
Abstract
This protocol is intended to provide chemists who discover or make new organic compounds with a valuable tool for validating the structural assignments of those new chemical entities. Experimental 1H and/or 13C NMR spectral data and its proper interpretation for the compound of interest is required as a starting point. The approach involves the following steps: (i) using molecular mechanics calculations (with, e.g., MacroModel) to generate a library of conformers; (ii) using density functional theory (DFT) calculations (with, e.g., Gaussian 09) to determine optimal geometry, free energies and chemical shifts for each conformer; (iii) determining Boltzmann-weighted proton and carbon chemical shifts; and (iv) comparing the computed chemical shifts for two or more candidate structures with experimental data to determine the best fit. For a typical structure assignment of a small organic molecule (e.g., fewer than ∼10 non-H atoms or up to ∼180 a.m.u. and ∼20 conformers), this protocol can be completed in ∼2 h of active effort over a 2-d period; for more complex molecules (e.g., fewer than ∼30 non-H atoms or up to ∼500 a.m.u. and ∼50 conformers), the protocol requires ∼3–6 h of active effort over a 2-week period. To demonstrate the method, we have chosen the analysis of the cis- versus the trans-diastereoisomers of 3-methylcyclohexanol (1- cis versus 1- trans). The protocol is written in a manner that makes the computation of chemical shifts tractable for chemists who may otherwise have only rudimentary computational experience.
Similar content being viewed by others

Introduction
NMR spectroscopy is the single most powerful and generally applicable spectroscopic tool for deducing the correct structure of newly isolated organic compounds. Of the three important classes of primary NMR data—chemical shifts, coupling constants and relative integrated signal intensity—the first is the most diagnostic of the local chemical and magnetic environment (i.e., the nuclei's structural surroundings) and, arguably, the most reliably addressable by computational methods1,2. Accordingly, this protocol deals only with computational aspects of chemical shifts, and only proton and carbon nuclei are considered.
The use of quantum chemical methods for predicting proton and carbon NMR chemical shifts has now evolved to the point where compounds of considerable (and ever-increasing) complexity and size are amenable for study. Over time, DFT3 has emerged as the most attractive method for predicting NMR properties of organic molecules4. The advent of improved computational methods, the substantial increase in access to sufficiently powerful computational resources and the increase in computer literacy more generally have lowered the barriers that impede the use of computation in many areas of science.
In spite of these advances, novices often view the use of a seemingly sophisticated computational study with some degree of trepidation. We once did also—the task can be daunting. The goal of this tutorial is to break down this barrier by describing protocols for computing chemical shifts of a set of candidate structures and then comparing the resulting set of theoretical values for each of those structures with the experimental data in question. Many excellent reviews5,6 exist that critically assess various theoretical aspects of the computation of NMR chemical shifts (e.g., improved functionals, improved basis sets, correction and scaling routines, multiple referencing); it is not our intent here to offer an in-depth discussion of those features. Instead, we aim to provide the typical experimentalist (who is, frequently, an inexperienced computationalist) with the guidance required to initiate such a study.
Our target audience member is a bench chemist who routinely prepares and/or isolates new, discrete organic compounds. There are (only) two prerequisites we assumed as we decided the point of departure for this protocol: (i) the reader will have access to and rudimentary familiarity with an appropriate computational chemistry package (e.g., Jaguar, GAMESS, Spartan, PCModel, etc., but preferably Gaussian and MacroModel) and the command-line interface (e.g., Terminal (in Mac OS X or Linux) or Command Prompt (in Windows)), and (ii) the reader will also have in hand quality 1H and/or 13C NMR spectral data for the compound(s) whose structure(s) is(are) of interest.
Both constitutional and configurational issues are central to correct structural assignments. Each of these features is highlighted by the natural product-based case studies summarized in Box 1. Because the synthetic chemist typically knows the structure of each substrate or reactant, the question of structure assignment for each new product more often involves correctly deducing the relative configuration of newly introduced stereocenters in the product rather than its constitution. In contrast, because there is less structural history to inform the analysis of newly isolated natural products, issues of constitution are more often important for the natural product isolation chemist7.
The need for something more sophisticated than an increment-based additivity approach for predicting chemical shifts
Introductory texts typically (and justifiably) teach students to use a substituent-based approach, especially when more than one substituent is present, in which tabulated data are incrementally applied to estimate chemical shifts8. Although this method certainly has value, the example described next shows that when one moves to the consideration of molecules bearing more than one stereocenter, this approach is no longer adequate.
Consider the case of the trans- versus the cis-diastereomers of 3-methylcyclohexanol (1- trans and 1- cis, respectively). The experimental 1H NMR spectra for 1- trans and 1- cis are shown in Figure 1a,b (see Supplementary Data 1 for a full listing of actual chemical shift values). There are substantial differences in these two spectra, especially within the upfield 0.7–2.1 p.p.m. range. Clearly, it would be valuable if computational approaches could reproduce these sorts of differences sufficiently well to allow confident assignment of structure.

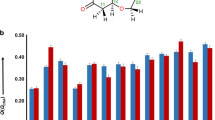
(a,b) Experimental 1H NMR spectra of 1- trans (a) and 1- cis (b) (CDCl3, 500 MHz). (c,d) Predicted 1H NMR spectra for 1- trans (c) and 1- cis (d) (CHCl3, ACD/Labs, C+H NMR predictors and DB)9. (e,f) MAEs (|Δδave|) for the proton (1H) chemical shifts between matched and mismatched pairs; experimental versus ACD-computed (e) or experimental versus DFT-computed (f).
Common software packages that use empirical (often increment-based) compilations of chemical shift information (e.g., tabulated shift increments or databases of known spectral data) allow users to predict the chemical shifts of a given input structure. These include 'ChemNMR' within ChemBioDraw (also known as ChemDraw) and 'C+H NMR Predictor and DB' within the ACD/Labs software suite. These methods sometimes can be sufficient for the task of resolving constitutional structural assignments. However, when issues associated with relative configuration are considered, increment-based methods are decidedly ill-equipped. Analysis of structures 1- trans and 1- cis by each of these programs quickly reveals these limitations, even for these simple structures. Namely, because ChemDraw (via the 'Predict 1H-NMR Shifts' command) treats diastereomeric structures as if they were the same, it predicts identical spectra (not shown) for the two, and it is therefore of no use for distinguishing between or among diastereomers.
We then used the ACD/Labs software to produce the spectra (see Supplementary Data 1 for the actual chemical shift values) for 1- trans and 1- cis presented in Figure 1c,d. One is struck by the high degree of similarity of these two empirically derived spectra in the aliphatic region of the spectrum of each (δ = 0.7–1.7 p.p.m.). Accordingly, neither is a particularly good match for the experimental spectrum of 1- trans or 1- cis. One commonly used method to quantitatively evaluate 'goodness of fit' is comparison of the mean absolute error (MAE) between computed/modeled versus experimental data sets

(where N is the number of unique chemical shifts used in the comparison).
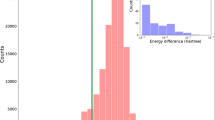
The MAEs for the chemical shifts of the two computed ACD spectra versus each of the two experimental sets of proton shift values are tabulated in Figure 1e. For comparison, the DFT-based approach described below in the PROCEDURE typically gives MAEs of ≤0.10 p.p.m. for 'correct' fits and ≥0.20 p.p.m. for 'incorrect' structure matches (cf. Fig. 1f). None of the four MAEexp/ACD values in Figure 1e is a particularly good fit, and, more strikingly, the ACD-derived shifts for each of these diastereomers show a better match for the same (trans) structure. Thus, the ACD approach is also inadequate for reliably distinguishing even this relatively simple pair of diastereomers.
