
Key Points
-
The huge number of genetic and epigenetic changes that are inherent to most cancer cells provide plenty of tumour-associated antigens that the host immune system can recognize, thereby requiring tumours to develop specific immune resistance mechanisms. An important immune resistance mechanism involves immune-inhibitory pathways, termed immune checkpoints, which normally mediate immune tolerance and mitigate collateral tissue damage.
-
A particularly important immune-checkpoint receptor is cytotoxic T-lymphocyte-associated antigen 4 (CTLA4), which downmodulates the amplitude of T cell activation. Antibody blockade of CTLA4 in mouse models of cancer induced antitumour immunity.
-
Clinical studies using antagonistic CTLA4 antibodies demonstrated activity in melanoma. Despite a high frequency of immune-related toxicity, this therapy enhanced survival in two randomized Phase III trials. Anti-CTLA4 therapy was the first agent to demonstrate a survival benefit in patients with advanced melanoma and was approved by the US Food and Drug Administration (FDA) in 2010.
-
Some immune-checkpoint receptors, such as programmed cell death protein 1 (PD1), limit T cell effector functions within tissues. By upregulating ligands for PD1, tumour cells block antitumour immune responses in the tumour microenvironment.
-
Early-stage clinical trials suggest that blockade of the PD1 pathway induces sustained tumour regression in various tumour types. Responses to PD1 blockade may correlate with the expression of PD1 ligands by tumour cells.
-
Multiple additional immune-checkpoint receptors and ligands, some of which are selectively upregulated in various types of tumour cells, are prime targets for blockade, particularly in combination with approaches that enhance the activation of antitumour immune responses, such as vaccines.
Abstract
Among the most promising approaches to activating therapeutic antitumour immunity is the blockade of immune checkpoints. Immune checkpoints refer to a plethora of inhibitory pathways hardwired into the immune system that are crucial for maintaining self-tolerance and modulating the duration and amplitude of physiological immune responses in peripheral tissues in order to minimize collateral tissue damage. It is now clear that tumours co-opt certain immune-checkpoint pathways as a major mechanism of immune resistance, particularly against T cells that are specific for tumour antigens. Because many of the immune checkpoints are initiated by ligand–receptor interactions, they can be readily blocked by antibodies or modulated by recombinant forms of ligands or receptors. Cytotoxic T-lymphocyte-associated antigen 4 (CTLA4) antibodies were the first of this class of immunotherapeutics to achieve US Food and Drug Administration (FDA) approval. Preliminary clinical findings with blockers of additional immune-checkpoint proteins, such as programmed cell death protein 1 (PD1), indicate broad and diverse opportunities to enhance antitumour immunity with the potential to produce durable clinical responses.
Similar content being viewed by others
Main
The myriad of genetic and epigenetic alterations that are characteristic of all cancers provide a diverse set of antigens that the immune system can use to distinguish tumour cells from their normal counterparts. In the case of T cells, the ultimate amplitude and quality of the response, which is initiated through antigen recognition by the T cell receptor (TCR), is regulated by a balance between co-stimulatory and inhibitory signals (that is, immune checkpoints)1,2 (Fig. 1). Under normal physiological conditions, immune checkpoints are crucial for the maintenance of self-tolerance (that is, the prevention of autoimmunity) and also to protect tissues from damage when the immune system is responding to pathogenic infection. As described in this Review, the expression of immune-checkpoint proteins can be dysregulated by tumours as an important immune resistance mechanism. T cells have been the major focus of efforts to therapeutically manipulate endogenous antitumour immunity owing to: their capacity for the selective recognition of peptides derived from proteins in all cellular compartments; their capacity to directly recognize and kill antigen-expressing cells (by CD8+ effector T cells; also known as cytotoxic T lymphocytes (CTLs)); and their ability to orchestrate diverse immune responses (by CD4+ helper T cells), which integrates adaptive and innate effector mechanisms. Thus, agonists of co-stimulatory receptors or antagonists of inhibitory signals (the subject of this Review), both of which result in the amplification of antigen-specific T cell responses, are the primary agents in current clinical testing (Table 1). Indeed, the blockade of immune checkpoints seems to unleash the potential of the antitumour immune response in a fashion that is transforming human cancer therapeutics.

Depicted are various ligand–receptor interactions between T cells and antigen-presenting cells (APCs) that regulate the T cell response to antigen (which is mediated by peptide–major histocompatibility complex (MHC) molecule complexes that are recognized by the T cell receptor (TCR)). These responses can occur at the initiation of T cell responses in lymph nodes (where the major APCs are dendritic cells) or in peripheral tissues or tumours (where effector responses are regulated). In general, T cells do not respond to these ligand–receptor interactions unless they first recognize their cognate antigen through the TCR. Many of the ligands bind to multiple receptors, some of which deliver co-stimulatory signals and others deliver inhibitory signals. In general, pairs of co-stimulatory–inhibitory receptors that bind the same ligand or ligands — such as CD28 and cytotoxic T-lymphocyte-associated antigen 4 (CTLA4) — display distinct kinetics of expression with the co-stimulatory receptor expressed on naive and resting T cells, but the inhibitory receptor is commonly upregulated after T cell activation. One important family of membrane-bound ligands that bind both co-stimulatory and inhibitory receptors is the B7 family. All of the B7 family members and their known ligands belong to the immunoglobulin superfamily. Many of the receptors for more recently identified B7 family members have not yet been identified. Tumour necrosis factor (TNF) family members that bind to cognate TNF receptor family molecules represent a second family of regulatory ligand–receptor pairs. These receptors predominantly deliver co-stimulatory signals when engaged by their cognate ligands. Another major category of signals that regulate the activation of T cells comes from soluble cytokines in the microenviron-ment. Communication between T cells and APCs is bidirectional. In some cases, this occurs when ligands themselves signal to the APC. In other cases, activated T cells upregulate ligands, such as CD40L, that engage cognate receptors on APCs. A2aR, adenosine A2a receptor; B7RP1, B7-related protein 1; BTLA, B and T lymphocyte attenuator; GAL9, galectin 9; HVEM, herpesvirus entry mediator; ICOS, inducible T cell co-stimulator; IL, interleukin; KIR, killer cell immunoglobulin-like receptor; LAG3, lymphocyte activation gene 3; PD1, programmed cell death protein 1; PDL, PD1 ligand; TGFβ, transforming growth factor-β; TIM3, T cell membrane protein 3.
T cell-mediated immunity includes multiple sequential steps involving the clonal selection of antigen-specific cells, their activation and proliferation in secondary lymphoid tissues, their trafficking to sites of antigen and inflammation, the execution of direct effector functions and the provision of help (through cytokines and membrane ligands) for a multitude of effector immune cells. Each of these steps is regulated by counterbalancing stimulatory and inhibitory signals that fine-tune the response. Although virtually all inhibitory signals in the immune response ultimately affect intracellular signalling pathways, many are initiated through membrane receptors, the ligands of which are either membrane-bound or soluble (cytokines). As a general rule, co-stimulatory and inhibitory receptors and ligands that regulate T cell activation are not necessarily overexpressed in cancers relative to normal tissues, whereas inhibitory ligands and receptors that regulate T cell effector functions in tissues are commonly overexpressed on tumour cells or on non-transformed cells in the tumour microenvironment. It is the soluble and membrane-bound receptor–ligand immune checkpoints that are the most druggable using agonist antibodies (for co-stimulatory pathways) or antagonist antibodies (for inhibitory pathways) (Table 1). Therefore, in contrast to most currently approved antibodies for cancer therapy, antibodies that block immune checkpoints do not target tumour cells directly, instead they target lymphocyte receptors or their ligands in order to enhance endogenous antitumour activity.
Another category of immune-inhibitory molecules includes certain metabolic enzymes, such as indoleamine 2,3-dioxygenase (IDO) — which is expressed by both tumour cells and infiltrating myeloid cells — and arginase, which is produced by myeloid-derived suppressor cells3,4,5,6,7,8,9. These enzymes inhibit immune responses through the local depletion of amino acids that are essential for anabolic functions in lymphocytes (particularly T cells) or through the synthesis of specific natural ligands for cytosolic receptors that can alter lymphocyte functions. Although this category is not covered in this Review, these enzymes can be inhibited to enhance intratumoral inflammation by molecular analogues of their substrates that act as competitive inhibitors or suicide substrates10,11,12.
