inhibitors15. PLD inhibition has previously been shown to block RAS–MAPK activation in SK-BR3 breast cancer cells46, and in in vitro assays with CD4+ T cells, treatment with the PLD1 inhibitor VU0155069 reduced extracellular signal-regulated kinases (ERK) phosphorylation after T cell activation15. ERK activation during T cell stimulation is required for MYC induction47, which, in turn, is a crucial event in the metabolic reprogramming that occurs during T cell activation. HIV targets these activated T cells and makes use of the increased metabolic stores that are generated to support rapid cell division and effector activity15. Consistent with this, treatment with the PLD1-selective compound VU0359595 reduced the induction of MYC in these T cells in in vitro experiments. Events that occur downstream of MYC activation, including the induction and phosphorylation of ribosomal protein S6 kinase, were also reduced after PLD1 inhibition15.
Deoxyribonucleotide triphosphate (dNTP) stores, which are necessary for the rapid replication of T cells during their expansion phase, are one of the key metabolic resources that accumulate after T cell activation and metabolic reprogramming, and HIV also makes use of these pools to complete its replication cycle48,49. To establish that the depletion of dNTP stores in activated T cells is a key mechanism downstream of PLD1 inhibition, dNTPs were added to VU0359595-treated CD4+ T cells and shown to rescue HIV replication in cell-based assays15. Thus, PLD1 inhibition in vitro restricts HIV infection by altering host-cell metabolic stores, which disrupts the virus life cycle.
New therapeutic compounds against HIV are increasingly essential owing to the emergence of resistant strains. There is also an unmet need to develop anti-HIV drugs that work in the CNS, where HIV has recently been shown to localize, mutate and possibly become more resistant to conventional antiretrovirus cocktails50. The ability of some PLD inhibitors to penetrate the CNS27,28 may provide a novel approach to treating otherwise difficult-to-reach reservoirs of the retrovirus. Interestingly, a pathway with a role that is strikingly similar to that of PLD1 in HIV infection was recently described; PLD activity was shown to promote RNA replication in a red clover necrotic mosaic plant virus51. This suggests that PtdOH modulation of virus replication is an evolutionarily conserved pathway and that this signalling lipid has a broad role in the infectious processes of pathogens.
Figure 10 depicts pathways downstream of PLD1 and PLD2 isoenzymes and illustrates their role in the infection process with pathogenic viruses. These pathways can be targeted for therapeutic purposes. The pathway involving PLD2, signalling via AKT, connects to autophagic flux, whereas PLD1 has recently been shown to modulate pyrimidine biosynthesis via the enzyme CAD (which contains carbamoyl aspartate synthase, aspartate transcarbamylase and dihydro-orotase domains), which makes CAD a potential target for future antiretroviral therapeutics52.

Left panel: phospholipase D2 (PLD2)-generated phosphatidic acid (PtdOH) recruits and binds to AKT and subsequently promotes autophagy via beclin 1, as detailed in Fig. 9. During the process of autophagy, autophagosomes fuse with endosomes and lysosomes, which leads to their maturation into autolysosomes. These display the autophagy markers LC3 (light chain 3, also known as MAP1LC3) and p62 (also known as sequestosome 1). This process results in acidification and allows the autolysosome to digest captured cellular material such as viruses. The respective details of PLD1 and PLD2 activation are discussed elsewhere3. Right panel: this shows a PLD1 pathway that affects pyrimidine biosynthesis and intracellular deoxyribonucleotide triphosphate (dNTP) levels via the enzyme CAD (which contains carbamoyl aspartate synthase, aspartate transcarbamylase and dihydro-orotase domains). PLD1 allosterically modulates the activity of mechanistic target of rapamycin (mTOR), which leads to the phosphorylation of CAD and enhances its catalytic activity. The subsequent production of pyrimidine nucleotides via the activation of CAD is independent of AKT. PLD1 inhibition, in turn, can lead to reductions in dNTP production.
Although the pathways downstream of PLD activity that can be exploited by viruses are distinct, they suggest a common mechanism for how inhibitors of particular PLD isoforms might serve as broad-spectrum inhibitors of infection. Short-term inhibition of a particular PLD enzyme can influence numerous cellular processes that affect viral infectivity and replication efficiency, and for many viruses, even a modest reduction in early replication efficiency can completely alter the subsequent infectious process and allow the host to successfully mount an immune response14. Balancing this effect, the redundancy of the lipid signalling products of the reaction catalysed by PLD enzymes might explain the limited in vivo toxicity of isoform-specific inhibitors.
The studies described above focused on mammalian (host) PLD enzymes as targets for disrupting pathogen replication. However, some bacterial and fungal pathogens contain their own evolutionarily distinct PLD enzymes, which can potentially be targeted with direct antimicrobials. Moreover, there is evidence that certain parasites can exploit pathways downstream of PLDs.
Bacteria and parasites. The activity of PLD enzymes in neutrophils and leukocytes is associated with antimicrobial activities such as phagocytosis, chemotaxis and membrane ruffling5, but an evolutionarily older function in bacteria may have been to promote bacterial fitness in the mammalian host. Several intracellular pathogens have genes encoding protein products that promote virulence through increases in pathogen internalization or intracellular survival. These genes are generally non-essential to the survival of the bacterial strain, but promote entry, immune evasion or inhibition of host-cell immune defences.
PLD enzymes that function as virulence factors are most common in obligate and facultative intracellular Gram-negative bacteria, such as Yersinia pestis, which is the causative agent of bubonic plague53. The Yersinia murine toxin (Ymt) contains two HKD motifs and is highly toxic when injected into mice54. Ymt is involved in transmission of the pathogen to fleas and is not required for virulence in mice55.
The exclusively human pathogen Neisseria gonorrhoeae is a Gram-negative bacterium that causes the sexually transmitted disease gonorrhoea. This bacterium invades cervical epithelial cells after initially binding to the complement CR3 receptor, which initiates membrane ruffling and endocytosis. A secreted 55 kDa PLD homologue, NgPLD, was identified following its release into cultures of host cells that were challenged with the bacterium56. Although the mechanism of secretion has not been identified, NgPLD has been shown to induce the recruitment of CR3 to the cell surface and to promote cytoskeletal rearrangement. The facilitation of infectivity by NgPLD is known to require its interaction with a human host-cell factor: namely, the serine–threonine kinase AKT57.