A more advanced strategy that uses more sophisticated computational methods is advantageous for successful analysis of most compounds of interest to the synthetic or natural product chemist. First, it is important and necessary to consider the conformational space to which the compound has access. A primer of fundamental concepts in conformational analysis is presented in Box 2. Second, the Boltzmann analysis of the relative contribution of each of the important conformers needs to be performed. The basic tenets of Boltzmann weighting to determine the mole fraction contribution to a multicomponent equilibrium are presented in Box 3. Third, superior computational methods need to be used for (i) defining the potential energy surface of the conformational space, (ii) determining high-quality energy values and geometries for all of the important conformers and (iii) computing reliable NMR chemical shift values. Although semiempirical computational approaches have been developed that have merit in this regard (e.g., CHARGE9, which is implemented in MestReNova or MNova), the use of quantum mechanics–based methods are generally accepted as the most reliable approach, especially so for characterizing new organic structures4.
As we have previously reported10, a DFT-based approach reliably provided a clear distinction between 1- cis versus 1- trans (as well as the remaining members of the family, cis/trans-2- and 4-methylcyclohexanol). In the course of that work we showed, among other things, the relative merits of using various functionals, including the hybrid variants WP04 and WC04, which we had previously developed specifically for computing proton and carbon chemical shifts11, to provide computed data that best distinguish the members of this family of albeit only modestly complex structures. We have chosen to present the details of this protocol in the context of the analysis of 1- cis versus 1- trans. Although the strategy is highly similar to that reported in 2006, we have made numerous improvements that have resulted in somewhat increased accuracy and, especially, greater convenience in implementation, and these are captured in the protocol below.


Experimental overview
The aim of this protocol is to serve as a practical guide for chemical shift computation by using DFT quantum mechanical calculations for comparison of the resulting shift values with experimental data and for drawing meaningful conclusions about structure assignment. Because often the most challenging aspect of small-molecule structure elucidation is assigning relative configuration, we have elected to demonstrate the method using (the relatively simple) cis- and trans-diastereomers of 3-methylcyclohexanol (1- cis and 1- trans). We recommend that new users carry out this protocol on these prototypical trial compounds to ensure that they obtain similar results, thereby validating their ability to properly implement the methodology in their local setting.
The general strategy (Fig. 2) involves five operations (operations I–V):
-
Operation I: conformational search. For each candidate structure, an input geometry is first drawn in Maestro (see Equipment) and a library of conformers is then generated by using a molecular mechanics conformational search in MacroModel.
-
Operation II: geometry optimization and frequency calculation. Each of these conformers is then subjected to geometry optimization and frequency calculation by using DFT in Gaussian 09.
-
Operation III: NMR shielding tensor calculations. NMR shielding tensor values, from which chemical shifts are derived, are computed for each conformer by using DFT in Gaussian 09.
-
Operation IV: Boltzmann-weighting of shielding tensors and conversion to chemical shifts. The computed shielding tensors for each nucleus in all conformers are Boltzmann-weighted and then converted to empirically scaled chemical shift values for each nucleus of the candidate structure.
-
Operation V: comparison of experimental and computed chemical shifts and assessment of goodness of fit. Operations I–IV are repeated for each candidate structure that one chooses to consider. Each set of computed data is compared with the experimental data set to reach a conclusion about structure.

Operation I: Steps 1–9 (and script A). Operation II: Steps 10–12 (and script B). Operation III: Steps 13 and 14 (and script C). Operation IV: Steps 15–21 (and script D) and Steps 17–21. Operation V: Steps 22–26. Repeat for each candidate structure and then evaluate the results by comparison with the experimental data set.
Operation I. Conformational search. MacroModel12 (part of the Schrödinger suite) has emerged as a widely used molecular mechanics software application for conformational searching of small organic molecules (Spartan is another commonly available and used package that is convenient for performing conformational searches). The software allows the use of several molecular mechanics force fields and several methods of conformational searching. In this protocol, we have used the Monte Carlo multiple minimum13 method, a stochastic approach to conformational searching that uses torsional sampling. We recommend that the value of the 'Energy window for saving structures' be kept at (the default value of) 5.02 kcal mol−1 (i.e., the maximum energy difference between any single conformer and the most stable conformer, the global minimum). Although this value is higher than energy ranges suggested elsewhere14, it ensures that conformers whose relative energies are overestimated by molecular mechanics remain 'in play' during the subsequent DFT geometry optimization (operation II).
For studying molecules similar in structural complexity to 3-methylcyclohexanol, the MacroModel default settings for the conformational search (maximum number of steps and other minimization criteria) typically result in a satisfactorily exhaustive search of the potential energy surface. For more complex structures, increasing the maximum number of steps (i.e., separate iterations) is advisable. Additional suggestions for improving the quality of a conformational search are discussed in the MacroModel User Manual and Reference Manual.
Operation II. Geometry optimization and frequency calculation. Although several software packages have been developed for quantum mechanical electronic structure calculations (e.g., Gaussian, GAMESS, Jaguar and Spartan), Gaussian is the most widely used software package for NMR computation. Thus, this protocol has been written using Gaussian 09 for all of the DFT calculations. These consist of geometry optimization and frequency calculation, the latter of which provides the free energies used in the Boltzmann analysis central to operation IV. We use the M06-2X functional (or the B3LYP functional if Gaussian 03 is used) with the 6-31+G(d,p) basis set for geometry optimization and frequency calculations because of its demonstrated ability to provide more accurate geometries and energetics15,16. We typically carry out geometry optimization by using the default parameters implemented in Gaussian 09 with the exception of specifying the use of a finer integration grid (i.e., Integral(UltraFineGrid)). Frequency calculations allow for structure validation by ensuring that each optimized geometry is not a local saddle point (i.e., transition structure) on the potential energy diagram, which, if present, is indicated by the presence of a negative (or imaginary) frequency. In addition, zero point and thermodynamic correction factors to the total electronic energy are calculated, giving rise to a net free-energy value for each optimized geometry. As an aside, we note that precisely the same approach described to this point would perfectly well serve the needs of a user interested only in computing energetics of a system (e.g., to predict or rationalize an experimental equilibrium value) rather than in continuing with NMR chemical shift calculations.
Operation III. NMR shielding tensor calculations and conversion to chemical shift values. NMR shielding tensors are computed with the GIAO (gauge-independent (or including) atomic orbitals) method17,18 in Gaussian. We typically use the B3LYP functional with the 6-311+G(2d,p) basis set. The simplest approach for converting the resulting set of tensor values to chemical shifts is to subtract the shielding tensor value of tetramethylsilane (TMS) (e.g., 31.88 for 1H at this level of theory) from each of the computed tensor values (analogous to setting the chemical shift of TMS to zero for the experimental data). A more reliable approach is to apply scaling and referencing factors (slope and intercept, respectively) that are derived from linear regression analysis of a test set of molecules19 to each of the computed tensor values. This has the effect of reducing some of the systematic error inherent in the theory used for the computation. After the use of scaling factors, the computed NMR shielding tensors are converted into referenced chemical shifts as defined by equation (2) below, where δ is the referenced chemical shift and σ is the computed NMR shielding tensor5.