In considering the mechanisms of action of inhibitors of various immune checkpoints, it is crucial to appreciate the diversity of immune functions that they regulate. For example, the two immune-checkpoint receptors that have been most actively studied in the context of clinical cancer immunotherapy, cytotoxic T-lymphocyte-associated antigen 4 (CTLA4; also known as CD152) and programmed cell death protein 1 (PD1; also known as CD279) — which are both inhibitory receptors — regulate immune responses at different levels and by different mechanisms. The clinical activity of antibodies that block either of these receptors implies that antitumour immunity can be enhanced at multiple levels and that combinatorial strategies can be intelligently designed, guided by mechanistic considerations and preclinical models. This Review focuses on the CTLA4 and PD1 pathways because these are the two immune checkpoints for which clinical information is currently available. However, it is important to emphasize that multiple additional immune checkpoints represent promising targets for therapeutic blockade based on preclinical experiments, and inhibitors for many of these are under active development (Table 1).
CTLA4: the godfather of checkpoints
The biology of CTLA4. CTLA4, the first immune-checkpoint receptor to be clinically targeted, is expressed exclusively on T cells where it primarily regulates the amplitude of the early stages of T cell activation. Primarily, CTLA4 counteracts the activity of the T cell co-stimulatory receptor, CD28 (Refs 13,14,15). CD28 does not affect T cell activation unless the TCR is first engaged by cognate antigen. Once antigen recognition occurs, CD28 signalling strongly amplifies TCR signalling to activate T cells. CD28 and CTLA4 share identical ligands: CD80 (also known as B7.1) and CD86 (also known as B7.2)16,17,18,19,20. Although the exact mechanisms of CTLA4 action are under considerable debate, because CTLA4 has a much higher overall affinity for both ligands, it has been proposed that its expression on the surface of T cells dampens the activation of T cells by outcompeting CD28 in binding CD80 and CD86, as well as actively delivering inhibitory signals to the T cell21,22,23,24,25,26. The specific signalling pathways by which CTLA4 blocks T cell activation are still under investigation, although a number of studies suggest that activation of the protein phosphatases, SHP2 (also known as PTPN11) and PP2A, are important in counteracting kinase signals that are induced by TCR and CD28 (Ref. 15). However, CTLA4 also confers 'signalling-independent' T cell inhibition through the sequestration of CD80 and CD86 from CD28 engagement, as well as active removal of CD80 and CD86 from the antigen-presenting cell (APC) surface27. The central role of CTLA4 for keeping T cell activation in check is dramatically demonstrated by the lethal systemic immune hyperactivation phenotype of Ctla4-knockout mice28,29.
Even though CTLA4 is expressed by activated CD8+ effector T cells, the major physiological role of CTLA4 seems to be through distinct effects on the two major subsets of CD4+ T cells: downmodulation of helper T cell activity and enhancement of regulatory T (TReg) cell immunosuppressive activity14,30,31 (Box 1). CTLA4 blockade results in a broad enhancement of immune responses that are dependent on helper T cells and, conversely, CTLA4 engagement on TReg cells enhances their suppressive function. CTLA4 is a target gene of the forkhead transcription factor FOXP3 (Refs 32,33), the expression of which determines the TReg cell lineage34,35, and TReg cells therefore express CTLA4 constitutively. Although the mechanism by which CTLA4 enhances the immunosuppressive function of TReg cells is not known, TReg cell-specific CTLA4 knockout or blockade significantly inhibits their ability to regulate both autoimmunity and antitumour immunity30,31. Thus, in considering the mechanism of action for CTLA4 blockade, both enhancement of effector CD4+ T cell activity and inhibition of TReg cell-dependent immunosuppression are probably important factors.
Clinical application of CTLA4-blocking antibodies — the long road from mice to FDA approval. Initially, the general strategy of blocking CTLA4 was questioned because there is no tumour specificity to the expression of the CTLA4 ligands (other than for some myeloid and lymphoid tumours) and because the dramatic lethal autoimmune and hyperimmune phenotype of Ctla4-knockout mice predicted a high degree of immune toxicity associated with blockade of this receptor. However, Allison and colleagues36 used preclinical models to demonstrate that a therapeutic window was indeed achieved when CTLA4 was partially blocked with antibodies. The initial studies demonstrated significant antitumour responses without overt immune toxicities when mice bearing partially immunogenic tumours were treated with CTLA4 antibodies as single agents. Poorly immunogenic tumours did not respond to anti-CTLA4 as a single agent but did respond when anti-CTLA4 was combined with a granulocyte–macrophage colony-stimulating factor (GM-CSF)-transduced cellular vaccine37. These findings suggested that, if there is an endogenous antitumour immune response in the animals after tumour implantation, CTLA4 blockade could enhance that endogenous response, which ultimately can induce tumour regression. In the case of poorly immunogenic tumours, which do not induce substantial endogenous immune responses, the combination of a vaccine and a CTLA4 antibody could induce a strong enough immune response to slow tumour growth and in some cases eliminate established tumours.
These preclinical findings encouraged the production and testing of two fully humanized CTLA4 antibodies, ipilimumab and tremelimumab, which began clinical testing in 2000. As with virtually all anticancer agents, initial testing was as a single agent in patients with advanced disease that were not responding to conventional therapy38. Both antibodies produced objective clinical responses in ∼10% of patients with melanoma, but immune-related toxicities involving various tissue sites were also observed in 25–30% of patients, with colitis being a particularly common event39,40,41 (Fig. 2). The first randomized Phase III clinical trial to be completed was for tremelimumab in patients with advanced melanoma. In this trial, 15 mg per kg tremelimumab was given every three months as a single agent and compared with dacarbazine (also known as DTIC), a standard melanoma chemotherapy treatment. The trial showed no survival benefit with this dose and schedule relative to dacarbazine42.

Depicted on the left of the figure are examples of regressions of lung (top two panels) and brain (lower panel) metastases in a patient with melanoma who was treated with the cytotoxic T-lymphocyte-associated antigen 4 (CTLA4) antibody, ipilimumab. 25–30% of patients treated with extended doses of anti-CTLA4 therapy can develop immune-related 'on-target' toxicities. However, the frequency of severe adverse toxicities was lower (10–15%) with the short course that was used in the Phase III trial that led to the approval of ipilimumab. This short-course regimen (4 doses at a cost of US$30,000 per dose) was recommended by the US Food and Drug Administration (FDA). As shown on the right of the figure, common tissues affected by immune-related toxicities from treatment with anti-CTLA4 therapy include the skin (dermatitis) and the colon (colitis). Tissues that do not undergo such rapid regeneration as the skin and colon, such as lung and liver and the pituitary and thyroid glands, are less frequently affected. Immune toxicities from anti-CTLA4 therapy are usually successfully mitigated by treatment with systemic steroids and tumour necrosis factor (TNF) blockers when systemic steroids are not effective. Ongoing tumour responses typically continue even after a course of steroids. Figure is reproduced, with permission, from Ref. 39 © (2003) National Academy of Sciences, USA.
However, ipilimumab fared better. Even though the intrinsic activity, response rates in Phase II trials and immune toxicity profiles were similar for both antibodies, ipilimumab was more carefully evaluated at different doses and schedules. Additionally, more careful definition of algorithms for improved clinical management of the immune toxicities (using steroids and tumour necrosis factor (TNF) blockers) mitigated the overall morbidity and mortality that were associated with immunological toxicities. Interestingly, although there is evidence that clinical responses might be associated with immune-related adverse events, this correlation is modest43. Finally, in a randomized three-arm clinical trial of patients with advanced melanoma that received either: a peptide vaccine of melanoma-specific gp100 (also known as PMEL) alone; the gp100 vaccine plus ipilimumab; or ipilimumab alone, there was a 3.5 month survival benefit for patients in both groups receiving ipilimumab (that is, with or without the gp100 peptide vaccine) compared with the group receiving the gp100 peptide vaccine alone44. As ipilimumab was the first therapy to demonstrate a survival benefit for patients with metastatic melanoma, it was approved by the US Food and Drug Administration (FDA) for the treatment of advanced melanoma in 2010 (dacarbazine was approved on the basis of response rate but has not been shown to provide a survival benefit in patients with melanoma).
More impressive than the mean survival benefit was the effect of ipilimumab on long-term survival: 18% of the ipilimumab-treated patients survived beyond two years (compared with 5% of patients receiving the gp100 peptide vaccine alone)44. In this and other studies, the proportion of long-term survivors was higher than the proportion of objective responders. The finding of ongoing responses and survival long after completion of a relatively short course of therapy (four doses of 10 mg per kg over 3 months) support the concept that immune-based therapies might re-educate the immune system to keep tumours in check after completion of the therapeutic intervention.
As with all oncology agents that benefit a limited proportion of treated patients, there has been much effort in defining biomarkers that predict clinical responses to anti-CTLA4 therapy. To date, no such pretreatment biomarker has been validated to the point at which it could be applied as part of standard-of-care therapeutic decision-making, although insights have emerged from the identification of certain post-treatment immune responses that seem to correlate with clinical outcome45,46,47.
