
Abstract
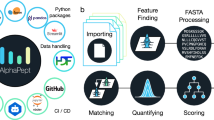
PatternLab for proteomics is an integrated computational environment that unifies several previously published modules for the analysis of shotgun proteomic data. The contained modules allow for formatting of sequence databases, peptide spectrum matching, statistical filtering and data organization, extracting quantitative information from label-free and chemically labeled data, and analyzing statistics for differential proteomics. PatternLab also has modules to perform similarity-driven studies with de novo sequencing data, to evaluate time-course experiments and to highlight the biological significance of data with regard to the Gene Ontology database. The PatternLab for proteomics 4.0 package brings together all of these modules in a self-contained software environment, which allows for complete proteomic data analysis and the display of results in a variety of graphical formats. All updates to PatternLab, including new features, have been previously tested on millions of mass spectra. PatternLab is easy to install, and it is freely available from http://patternlabforproteomics.org.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout









Similar content being viewed by others


References
Hebert, A.S. et al. The one-hour yeast proteome. Mol. Cell. Proteomics 13, 339–347 (2014).
Yates, J.R. Mass spectrometry and the age of the proteome. J. Mass Spectrom. 33, 1–19 (1998).
Zhang, B., Chambers, M.C. & Tabb, D.L. Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J. Proteome Res. 6, 3549–3557 (2007).
Hwang, S.-I. et al. Systematic characterization of nuclear proteome during apoptosis: a quantitative proteomic study by differential extraction and stable isotope labeling. Mol. Cell. Proteomics 5, 1131–1145 (2006).
Aquino, P.F. et al. Exploring the proteomic landscape of a gastric cancer biopsy with the shotgun imaging analyzer. J. Proteome Res. 13, 314–320 (2014).
Calvete, J.J., Sanz, L., Angulo, Y., Lomonte, B. & Gutiérrez, J.M. Venoms, venomics, antivenomics. FEBS Lett. 583, 1736–1743 (2009).
Valente, R.H., Dragulev, B., Perales, J., Fox, J.W. & Domont, G.B. BJ46a, a snake venom metalloproteinase inhibitor. Isolation, characterization, cloning and insights into its mechanism of action. Eur. J. Biochem 268, 3042–3052 (2001).
Eng, J.K., McCormack, A.L. & Yates, J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 (1994).
Washburn, M.P., Wolters, D. & Yates, J.R. III. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247 (2001).
Köcher, T., Pichler, P., Swart, R. & Mechtler, K. Analysis of protein mixtures from whole-cell extracts by single-run nanoLC-MS/MS using ultralong gradients. Nat. Protoc. 7, 882–890 (2012).
Keller, A., Nesvizhskii, A.I., Kolker, E. & Aebersold, R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 (2002).
Cociorva, D., L Tabb, D. & Yates, J.R. Validation of tandem mass spectrometry database search results using DTASelect. Curr. Protoc. Bioinformatics 16 74, 13.4.1–13.4.14 (2007).
Ross, P.L. et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 3, 1154–1169 (2004).
Oda, Y., Huang, K., Cross, F.R., Cowburn, D. & Chait, B.T. Accurate quantitation of protein expression and site-specific phosphorylation. Proc. Natl. Acad. Sci. USA 96, 6591–6596 (1999).
Carvalho, P.C., Hewel, J., Barbosa, V.C. & Yates, J.R. III. Identifying differences in protein expression levels by spectral counting and feature selection. Genet. Mol. Res. 7, 342–356 (2008).
Liu, H., Sadygov, R.G. & Yates, J.R. III. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 76, 4193–4201 (2004).
Neilson, K.A. et al. Less label, more free: approaches in label-free quantitative mass spectrometry. Proteomics 11, 535–553 (2011).
Shevchenko, A., Valcu, C.-M. & Junqueira, M. Tools for exploring the proteomosphere. J. Proteomics 72, 137–144 (2009).