To address solvation, we typically use the integrated equation formalism polarized continuum model (IEFPCM)20 during the DFT calculations for both operations II and III. Other approaches for the treatment of solvation effects have been summarized and discussed elsewhere5,10,19. Because experimental NMR data are most often recorded in CDCl3, chloroform solvation has been used as the default in the calculations, but this can be readily changed if the experimental spectral data have been recorded in a different solvent. Substrate solvation cavities are modeled by united-atomic radii (i.e., UA0)21 for the geometry optimization/frequency calculations and individual atomic radii (i.e., Bondi)22 for the NMR calculations.
Operation IV. Boltzmann analysis of DFT NMR data. Energetic data resulting from operation II and NMR shielding tensor data from operation III are manipulated by the use of a script (nmr-data_compilation.py, script D, Fig. 2). Boltzmann weighting factors (Box 3) for each conformer are determined at 25 °C by using the relative free energies obtained from the frequency calculations (i.e., the 'sum of the electronic and thermal free energy' values). The resulting weighting factors (mole fraction contributions) are applied to the computed NMR shielding tensors for each nucleus of each individual conformer. Summation of the weighted tensors across all conformers gives the Boltzmann-weighted average NMR shielding tensors for the candidate structure. En route, the user is asked to input a value for the linear regression intercept and slope values to reference and scale (respectively) the computed shielding tensor data. Alternatively, the computed shielding tensor data of a single molecule (e.g., TMS) could be used in place of the linear regression intercept, and scaling could be omitted. Both the reference and scaling data need to have been computed at the same level of theory as used for the candidate structure (see Step 14 in the PROCEDURE).
Operation V. Comparison of experimental and computed chemical shifts. Several methods can be used to compare the experimental and computed chemical shifts of a candidate structure to determine a goodness of fit. The procedure describes the use of MAE. Regression analysis (R2) and corrected MAE (CMAE) are popular alternatives. The recent review by Tantillo et al.5 describes these and additional methods that have been used for judging the comparisons between experimental and computed NMR chemical shifts. In addition, other methods that deal more explicitly with the challenges associated with identifying the best fit14,23,24 and that are complementary to the approach described here are presented below (under 'Alternatives').
We have created (and provided in Supplementary Data 2) four Python scripts, A–D, which are helpful in automating aspects of the procedure that are otherwise laborious and tedious. These are used to do the following:
-
Script A. Write a Gaussian input file for each structure (local minimum) obtained from operation I (write-g09-inputs.py and variants thereof that use different functional and basis set combinations).
-
Script B. Check for redundant conformers or any that has an imaginary frequency (duplicate_conf_and_imag_freq-check.py).
-
Script C. Extract the NMR shielding tensor for a DFT-computed reference compound of interest (get-ref-shifts.py).
-
Script D. Perform the Boltzmann analysis to generate the weighted-averaged, referenced chemical shifts for each proton and carbon in the candidate structure (nmr-data_compilation.py).
Practical considerations
This protocol is applicable to structures that have a considerable breadth of complexity, but, inevitably, there are limitations. Molecular size, the number of degrees of freedom inherent to the candidate structures (which directly affects the number of conformers) and the level of computational horsepower to which one has access are three important issues that affect the feasibility of tackling a given problem in a given setting. Representative structures that we have studied with this protocol are shown in Figure 3. All have proven to be accessible in our setting; some of these studies have been published (e.g., 8 (refs. 11,25) and 11 (ref. 26)). These serve as indicators of the types of structures that can be addressed. In instances when the structural complexity of the molecule of interest becomes intractable, there are recourses. These include (i) the use of only the global minimum from a molecular mechanics conformational search to proceed with the more computationally intensive ab initio phases of the calculations5,27, (ii) the use of a judiciously chosen small subset of structures from the conformer library generated in a molecular mechanics search28,29 or (iii) the use of a judiciously chosen substructure (e.g., through truncation) as a model for the chemical shifts in question in the structurally ambiguous portion of the molecule (e.g., 3, Box 1 and 11, Fig. 3)26,27.

Structures that we have studied by computing their chemical shifts with DFT.
A final caveat should be emphasized. It is important to use only those protons (or carbons) for which the experimental chemical shift values have been unambiguously established. One does not need to know the experimental chemical shift of every nucleus in the structure under study; however, it is imperative to use in the analysis only nuclei for which the chemical shifts have been interpreted and assigned with absolute certainty.
Alternatives
Alternative approaches, especially those describing other strategies for comparison of experimental with computed data sets (operation V), have been reported. Goodman and co-workers have developed highly effective statistical treatments for use in such comparisons14,23. Their parameters are called CP3 (when experimental data are available for more than one isomeric candidate structure)23 and DP4 (when experimental data are available for a single isomeric candidate structure)14. These allow the assignment of a specific numerical probability to the goodness-of-fit question. Sarotti24 has advanced the use of a training set as an artificial neural network and a pattern recognition protocol for distinguishing good from poor fits between the computed and experimental data25. These approaches use molecular mechanics (MM) to identify conformer geometries and then either MM or semiempirical theory (Austin model 1; AM1) to compute energies. DFT [B3LYP/6-31+g(d) or mPW1PW91/6-31G(d)] is then used to compute the GIAO NMR chemical shifts. Overall, this reduces the demands on computational power.
For the protocol detailed here, we have described the use of the following software: MacroModel and Maestro for molecular mechanics calculations, Gaussian 09 for the subsequent ab initio computations and Python scripts for managing the output data. Alternative packages and modules certainly could be substituted according to availability, familiarity and so on. Similarly, the choice of basis set and DFT functional (i.e., the level of theory) could be modified from what is specified here, which in some cases might better serve some users' needs (e.g., speed or expense versus quality considerations). We have written complementary Python scripts to accommodate the use of (the earlier) Gaussian 03. Each script is a '.py' file that has been archived within the '.zip' file named Supplementary Data 2.

Materials
EQUIPMENT
Spectral data
-
1H and/or 13C NMR spectral data for the compound under study
Software requirements for calculations
-
Structure generation program. This should be capable of providing file types that can be read by Maestro30 (e.g., '.sdf'). This could be Maestro itself or, e.g., ChemBio3D
-
X-ray structure conversion program. For example, Mercury CSD31, for converting a '.cif' into a '.sdf' file format (for (optional) Step 1C, freely downloadable; e.g., at: http://www.ccdc.cam.ac.uk/products/mercury/)
-
Molecular mechanics program. We use MacroModel (version 10.0)13 and Maestro (version 9.4) within the Schrödinger package of software (Schrödinger Suite 2011), and we have written the procedure below accordingly. Maestro is a graphical interface that communicates with MacroModel. It is freely available to academic users https://www.schrodinger.com/downloadcenter/10/)
-
Quantum mechanics software capable of performing DFT geometry optimizations, thermochemistry (frequency) and NMR shielding tensor calculations. We use Gaussian 09 (ref. 32), and we have written the procedure below accordingly
Critical
The PROCEDURE provides considerable information that will guide those using earlier versions of MacroModel, Maestro and Gaussian (e.g., Gaussian 03).