Several other bacterial pathogens have been shown to express Pld genes that function as virulence factors, including P. aeruginosa, Legionella monocytogenes, Chlamydia trachomatis and Acinetobacter baumannii, among others5,58,59,60,61,62,63. P. aeruginosa is an opportunistic pathogen that afflicts immunocompromised individuals and is a major cause of hospital-acquired infections. Patients with cystic fibrosis are particularly susceptible to chronic infection with P. aeruginosa, and infection ultimately leads to respiratory failure. Unlike other bacterial Pld enzymes, P. aeruginosa PLD (PldA) is comparatively large at 122 kDa and has a much higher homology to eukaryotic PLD enzymes than to other bacterial isoforms, probably as a result of a horizontal gene transfer62. In P. aeruginosa, PldA is a secreted effector of the H2 type VI secretion system. Similarly to many other bacterial Pld virulence factors, PldA is internalized and induces changes in vesicle trafficking in host cells, but it has the unusual property that its catalytic activity is stimulated by polyphosphatidylinositols, which may explain the observed localization of PldA in intracellular vesicles63.
The conventional benzimidazolone and triazaspirone series compounds have little to no effect on P. aeruginosa PldA and are likely to have minimal efficacy against other bacterial PLD enzymes. However, a series of desketoraloxifene-based compounds, such as compound 21, that are effective inhibitors of PldA, was recently identified18 and may provide novel pharmacological interventions. The most potent compound in this study was raloxifene, which is a SERM that had previously been identified as a direct inhibitor of mammalian PLD activity39. A series of analogues in which oestrogen receptor binding was eliminated have been characterized (Fig. 8); yet analogues such as compound 21 retain the ability to inhibit human PLD1 and human PLD2, as well as P. aeruginosa PldA18. This new compound series provides an excellent starting point for the development of more potent and selective inhibitors of PLD enzymes. It also includes compounds that have been used in the clinic for decades that unknowingly suppressed the activity of particular PLD enzymes when used at therapeutic doses.
Recently, it was found that certain parasites might also utilize pathways downstream of PLDs. This was illustrated by in vitro experiments showing that PLD inhibitors such as VU0285655 can block the proliferation of Plasmodium falciparum (one of the causal agents of malaria) and that of Toxoplasma gondii (which causes toxoplasmosis)64.
PLD in cancer signalling pathways
Increased expression of PLD enzymes, their subcellular mislocalization and altered PLD catalytic activity have been implicated as contributing factors in several types of human cancer, and recent reviews have examined the role of PLD1 and PLD2 in pathways involved in cancer progression and tumorigenesis3,65,66,67,68. Numerous reports have directly linked PLD enzymes to transformation and tumorigenesis in a variety of cancer types, including breast16,69,70,71, ovarian72,73, lung74, colon75,76, renal77, pancreatic78, prostate79 and brain cancer3,36,52, among others. Such a widespread occurrence of elevated PLD activity and expression in human cancers suggests a connection between processes that are central to cellular transformation and PtdOH signalling.
RTKs, which can act as oncogenic drivers in cancer, are associated with the activation of PLD2, and there is evidence that PLD1 is also a downstream signalling component of RTK-induced signalling cascades3,5. RTKs such as epidermal growth factor receptor (EGFR) initiate signalling cascades that are known to have roles in cellular transformation, including signalling pathways that lead to the production of lipid second messengers. Binding of the hormone epidermal growth factor leads to EGFR dimerization, which is followed by the autophosphorylation of multiple tyrosine residues that form binding sites for Src homology 2 (SH2) domains, including those of growth factor receptor-bound protein 2 (GRB2), PLCγ and the p85 subunit of phosphoinositide 3-kinase. PLD2 binds to an SH3 domain of GRB2, and this is an important step in the catalytic activation of PLD2 and in the generation of PtdOH; this step also facilitates the recruitment of the guanine nucleotide-exchange protein son-of-sevenless (SOS) to the membrane80. This event promotes a feedforward mechanism by which PLD2 activates RAS GTPase; this, in turn, leads to RALA GTPase activation, which further modulates PLD activity3.
A seminal observation was that in vivo tumour formation by HRAS-driven cancer cells can be blocked by the co-expression of a catalytically inactive form of PLD1 (Ref. 81). This compelling report boosted the search for isoenzyme-selective, small-molecule inhibitors of PLD activity. This effect of PLD1 inhibition on HRAS-driven tumour formation probably represents just one of several key roles of PLD1 in RAS-mediated oncogenesis; the role of PLD1 in this process was reiterated by the recent observation that the novel oncogene FAM83 acts downstream of PLD catalytic activity to promote mammary epithelial cell transformation via a RAS-dependent and EGFR-dependent mechanism82,83.
One of the hallmarks of cancer is the elevated use of glucose via glycolysis. Historically, this was thought to be primarily in response to the higher energy needs of cancer cells relative to those of normal cells, but it is now appreciated that most cancer cells obtain their cellular energy through oxidative phosphorylation3. Recent evidence suggests that rapidly dividing cells with dramatic growth rates use the glycolytic pathway as a means of biosynthesizing lipids, amino acids and nucleotides3. These intermediate molecules may be viewed as 'metabolic currency', and dysfunctional regulation of signalling pathways is a frequent cause of the abnormal metabolic rates of cancer cells. This phenomenon of higher metabolic rates of cancer cells relative to healthy cells and the preference of cancer cells to use aerobic glycolysis is known as the Warburg effect, and recent insights into cancer cell metabolism have led to the development of small-molecule inhibitors that disrupt these cellular pathways. Both PLD1 and PLD2 have been shown to at least partially modulate essential pathways that are involved in metabolic reprogramming67. Small-molecule inhibitors of PLD isoenzymes may, therefore, be better tolerated than conventional chemotherapeutic agents because they only partially suppress the stress-mediated responses of metabolic pathways as opposed to shutting them down completely36.