Beausoleil, S.A., Villén, J., Gerber, S.A., Rush, J. & Gygi, S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24, 1285–1292 (2006).
Carvalho, P.C. et al. YADA: a tool for taking the most out of high-resolution spectra. Bioinformatics 25, 2734–2736 (2009).
Keller, A., Eng, J., Zhang, N., Li, X. & Aebersold, R. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol. 1, 2005.0017 (2005).
Deutsch, E.W. et al. Trans-Proteomic Pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteomics Clin. Appl. 9, 745–754 (2015).
Kohlbacher, O. et al. TOPP–the OpenMS proteomics pipeline. Bioinformatics 23, e191–e197 (2007).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
Cox, J. et al. A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 4, 698–705 (2009).
Carvalho, P.C., Fischer, J.S.G., Chen, E.I., Yates, J.R. & Barbosa, V.C. PatternLab for proteomics: a tool for differential shotgun proteomics. BMC Bioinformatics 9, 316 (2008).
MacLean, B. et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010).
Giardine, B. et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 15, 1451–1455 (2005).
Boekel, J. et al. Multi-omic data analysis using Galaxy. Nat. Biotechnol. 33, 137–139 (2015).
Egertson, J.D., MacLean, B., Johnson, R., Xuan, Y. & MacCoss, M.J. Multiplexed peptide analysis using data-independent acquisition and Skyline. Nat. Protoc. 10, 887–903 (2015).
Carvalho, P.C., Yates, J.R. III. & Barbosa, V.C. Improving the TFold test for differential shotgun proteomics. Bioinformatics 28, 1652–1654 (2012).
Leprevost, F.V. et al. Pinpointing differentially expressed domains in complex protein mixtures with the cloud service of PatternLab for Proteomics. J. Proteomics 89, 179–182 (2013).
Leprevost, F.V. et al. PepExplorer: A similarity-driven tool for analyzing de novo sequencing results. Mol. Cell. Proteomics 13, 2480–2489 (2014).
Fischer, J. et al. A scoring model for phosphopeptide site localization and its impact on the question of whether to use MSA. J. Proteomics 129, 42–50 (2015).
Fischer, J. et al. Dynamic proteomic overview of glioblastoma cells (A172) exposed to perillyl alcohol. J. Proteomics 73, 1018–1027 (2010).
Carvalho, P.C. et al. GO Explorer: a gene-ontology tool to aid in the interpretation of shotgun proteomics data. Proteome Sci. 7, 6 (2009).
Lima, D.B. et al. SIM-XL: a powerful and user-friendly tool for peptide cross-linking analysis. J. Proteomics 129, 51–55 (2015).
Borges, D. et al. Using SIM-XL to identify and annotate cross-linked peptides analyzed by mass spectrometry. Protoc. Exch. doi:10.1038/protex.2015.015 (2015).
Carvalho, P.C., Yates Iii, J.R. & Barbosa, V.C. Analyzing shotgun proteomic data with PatternLab for proteomics. Curr. Protoc. Bioinformatics 30, 13.13.1–13.13.15 (2010).
Carvalho, P.C. et al. Search engine processor: filtering and organizing peptide spectrum matches. Proteomics 12, 944–949 (2012).
Carvalho, P.C., Fischer, J.S.G., Xu, T., Yates, J.R. III. & Barbosa, V.C. PatternLab: from mass spectra to label-free differential shotgun proteomics. Curr. Protoc. Bioinformatics 40, 13.19.1–13.19.18 (2012).
Eng, J.K., Jahan, T.A. & Hoopmann, M.R. Comet: an open-source MS/MS sequence database search tool. Proteomics 13, 22–24 (2013).
Richards, A.L. et al. One-hour proteome analysis in yeast. Nat. Protoc. 10, 701–714 (2015).
UniProt Consortium. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res. 41, D43–D47 (2013).
Elias, J.E. & Gygi, S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4, 207–214 (2007).