An important feature of the anti-CTLA4 clinical responses that distinguishes them from conventional chemotherapeutic agents and oncogene-targeted small molecule drugs is their kinetics. Although responses to chemotherapies and tyrosine kinase inhibitors (TKIs) commonly occur within weeks of initial administration, the response to immune-checkpoint blockers is slower and, in many patients, delayed (up to 6 months after treatment initiation). In some cases, metastatic lesions actually increase in size on computed tomography (CT) or magnetic resonance imaging (MRI) scans before regressing, which seems to occur owing to increased immune cell infiltration. These findings demand a re-evaluation of response criteria for immunotherapeutics away from the conventional time-to-progression or Response Evaluation Criteria in Solid Tumours (RECIST) objective response criteria, which were developed on the basis of experiences with chemotherapeutic agents and as the primary measure of drug efficacy48.
Blockade of the PD1 pathway
Another immune-checkpoint receptor, PD1, is emerging as a promising target, thus emphasizing the diversity of potential molecularly defined immune manipulations that are capable of inducing antitumour immune responses by the patient's own immune system.
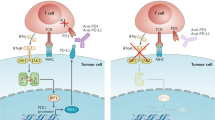
The biology of the PD1 pathway. In contrast to CTLA4, the major role of PD1 is to limit the activity of T cells in peripheral tissues at the time of an inflammatory response to infection and to limit autoimmunity49,50,51,52,53,54,55 (Fig. 3). This translates into a major immune resistance mechanism within the tumour microenvironment56,57,58. PD1 expression is induced when T cells become activated49. When engaged by one of its ligands, PD1 inhibits kinases that are involved in T cell activation through the phosphatase SHP250, although additional signalling pathways are also probably induced. Also, because PD1 engagement inhibits the TCR 'stop signal', this pathway could modify the duration of T cell–APC or T cell–target cell contact59. Similarly to CTLA4, PD1 is highly expressed on TReg cells, where it may enhance their proliferation in the presence of ligand60. Because many tumours are highly infiltrated with TReg cells that probably further suppress effector immune responses, blockade of the PD1 pathway may also enhance antitumour immune responses by diminishing the number and/or suppressive activity of intratumoral TReg cells.

a | The cytotoxic T-lymphocyte-associated antigen 4 (CTLA4)-mediated immune checkpoint is induced in T cells at the time of their initial response to antigen. The level of CTLA4 induction depends on the amplitude of the initial T cell receptor (TCR)-mediated signalling. High-affinity ligands induce higher levels of CTLA4, which dampens the amplitude of the initial response. The key to the regulation of T cell activation levels by the CD28–CTLA4 system is the timing of surface expression. Naive and memory T cells express high levels of cell surface CD28 but do not express CTLA4 on their surface. Instead, CTLA4 is sequestered in intracellular vesicles. After the TCR is triggered by antigen encounter, CTLA4 is transported to the cell surface. The stronger the stimulation through the TCR (and CD28), the greater the amount of CTLA4 that is deposited on the T cell surface. Therefore, CTLA4 functions as a signal dampener to maintain a consistent level of T cell activation in the face of widely varying concentrations and affinities of ligand for the TCR. b | By contrast, the major role of the programmed cell death protein 1 (PD1) pathway is not at the initial T cell activation stage but rather to regulate inflammatory responses in tissues by effector T cells recognizing antigen in peripheral tissues. Activated T cells upregulate PD1 and continue to express it in tissues. Inflammatory signals in the tissues induce the expression of PD1 ligands, which downregulate the activity of T cells and thus limit collateral tissue damage in response to a microorganism infection in that tissue. The best characterized signal for PD1 ligand 1 (PDL1; also known as B7-H1) induction is interferon-γ (IFNγ), which is predominantly produced by T helper 1 (TH1) cells, although many of the signals have not yet been defined completely. Excessive induction of PD1 on T cells in the setting of chronic antigen exposure can induce an exhausted or anergic state in T cells. MHC, major histocompatibility complex.
The two ligands for PD1 are PD1 ligand 1 (PDL1; also known as B7-H1 and CD274) and PDL2 (also known as B7-DC and CD273)50,61,62,63. These B7 family members share 37% sequence homology and arose through gene duplication, which has positioned them within 100 kb of each other in the genome63. Recently, an unexpected molecular interaction between PDL1 and CD80 was discovered64, whereby CD80 expressed on T cells (and possibly APCs) can potentially behave as a receptor rather than a ligand by delivering inhibitory signals when engaged by PDL1 (Refs 65,66). The relevance of this interaction in tumour immune resistance has not yet been determined. Finally, genetic evidence from PD1-deficient T cells suggests that both PDL1 and PDL2 may bind to a co-stimulatory receptor that is expressed on T cells67. These complex binding interactions are reminiscent of the CD80 and CD86 ligand pair, each of which binds the co-stimulatory receptor CD28 that is expressed on resting T cells and the inhibitory receptor CTLA4 that is expressed on activated T cells. However, as stated above, PD1 predominantly regulates effector T cell activity within tissue and tumours, whereas CTLA4 predominantly regulates T cell activation (Fig. 3). Understanding the role of these various interactions in different cancer settings is highly relevant for the selection of both antibodies and recombinant ligands for use in the clinic.
PD1 is more broadly expressed than CTLA4: it is induced on other activated non-T lymphocyte subsets, including B cells and natural killer (NK) cells68,69, which limits their lytic activity. Therefore, although PD1 blockade is typically viewed as enhancing the activity of effector T cells in tissues and in the tumour microenvironment, it also probably enhances NK cell activity in tumours and tissues and may also enhance antibody production either indirectly or through direct effects on PD1+ B cells70.
In addition, chronic antigen exposure, such as occurs with chronic viral infection and cancer, can lead to high levels of persistent PD1 expression, which induces a state of exhaustion or anergy among cognate antigen-specific T cells. This state, which has been demonstrated in multiple chronic viral infections in mice and humans, seems to be partially reversible by PD1-pathway blockade71. Finally, although the major role of the PD1 pathway is in limiting immune effector responses in tissues (and tumours), it can also shift the balance from T cell activation to tolerance at the early stages of T cell responses to antigens within secondary lymphoid tissues (that is, at a similar stage as CTLA4). Taken together, these findings imply a complex set of mechanisms of action for PD1-pathway blockade.
Regulation of expression of PD1 and its ligands in tumours: constitutive versus adaptive immune resistance. PD1 is expressed on a large proportion of tumour-infiltrating lymphocytes (TILs) from many different tumour types72,73. Some of the enhanced PD1 expression among CD4+ TILs reflects a generally high level of PD1 expression on TReg cells, which, as noted above, can represent a large proportion of intratumoral CD4+ T cells. Increased PD1 expression on CD8+ TILs may either reflect an anergic or exhausted state, as has been suggested by decreased cytokine production by PD1+ compared with PD1− TILs from melanomas73.
Just as PD1 is highly expressed on TILs from many cancers, the PD1 ligands are commonly upregulated on the tumour cell surface from many different human tumours2,56. On cells from solid tumours, the major PD1 ligand that is expressed is PDL1. Forced expression of PDL1 on mouse tumour cells inhibits local antitumour T cell-mediated responses56,74,75. Indeed, this combination of findings provides the basis for PD1-pathway blockade to enhance antitumour effector functions in the tumour microenvironment. Immunohistochemistry (IHC) techniques and flow cytometry-based analyses of surface expression have shown that the selective upregulation of PD1 ligands in various types of human tumour is heterogeneous at a number of levels58. Expression patterns of PD1 ligands may be crucial for determining the suitability of therapeutic blockade of this pathway because its primary role in cancer is thought to be immune inhibition within the tumour microenvironment and because PD1 only inhibits lymphocyte function when it is engaged by its ligands, PDL1 and PDL2.
Initially, most melanoma, ovarian and lung cancer samples were reported to have high expression levels of PDL156,75,76 and, subsequently, many other human cancers were reported to upregulate PDL1 expression (reviewed in Ref. 2). In addition to tumour cells, PDL1 is commonly expressed on myeloid cells in the tumour microenvironment77,78,79. An initial report in renal cancer demonstrated that the expression of PDL1 on either tumour cells or TILs in primary tumours predicted a worse prognosis (decreased overall survival) relative to PDL1− tumours80. Since that report, analyses of various tumours have suggested that the PDL1 status can either correlate with poor prognosis, better prognosis or show no correlation with prognosis58,81,82,83,84,85. Probable contributing factors to the wide range of reported outcomes among different patient cohorts are variations in IHC technique, cancer type, stage of cancer analysed (most analyses are of primary, not metastatic, lesions) and treatment history.