Software requirements for use of scripts and final processing of NMR data
-
Command-line interface application (Terminal (in Mac OS X or Linux) or Command Prompt (in Windows))
-
Python, version 2 or 3 (included with Mac OS X and Linux operating systems)
-
IDLE Python script editor (freely downloadable; e.g., at: http://www.python.org/download/)
-
GaussView 5 (ref. 33) (or equivalent molecular visualization application)
-
Microsoft Excel (or equivalent spreadsheet application)
Hardware requirements for calculations
-
A computer (or supercomputing node) having at least 4 GB of RAM and a dual-core processor is readily sufficient to perform the conformational searches, geometry optimizations, frequency calculations and NMR chemical shift calculations on the 3-methylcyclohexanol trial compounds 1- cis and 1- trans. Of course, the power and speed of the computational resources to which the user has access will affect the timing estimates provided for some of the steps in the PROCEDURE. Alternatively, as the structural complexity of the molecules under study increases, access to more powerful computing hardware will be advantageous
Hardware requirements for the use of scripts and final processing of NMR data
-
Execution of Python scripts for data processing and manipulation can be carried out on any standard personal computer having the necessary software.
Procedure
Create input geometry and carry out a conformational search with molecular mechanics in MacroModel
Timing 30 min
-
1
Creation of an input structure. Choose one of the following options to input a structure into Maestro: (option A) draw the structure with the structure editor native to Maestro; (option B) import a set of atomic coordinates created with a different structure editor (e.g., ChemBio3D, GaussView, PCModel or MacMolPlt) in a format readable by Maestro; or (option C) import coordinates from a relevant crystal structure data file (e.g., '.cif' from the Cambridge Crystallographic Data Centre (CCDC)).
-
A
Draw the structure in Maestro
-
i
Launch Maestro.
-
ii
Create and set the working directory. We recommend creating a master directory (e.g., entitled '3-methylcyclohexanol-master_dir') within which to store the subdirectories that contain the computational files associated with each candidate structure (e.g., 'cis-3-methylcyclohexanol-sub_dir'). Select Project → Change Directory to open a file chooser window. Click the Create New Folder button and change the directory label to '3-methylcyclohexanol-master_dir.' Open the 3-methylcyclohexanol master directory and again click the Create New Folder button and change the directory label to 'cis-3-methylcyclohexanol-sub_dir.' Select the 'cis-3-methylcyclohexanol-sub_dir' directory and click choose to set the working directory for computational files associated with the cis-diastereomer of 3-methylcyclohexanol.
-
iii
Create an input geometry for cis-3-methylcyclohexanol (1- cis). Draw cis-3-methylcyclohexanol in the Maestro workspace (Fig. 4) by using the build and edit toolbars. For Maestro version 9.2 (and later), display the build toolbar by clicking the Build button and display the edit toolbar by clicking the Edit button. For Maestro version 9.1 or earlier, display the build toolbar by clicking the Show/Hide the build toolbar button. Draw the carbon skeleton with the Draw structures button (Fig. 4a) on the Build toolbar. Add nonhydrogen (non-H) atoms by clicking the drop-down arrow on the pencil button or the Set element button (Fig. 4b) on the build toolbar. Add all implicit hydrogen atoms by double-clicking the Add hydrogens button on the edit toolbar (Fig. 4c) (located on the main toolbar in Maestro version 9.1 or earlier).
Figure 4: The window and toolbars used to create a structure in Maestro version 9.2 (and later). 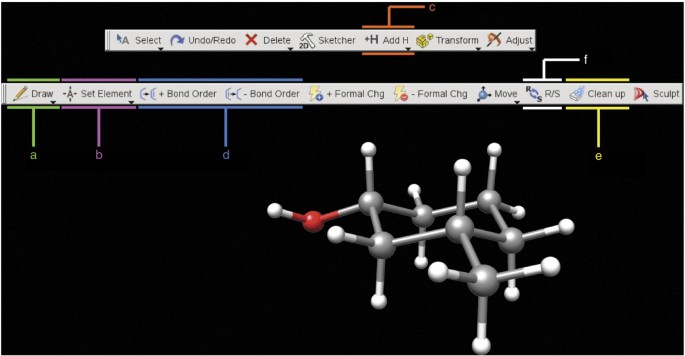
(a–f) The sequence of a–f represents a logical order in which to carry out the operations: use a to create the heavy atom skeleton, b to define which atoms are not carbon, c to fill the remaining open valencies with hydrogen atoms, d to adjust the bond order, e to neaten the structure and f to isomerize stereocenters, if needed. These operations are described in further detail in Steps 1A(iii–v) and 19.
-
iv
(Optional) Use the following tools to draw more advanced structures. Change the explicit bond order with the Increment/Decrement bond order buttons (Fig. 4d) on the build toolbar. Buttons for predrawn functional groups and cyclic hydrocarbon templates are located on the fragment toolbar, which can be displayed by clicking the Fragment button. In addition, the invert chirality button (Fig. 4f) is useful for quickly drawing other (diastereomeric) candidate structures.
-
v
Neaten this initial (and perhaps crudely drawn) structure by clicking the Clean Up Geometry button on the build toolbar (Fig. 4e). The geometry of the input structure will be quickly minimized with a molecular mechanics routine, and then it will be redrawn in the workspace.
-
vi
Select Workspace → Create Project Entry. A window will open. Change the title to 'cis-3-methylcyclohexanol-initial_geometry' and click Create to add the drawn geometry to the Project Table.
-
i
-
B
Import the atomic coordinates into Maestro as an .sdf file
-
i
Import a set of atomic coordinates for cis-3-methylcyclohexanol (1- cis) that were created in, for example, ChemBio3D Ultra (also known as Chem3D). Maestro reliably handles '.sdf' files, so we recommend creating this input file type. This can be accomplished in most software packages capable of producing a chemical structure. In ChemBio3D, draw cis-3-methylcyclohexanol and select File → Save As to open a save as window. Enter 'cis-3-methylcyclohexanol_Chem3D' as the filename, change the file type to SDFile (*.sdf) and select Save.
-
ii
Launch Maestro and create a working directory as described in Step 1A(i,ii).
-
iii
Select Project → Import Structures to open a file chooser window. Change the Files of type: to Any (*.*). Locate and select the file 'cis-3-methylcyclohexanol_Chem3D.sdf' created in Step 1B(i).
-
iv
Perform Step 1A(vi) to finish inputting the geometry to Maestro.
-
i
-
C
Import the atomic coordinates from an X-ray crystal structure (e.g., a .cif file)
-
i
For more complex structures, we recommend using this method, if possible, because the risk of error in drawing the input geometry is reduced. In this example, we will demonstrate how to input the crystal structure geometry from a .cif file of maoecrystal V into Maestro.
-
ii
Go to the CCDC homepage (http://www.ccdc.cam.ac.uk/products/mercury/) and click the Request Structure link. Request the crystal structure of maoecrystal V (its CCDC deposition number is 249099). A .txt file that contains coordinates for the crystal structure of maoecrystal V will be attached to (or embedded in) an e-mail from the CCDC. Save the attachment as 'maoecrystal-V.cif' to a local hard drive.
-
iii
Start Mercury.
-
iv
Locate and open the 'maoecrystal-V.cif' file within Mercury. Select File → Save As, change the file format to PDB files (*.pdb *.ent) and click OK. A save as window will open; change the filename to 'maoecrystal-V_Xray.pdb,' specify a directory and Save.
-
v
Launch Maestro and create a working directory as described in Step 1A(i and ii).
-
vi
Select Project → Import Structures to open a file chooser window. Change the Files of type: to Any (*.*). Locate and select the 'maoecrystal-V_Xray.pdb' file created in Step 1C(iv).
-
vii
Perform Step 1A(vi) to finish inputting the geometry to Maestro.
Critical Step
Carefully check that both the constitution and relative configuration of the input structure are correct.