Many extracellular signals, such as hormonal stimulation and acute removal of nutrients, can upregulate the production of certain metabolic species. Recently, it was demonstrated in vitro that the PLD1-selective inhibitor VU0359595 and the PLD2-selective inhibitor VU0364739 transiently reduce pyrimidine biosynthesis in human glioblastoma-derived cells52. A detailed comparison of parallel metabolomic and phosphoproteomic analyses suggested that these effects were mediated through the modulation of the enzyme CAD, which acts downstream of mTOR. This is consistent with previous reports that demonstrated the modulation of mTOR pathways by PLD-generated PtdOH68,84, and may explain why some PLD inhibitors, via their indirect effect on CAD function, can have activity against both cancer cells and viruses15,16,46 (see also Fig. 10).
PLD and the CNS
Since the first description of mammalian PLD activity in 1973 in rat brain extracts85, the role of PLD enzymes in the CNS has been of intense interest86,87,88. It is now appreciated that signal-dependent activation of PLD enzymes is observed in a diverse array of brain-derived and neuron-derived cells (for example, glial cells, primary neurons, and glioma and neuroblastoma cells), and various neurotransmitters and neuromodulators have been shown to activate PLD enzymes86,87,88. In the CNS, PLD1 and PLD2 display a distinct differential distribution and function, and numerous studies in the past 10 years have begun to delineate the potential of both isoenzymes to represent therapeutic targets in CNS disorders. Previous efforts relied on the overexpression of either catalytically active or inactive forms of PLD1 or PLD2 in cells; on mice in which particular PLD isoenzymes were genetically deleted; or on the use of RNAi to knock down individual PLD isoforms in an effort to discern their discrete roles in brain disorders8,9,86,87,88,89,90,91,92,93,94.
In 2001, it was found that upon inducing experimental autoimmune encephalomyelitis (EAE) in rats, PLD1 expression increased in the spinal cord and in EAE lesions95. This study suggested that PLD1 has a role in autoimmune CNS inflammation, and is involved in the activation of macrophages and astrocytes during the formation of EAE lesions, which suggests that PLD1 inhibitors are possible therapeutics for the treatment of multiple sclerosis and other neuroinflammatory diseases. PLD1 was also shown to be upregulated in the spinal cords of rats with clip compression injuries, which suggests that PLD1 inhibitors might also be useful for the treatment of spinal cord injury89. In addition, PLD1 has been implicated in arterial thrombosis and ischaemic stroke8. In 2010, Pld1−/− mice were shown to display impaired α2bβ3 integrin activation and defective glycoprotein 1b-dependent aggregate formation, which led to protection from thrombosis and ischaemic brain injury without increasing bleeding times8. This finding was recapitulated in 2013 in a study that used the PLD inhibitor FIPI and demonstrated that pharmacological inhibition of PLD also reduced occlusive thrombosis, led to smaller infarct sizes, and improved motor and cognition function90.
Among the exciting discoveries relevant to CNS disorders is the identification of PLD as a potential therapeutic target in Alzheimer disease. Studies reporting that PLD1 was upregulated in the mitochondrial fraction of brain tissue from patients with Alzheimer disease, and that the β-amyloid (Aβ) region of amyloid precursor protein (APP) binds to and stimulates PLD91,92, contributed to recent interest in the development of lead compounds. In 2010, using Pld1−/− and Pld2−/− mice, the non-overlapping roles of PLD1 and PLD2 in the pathogenesis of Alzheimer disease were demonstrated, along with the potential of therapeutically targeting these enzymes9. Overexpression and biochemical studies showed that PLD1 (but not PLD2) regulates the trafficking of APP and the assembly of the γ-secretase complex via a direct interaction with presenilin 1 (PS1)9,93. In in vivo experiments, PLD2 was shown to be required for the synaptotoxic action of Aβ, and Pld2 ablation rescued memory deficits and promoted synaptic protection in SwAPP mice, despite a high Aβ load9. Finally, in 2014, a genome-wide association study identified a rare variant in PLD3 that doubled the risk for late-onset Alzheimer disease (LOAD); however, less than 1% of individuals with LOAD carry the PLD3 mutation94. PLD3 is highly expressed in brain regions that are prone to Alzheimer disease, such as the cortex and hippocampus, and is expressed at significantly lower levels in neurons from the brains of patients with Alzheimer disease than in control brains94. Unfortunately, it is unknown whether any of the existing PLD-inhibitor chemotypes modulate PLD3 catalytic activity, and the precise biological function of PLD3 in the human CNS is also unknown. It is also noteworthy that several recent reports have questioned some of the initial findings regarding the molecular association between rare alleles of PLD3 and the occurrence of early onset Alzheimer disease96,97,98.
With the next generation of potent, isoenzyme-selective and highly brain-penetrant PLD inhibitors now available, the next few years should provide promising advances in targeting PLD isoenzymes in multiple CNS disorders and should identify new targets in cellular lipid signalling cascades.
Looking ahead and moving forward
Lipid signalling metabolite intermediates provide potential targets for therapeutic intervention in cancer, infectious diseases, and CNS disorders. Here, it is of note that the production of newly synthesized PtdOH, as a consequence of infection-induced activation of cellular signalling pathways, may be under-reported. PtdOH accumulation may be highly transient owing to the rapid flux of these lipid species, as enzymes convert PtdOH into DAG, lysophosphatidic acid (lysoPtdOH) and constituents of the Kennedy pathway (see Fig. 2). Investigations in HCMV-infected cell lines suggested that PtdOH generation in response to infection occurred via de novo biosynthesis and not as a result of the activity of PLD enzymes13. However, we observed increases in PtdOH mass following the infection of cell lines with influenza virus, an effect that was reduced after treatment with isoform-selective PLD inhibitors14. This leads us to propose the 'PtdOH hypothesis of infection', which postulates that PtdOH accumulation may be a component of infection that is broadly relevant to viral, bacterial and fungal pathogens. Moreover, although the roles of PLD enzymes in cancer, and infectious and neurodegenerative diseases are undoubtedly complex and multifaceted, it is tempting to speculate that this reflects a shared role of intermediate metabolism in each of these diseases.