Although most of the analyses of PD1 ligand expression have focused on PDL1, PDL2 has also been reported to be upregulated in various tumours. PDL2 is highly upregulated on cells from certain B cell lymphomas, such as primary mediastinal B cell lymphoma, follicular cell B cell lymphoma and Hodgkin's disease86. Upregulation of PDL2 on these lymphomas is commonly associated with gene amplification or rearrangement with the class II major histocompatibility complex (MHC) transactivator (CIITA) locus, which is highly transcriptionally active in B cell lymphomas87.
Given the heterogeneity of the expression levels of PD1 ligands and their potential relevance as biomarkers for blockade of the PD1 pathway, it is important to understand the signals that induce the expression of PD1 ligands on tumour cells and also haematopoietic cells within the tumour microenvironment. Two general mechanisms for the regulation of PDL1 by tumour cells have emerged: innate immune resistance and adaptive immune resistance (not to be confused with innate and adaptive immunity) (Fig. 4). For some tumours, such as glioblastomas, it has been demonstrated that PDL1 expression is driven by constitutive oncogenic signalling pathways in the tumour cell (innate immune resistance). The expression on glioblastomas is enhanced on deletion or silencing of PTEN, which implicates the involvement of the PI3K–AKT pathway88. Similarly, constitutive anaplastic lymphoma kinase (ALK) signalling, which is observed in certain lymphomas and occasionally in lung cancer, has been reported to drive PDL1 expression through signal transducer and activator of transcription 3 (STAT3) signalling89.

The examples in this figure use the programmed cell death protein 1 (PD1) ligand, PDL1 (also known as B7-H1), for illustrative purposes, although the concept probably applies to multiple immune-checkpoint ligands, including PDL2 (also known as B7-DC). a | Innate immune resistance. In some tumours, constitutive oncogenic signalling can upregulate PDL1 expression on all tumour cells, independently of inflammatory signals in the tumour microenvironment. Activation of the AKT and signal transducer and activator of transcription 3 (STAT3) pathways has been reported to drive PDL1 expression. b | Adaptive immune resistance. In some tumours, PDL1 is not constitutively expressed, but rather it is induced in response to inflammatory signals that are produced by an active antitumour immune response. The non-uniform expression of PDL1, which is commonly restricted to regions of the tumour that have tumour-infiltrating lymphocytes, suggests that PDL1 is adaptively induced as a consequence of immune responses within the tumour microenvironment. Adaptive induction may be a common mechanism for the expression of multiple immune-checkpoint molecules in tumours. IFNγ, interferon-γ; MHC, major histocompatibility complex; TCR, T cell receptor.
The alternative mechanism for PDL1 upregulation on tumours that has emerged from both clinical and preclinical studies reflects their adaptation to endogenous tumour-specific immune responses — a process termed adaptive immune resistance58 (Fig. 4). In adaptive immune resistance, the tumour uses the natural physiology of PD1 ligand induction that normally occurs to protect a tissue from infection-induced immune-mediated damage in order to protect itself from an antitumour immune response. Expression of PDL1 as an adaptive response to endogenous antitumour immunity can occur because PDL1 is induced on most tumour cells in response to interferons (IFNs) — predominantly IFNγ — which also occurs in epithelial and stromal cells in normal tissues90,91,92. This mechanism represents an alternative to the conventional drug resistance mechanisms that involve the mutation of drug targets. It also contrasts with mechanisms of viral immune escape that involve the mutation of immunodominant epitopes. The mechanism of adaptive immune resistance intrinsically implies that immunosurveillance exists even in advanced cancers. However, the tumour ultimately resists immune elimination by upregulating the expression of ligands for inhibitory receptors on tumour-specific lymphocytes that consequently inhibit antitumour immune responses in the tumour microenvironment.
Various preclinical and clinical studies support the adaptive immune resistance hypothesis. Gajewski and colleagues93 have shown that melanomas can be approximately divided into inflammatory and non-inflammatory categories, which were defined by the expression of multiple inflammatory genes, including those involved in IFN pathways. A recent study of melanoma demonstrated a strong correlation between cell surface PDL1 expression on tumour cells and both lymphocytic infiltration and intratumoral IFNγ expression. This correlation was not only seen among tumours but within individual PDL1+ tumours at the regional level, in which regions of lymphocyte infiltration were also exactly where PDL1 was expressed on both tumour cells and TILs58. These findings suggest that the occurrence of a negative feedback loop whereby IFNγ induces PDL1 expression, which in turn suppresses the activity of PD1+ T cells.
Evidence of clinical activity for PD1 blockade. Taken together, the general findings of increased PD1 expression by TILs and the increased PD1 ligand expression by tumour cells provided an important rationale for the capacity of antibody blockade of this pathway to enhance intratumoral immune responses. This was validated through many studies using mouse models of cancer, which demonstrated enhanced antitumour immunity through antibody blockade of PD1 or its ligands56,57,74. Furthermore, the relatively mild phenotypes of Pd1 (also known as Pdcd1), Pdl1 (also known as Cd274 and Pdcd1lg1) and Pdl2 (also known as Cd273 and Pdcd1lg2) knockout mice suggest that blockade of this pathway would result in less collateral immune toxicity than for CTLA4 blockade, which seems to be the case in clinical trials.
Although the clinical experience with PD1 antibodies is currently much less extensive than with CTLA4 antibodies, the initial results look extremely promising. In the first Phase I clinical trial with a fully human IgG4 PD1 antibody, there were somecases of tumour regression, including mixed responses, partial responses and a complete response94. Tumour regressions were observed in four of the five histologies examined: colon, renal and lung cancers and melanoma, which were also associated with significant increases in lymphocyte infiltration into metastatic tumours. In initial results from a second clinical trial, extending the treatment with anti-PD1 to 2 years, objective responses were observed in 16 out of 39 patients with advanced melanoma, and an additional 14 patients achieved either a mixed response or disease stabilization95. Similar response rates have been observed in renal cancer96, and there is ongoing evaluation of anti-PD1 in lung cancer. Follow-up on the initial clinical trial suggests that the responses are durable. In fact, all objective responders from the initial Phase I trial continued to be in remission more than a year after the cessation of therapy (E. Lipson and S. Topalian, Johns Hopkins University School of Medicine, personal communication).
As predicted by the distinct phenotypes of Pd1-knockout mice versus Ctla4-knockout mice, the frequency of immune-related toxicities from anti-PD1 treatment seems to be less than anti-CTLA4 treatment. For example, only 1 of 39 patients in the initial Phase I trial of anti-PD1 had a severe immune-related adverse event. When immune-related toxicities owing to anti-PD1 treatment occur, they seem to be grossly similar to those caused by anti-CTLA4 treatment and can affect different organs in different patients. Ultimately, the efficacy of anti-PD1 treatment awaits evaluation in many ongoing trials.
It is logical to imagine that the enhancement of antitumour immune responses on blockade of the PD1 pathway would substantially depend on the expression of a PD1 ligand by tumour cells. Analyses of 9 patients that were treated with anti-PD1 in the initial Phase I trial demonstrated a strong correlation between PDL1 expression and response: 0 of 5 patients with no membrane PDL1 expression on pretreatment biopsies responded to anti-PD1 therapy, whereas 3 of 4 patients with >5% of tumour cells expressing PDL1 on the cell membrane had either an objective response or a mixed response. The lack of response in patients whose tumour cells exclusively had cytosolic PDL1 is notable because cytosolic PDL1 would fail to activate the PD1 pathway. If validated in larger series of patients, this finding sets the stage for a broader assessment of immune-checkpoint ligands and receptors as targets for antibody blockade, as well as for the assessment of ligand expression in tumours as biomarkers for predicting the success of strategies that involve the blockade of specific immune-checkpoint pathways.
There are various companies that are developing and testing antibodies targeted to either PD1 or PDL1 but currently there is no published information on their clinical performance. Based on the known interactions of the PD1 ligands, it is theoretically possible that a PD1 antibody would have distinct biological activity from a PDL1 antibody. A PD1 antibody would block PD1 from interacting with both PDL1 and PDL2 but not the interaction between PDL1 and CD80. By contrast, most PDL1 antibodies block the interaction between PDL1 and CD80 and between PDL1 and PD1 but would not block PD1 from interacting with PDL2. Thus, it is possible that, depending on which interactions dominate in a particular cancer, PD1 and PDL1 antibodies might not have redundant activity.