-
i
-
A
-
2
(Optional) Force-field evaluation. To determine the molecular mechanics force field best parameterized for the structure of interest, submit the input geometry to a molecular mechanics 'current energy calculation' (i.e., no energy minimization is necessary) by selecting Applications → MacroModel → Current Energy. A window will open that displays the possible choices of force field for the energy calculation. Change the force field to OPLS_2005, append 'OPLS_2005' to the job name and click Run. Click the Jobs → Monitor buttons, which are located in the lower left corner of the workspace. A monitor window will open that logs information related to the energy calculation. Double-click on the JobID associated with the energy calculation and scroll through the logs. Note the number of 'medium- and low-quality' stretch, bend and torsion parameters. Repeat this step by using the Merck molecular force field (MMMF). In general, the force field that is parameterized with the greatest number of high-quality parameters should be used for the subsequent conformational search (Step 3). We find that the OPLS_2005 force field (versus, e.g., MMFFs) has fewer than or an equal number of low- and/or medium-quality parameters for CHNO-containing molecules, and we typically use OPLS_2005.
-
3
Conformational search. Choose from the following two options to run a conformational search of the input geometry, either interactively within Maestro (option A) or from the command-line interface (option B). The choice of either option depends on the user′s computational resources.
-
A
Conformational search interactively within Maestro
-
i
While in Maestro, select Applications → MacroModel → Conformational Search. A window will open that displays the possible options that can be modified for the conformational search.
-
ii
Select the Potential tab, select OPLS_2005 (or the most well-parameterized force field from Step 2) and change the solvent from Water to CHCl3.
-
iii
Select the CSearch tab. Change the method to Torsional Sampling (MCMM). Uncheck the Multiligand box and the Perform automatic setup during calculation box. Click the Perform Automatic Setup button. Change the torsional sampling options to Extended.
-
iv
Delete the default name and enter 'cis-3-methylcyclohexanol-csearch.' Do not use spaces, back-slashes or forward slashes in file names. Select Run to begin the conformational search.
-
v
Click the Jobs → Monitor buttons (located in the lower left corner of the workspace), and a monitor window will open that displays the progress of the conformational search. When the conformational search is finished, the monitor window will read 'BatchMin: normal termination' and display the date and time of completion. A conformational search in Maestro that has completed successfully will result in the generation of an output file with '-out.maegz' ending the filename. This file contains data for all conformers generated in the conformational search, and it can be accessed at anytime within Maestro. Ideally, there will be no medium- or low-quality parameters for the MM force field used for the conformational search. MacroModel reports these values as 'the number of high-, medium-, low-quality stretch/bend/torsion parameters = x y z' in the monitor window (also reported in the '.log' file).
-
vi
View the structures of the family of unique conformers by selecting Project → Show Table to display the Project Table window. The Project Table window displays all structures that have been inputted into Maestro during the current session as rows in a table. To display the structures in the workspace, check the box in the In column for the row(s) of the Project Table. Multiple structures can be displayed at once.
-
vii
In the Project Table, select Show → All to display information from the conformational search. For each conformer in the Project Table, inspect the column named 'Minimization Converged-(force field)' to ensure that each conformer has converged, as evidenced by a checked box for each conformer.
-
viii
For each conformer reported in the Project Table, inspect the column named 'Times Found-(force field)' to ensure that each conformer was found at least ten times.
-
ix
(Optional) If the conformational search yields a tractable number of local minima, we recommend examining the geometry and energy of each structure to ensure that there are no redundant conformers and that all logically anticipated conformers (rotamers and invertamers) have been located.
-
i
-
B
Conformational search within the (Unix or Windows) command-line interface
-
i
Perform Step 3A(i–iii) to input the preferences for the conformational search.
-
ii
Click the settings button to the right of the Job name: input box and a window will open. Change 'Append new entries as a new group' setting to 'Do not incorporate.' Change the filename to 'cis-3-methylcyclohexanol-csearch.' Avoid using spaces, back-slashes or forward slashes in file names. Select Save to save the job input file for the conformational search.
-
iii
Open the command-line interface (Terminal in Mac OS X or Linux or Command Prompt in Windows). Load the module that contains MacroModel (e.g., Schrödinger). Navigate to the directory that contains the input file for the conformational search.
-
iv
Run the conformational search from the command-line interface by entering the following command:> bmin cis-3-methylcyclohexanol-csearch
-
v
Check for completion of the conformational search job by entering the following command (replace 'ls' with 'dir' in the following command if Command Prompt in Windows is used):> ls *.log
If the conformational search job was executed successfully, the following will be displayed:> cis-3-methylcyclohexanol-csearch.log
-
vi
Check for completion of the conformational search job by opening the log file by using the following command:> less cis-3-methylcyclohexanol-csearch.log
Scroll to the end of the file by using the Shift+Page down key combination. If the conformational search job has been completed, the following will be displayed:BatchMin: normal termination
A conformational search in the Command-line interface that has completed successfully will also result in the generation of an output file with '-out.maegz' ending the filename. This file contains data for all conformers generated in the conformational search, and it can be accessed at anytime within Maestro.
-
vii
In Maestro, import the structures resulting from the conformational search by selecting Project → Import Structures and locating the 'cis-3-methylcyclohexanol-csearch-out.maegz' file.
-
viii
Perform Step 3A(vi–ix) to check the results of the conformational search.
-
i
-
A

Create Gaussian input files for each conformer
Timing 15 min
-
4
Download to a local hard drive those Python scripts from Supplementary Data 2 that correspond to the version of Python being used. Move the 'write-g09-inputs.py' (for Gaussian 03, use 'write-g03-inputs.py') script to the directory named 'cis-3-methylcylohexanol-sub_dir' (created in Step 1A(ii)), which contains the output file resulting from the conformational search (i.e., 'cis-3-methylcylohexanol-csearch-out.maegz').
-
5
(Optional) Allocation of the number of core processors and the amount of memory used for a given job has a substantial effect on the computational run time of DFT calculations. The default settings in the above scripts are 8 processors and 8 GB of RAM. To change these settings, open the 'write-g09-inputs.py' (or 'write-g03-inputs.py') Python script within the Python script editor (i.e., IDLE). Edit the memory allocation values ('%mem=8gb') on lines 111 and 138 and/or the number of core processors ('%nproc=8') on lines 112 and 139 of the script.
-
6
In Maestro, clear the contents of the Project Table by selecting Project → Close. (You will be asked if you would like to Save in the window that opens, and you can choose to do so or not; if so, a '.prj' file will be created). Select Project → Import Structures and locate the 'cis-3-methylcyclohexanol-csearch-out.maegz' file. This will import only the structures from the conformational search. This step is necessary because the 'write-g09-inputs.py' Python script extracts all contents of the Maestro Workspace and Project Table when executed (Step 8); it is necessary that no extraneous structures be present in the Table or Workspace.
-
7
Conformer name standardization (optional here, but necessary for earlier versions of Maestro). In the Project Table, provide a name for each conformer by clicking in the title cell of the lowest-energy conformer. Change the entry name to 'cis-3-methylcyclohexanol' and hold the control key while pressing the enter key, which will enter this name for all of the conformers in the Project Table.
Critical Step
All conformers need to have the same name; otherwise, the 'write-g09-inputs.py' Python script will not sort the Gaussian input files properly.
-
8
Creation of Gaussian input files for geometry optimization, frequency calculation and NMR shielding tensor calculation. In Maestro, select Window and ensure that the Command Input Area is checked. To execute the script, enter the command 'pythonimport write-g09-inputs' in the dialog box to the right of Commands. For each conformer, the script will automatically create two Gaussian input files—the geometry optimization and frequency calculation input file (named 'filename-opt_freq-conf_#.com') and the NMR magnetic shielding tensor calculation file (named 'filename-nmr-conf_#.com'). The script will also automatically create a new directory (named 'filename-gaussian_files') and move the Gaussian input files to that directory.