Cottrell, J.S. & Creasy, D.M. Response to: the problem with peptide presumption and low mascot scoring. J. Proteome Res. 10, 5272–5273 (2011).
Bandeira, N. Spectral networks: a new approach to de novo discovery of protein sequences and posttranslational modifications. BioTechniques 42 687 (2007).
Na, S., Bandeira, N. & Paek, E. Fast multi-blind modification search through tandem mass spectrometry. Mol. Cell. Proteomics 11, M111.010199 (2012).
Shevchenko, A. et al. Charting the proteomes of organisms with unsequenced genomes by MALDI-quadrupole time-of-flight mass spectrometry and BLAST homology searching. Anal. Chem. 73, 1917–1926 (2001).
Xu, T. et al. ProLuCID, a fast and sensitive tandem mass spectra-based protein identification program. Mol. Cell Proteomics 5, S174 (2006).
Borges, D. et al. Effectively addressing complex proteomic search spaces with peptide spectrum matching. Bioinformatics 29, 1343–1344 (2013).
Zybailov, B. et al. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res. 5, 2339–2347 (2006).
Thompson, A. et al. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 75, 1895–1904 (2003).
McAlister, G.C. et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 86, 7150–7158 (2014).
Picotti, P. & Aebersold, R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat. Methods 9, 555–566 (2012).
Vizcaíno, J.A. et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069 (2013).
Chambers, M.C. et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 (2012).
Martens, L. et al. mzML–a community standard for mass spectrometry data. Mol. Cell. Proteomics 10, R110.000133 (2011).
McDonald, W.H. et al. MS1, MS2, and SQT-three unified, compact, and easily parsed file formats for the storage of shotgun proteomic spectra and identifications. Rapid Commun. Mass Spectrom. 18, 2162–2168 (2004).
Nesvizhskii, A.I. Proteogenomics: concepts, applications and computational strategies. Nat. Methods 11, 1114–1125 (2014).
de Miguel, N. et al. Proteome analysis of the surface of Trichomonas vaginalis reveals novel proteins and strain-dependent differential expression. Mol. Cell. Proteomics 9, 1554–1566 (2010).
Clair, G., Armengaud, J. & Duport, C. Restricting fermentative potential by proteome remodeling: an adaptive strategy evidenced in Bacillus cereus. Mol. Cell. Proteomics 11, M111.013102 (2012).
Webb, K.J., Xu, T., Park, S.K. & Yates, J.R. Modified MuDPIT separation identified 4488 proteins in a system-wide analysis of quiescence in yeast. J. Proteome Res. 12, 2177–2184 (2013).
Christie-Oleza, J.A., Piña-Villalonga, J.M., Bosch, R., Nogales, B. & Armengaud, J. Comparative proteogenomics of twelve Roseobacter exoproteomes reveals different adaptive strategies among these marine bacteria. Mol. Cell. Proteomics 11, M111.013110 (2012).
Christie-Oleza, J.A., Fernandez, B., Nogales, B., Bosch, R. & Armengaud, J. Proteomic insights into the lifestyle of an environmentally relevant marine bacterium. ISME J. 6, 124–135 (2012).
Chaves, D.F.S. et al. Comparative proteomic analysis of the aging soleus and extensor digitorum longus rat muscles using TMT labeling and mass spectrometry. J. Proteome Res. 12, 4532–4546 (2013).
Shah, M. et al. Proteomic analysis of the endosperm ontogeny of Jatropha curcas L. seeds. J. Proteome Res. 14, 2557–2568 (2015).
Acknowledgements
We thank Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação do Câncer, Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) for its BBP grant and Programa de Apoio à Pesquisa Estratégica em Saúde da Fiocruz (PAPES VII). J.R.Y. acknowledges funding from the US National Institutes of Health (P41 GM103533, R01 MH067880, and R01 MH100175) and the National Heart, Lung and Blood Institute (NHBLI) Proteomics Center at the University of California at Los Angeles (UCLA) (HHSN268201000035C). J.J.M. acknowledges NIH research resources (5P41RR011823) and funding from the National Institute of General Medical Sciences (8 P41 GM103533).