Multiple promising immune checkpoints
Basic immunological studies have demonstrated that various immune-checkpoint receptors are expressed coordinately under circumstances of tolerance to self-antigens and chronic infections, as well as in inflammatory settings. In addition to defined lymphocyte inhibitory receptors, numerous B7 family inhibitory ligands — in particular B7-H3 (also known as CD276) and B7-H4 (also known as B7-S1, B7x and VCTN1) — do not yet have defined receptors, but mouse knockout experiments support an immune inhibitory role for these ligands97. In addition, B7-H3 and B7-H4 are upregulated on tumour cells or tumour-infiltrating cells98. B7-H3 seems to be upregulated on endothelial cells of the tumour vasculature, and B7-H4 has been reported to be expressed on tumour-associated macrophages97. Preclinical mouse models of cancer have shown that blockade of many of these individual immune-checkpoint ligands or receptors can enhance antitumour immunity, and dual blockade of coordinately expressed receptors can produce additive or synergistic antitumour activities. Inhibitors for a number of these immune-checkpoint targets are either entering the clinic or are under active development. Those described below are targets for which blocking antibodies or small molecule inhibitors are currently available but do not represent a comprehensive list.
Lymphocyte activation gene 3 (LAG3; also known as CD223), 2B4 (also known as CD244), B and T lymphocyte attenuator (BTLA; also known as CD272), T cell membrane protein 3 (TIM3; also known as HAVcr2), adenosine A2a receptor (A2aR) and the family of killer inhibitory receptors have each been associated with the inhibition of lymphocyte activity and in some cases the induction of lymphocyte anergy. Antibody targeting of these receptors, either alone or in combination with a second immune-checkpoint blocker, has been shown to enhance antitumour immunity in animal models of cancer. Because many tumour cells express multiple inhibitory ligands, and TILs express multiple inhibitory receptors, there are many opportunities to enhance antitumour immunity through dual or triple blockade of immune checkpoints. Although human blocking antibodies that are specific for a number of these 'second generation' inhibitory receptors are under development, none have yet entered the clinic. Most of these receptors are induced on T cell activation, in keeping with the biological theme that they have roles in feedback inhibition of T cell responses when their cognate ligands are present. In addition to providing inhibitory signals to activated effector T cells, some of these receptors, such as LAG3, are highly expressed on TReg cells, where they are important for amplifying the immunosuppressive activity of TReg cells99. This implies that as with CTLA4 and PD1, these receptors have dual roles in inhibiting effector immune responses, and blocking antibodies therefore have multiple potential mechanisms of action.
LAG3 was cloned over 20 years ago as a CD4 homologue100, but its function in the immune checkpoint was only defined in 2005 when it was shown to have a role in enhancing the function of TReg cells99,101. LAG3 also inhibits CD8+ effector T cell functions independently of its role on TReg cells102. The only known ligand for LAG3 is MHC class II molecules, which are upregulated on some epithelial cancers (generally in response to IFNγ) but are also expressed on tumour-infiltrating macrophages and dendritic cells. The role of the LAG3–MHC class II interaction in the LAG3-mediated inhibition of T cell responses is unclear because LAG3 antibodies that do not block the LAG3–MHC class II interaction nonetheless enhance T cell proliferation and effector cell functions in vitro and in vivo. This interaction may be most important for the role of LAG3 in enhancing TReg cell function. LAG3 is one of various immune-checkpoint receptors that are coordinately upregulated on both TReg cells and anergic T cells, and simultaneous blockade of these receptors can result in enhanced reversal of this anergic state relative to blockade of one receptor alone. In particular, PD1 and LAG3 are commonly co-expressed on anergic or exhausted T cells103,104. Dual blockade of LAG3 and PD1 synergistically reversed anergy among tumour-specific CD8+ T cells and virus-specific CD8+ T cells in the setting of chronic infection. Dramatic evidence of the effects of coordinate T cell inhibition by PD1 and LAG3 comes from Pd1−/−Lag3−/− double-knockout mice, which completely reject even poorly immunogenic tumours in a T cell-dependent manner but also develop autoimmune syndromes much more quickly than Pd1−/− or Lag3−/− single-knockout mice. The autoimmune syndromes in Pd1−/−Lag3−/− double-knockout mice are ultimately fatal, although they do not develop as quickly as in Ctla4-knockout mice105. These findings emphasize the balance between antitumour effects and autoimmune side effects that must be considered in all of the immune-checkpoint-blockade strategies.
TIM3, the ligand of which is galectin 9 (a galectin that is upregulated in various types of cancer, including breast cancers) inhibits T helper 1 (TH1) cell responses106, and TIM3 antibodies enhance antitumour immunity107. TIM3 has also been reported to be co-expressed with PD1 on tumour-specific CD8+ T cells, and dual blockade of both molecules significantly enhances the in vitro proliferation and cytokine production of human T cells when stimulated by the cancer–testes antigen, NY-ESO-1. In animal models, coordinate blockade of PD1 and TIM3 was reported to enhance antitumour immune responses and tumour rejection in circumstances in which only modest effects from blockade of each individual molecule were observed108,109,110.
BTLA was first identified as an inhibitory receptor on T cells on the basis of the enhanced T cell responses that were observed in Btla-knockout mice111. Subsequently, herpesvirus entry mediator (HVEM; also known as TNFRSF14) — which is expressed on certain tumour cell types (for example, melanoma) as well as on tumour-associated endothelial cells — was shown to be the BTLA ligand112. This is a rare case in which a TNF family member interacts with an immunoglobulin supergene family member. BTLA expression levels on activated virus-specific CD8+ T cells are quite low, but can be much higher on TILs from patients with melanoma. BTLAhi T cells are inhibited in the presence of its ligand, HVEM. Thus, BTLA may also be a relevant inhibitory receptor for T cells in the tumour microenvironment113. The system of HVEM-interacting molecules is complex: two additional interacting molecules, CD160 (an immunoglobulin superfamily member) and LIGHT (also known as TNFSF14, a TNF family member), seem to mediate inhibitory and co-stimulatory activity, respectively. It also seems that signalling can be bidirectional, depending on the specific combination of interactions. The complexity of this system makes therapeutic inhibition strategies less straightforward than for other inhibitory receptors or ligands, although dual blockade of BTLA and PD1 clearly enhances antitumour immunity114.
A2aR, the ligand of which is adenosine, inhibits T cell responses, in part by driving CD4+ T cells to express FOXP3 and hence to develop into TReg cells115. Deletion of this receptor results in enhanced and sometimes pathological inflammatory responses to infection. This receptor is particularly relevant to tumour immunity because the rate of cell death in tumours from cell turnover is high, and dying cells release adenosine. In addition, TReg cells express high levels of the exoenzymes CD39 (also known as NTPDase 1), which converts extracellular ATP to AMP, and CD73 (also known as 5′-NT), which converts AMP to adenosine116. Given that A2aR engagement by adenosine drives T cells to become TReg cells, this can produce a self-amplifying loop within the tumour. Indeed, tumours grow more slowly in A2aR (also known as Adora2a)-knockout mice, and tumour vaccines are much more effective against established tumours in these mice117. A2aR can be inhibited either by antibodies that block adenosine binding or by adenosine analogues, some of which are fairly specific for A2aR. Although these drugs have been used in clinical trials for Parkinson's disease, they have not yet been tested clinically in patients with cancer.
Killer inhibitory receptors are a broad category of inhibitory receptors that can be divided into two classes based on structure: killer cell immunoglobulin-like receptors (KIRs) and C-type lectin receptors, which are type II transmembrane receptors118,119,120. These receptors were originally described as crucial regulators of the killing activity of NK cells, although many are expressed on T cells and APCs121. The importance of their inhibitory role on T cells and APCs (for example, dendritic cells) is less well studied but the resulting activation of NK cells can provide potent antitumour activity. Many of the killer inhibitory receptors are specific for subsets of human leukocyte antigens (HLAs; the human MHC molecules) and possess allele-specificity. However, other receptors recognize broadly expressed molecules; for example, the C-type lectin receptor KLRG1 recognizes E-cadherin. The potential value of NK cells in antitumour immune responses when their inhibitory receptors are not appropriately engaged is best exemplified by the significantly enhanced graft-versus-tumour effects in allogeneic bone marrow transplants elicited by mismatches between donor NK inhibitory receptors and recipient HLA alleles. The big question in therapeutic blockade of NK inhibitory receptors is which among the >20 receptors should be targeted?