-
9
Select Project → Import Structures to open a file chooser window. Change the Files of type: to Any (*.*). Open the 'cis-3-methylcyclohexanol-gaussian_files' directory that was created in Step 8. Verify that the number of 'cis-3-methylcyclohexanol-opt_freq-conf_#.com' and 'cis-3-methylcyclohexanol-nmr-conf_#.com' files is equal to the number of unique conformers found in the molecular mechanics conformational search (Step 3).
Pause point
After the 'write-g09-inputs.py' Python script has been run, all Gaussian input files are saved, and they can be submitted to Gaussian at any later time.
Carry out DFT geometry optimization and frequency calculations for each conformation in Gaussian 09
Timing variable, depending on the molecule under study
-
10
Geometry optimization and frequency calculation in Gaussian. Submit each optimization and frequency Gaussian '.com' input file to be run in Gaussian. A frequency and geometry calculation will be run simultaneously, and it will generate a single '.out' Gaussian output file. There are a number of ways in which this can be done (individually, batch and so on) depending on factors such as the operating system of the host computer, parallelization protocols or local administrative protocols. Discussion of many of these issues can be found at the Gaussian website (http://www.gaussian.com/g_tech/g_ur/g09help.htm). The files resulting from the Maestro, MacroModel and Gaussian jobs (including '.chk' and '.out') are included in Supplementary Data 3 and 4 for 1- cis and 1- trans, respectively.
Pause point
The '.out' and '.chk' files can be accessed any time after the Gaussian jobs are completed.
-
11
Validation of the DFT geometry-optimized (and energy-minimized) conformers. Check for duplicate conformers or for saddle-point geometries (as evidenced by the presence of a negative (or imaginary) frequency) by moving the 'duplicate_conf_and_imag_freq-check.py' Python script to the directory containing the Gaussian '.out' files (e.g., 'cis-3-methylcyclohexanol'). Execute the 'duplicate_conf_and_imag_freq-check.py' Python script in the command-line interface (e.g., with Terminal (for Linux or Mac OS X) or with Command Prompt (for Windows)) by entering the following command:> python duplicate_conf_and_imag_freq-check.py
The script will request the name of the candidate structure by displaying the following prompt:Enter the name of the candidate structure:
Enter 'cis-3-methylcyclohexanol,' and if the script then executes successfully, the following message will be displayed in the command-line interface:The script successfully performed the task of creating the cis-3-methylcyclohexanol-conf_energy_and_imag_freq.csv file that shows the conformer number, conformer filename, total electronic energy, free energy, and total number of imaginary frequencies for each conformer.
-
12
Open the 'cis-3-methylcyclhexanol-conf_energy_and_imag_freq.csv' file (a .csv file is a simple, Microsoft Excel-readable text file that uses a 'comma separated values' format) in Excel (or equivalent spreadsheet application). Verify that there are no imaginary frequencies in any of the conformer structures by viewing the column labeled 'Number of Imaginary Frequencies.' Compare the energies of the conformers by viewing the column labeled 'Relative Energy.' If any conformers have nearly the same energy (e.g., within 0.01 kcal mol−1 of one another), open the corresponding '.out' files of each in GaussView. If the geometries are the same, then remove all but one from the 'cis-3-methylcyclohexanol-gaussian_files' directory to ensure that the conformer is not double-counted during the Boltzmann weighting of the computed NMR chemical shifts.
Compute NMR chemical shift data in Gaussian
Timing <10 min of active effort and ∼30 min of computational wall time per conformer for the 3-methylcyclohexanols
-
13
NMR shielding tensor calculations in Gaussian. Submit each 'cis-3-methylcyclohexanol-nmr-conf-#.com' input file to be run in Gaussian. See comment in Step 10 (and http://www.gaussian.com/g_tech/g_ur/k_nmr.htm). A GIAO magnetic shielding tensor calculation will be run and a '.out' output file will be created for each conformer.
-
14
(Optional) Computation of the NMR shielding tensors of TMS for use as a single reference compound. We have provided reference and scaling parameters for use with this protocol, but if a different level of theory is implemented, then the user must calculate new reference and scaling parameters. Application of scaling factors will reduce the systematic error, but they are cumbersome and tedious to calculate. Instead, we recommend that the user compute the NMR shielding tensors of TMS to reference the computed chemical shifts without scaling. Download the 'tms_std-g09.com' (or '...g03.com' for Gaussian 03) input file from Supplementary Data 2, modify the functional and/or basis set and submit it to be run in Gaussian. For guidance on submitting Gaussian input files, see Step 10 (and http://www.gaussian.com/g_tech/g_ur/k_nmr.htm). A geometry optimization, frequency and NMR shielding tensor calculation will be run and the 'tms_std-g09.out' (or '...g03.com' for Gaussian 03) file will be created. Move the 'get-ref-shifts.py' Python script to the directory containing the 'tms_std-g09.out' file. In the command-line interface (e.g., with Terminal for Linux or Mac OS X or with Command Prompt for Windows), execute the script to extract the computed carbon and proton NMR shielding tensors of TMS by entering the following command:> python get-ref-shifts.py tms-std-g09.out
This script reads the '.out' Gaussian output file and extracts the carbon and proton NMR shielding tensors and prints the data in.csv files. If the Pyton script has been executed successfully, the following command will be printed in the output window (actual numbers represent those using the level of theory recommended in this protocol):The average NMR shielding tensor of the hydrogen atoms = 31.8819The average NMR shielding tensor of the carbon atoms = 183.7949Additionally, the script has performed the task of creating the following files, which contain the NMR shielding tensors of the individual proton and carbon atoms of the reference standard:tms_std-nmr-protons.csvtms_std-nmr-carbons.csv
Compile the NMR magnetic shielding tensor and free-energy data for each conformer, determine the Boltzmann-weighted average magnetic shielding tensors for each proton and carbon atom and reference the tensor data to create the chemical shift values for the candidate structure under study
Timing <5 min
-
15
Assembling the NMR and free-energy data by using the 'nmr-data_compilation.py' Python script. Move the 'nmr-data_compilation.py' script to the directory containing the Gaussian '.out' files (e.g., 'cis-3-methylcyclohexanol-sub_dir'). Execute the 'nmr-data_compilation.py' script in the command-line interface (e.g., with Terminal (for Linux or Mac OS X) or with Command Prompt (for Windows)) by entering the following command:> python nmr-data_compilation.py
This command will run the 'nmr-data_compilation.py' script in Python and analyze all '.out' files in the current working directory. The script will then prompt for scaling and/or reference parameters by displaying the following list of options:A. Enter reference and scaling factor data from regression analysis of a test set of molecules.B. Enter reference data from computation of a reference standard (e.g., TMS) NMR shielding tensors.C. Do not reference or scale NMR shielding tensor data.
Select one of these options to scale and/or reference the computed chemical shifts by entering the corresponding letter: scale and reference chemical shifts by using regression analysis parameters (option A), reference chemical shifts to a reference standard (e.g., TMS) without applying empirical scaling (option B) or omit reference and scaling entirely (option C). We have provided referencing and scaling factor data below. See Step 14 for generating alternative referencing and scaling factor parameters.
-
A
Scale and reference chemical shifts using regression analysis parameters
-
i
Select option A by entering A (not case-sensitive) in the option list.