The recent discovery that PLD1 influences dNTP stores through effects on mTOR and CAD15,52 provides new insights into how small-molecule inhibitors of PLD1 affect retroviral replication. Since abnormalities in mTOR signalling also occur in the early stages of Alzheimer disease99 and in cancer, PLD1 inhibitors may have similar mechanisms of action in these different conditions. Moreover, the PLD2–AKT signalling axis seems to be a common node of importance in several pathogenic processes. This pathway lies upstream of the beclin 1–rubicon complex36 and other key components that regulate cellular autophagy100, illustrating a shared role of lipid metabolism in cancer, infection and CNS conditions.
Targeting distinct PLD isoenzymes may have broad therapeutic potential and it is likely that small-molecule inhibitors of PLD isoenzymes will be tested for efficacy in diseases for which there is currently an unsatisfactory standard of care. The absence of toxic effects in animal models is highly encouraging. Many details remain to be discovered about the roles of PLD isoenzymes, but the development of multiple chemical scaffolds that function as small-molecule, isoenzyme-selective inhibitors of PLD provides an important step in defining the therapeutic value of targeting both mammalian and bacterial forms of PLD. It will be essential to continue and extend quantitative measurements of PtdOH and its metabolic products in various disease states, and to identify whether the source of disease-associated alterations in levels of PtdOH is the misregulation of PLD1, PLD2 or some other enzyme that is involved in the production of this important signalling lipid. With current advances in mass spectrometry-based metabolomic, lipidomic and phosphoproteomic analyses, new participants in established signalling and metabolic pathways are being revealed, which provide exciting opportunities for therapeutic targeting.
A key question to be answered is whether PtdOH predominantly serves a biophysical role (for example, in entry and egress of viruses), or whether it primarily affects nuclear transcriptional events or other innate immune processes that are also exploited by bacteria and parasites. The structural roles of lipids as components of the viral envelope also remain an area for further scrutiny101. PtdOH generation occurring via de novo synthesis in the Kennedy pathway, the PLD pathway and the PLC–DGK pathway needs to be further interrogated experimentally in other pathogenic microorganisms. This will allow to determine whether inhibitors of these pathways could serve as broad-spectrum anti-infectious agents, which are crucially needed as drug-resistant strains continue to emerge.
References
Ponting, C. P. & Kerr, I. D. A novel family of phospholipase D homologues that includes phospholipid synthases and putative endonucleases: identification of duplicated repeats and potential active site residues. Protein Sci. 5, 914–922 (1996). This is the original report that identified the sequences that define the PLD superfamily.
Sato, T., Hongu, T., Sakamoto, M., Funakoshi, Y. & Kanaho, Y. Molecular mechanism of N-formyl-methionyl-leucyl-phenylalanine-induced superoxide generation and degranulation in mouse neutrophils: phospholipase D is dispensable. Mol. Cell. Biol. 33, 136–145 (2013).
Bruntz, R. C., Lindsley, C. W. & Brown, H. A. Phospholipase D and phosphatidic acid signaling pathways as targets for therapeutics: roles in cancer. Pharmacol. Rev. 66, 1033–1079 (2014).
Peng, X. & Frohman, M. A. Mammalian phospholipase D physiological and pathological roles. Acta. Physiol. (Oxf). 204, 219–226 (2012).
Selvy, P. E., Lavieri, R. L., Lindsley, C. W. & Brown, H. A. Phospholipase D: enzymology, signaling, and chemical modulation. Chem. Rev. 111, 6064–6119 (2011).
Oude Weernink, P. A., Han, L., Jakobs, K. H. & Schmidt, M. Dynamic phospholipid signaling by G protein-coupled receptors. Biochim. Biophys. Acta. 1768, 880–900 (2007).
DeYonker, N. J. & Webster, C. E. Phosphoryl transfers of the phospholipase D superfamily: a quantum mechanical theoretical study. J. Am. Chem. Soc. 135, 13764–13774 (2013).
Elvers, M. et al. Impaired αIIbβ3 integrin activation and shear-dependent thrombus formation in mice lacking phospholipase D1. Sci. Signal. 3, ra1 (2010).
Oliveira, T. G. et al. Phospholipase D2 ablation ameliorates Alzheimer's disease-linked synaptic dysfunction and cognitive deficits. J. Neurosci. 30, 16419–16428 (2010).
Norton, L. J. et al. PLD1, rather than PLD2, regulates phorbol ester-, adhesion dependent-, and Fcγ-receptor-stimulated reactive oxygen species production in neutrophils. J. Cell Sci. 124, 1973–1983 (2011).
Scott, S. A. et al. Regulation of phospholipase D activity and phosphatidic acid production after purinergic (P2Y6) receptor stimulation. J. Biol. Chem. 288, 20477–20487 (2013).
Yoon, M. S. et al. Rapid mitogenic regulation of the mTORC1 inhibitor, DEPTOR, by phosphatidic acid. Mol. Cell 58, 549–556 (2015).
Liu, S. T. et al. Synaptic vesicle-like lipidome of human cytomegalovirus virions reveals a role for SNARE machinery in virion egress. Proc. Natl Acad. Sci. USA 108, 12869–12874 (2011).
Oguin, T. H. et al. Phospholipase D facilitates efficient entry of influenza virus, allowing escape from innate immune inhibition. J. Biol. Chem. 289, 25405–25417 (2014). This paper demonstrates that viruses can activate PLD enzymes in the host cell as part of infection.
Taylor, H. E. et al. Phospholipase D1 couples CD4+ T cell activation to c-Myc-dependent deoxyribonucleotide pool expansion and HIV-1 replication. PLoS Pathog. 11, e1004864 (2015).
Scott, S. A. et al. Design of isoform-selective phospholipase D inhibitors that modulate cancer cell invasiveness. Nat. Chem. Biol. 5, 108–117 (2009). This was the first paper to show that isoform-selective inhibition of PLD enzymes with small-molecule ligands is possible.