Future prospects: biomarkers and combinations
There is nothing like a clinical success to open up a new area of therapeutics. The FDA approval of anti-CTLA4 therapy, quickly followed by reports of encouraging preliminary clinical data for anti-PD1 therapy, has engendered a new-found awareness among oncologists of the potential antitumour activity of a patient's endogenous immune system once the 'brakes' elicited by the immune system have been released. As described above, these immune checkpoints are a tiny fraction of the receptors and ligands that have been defined by genetic and biological analyses to inhibit specific types of immune responses at various levels. The opportunities to explore therapeutically the plethora of potential immune-checkpoint targets bring forth two challenges. First is the definition of potential biomarkers that can determine which immune-checkpoint pathway or pathways dominate in a particular tumour — this will be crucial to guide the choice of inhibitor. In the case of immune-checkpoint pathways that primarily operate in the tumour microenvironment, such as the PD1 pathway, the expression of the key ligands and receptors in tumour biopsies are certainly the most obvious potential determinants of responsiveness to pathway blockade. It is also possible that specific oncogenic pathways, such as PI3K–AKT or STAT3 (which are constitutively activated in some tumours), may induce the expression of specific immune-inhibitory molecules and could thus be used as surrogate biomarkers.
The second challenge is the clinical development of combinatorial approaches. Preclinical models validate dramatic synergy between tumour vaccines and inhibition of most of the immune checkpoints described here. Anti-CTLA4 therapy strongly enhances the amplitude of vaccine-induced antitumour responses in many poorly immunogenic tumour models, as does anti-PD1 therapy37,122. The concept of adaptive immune resistance — whereby immune-checkpoint ligands such as PDL1 are induced in tumours in response to an endogenous antitumour immune response — suggests that PD1-pathway blockade as a monotherapy will only succeed in the setting of a pre-existing antitumour immune response in the patient. However, expanded efficacy might be achieved when PD1-pathway blockade is combined with a vaccine or any other therapy that induces de novo antitumour immune responses (Fig. 5). As is covered in detail elsewhere in this issue123, emerging studies demonstrate that targeted cancer therapies that are not conventionally thought of as immunotherapies can elicit or enhance antitumour immunity. Such therapies include: vascular endothelial growth factor (VEGF)–VEGF receptor (VEGFR) inhibitors; RAF inhibitors; certain chemotherapeutic agents; antibodies targeted to receptor tyrosine kinases that are overexpressed in tumours; and epigenetic therapies. These therapies may therefore force the tumours to upregulate immune checkpoints that consequently can be blocked as part of a combinatorial strategy.

The adaptive immune resistance mechanism implies that the blockade of an induced immune-checkpoint protein, such as programmed cell death protein 1 (PD1), as a single intervention will only induce tumour regressions when there is a pre-existing antitumour immune response to be 'unleashed' when the pathway is blocked. Multiple interventions, such as vaccines, that activate a de novo antitumour immune response may not induce tumour regressions because tumours respond by upregulating immune-checkpoint ligands. Therefore, combining the two approaches may induce tumour regressions in patients that would not have responded to either treatment alone. PDL1, PD1 ligand 1; TAM, tumour-associated macrophage.
References
Greenwald, R. J., Freeman, G. J. & Sharpe, A. H. The B7 family revisited. Annu. Rev. Immunol. 23, 515–548 (2005).
Zou, W & Chen, L. Inhibitory B7-family molecules in the tumour microenvironment. Nature Rev. Immunol. 8, 467–477 (2008). A good review of the basic biology of the expanding B7 family of regulatory molecules in the context of cancer immunology.
Mellor, A. L., Keskin, D. B., Johnson, T., Chandler, P. & Munn, D. H. Cells expressing indoleamine 2,3-dioxygenase inhibit T cell responses. J. Immunol. 168, 3771–3776 (2002).
Friberg, M. et al. Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. J. Cancer 101, 151–155 (2002).
Hou, D. Y. et al. Inhibition of indoleamine 2,3-dioxygenase in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with antitumor responses. Cancer Res. 67, 792–801 (2007).
Munn, D. H. & Mellor, A. L. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Invest. 17, 1147–1154 (2007).
Bak, S. P., Alonso, A., Turk, M. J. & Berwin, B. Murine ovarian cancer vascular leukocytes require arginase-1 activity for T cell suppression. Mol. Immunol. 46, 258–268 (2008).
Ochoa, A. C., Zea, A. H., Hernandez, C. & Rodriguez, P. C. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin. Cancer Res. 13, 721–726 (2007).
Rodríguez, P. C. & Ochoa, A. C. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol. Rev. 222, 180–191 (2008).
Löb, S. et al. IDO1 and IDO2 are expressed in human tumors: levo- but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer Immunol. Immunother. 58, 153–157 (2009).
Qian, F. et al. Efficacy of levo-1-methyl tryptophan and dextro-1-methyl tryptophan in reversing indoleamine-2, 3-dioxygenase-mediated arrest of T-cell proliferation in human epithelial ovarian cancer. Cancer Res. 69, 5498–5504 (2009).
Reisser, D., Onier-Cherix, N. & Jeannin, J. F. Arginase activity is inhibited by L-NAME, both in vitro and in vivo. J. Enzyme Inhib. Med. Chem. 17, 267–270 (2002).
Schwartz, R. H. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell 71, 1065–1068 (1992).
Lenschow, D. J., Walunas, T. L. & Bluestone, J. A. CD28/B7 system of T cell costimulation. Annu. Rev. Immunol. 14, 233–258 (1996).
Rudd, C. E., Taylor, A. & Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 229, 12–26 (2009).
Hathcock, K. S. et al. Identification of an alternative CTLA-4 ligand costimulatory for T cell activation. Science 262, 905–907 (1993).
Freeman, G. J. et al. Cloning of B7–2: a CTLA-4 counter-receptor that costimulates human T cell proliferation. Science 262, 909–911 (1993).
Azuma, M. et al. B70 antigen is a second ligand for CTLA-4 and CD28. Nature 366, 76–79 (1993).
Linsley, P. S., Clark, E. A. & Ledbetter, J. A. T-cell antigen CD28 mediates adhesion with B cells by interacting with activation antigen B7/BB-1. Proc. Natl Acad. Sci. USA 87, 5031–5035 (1990).
Linsley, P. S. et al. CTLA-4 is a second receptor for the B cell activation antigen B7. J. Exp. Med. 174, 561–569 (1991).
Linsley, P. S. et al. Human B7–1 (CD80) and B7–2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1, 793–801 (1994).
Riley, J. L. et al. Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc. Natl Acad. Sci. USA 99, 11790–11795 (2002).
Schneider, H. et al. Reversal of the TCR stop signal by CTLA-4. Science 313, 1972–1975 (2006).
Egen, J. G. & Allison, J. P. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity 16, 23–35 (2002).
Parry, R. V. et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell Biol. 25, 9543–9553 (2005).
Schneider, H. et al. Cutting edge: CTLA-4 (CD152) differentially regulates mitogen-activated protein kinases (extracellular signal-regulated kinase and c-Jun N-terminal kinase) in CD4+ T cells from receptor/ligand-deficient mice. J. Immunol. 169, 3475–3479 (2002).
Qureshi, O. S. et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332, 600–603 (2011).
Tivol, E. A. et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3, 541–547 (1995).
Waterhouse, P. et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 270, 985–988 (1995). References 28 and 29 show the dramatic immune phenotype of Ctla4 -knockout mice, proving that CTLA4 is a crucial immune-checkpoint receptor in the immune system.
Wing, K. et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275 (2008). A T Reg cell-specific Ctla4 -knockout approach to prove that CTLA4 has a role in T Reg cell function that is independent of its role in modulating the amplitude of effector T cell activation.
Peggs, K. S., Quezada, S. A., Chambers, C. A., Korman, A. J. & Allison, J. P. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J. Exp. Med. 206, 1717–1725 (2009). A transgenic approach to prove that CTLA4 has independent roles in T Reg cell and effector T cell function.
Hori, S., Nomura, T. & Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 299, 1057–1061 (2003).
Fontenot, J. D., Gavin, M. A. & Rudensky, A. Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nature Immunol. 4, 330–336 (2003). References 32 and 33 define FOXP3 as a crucial transcription factor that mediates T Reg cell function.
Hill, J. A. et al. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity 27, 786–800 (2007).
Gavin, M. A. et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature 445, 771–775 (2007).
Leach, D. R., Krummel, M. F. & Allison, J. P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 271, 1734–1736 (1996). The first demonstration in mouse models of the ability of CTLA4 antibodies to induce therapeutic antitumour immunity.
van Elsas, A., Hurwitz, A. A. & Allison, J. P. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J. Exp. Med. 190, 355–366 (1999).
Hodi, F. S. et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc. Natl Acad. Sci. USA 100, 4712–4717 (2003). The first clinical report of CTLA4 antibody treatment of cancer patients.