-
ii
The script will then prompt for computed and experimental data of the reference standard by displaying the following messages one at a time:Enter the 1H scaling factor INTERCEPT:Enter the 1H scaling factor SLOPE:Enter the 13C scaling factor INTERCEPT:Enter the 13C scaling factor SLOPE:
Enter the desired input in response to each of these four commands. Enter the linear regression scaling parameters (i.e., scaling factors) provided in the table below. These are needed for scaling and referencing the computed NMR data for the level of theory described in this protocol (i.e., B3LYP/6-311+G(2d,p)//M06-2X/6-31+G(d,p)). These scaling parameters were created by the method of Tantillo et al.5. These values are unique to this specific functional and basis set combination, but they are independent of the structure under study. In other words, these values should be used for any compound, not just for 3-methylcyclohexanol.
Table 2
Scaling factors
Slope
Intercept
1H
−1.0767
31.9477
13C
−1.0522
181.2412
-
i
-
B
Reference chemical shifts to a reference standard (e.g., TMS) without applying empirical scaling
-
i
Select option B by entering B (not case-sensitive) in the option list.
-
ii
The script will then prompt for computed and experimental data of the reference standard by displaying the following messages one at a time:Enter the computed 1H NMR shielding tensor of the reference standard:Enter the experimental 1H chemical shift of the reference standard:Enter the computed 13C NMR shielding tensor of the reference standard:Enter the experimental 13C chemical shift of the reference standard:
Enter the desired input after each message has been displayed. If the recommended level of theory has been used, then enter the TMS NMR shielding tensors in the table below. See Step 14 to obtain NMR shielding tensors of TMS at a different level of theory.
Table 3
Tensors
1H
31.8819
13C
183.7949
-
i
-
C
Omit reference and scaling
-
i
Select option C by entering C (not case-sensitive) in the option list to omit application of any scaling and referencing factors. This would provide one with the unscaled and unreferenced Boltzmann-weighted NMR shielding tensors, which could be of value to someone developing a new method for scaling or referencing.
The script will request the name of the candidate structure by displaying the following prompt:Enter the name of the candidate structure:
If acceptable values have been entered after and the python script has executed successfully, the following message will be displayed in the command-line interface:The script successfully performed the Boltzmann weighting, compiled the results of the NMR computation, assembled, scaled, and/or referenced these data in the following '.csv' files:cis-3-methylcyclohexanol-nmr_data_compilation-master_proton.csvcis-3-methylcyclohexanol-nmr_data_compilation-avg_proton.csvcis-3-methylcyclohexanol-nmr_data_compilation-master_carbon.csvcis-3-methylcyclohexanol-nmr_data_compilation-avg_carbon.csv
This script parses NMR shielding tensor and free-energy data, calculates the Boltzmann-weighted average shielding tensor data set, converts tensor data into scaled and/or referenced chemical shifts and exports these data to several different Excel-readable files (.csv) for the candidate structure (e.g., cis-3-methylcyclohexanol). The 'filename-nmr_data_compilation-master_nucleus.csv' file includes the Boltzmann analysis of the set of conformers and the scaled and/or referenced computed NMR chemical shift values for all nuclei of each conformer. The 'filename-nmr_data_compilation-avg_nucleus.csv' file includes the Boltzmann-weighted average chemical shifts for the respective nuclei of the candidate structure, scaled and/or referenced.
-
i
-
A
-
16
Verify that the script has worked properly by entering the following command in the command-line interface (replace 'ls' with 'dir' in the following command if Command Prompt in Windows is used):> ls *nmr_data_compilation*.csv
This command will search the current directory for .csv files that contain 'nmr_data_compilation' in their title. If the script executed successfully, the four files named in Step 15 will be listed.
Compare computed chemical shifts for the candidate structure with the available experimental data for the compound(s) under study
Timing 30–60 min per candidate structure
-
17
Correlating the (often non-intuitive) atom numbers assigned in the Gaussian output structures with conventional (and more convenient) atom numbering. Start GaussView and then open any one of the Gaussian NMR output files (e.g., 'cis-3-methylcyclohexanol-nmr-conf-1.out').
-
18
Select View → Labels in GaussView to display the atom numbers (automatically assigned during structure creation in Maestro) on the structure. These atom numbers are those present in the .csv files created in Step 15, and they are displayed in column A, labeled as 'Gaussian atom numbers' when opened in Excel.
-
19
Start Excel and use it to open the 'cis-3-methylcyclohexanol-nmr_data_compilation-avg_proton.csv' file. Column B bears the header 'logical atom numbers,' but the cells below are empty. Enter an atom number, label or name (e.g., H1, H2ax or Me; and C1, C2, etc.) for each nucleus. For maximal logic and convenience, these entries should follow the same labeling scheme used for the experimental chemical shift assignments. Save the file as, e.g., 'cis-3-methylcyclohexanol_comp-vs-exp_proton.xlsx.'
Critical Step
Each conformer of a single candidate structure will be labeled with the same Gaussian atom numbers, but atom numbers of different candidate structures might be different, depending on how the input structure was created. One trick for minimizing the complications that can arise from inconsistent numbering schemes for different diastereomeric isomers is to open the structure in Maestro of any conformer resulting from the conformational search of one candidate structure (i.e., a '-out.maegz' file, Step 3A(v)), and use the R/S button (Fig. 4f) to invert the configuration of a stereocenter in the existing structure. Use that newly created structure to begin the conformational search of the new (diastereomeric) candidate structure. This will ensure that the same (albeit still nonintuitive) atom numbering scheme is applied to each candidate structure.
-
20
Averaging the individual shifts of chemically equivalent (and rapidly interconverting) nuclei in relevant structures. The need for this step is encountered most often in structures containing a methyl group. It is necessary because the three methyl protons in any single conformer/rotamer of, e.g., cis-3-methylcyclohexanol are inequivalent. That is, methyl group rotation is fixed, of course, during the computation. However, rapid rotation averages the experimentally observed shift to a single value. In the Excel spreadsheet created in Step 19, identify the three methyl proton shift values (column C, labeled 'chemical shift'). Average the chemical shift of the three individual methyl protons and enter that new value in a separate/new row as a replacement for the three individual protons of that methyl group.
Critical Step
Do not include the three individual degenerate shift values during the data analysis described next (Steps 21–26). These must be averaged as described immediately above in order to take into account the chemical equivalence of the three methyl protons on the NMR time scale. (Other somewhat common substituents that warrant analogous treatment because of their symmetry properties include the 2,6- and 3,5-protons and carbons in symmetrically substituted arenes, methylene protons in achiral candidate structures or methyl carbons in t-butyl or trimethylsilyl groups.)
-
21
Repeat Steps 19 and 20 for the computed carbon NMR data.
-
22
MAE. There is no universally accepted best practice for carrying out the evaluation of the computed versus experimental data for determining the goodness of fit. Nonetheless, comparison of the MAE, which is simply the error between δDFT and δexp, averaged across all nuclei for which an experimental value is unequivocally known, is the most commonly used criterion. This comparison is most easily carried out by spreadsheet analysis. A spreadsheet will have been created at the end of Step 20 (or 21) that contains all of the computed (and conformationally averaged) chemical shifts for the candidate structure. Again, there are many different ways to manipulate the data to yield the MAE value, but all methods involve entering the experimental chemical shift values into a spreadsheet file. An example file ('cis-3-methylcyclohexanol_comp-vs-exp_proton.xlsx') is provided in Supplementary Data 2. This file allows one to compare the computed with experimental chemical shifts, and it shows the resulting MAE values. By using the keyboard shortcut toggle–Ctrl-∼ (Ctrl-tilde) in Excel, one is able to view the underlying mathematical formulas for the entire spreadsheet. This is an instructive way to learn the logic used to create files such as 'MAE-analysis-3-methylcyclohexanol.xlsx'.