Author information
Authors and Affiliations
Contributions
P.C.C., J.R.Y. and V.C.B. have participated in the PatternLab project since its beginning in 2008. D.B.L. participated in updating features from several modules and the graphical user interface, as well as in helping migrate to the new PatternLab project file format. F.V.L. developed the PepExplorer module together with P.C.C. M.D.M.S. developed several functions in PepExplorer and had a major participation in the development of the isobaric quantification module. J.S.G.F., P.F.A. and J.J.M. have been participating in PatternLab since early versions by continuously performing beta testing, pointing out required features and providing suggestions on how to make the software more user-friendly. P.C.C. and D.B.L. created the supplementary video. P.C.C. and V.C.B. wrote the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 PatternLab’s target-decoy sequence database generation module.
This module provides options for parsing data from UniProt, NCBI, IPI, and a Generic Format. The module can automatically include the sequences of 127 common contaminants to proteomics and simplify datasets by eliminating subset sequences or sequences having an identification threshold above a given user specification. In these cases, a note is appended to the description of the remaining sequence to indicate the eliminated sequence(s).
Supplementary Figure 2 The modification library window.
New modifications can be included by typing the data in the corresponding cells and then clicking on the ‘Update my lib’ button. Modifications can be included in the search by selecting the desired rows and then clicking on the ‘Add selected row to my search.xml’ button.
Supplementary Figure 3 SEPro’s Entry Screen.
PatternLab for proteomics 4.0 makes available preset configurations for filtering results from high-resolution and low-resolution MS1 acquisitions. Regardless, all SEPro filtering parameters are made available in the ‘Advanced Parameters’ tab.
Supplementary Figure 4 XICs quantitation histogram.
A histogram of minus the logarithm of the label-free quantitation values for all the XICs obtained by simultaneously analyzing 26 3-hour LC/MS/MS shotgun proteomic experiments on an Orbitrap Elite (Thermo, San Jose).
Supplementary Figure 5 PatternLab’s Isobaric Analyzer.
The lower-right panel contains three plots. The topmost one shows the total signal, obtained only from identified spectra of a given run, for each isobaric marker before normalization. The middle one shows the signals after applying the Channel Signal normalization. The bottommost plot shows the total signal, obtained from all mass spectra of a given run, regardless of identification status, for each isobaric marker before normalization.
Supplementary Figure 6 PatternLab’s XD Scoring module.
This module relies on the delta XCorr distribution to fit a Gaussian mixture model that ultimately results in p-values for the phosphosites.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–6 (PDF 994 kb)
PatternLab 4.0 in action
An overview of the main modules in action. (MP4 51995 kb)
Rights and permissions
About this article

Cite this article
Carvalho, P., Lima, D., Leprevost, F. et al. Integrated analysis of shotgun proteomic data with PatternLab for proteomics 4.0. Nat Protoc 11, 102–117 (2016). https://doi.org/10.1038/nprot.2015.133
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2015.133
This article is cited by
-
Mitofusin 1 silencing decreases the senescent associated secretory phenotype, promotes immune cell recruitment and delays melanoma tumor growth after chemotherapy
Scientific Reports (2024)
-
Deficiency of endothelial sirtuin1 in mice stimulates skeletal muscle insulin sensitivity by modifying the secretome
Nature Communications (2023)
-
Improving quantitation accuracy in isobaric-labeling mass spectrometry experiments with spectral library searching and feature-based peptide-spectrum match filter
Scientific Reports (2023)
-
High salt-induced PSI-supercomplex is associated with high CEF and attenuation of state transitions
Photosynthesis Research (2023)
-
Aberrant astrocyte protein secretion contributes to altered neuronal development in multiple models of neurodevelopmental disorders
Nature Neuroscience (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