Phan, G. Q. et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc. Natl Acad. Sci. USA 100, 8372–8377 (2003). The first clear demonstration of tumour regressions induced by anti-CTLA4 therapy in patients with melanoma.
Ribas, A. et al. Antitumor activity in melanoma and anti-self responses in a phase I trial with the anti-cytotoxic T lymphocyte-associated antigen 4 monoclonal antibody CP-675,206. J. Clin. Oncol. 23, 8968–8977 (2005).
Beck, K. E. et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J. Clin. Oncol. 24, 2283–2289 (2006).
Ribas, A. Clinical development of the anti-CTLA-4 antibody tremelimumab. Semin. Oncol. 37, 450–454 (2010).
Downey, S. G. et al. Prognostic factors related to clinical response in patients with metastatic melanoma treated by CTL-associated antigen-4 blockade. Clin. Cancer Res. 13, 6681–6688 (2007).
Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010). A crucial Phase III trial demonstrating statistical survival benefit in patients with melanoma that were treated with the anti-CTLA4 therapy, ipilimumab.This was the first drug to demonstrate survival benefit in patients with advanced melanoma in a randomized trial.
Yuan, J. et al. Integrated NY-ESO-1 antibody and CD8+ T-cell responses correlate with clinical benefit in advanced melanoma patients treated with ipilimumab. Proc. Natl Acad. Sci. USA. 108, 16723–16728 (2011).
Liakou, C. I. et al. CTLA-4 blockade increases IFNγ-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc. Natl Acad. Sci. USA 105, 14987–14992 (2008).
Ji, R. R et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother. 07 Dec 2011 (doi:10.1007/s00262-011-1172-6).
Hoos, A. et al. Improved endpoints for cancer immunotherapy trials. J. Natl Cancer Inst. 102, 1388–1397 (2010).
Ishida, Y., Agata, Y., Shibahara, K. & Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 11, 3887–3895 (1992).
Freeman, G. J. et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192, 1027–1034 (2000). The demonstration that PD1 is the receptor for PDL1.
Keir, M. E. et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J. Exp. Med. 203, 883–895 (2006).
Nishimura, H. et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 291, 319–322 (2001).
Nishimura, H., Nose, M., Hiai, H., Minato, N. & Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 11, 141–151 (1999). The first demonstration that PD1 downmodulates immune responses.
Okazaki, T. & Honjo, T. PD-1 and PD-1 ligands: from discovery to clinical application. Int. Immunol. 19, 813–824 (2007).
Keir, M. E., Butte, M. J., Freeman, G. J. & Sharpe, A. H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 26, 677–704 (2008).
Dong, H. et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nature Med. 8, 793–800 (2002). The crucial demonstration that PDL1 is upregulated on tumour cells and that PDL1 expression on tumour cells mediates immune resistance.
Blank, C. et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 64, 1140–1145 (2004).
Taube, J. M. et al. B7-H1 expression co-localizes with inflammatory infiltrates in benign and malignant melanocytic lesions: implications for immunotherapy. Sci. Transl. Med. (In the press, 2012). Introduction of the adaptive immune resistance hypothesis that proposes that the expression of immune-checkpoint ligand by tumours can be induced by an antitumour immune response as an adaptive immune-protective mechanism.
Fife, B. T. et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nature Immunol. 10, 1185–1192 (2009).
Francisco, L. M. et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 206, 3015–3029 (2009).
Dong, H., Zhu, G., Tamada, K. & Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nature Med. 5, 1365–1369 (1999). The discovery of PDL1.
Latchman, Y. et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nature Immunol. 2, 261–268 (2001).
Tseng, S. Y. et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J. Exp. Med. 193, 839–846 (2001).
Butte, M. J., Keir, M. E., Phamduy, T. B., Sharpe, A. H. & Freeman, G. J. Programmed death-1 ligand 1 interacts specifically with the B7–1 costimulatory molecule to inhibit T cell responses. Immunity 27, 111–122 (2007).
Paterson, A. M. et al. The programmed death-1 ligand 1:B7–1 pathway restrains diabetogenic effector T cells in vivo. J. Immunol. 187, 1097–1105 (2011).
Park, J. J. et al. B7-H1/CD80 interaction is required for the induction and maintenance of peripheral T-cell tolerance. Blood 116, 1291–1298 (2010).
Shin, T. et al. In vivo costimulatory role of B7-DC in tuning T helper cell 1 and cytotoxic T lymphocyte responses. J. Exp. Med. 201, 1531–1541 (2005).
Terme, M. et al. IL-18 induces PD-1-dependent immunosuppression in cancer. Cancer Res. 71, 5393–5399 (2011).
Fanoni, D. et al. New monoclonal antibodies against B-cell antigens: possible new strategies for diagnosis of primary cutaneous B-cell lymphomas. Immunol. Lett. 134, 157–160 (2011).
Velu, V. et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature 458, 206–210 (2009).
Barber, D. L. et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687 (2006). The demonstration that the PD1 pathway mediates T cell 'exhaustion'. This paper investigated chronic viral infection but the mechanism is probably applicable to tumour-induced immune tolerance.
Sfanos, K. S. et al. Human prostate-infiltrating CD8+ T lymphocytes are oligoclonal and PD-1+. Prostate 69, 1694–1703 (2009).
Ahmadzadeh, M. et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 114, 1537–1544 (2009).
Iwai, Y. et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl Acad. Sci. USA 99, 12293–12297 (2002).
Konishi, J. et al. B7-H1 expression on non-small cell lung cancer cells and its relationship with tumor-infiltrating lymphocytes and their PD-1 expression. Clin. Cancer Res. 10, 5094–5100 (2004).
Brown, J. A. et al. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J. Immunol. 170, 1257–1266 (2003).
Curiel, T. J. et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nature Med. 9, 562–567 (2003).
Kuang, D. M. et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med. 206, 1327–1337 (2009).
Liu, Y., Zeng, B., Zhang, Z., Zhang, Y. & Yang, R. B7-H1 on myeloid-derived suppressor cells in immune suppression by a mouse model of ovarian cancer. Clin. Immunol. 129, 471–481 (2008).
Thompson, R. H. et al. Costimulatory B7-H1 in renal cell carcinoma patients: indicator of tumor aggressiveness and potential therapeutic target. Proc. Natl Acad. Sci. USA 101, 17174–17179 (2004).
Hamanishi, J. et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl Acad. Sci. USA 104, 3360–3365 (2007).
Ohigashi, Y. et al. Clinical significance of programmed death-1 ligand-1 and programmed death-1 ligand-2 expression in human esophageal cancer. Clin. Cancer Res. 11, 2947–2953 (2005).
Wu, C. et al. Immunohistochemical localization of programmed death-1 ligand-1 (PD-L1) in gastric carcinoma and its clinical significance. Acta Histochem. 108, 19–24 (2006).
Ghebeh, H. et al. The B7-H1 (PD-L1) T lymphocyte-inhibitory molecule is expressed in breast cancer patients with infiltrating ductal carcinoma: correlation with important high-risk prognostic factors. Neoplasia 8, 190–198 (2006).
Hino, R. et al. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer 116, 1757–1766 (2010).
Rosenwald, A. et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J. Exp. Med. 198, 851–862 (2003).
Steidl, C. et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature 471, 377–381 (2011).
Parsa, A. T. et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nature Med. 13, 84–88 (2007). The first evidence of an oncogenic pathway causing the induction of PDL1 expression on tumour cells.
Marzec, M. et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl Acad. Sci. USA 105, 20852–20857 (2008).
Kim, J. et al. Constitutive and inducible expression of b7 family of ligands by human airway epithelial cells. Am. J. Respir. Cell. Mol. Biol. 33, 280–289 (2005).
Lee, S. K. et al. IFN-γ regulates the expression of B7-H1 in dermal fibroblast cells. J. Dermatol. Sci. 40, 95–103 (2005).
Wilke, C. M. et al. Dual biological effects of the cytokines interleukin-10 and interferon-γ. Cancer Immunol. Immunother. 60, 1529–1541 (2011).
Gajewski, T. F., Louahed, J. & Brichard, V. G. Gene signature in melanoma associated with clinical activity: a potential clue to unlock cancer immunotherapy. Cancer J. 16, 399–403 (2010).
Brahmer, J. R. et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 28, 3167–3175 (2010). The first report of the treatment of patients and preliminary clinical efficacy of PD1 antibodies in multiple cancer histologies.
Sznol, M. et al. Safety and antitumor activity of biweekly MDX 1106 (Anti PD 1) in patients with advanced refractory malignancies. J. Clin. Oncol. 28 (Suppl.), Abstract 2506 (2010).