Scott, S. A., Mathews, T. P., Ivanova, P. T., Lindsley, C. W. & Brown, H. A. Chemical modulation of glycerolipid signaling and metabolic pathways. Biochim. Biophys. Acta. 1841, 1060–1084 (2014).
Scott, S. A. et al. Discovery of desketoraloxifene analogs as inhibitors of mammalian, Pseudomonas aeruginosa, and NAPE phospholipase D enzymes. ACS Chem. Biol. 10, 421–432 (2015).
Oh, M. K., Yang, H. & Roberts, M. F. Using O-(n-alkyl)-N-(N,N'-dimethylethyl)phosphoramidates to investigate the role of Ca2+ and interfacial binding in a bacterial phospholipase D. Biochim. Biophys. Acta. 1649, 146–153 (2003).
Egertová, M., Simon, G. M., Cravatt, B. F. & Elphick, M. R. Localization of N-acyl phosphatidylethanolamine phospholipase D (NAPE-PLD) expression in mouse brain: a new perspective on N-acylethanolamines as neural signaling molecules. J. Comp. Neurol. 506, 604–615 (2008).
Monovich, L. et al. Optimization of halopemide for phospholipase D2 inhibition. Bioorg. Med. Chem. Lett. 17, 2310–2311 (2007). This report from Novartis catalysed current research into PLD inhibitors by showing that halopemide, an antipsychotic agent used in humans, is also an inhibitor of PLD2.
Loonen, A. J., Soe-Agnie, C. J. & Soudijn, W. Effects of halopemide on GABA receptor binding, uptake and release. Brain Res. 210, 485–492 (1981).
Loonen, A. J. & Soudijn, W. Halopemide, a new psychotropic agent. Cerebral distribution and receptor interactions. Pharm. Weekbl. Sci. 7, 1–9 (1985).
Su, W. et al. 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a phospholipase D pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol. Pharmacol. 75, 437–446 (2009).
Lewis, J. A. et al. Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part I: impact of alternative halogenated privileged structures for PLD1 specificity. Bioorg. Med. Chem. Lett. 19, 1916–1920 (2009). This paper demonstrates that highly selective PLD1 inhibitors can be developed.
Lavieri, R. et al. Design and synthesis of isoform-selective phospholipase D (PLD) inhibitors. Part II: identification of the 1,3,8-triazaspiro[4,5]decan-4-one privileged structure that engenders PLD2 selectivity. Bioorg. Med. Chem. Lett. 19, 2240–2243 (2009).
Lavieri, R. R. et al. Design, synthesis and biological evaluation of halogenated N-(2-(4-oxo-1-phenyl-1,3,8-triazasprio[4.5]decan-8-yl)ethyl)benzamides: discovery of an isoform-selective small molecule phospholipase D2 (PLD2) inhibitor. J. Med. Chem. 53, 6706–6719 (2010). This paper demonstrates that highly selective PLD2 inhibitors can be developed.
O'Reilly, M. C. et al. Development of dual PLD1/2 and PLD2 selective inhibitors from a common 1,3,8-triazaspiro[4.5]decane core: discovery of ML298 and ML299 that decrease invasive migration in U87-MG glioblastoma cells. J. Med. Chem. 56, 2695–2699 (2013).
O'Reilly, M. C. et al. Discovery of a highly selective PLD2 inhibitor, VU0468809 (ML395): a new probe with improved physiochemical properties and broad spectrum antiviral activity against influenza strains. ChemMedChem. 9, 2633–2637 (2014). This study reports the seminal finding that selective PLD2 inhibition is efficacious across a broad panel of influenza viruses, including pandemic strains.
O'Reilly, M. C., Scott, S. A., Brown, H. A. & Lindsley, C. W. Further evaluation of novel structural modifications to scaffolds that engender PLD isoform selective inhibition. Bioorg. Med. Chem. Lett. 24, 5553–5557 (2014).
Lindsley, C. W. et al. Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorg. Med. Chem. Lett. 15, 761–764 (2005).
DeFeo-Jones, D. et al. Tumor cell sensitization to apoptotic stimuli by selective inhibition of specific Akt/PKB family members. Mol. Cancer Ther. 4, 271–279 (2005).
Bornhop, D. J., Baksh, M., Finn, M. G. & Kussrow, A. Backscattering interferometric analysis of membrane materials. US Patent 20130040306 (2013).
Lavieri, R. R. Synthesis, Development and Biochemical Characterization of Small Molecule, Isoform-Selective Phospholipase D Inhibitors and Photoactivatable Probes. Ph.D. thesis, Vanderbilt Univ. (2014).
Ganesan, R., Mahankali, M. Alter, G. & Gomez-Cambronero, J. Two sites of action for PLD2 inhibitors: the enzyme catalytic center and an allosteric, phosphoinositide binding pocket. Biochim. Biophys. Acta. 1851, 261–272 (2015).
Bruntz, R. C., Taylor, H. E., Lindsley, C. W. & Brown, H. A. Phospholipase D2 mediates survival signaling through direct regulation of Akt in glioblastoma cells. J. Biol. Chem. 289, 600–616 (2014). This study establishes a link between PLD2 activity and AKT in survival pathways that are crucial in cancer.
Cummings, S. R. et al. The effect of raloxifene on risk of breast cancer in postmenopausal women: results from the MORE randomized trial. Multiple Outcomes of Raloxifene Evaluation. JAMA 281, 2189–2197 (1999).
Plowman, P. N. Tamoxifen as adjuvant therapy in breast cancer. Current status. Drugs 46, 819–833 (1993).
Eisen, S. F. & Brown, H. A. Selective estrogen receptor modulators differentially regulate phospholipase D catalytic activity in ER-negative breast cancer cells. Mol. Pharmacol. 62, 911–920 (2002). This is the first study to report that drugs used in humans directly inhibit PLD activity.
Grese, T. A. et al. Molecular determinants of tissue selectivity in estrogen receptor modulators. Proc. Natl Acad. Sci. USA 94, 14105–14110 (1997).