McDermott, D. F. et al. A phase I study to evaluate safety and antitumor activity of biweekly MDX 1106 (Anti PD 1) in patients with RCC and other advanced refractory malignancies. J. Clin. Oncol. 29 (Suppl. 7), Abstract 331 (2011).
Yi, K. H. & Chen, L. Fine tuning the immune response through B7-H3 and B7-H4. Immunol. Rev. 229, 145–151 (2009).
He, C., Qiao, H., Jiang, H. & Sun, X. The inhibitory role of B7-H4 in antitumor immunity: association with cancer progression and survival. Clin. Dev. Immunol. 2011, 695834 (2011).
Huang, C. T. et al. Role of LAG-3 in regulatory T cells. Immunity 21, 503–513 (2004). The identification of LAG3 as an immune-checkpoint receptor.
Triebel, F. et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 171, 1393–1405 (1990).
Goldberg, M. V. & Drake, C. G. LAG-3 in cancer immunotherapy. Curr. Top. Microbiol. Immunol. 344, 269–278 (2011).
Grosso, J. F. et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J. Clin. Invest. 117, 3383–3392 (2007).
Blackburn, S. D. et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nature Immunol. 10, 29–37 (2009).
Grosso, J. F. et al. Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. J. Immunol. 182, 6659–6669 (2009).
Woo, S. R. et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 72, 917–927 (2012).
Zhu, C. et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nature Immunol. 6, 1245–1252 (2005). The demonstration that TIM3 is an immune-checkpoint receptor.
Ngiow, S. F. et al. Anti-TIM3 antibody promotes T cell IFN-γ-mediated antitumor immunity and suppresses established tumors. Cancer Res. 71, 3540–3551 (2011).
Sakuishi, K. et al. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 207, 2187–2194 (2010).
Fourcade, J. et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 207, 2175–2186 (2010).
Baitsch, L. et al. Extended co-expression of inhibitory receptors by human CD8 T-cells depending on differentiation, antigen-specificity and anatomical localization. PLoS ONE 7, e30852. (2012).
Watanabe, N. et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nature Immunol. 4, 670–679 (2003).
Sedy, J. R. et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nature Immunol. 6, 90–98 (2005).
Lasaro, M. O. et al. Active immunotherapy combined with blockade of a coinhibitory pathway achieves regression of large tumor masses in cancer-prone mice. Mol. Ther. 19, 1727–1736 (2011).
Fourcade, J. et al. CD8+ T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res. 72, 887–896 (2012).
Zarek, P. E. et al. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood 111, 251–259 (2008).
Deaglio, S. et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 204, 1257–1265 (2007).
Waickman, A. T. et al. Enhancement of tumor immunotherapy by deletion of the A(2A) adenosine receptor. Cancer Immunol. Immunother. 25 Nov 2011 (doi:10.1007/s00262-011-1155-7).
Lanier, L. L. Up on the tightrope: natural killer cell activation and inhibition. Nature Immunol. 9, 495–502 (2008).
Plougastel, B. F. & Yokoyama, W. M. Extending missing-self? Functional interactions between lectin-like NKrp1 receptors on NK cells with lectin-like ligands. Curr. Top. Microbiol. Immunol. 298, 77–89 (2006).
Raulet, D. H. Missing self recognition and self tolerance of natural killer (NK) cells. Semin. Immunol. 18, 145–150 (2006).
Mingari, M. C., Pietra, G. & Moretta, L. Human cytolytic T lymphocytes expressing HLA class-I-specific inhibitory receptors. Curr. Opin. Immunol. 17, 312–319 (2005).
Li, B. et al. Anti-programmed death-1 synergizes with granulocyte macrophage colony-stimulating factor—secreting tumor cell immunotherapy providing therapeutic benefit to mice with established tumors. Clin. Cancer Res. 15, 1623–1634 (2009).
Vanneman, M. & Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nature Rev. Cancer 12, 237–251 (2012).
Acknowledgements
Work in the author's laboratory is supported by the Melanoma Research Alliance, the Hussman Foundation and the Seraph Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no competing financial interests.
Related links
Glossary
- Amplitude
-
In immunology, this refers to the level of effector output. For T cells, this can be levels of cytokine production, proliferation or target killing potential.
- Quality
-
In immunology, this refers to the type of immune response generated, which is often defined as the pattern of cytokine production. This, in turn, mediates responses against specific types of pathogen. For example, CD4+ T cells can be predominantly: TH1 cells (characterized by IFNγ production; these cells are important for antiviral and antitumour responses); TH2 cells (characterized by IL-4 and IL-13 production; these cells are important for antihelminth responses); or TH17 cells (characterized by IL-17 and IL-22 production; these cells are important for mucosal bacterial and fungal responses).
- Autoimmunity
-
Immune responses against an individual's normal cells or tissues.
- CD8+ effector T cells
-
T cells that are characterized by the expression of CD8. They recognize antigenic peptides presented by MHC class I molecules and are able to directly kill target cells that express the cognate antigen.
- CD4+ helper T cells
-
T cells that are characterized by the expression of CD4. They recognize antigenic peptides presented by MHC class II molecules. This type of T cell produces a vast range of cytokines that mediate inflammatory and effector immune responses. They also facilitate the activation of CD8+ T cells and B cells for antibody production.
- Myeloid cells
-
Any white blood cell (leukocyte) that is not a lymphocyte: macrophages, dendritic cells and granulocytic cells.
- Suicide substrates
-
Molecules that inhibit an enzyme by mimicking its substrate and covalently binding to the active site.
- Antigen-presenting cell
-
(APC). Any cell that displays on its surface an MHC molecule with a bound peptide antigen that a T cell recognizes through its TCR. This can be a dendritic cell or a macrophage, or any cell that expresses antigen and would be killed by an activated CD8+ effector T cell-specific response (such as a tumour cell or virally infected cell).
- Regulatory T (TReg) cell
-
A type of CD4+ T cell that inhibits, rather than promotes, immune responses. They are characterized by the expression of the forkhead transcription factor FOXP3, the lack of expression of effector cytokines such as IFNγ and the production of inhibitory cytokines such as TGFβ, IL-10 and IL-35.
- Immunogenic tumours
-
In the case of tumours in mice, this refers to a tumour that naturally elicits an immune response when growing in a mouse. With regard to human tumours, melanoma is typically considered immunogenic because patients with melanoma often have increased numbers of T cells that are specific for melanoma antigens.
- Objective clinical responses
-
A diminution of total cross-sectional area of all metastatic tumours — as measured by a CT or MRI scan — by >30% (corresponding to ∼50% decrease in volume) with no growth of any metastatic tumours.
- Response rate
-
The proportion of treated patients that achieve an objective response.
- Natural killer (NK) cells
-
Immune cells that kill cells using mechanisms similar to CD8+ effector T cells but do not use a clonal TCR for recognition. Instead, they are activated by receptors for stress proteins and are inhibited through distinct receptors, many of which recognize MHC molecules independently of the bound peptide.
- Anergy
-
A form of T or B cell inactivation in which the cell remains alive but cannot be activated to execute an immune response. Anergy is a reversible state.
- Mixed response
-
Response to a therapy whereby some metastatic tumours shrink and others grow.
- Partial response
-
An objective response in which some tumours remain visible on CT or MRI scans.
- Complete response
-
An objective response in which all measurable tumours completely disappear.
- Macrophages
-
Specialized immune cells that, on stimulation by pathogen-derived molecules or T cells, will engulf pathogens (particularly those that have antibodies or complement bound to them). They can also present antigen to T cells, but not as efficiently as dendritic cells.
- Dendritic cells
-
Specialized immune cells that, when activated, present antigens to and activate T cells to initiate adaptive immune responses.
- Graft-versus-tumour effects
-
An immune attack against tumour cells in the host mediated by transplanted allogeneic T cells.
- Allogeneic
-
Cells from a different individual that express different MHC alleles.
Rights and permissions
About this article
Cite this article
Pardoll, D. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12, 252–264 (2012). https://doi.org/10.1038/nrc3239
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrc3239
This article is cited by
-
Identification of breast cancer subgroups and immune characterization based on glutamine metabolism-related genes
BMC Medical Genomics (2024)
-
Immune evasion in cell-based immunotherapy: unraveling challenges and novel strategies
Journal of Biomedical Science (2024)
-
Comprehensive analysis the prognostic and immune characteristics of mitochondrial transport-related gene SFXN1 in lung adenocarcinoma
BMC Cancer (2024)
-
ImmunoPET provides a novel way to visualize the CD103+ tissue-resident memory T cell to predict the response of immune checkpoint inhibitors
EJNMMI Research (2024)
-
Immune modulation in malignant pleural effusion: from microenvironment to therapeutic implications
Cancer Cell International (2024)