Sun, X. & Whittaker, G. R. Entry of influenza virus. Adv. Exp. Med. Biol. 790, 72–82 (2013).
Blaising, J. et al. Silibinin inhibits hepatitis C virus entry into hepatocytes by hindering clathrin-dependent trafficking. Cell. Microbiol. 15, 1866–1882 (2013).
Kolokoltsov, A. A. et al. Small interfering RNA profiling reveals key role of clathrin-mediated endocytosis and early endosomes formation for infection by respiratory syncytial virus. J. Virol. 81, 7786–7800 (2007).
Burkard, C. et al. Coronavirus cell entry occurs through the endo-/lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog. 10, e1004502 (2014).
Yang, L. et al. Role of phospholipase D2/phosphatidic acid signal transduction in μ- and δ-opioid receptor endocytosis. Mol. Pharmacol. 78, 105–112 (2010).
Kang, D. W. et al. Autoregulation of phospholipase D activity is coupled to selective induction of phospholipase D1 expression to promote invasion of breast cancer cells. Int. J. Cancer 128, 805–816 (2011).
Wang, R. et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882 (2011).
Korin, Y. D. & Zack, J. A. Nonproductive human immunodeficiency virus type 1 infection in nucleoside-treated G0 lymphocytes. J. Virol. 73, 6526–6532 (1999).
Plesa, G. et al. Addition of deoxynucleosides enhances human immunodeficiency virus type 1 integration and 2LTR formation in resting CD4+ T cells. J. Virol. 81, 13938–13942 (2007).
Sturdevant, C. B. et al. Compartmentalized replication of R5 T cell-tropic HIV-1 in the central nervous system early in the course of infection. PLoS Pathog. 11, e1004720 (2015).
Hyodo, K. et al. Phosphatidic acid produced by phospholipase D promotes RNA replication of a plant RNA virus. PLoS Pathog. 11, e1004909 (2015).
Mathews, T. P. et al. Human phospholipase D activity transiently regulates pyrimidine biosynthesis in malignant gliomas. ACS Chem. Biol. 10, 1258–1268 (2015).
Ajl, S. J., Reedal, J. S., Durrum, E. L. & Warren, J. J. Studies on plague. I. Purification and properties of the toxin of Pasteurella pestis. J. Bacteriol. 70, 158–169 (1955).
Rudolph, A. E. et al. Expression, characterization, and mutagenesis of the Yersinia pestis murine toxin, a phospholipase D. J. Biol. Chem. 274, 11824–11831 (1999).
Hinnebusch, B. J. et al. Role of Yersinia murine toxin in survival of Yersinia pestis in the midgut of the flea vector. Science 296, 733–735 (2002).
Edwards, J. L., Entz, D. D. & Apicella, M. A. Gonococcal phospholipase D modulates the expression and function of complement receptor 3 in primary cervical epithelial cells. Infect. Immun. 71, 6381–6391 (2003).
Edwards, J. L. & Apicella, M. A. Neisseria gonorrhoeae PLD directly interacts with Akt kinase upon infection of primary, human, cervical epithelial cells. Cell Microbiol. 8, 1253–1257 (2006).
Taylor, L. D., Nelson, D. E., Dorward, D. W., Whitmire, W. M. & Caldwell, H. D. Biological characterization of Chlamydia trachomatis plasticity zone MACPF domain family protein CT153. Infect. Immun. 78, 2691–2699 (2010).
Schroeder, G. N. et al. Legionella pneumophila effector LpdA is a palmitoylated phospholipase D virulence factor. Infect. Immun. 83, 3898–4002 (2015).
Jacobs, A. C. et al. Inactivation of phospholipase D diminishes Acinetobacter baumannii pathogenesis. Infect. Immun. 78, 1952–1962 (2010).
Russell, A. B. et al. Diverse type VI secretion phospholipases are functionally plastic antibacterial effectors. Nature 496, 508–512 (2013).
Wilderman, P. J., Vasil, A. I., Martin, W. E., Murphy, R. C. & Vasil, M. L. Pseudomonas aeruginosa synthesizes phosphatidylcholine by use of the phosphatidylcholine synthase pathway. J. Bacteriol. 184, 4792–4799 (2002). This paper reports the identification of a novel bacterial virulence factor.
Spencer, C. & Brown, H. A. Biochemical characterization of Pseudomonas aeruginosa phospholipase D. Biochemistry 54, 1208–1218 (2015).
Saïdani, N. et al. Discovery of compounds blocking the proliferation of Toxoplasma gondii and Plasmodium falciparum in a chemical space based on piperidinyl-benzimidazolone analogs. Antimicrob. Agents Chemother. 58, 2586–2597 (2014).
Gomez-Cambronero, J. Phospholipase D in cell signaling: from myriad of cell functions to cancer growth and metastasis. J. Biol. Chem. 289, 22557–22566 (2014).
Zhang, Y. & Frohman, M. A. Cellular and physiological roles of phospholipase D1 in cancer. J. Biol. Chem. 289, 22567–22574 (2014).
Kang, D. W., Choi, K. Y. & Min do, S. Functional regulation of phospholipase D expression in cancer and inflammation. J. Biol. Chem. 289, 22575–22582 (2014).
Foster, D. A., Salloun, D., Mennon, D. & Frias, M. A. Phospholipase D and the maintenance of phosphatidic acid levels for regulation of mammalian target of rapamycin (mTOR). J. Biol. Chem. 289, 22583–22588 (2014).
Uchida, N., Okamura, S., Nagamachi, Y. & Yamashita, S. Increased phospholipase D activity in human breast cancer. J. Cancer Res. Clin. Oncol. 123, 280–285 (1997).
Noh, D. Y. et al. Overexpression of phospholipase D1 in human breast cancer tissues. Cancer Lett. 161, 207–214 (2000).
Chen, Y., Zheng, Y. & Foster, D. A. Phospholipase D confers rapamycin resistance in human breast cancer cells. Oncogene 22, 3937–3942 (2003).
Eder, A. M., Sasagawa, T., Mao, M., Aoki, J. & Mills, G. B. Constitutive and lysophosphatidic acid (LPA)-induced LPA production role of phospholipase D and phospholipase A2. Clin. Cancer Res. 6, 2482–2491 (2000).
Iorio, E. et al. Activation of phosphatidylcholine cycle enzymes in human epithelial ovarian cancer cells. Cancer Res. 70, 2126–2135 (2010).
Hsu, Y. L. et al. Phospholipase D signaling pathway is involved in lung cancer-derived IL-8 increased osteoclastogenesis. Carcinogenesis 31, 587–596 (2010).
Yoshida, M., Okamura, S., Kodaki, T., Mori, M. & Yamashita, S. Enhanced levels of oleate-dependent and Arf-dependent phospholipase D isoforms in experimental colon cancer. Oncol. Res. 10, 399–406 (1998).
Yamada, Y. et al. Association of a polymorphism of the phospholipase D2 gene with the prevalence of colorectal cancer. Mol. Med. 81, 126–131 (2003).
Zhao, Y. et al. Increased activity and intranuclear expression of phospholipase D2 in human renal cancer. Biochem. Biophys. Res. Commun. 278, 140–143 (2000).
Douziech, N. et al. Growth effects of regulatory peptides and intracellular signaling routes in human pancreatic cancer cell lines. Endocrine. 9, 171–183 (1998).
Qi, C. et al. Lysophosphatidic acid stimulates phospholipase D activity and cell proliferation in PC-3 human prostate cancer cells. J. Cell. Physiol. 174, 261–272 (1998).
Zhao, C., Du, G., Skowronek, K., Frohman, M. A. & Bar-Sagi, D. Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by SOS. Nat. Cell Biol. 9, 706–712 (2007).
Buchanan, F. G. et al. Requirement of phospholipase D1 activity in H-RasV12-induced transformation. Proc. Natl Acad. Sci. USA 102, 1638–1642 (2005).
Cipriano, R. et al. FAM83B mediates EGFR- and RAS-driven oncogenic transformations. J. Clin. Invest. 122, 3197–3210 (2012).
Cipriano, R. et al. Hyperactivation of EGFR and downstream effector phospholipase D1 by oncogenic FAM83B. Oncogene 33, 3298–3306 (2014).
Sun, Y. & Chen, J. mTOR signaling: PLD takes center stage. Cell Cycle 7, 3118–3123 (2008).
Saito, M. & Kanfer, J. Solubilization and properties of a membrane-bound enzyme from rat brain catalyzing a base-exchange reaction. Biochem. Biophys. Res. Commun. 53, 391–398 (1973).
Klein, J., Chalifa, V., Liscovitch, M. & Löffelholz, K. Role of phospholipase D activation in nervous system physiology and pathophysiology. J. Neurochem. 65, 1445–1455 (1995).
Oliveira, T. G. & Di Paolo, G. Phospholipase D in brain function and Alzheimer's disease. Biochim. Biophys. Acta. 1801, 799–805 (2010).
Lindsley, C. W. & Brown, H. A. Phospholipase D as a therapeutic target in brain disorders. Neuropsychopharmacology 37, 301–302 (2012).
Jung, K. et al. Upregulation of phospholipase D1 in the spinal cords of rats with clip compression injury. Neurosci. Lett. 336, 126–130 (2003).
Stegner, D. et al. Pharmacological inhibition of phospholipase D protects mice from occlusive thrombus formation and ischemic stroke — brief report. Arterioscler. Thromb. Vasc. Biol. 33, 2212–2217 (2013).
Jin, J.-K. et al. Phospholipase D1 is up-regulated in the mitochondrial fraction from the brains of Alzheimer's disease patients. Neurosci. Lett. 407, 263–267 (2006).
Jin, J.-K. et al. Phospholipase D1 is associated with amyloid precursor protein in Alzheimer's disease. Neurobiol. Aging 28, 1015–1027 (2007).
Cai, D. et al. Presenilin-1 uses phospholipase D as a negative regulator of β-amyloid formation. Proc. Natl Acad. Sci. USA 103, 1941–1946 (2006).
Cruchaga, C. et al. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer's disease. Nature 505, 550–554 (2014).
Ahn, M., Min, D. S., Kang, J., Jung, K. & Shin, T. Increased expression of phospholipase D1 in the spinal cords of rats with experimental autoimmune encephalomyelitis. Neurosci. Lett. 316, 95–98 (2001).
Lambert, J. C. et al. PLD3 and sporadic Alzheimer's disease risk. Nature 520, E1 (2015).
Van der Lee, S. J. et al. PLD3 variants in population studies. Nature 520, E2–E3 (2015).
Hooli, B. V. et al. PLD3 gene variants and Alzheimer's disease. Nature 520, E7–E8 (2015).
Tramutola, A. et al. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J. Neurochem. 133, 739–749 (2015).
Jang, Y. H., Choi, K. Y. & Min, D. S. Phospholipase D-mediated autophagic regulation is a potential target for cancer therapy. Cell Death Differ. 21, 533–546 (2014).
Ivanova, P. T. et al. Lipid composition of viral envelope of three strains of influenza virus — not all viruses are created equal. ACS Infect. Dis. 1, 399–452 (2015).
Powner, D. J., Pettitt, T. R., Anderson, R., Nash, G. B. & Wakelam, M. J. Stable adhesion and migration of human neutrophils requires phospholipase D-mediated activation of the integrin CD11b/CD18. Mol. Immunol. 44, 3211–3221 (2007).
Brown, H. A., Gutowski, S., Moomaw, C. R., Slaughter, C. & Sternweis, P. C. ADP-ribosylation factor (ARF), a small GTP-dependent regulatory protein, stimulates phospholipase D activity. Cell 75, 1137–1144 (1993). This paper identifies the first protein and lipid modulators of PLD using a novel in vitro biochemical assay.
Brown, H. A., Gutowski, S., Kahn, R. A. & Sternweis, P. C. Partial purification and characterization of Arf-sensitive phospholipase D from porcine brain. J. Biol. Chem. 270, 14935–14943 (1995).
Jiang, X., Gutowski, S., Singer, W. D. & Sternweis, P. C. Assays and characterization of mammalian phosphatidylinositol-4,5-bisphosphate-sensitive phospholipase D. Methods Enzymol. 345, 328–334 (2002).
Morris, A. J., Engelbrecht, J. & Frohman, M. A. Structure and regulation of phospholipase D. Trends Pharmacol. Sci. 17, 182–185 (1996).
Cockcroft, S. et al. Phospholipase D: a downstream effector of ARF in granulocytes. Science 263, 523–526 (1994).
Brown, H. A. & Sternweis, P. C. Stimulation of phospholipase D by ADP-ribosylation factor. Methods Enzymol. 257, 313–324 (1995).
Lambeth, J. D. et al. ADP-ribosylation factor functions synergistically with a 50-kDa cytosolic factor in cell-free activation of human neutrophil phospholipase D. J. Biol. Chem. 270, 2431–2434 (1995).
Kwak, J. Y., Lopez, I., Uhlinger, D. J., Ryu, S. H. & Lambeth, J. D. RhoA and a cytosolic 50-kDa factor reconstitute GTPγS-dependent phospholipase D activity in human neutrophil subcellular fractions. J. Biol. Chem. 270, 27093–27098 (1995).
Park, S. K., Min, D. S. & Exton, J. H. Definition of the protein kinase C interaction site of phospholipase D. Biochim. Biophys. Res. Commun. 244, 364–367 (1998).
Exton, J. H. Regulation of phospholipase D. Biochim. Biophys. Acta. 1439, 121–136 (1999).
Brown, H. A., Henage, L. G., Preininger, A. M., Xiang, Y. & Exton, J. H. Biochemical analysis of phospholipase D. Methods Enzymol. 434, 49–87 (2007).
Honda, A. et al. Phosphatidylinositol 4-phosphate 5-kinase α is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell 99, 521–532 (1999).
Kooijman, E. E., Chupin, V., de Kruijff, B. & Burger, K. N. Modulation of membrane curvature by phosphatidic acid and lysophosphatidic acid. Traffic. 4, 162–174 (2002).
Kooijman, E. E. & Burger, K. N. Biophysics and function of phosphatidic acid: a molecular perspective. Biochim. Biophys. Acta. 1791, 881–888 (2009).
Acknowledgements
The authors are grateful for generous support from the US National Institutes of Health (NIGMS 1R01GM121629; HAB and HHSN272200800058C; H.A.B. and P.G.T.), the Centers of Excellence for Influenza Research and Surveillance (CEIRS; HHSN272201400006C; P.G.T.) and the Molecular Libraries Probe Production Centers Network (MLPCN; C.W.L.), in addition to Voices Against Brain Cancer (H.A.B.), the Vanderbilt Institute of Chemical Biology (H.A.B.), the Vanderbilt University Bixler-Johnson-Mayes Endowed Chair (H.A.B.), the William K. Warren Jr. Endowment (C.W.L.), and The Hartwell Foundation (P.G.T.). The authors thank T. H. Oguin 3rd for helpful discussions, as well as M. D. Armstrong and P. T. Ivanova for invaluable discussions and assistance with editorial and graphical content.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
H.A.B., C.W.L. and P.G.T. are inventors on patents that protect allosteric phospholipase D inhibitors, as well as a method of treatment using these inhibitors.
Related links
FURTHER INFORMATION
Glossary
- Phospholipase D
-
(PLD). A class of phosphodiesterases that hydrolyse phosphatidylcholine and other amine-containing glycerophospholipids to generate phosphatidic acid and a free head group.
- Phosphatidic acid
-
(PtdOH). A molecule that functions as both a lipid second messenger and a key intermediate in glycerophospholipid metabolism. The phospholipase D pathway is one pathway that is used to generate receptor-mediated PtdOH. A major pathway of PtdOH metabolism is conversion to diacylglycerol by lipid phosphatases.
- Transphosphatidylation
-
An alternative reaction in which a short-chain alcohol competes with water as a nucleophile to generate a phosphatidyl alcohol product instead of phosphatidic acid. This naturally occurring reaction, in which alcohols function as alternative substrates of phospholipase D (PLD) enzymes, has an inhibitory effect by diverting the primary catalytic reaction to form new reaction products, but alcohols themselves are not inhibitors of PLD enzymes.
- Drug metabolism and pharmacokinetics
-
(DMPK). Refers to all aspects of the absorption, distribution, metabolism and excretion of a drug.
- Phosphatidylinositol 4,5-bisphosphate
-
(PI(4,5)P2). A glycerophospholipid that is generally found in low levels within cells and functions as a lipid messenger as well as a substrate for other second messengers in many cell-signalling pathways.
- Lipidomics, metabolomics, phosphoproteomics
-
A subset of systems biology-level analyses of the constituents of cells and tissues that are usually conducted as a quantitative analysis between two conditions, such as healthy versus diseased tissue, or treatment with or without a particular drug. The analysis of how lipids, aqueous metabolites and peptide phosphorylation change in response to a defined input provides insights into the mechanistic processes that constitute a complex cellular function or pathophysiological state.
Rights and permissions
About this article

Cite this article
Brown, H., Thomas, P. & Lindsley, C. Targeting phospholipase D in cancer, infection and neurodegenerative disorders. Nat Rev Drug Discov 16, 351–367 (2017). https://doi.org/10.1038/nrd.2016.252
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrd.2016.252
This article is cited by
-
Exercise mimetics: a novel strategy to combat neuroinflammation and Alzheimer’s disease
Journal of Neuroinflammation (2024)
-
SL-scan identifies synthetic lethal interactions in cancer using metabolic networks
Scientific Reports (2023)
-
Systemic impact of heavy metals and their role in cancer development: a review
Environmental Monitoring and Assessment (2023)
-
Incorporation of DNA methylation quantitative trait loci (mQTLs) in epigenome-wide association analysis: application to birthweight effects in neonatal whole blood
Clinical Epigenetics (2022)
-
Structural insights into PA3488-mediated inactivation of Pseudomonas aeruginosa PldA
Nature Communications (2022)