-
23
Repeat Step 22 for the carbon nuclei.
-
24
(Optional) Other methods for data analysis: Alternative approaches for analyzing the goodness of fit include regression analysis and evaluation of R2 (coefficient of determination); the determination of the corrected MAE could also be used at this point. The recent methods of Smith and Goodman14,23 and of Sarotti24 are particularly notable. Each uses a sophisticated statistical treatment of NMR shift data that has been computed at lower levels of theory than described here to arrive at the best fit for structure assignment. It is beyond the scope of this protocol to present these alternative methods in detail, but readers should be aware of those approaches. Suffice it to say that these approaches, although less demanding of computational time (roughly half), require a comparable amount of active effort by the researcher to the procedure described in detail here.
-
25
Repeat Steps 1–24 for each candidate structure (i.e., diastereomer or constitutional isomer).
-
26
Decide which computed structure shows the best correlation(s) (e.g., lowest MAE) with the available experimental spectral data for the compound under study.
Troubleshooting
Troubleshooting advice can be found in Table 1.
Timing
Most of the operations that require active effort by the user have been automated with Python scripts. The computations themselves comprise the majority of total time required to complete the protocol, and they do not require active effort by the user. The computational time (Steps 10–14) will vary with the molecular size, the number of degrees of freedom in the candidate structure and the level of computational horsepower available to the user. The times estimated below are active effort on the part of the researcher.
Steps 1–3: 30 min
Steps 4–9: 15 min
Steps 10–12: <10 min of active effort and ∼1 h of computational wall time per conformer for the 3-methylcyclohexanols. The time for computation depends on the complexity of the molecule under study; the demands scale nonlinearly with issues such as the number of atoms and the number of degrees of freedom (e.g., rotatable bonds)
Steps 13 and 14: <10 min of active effort (and ∼30 min of computational wall time per conformer for the 3-methylcyclohexanols)
Steps 15 and 16: <5 min
Steps 17–24: 30–60 min per candidate structure
Anticipated results
We recommend that new users first reproduce this procedure and analyze cis-3-methylcyclohexanol (1- cis) at the level of theory recommended in this protocol. Specifically, we have (i) optimized the geometries and computed the free energies with the M06-2X functional and the 6-31+G(d,p) basis set and (ii) computed the NMR chemical shifts with the B3LYP functional and 6-311+G(2d,p) basis set. By convention, this is denoted as B3LYP/6-311+G(2d,p)//M06-2X/6-31+G(d,p). The expected outcomes after the conformational analysis, geometry optimizations and frequency calculations are shown in Figures 5 and 6. Six conformers are obtained from the conformational search, with 99.8% of the contribution coming from the three diequatorial conformers, which are C–OH rotamers of one another.

(a) The relative Gibbs (or free) energy (in kcal mol−1) of each conformer with respect to the global minimum. (b) The Boltzmann mole fraction (in %) for each conformer.

(a) Computed (with B3LYP/6-311+G(2d,p)//M06-2X/6-31+G(d,p)) and experimental 1H and 13C chemical shift values of 1- cis (atom labels correspond to those shown in the diequatorial conformer shown at the top). (b,c) MAEs for the 1H and 13C chemical shifts, respectively, between matched and mismatched pairs of 1- cis versus 1- trans. *Chemical shifts were derived from application of scaling factors (slope = −1.0767, intercept = 31.9477) to the 1H NMR shielding tensors computed at the B3LYP/6-311+G(2d,p)//M06-2X/6-31+G(d,p) level of theory. #Chemical shifts were derived from application of scaling factors (slope = −1.0522, intercept = 181.2412) to the 13C NMR shielding tensors computed at the B3LYP/6-311+G(2d,p)//M06-2X/6-31+G(d,p) level of theory.
The computed values (referenced and scaled) from the NMR chemical shift calculations and their comparisons with the experimental values for 1- cis are shown in Figure 6a. The computed 1H and 13C chemical shifts are all within 0.20 and 5.0 p.p.m., respectively, of the experimental values. The analogous data for 1- trans are provided in Supplementary Data 1. The MAEs for the 1H and 13C chemical shifts of the computed chemical shifts versus the experimental values for each of the two diastereomers are tabulated in Figure 6b,c. The 1H-matched MAEs are <0.10 p.p.m. and the mismatched MAEs are >0.20 p.p.m. for both diastereomers of 3-methylcyclohexanol. These differences in MAEs clearly allow one to match each set of computed chemical shifts with the correct experimental data. The 13C-matched MAEs are both <2.0 p.p.m. The 13C-mismatched MAEs are both higher than the corresponding matched set of MAEs. Consistent with our previous observations, this indicates that 1H are more effective than 13C chemical shifts for discriminating stereochemical differences in these types of systems10,34.
It can be anticipated that once readers have successfully navigated this introductory example, they will be interested in extending their studies to compounds having somewhat more complex structures. New issues arise as the level of structural complexity increases (e.g., increased number of conformational minima that must be identified via the initial conformational search, increased computational time required for the DFT geometry optimizations and NMR calculations, potential for a greater number of (diastereomeric) candidate structures that must be considered and potential compression of the goodness-of-fit parameters). Dealing with these in a detailed manner is beyond the scope of this protocol. Many of these factors have been mentioned above in the introductory commentary and in the TROUBLESHOOTING table. In this regard, we also again call attention to our earlier studies of the tricyclic compounds 8 (refs. 26,35) and 11 (ref. 29) (cf. Fig. 3), from which useful guidance can be taken to assist users wanting to extend their investigations.
Finally, one might ask when the approach described in this protocol should be implemented. There is no simple, single answer because many variables come into play. How essential is it that the structure be unambiguously known? What is the level of NMR expertise of the investigator(s)? How well-established are other known closely related structures and what computational resources and expertise are available locally? Hopefully, working through this protocol will give each researcher, at the very least, a better ability to answer these questions.
References
Casabianca, L.B. & de Dios, A.C. Ab initio calculations of NMR chemical shifts. J. Chem. Phys. 128, 052201-1–052201-10 (2008).
Bally, T. & Rablen, P.R. Quantum-chemical simulation of 1H NMR spectra. 2. Comparison of DFT-based procedures for computing proton-proton coupling constants in organic molecules. J. Org. Chem. 76, 4818–4830 (2011).
Cramer, C.J. Chapter 8 (density functional theory) in Essentials of Computational Chemistry 2nd edn. (John Wiley & Sons, 2004).
Jain, R., Bally, T. & Rablen, P.R. Calculating accurate proton chemical shifts of organic molecules with density functional methods and modest basis sets. J. Org. Chem. 74, 4017–4023 (2009).
Lodewyk, M.W., Siebert, M.R. & Tantillo, D.J. Computational prediction of 1H and 13C chemical shifts: a useful tool for natural product, mechanistic and synthetic organic chemistry. Chem. Rev. 112, 1839–1862 (2011).
Tantillo, D.J. Walking in the woods with quantum chemistry–applications of quantum chemical calculations in natural products research. Nat. Prod. Rep. 30, 1079–1086 (2013).
Nicolaou, K.C. & Snyder, S.A. Chasing molecules that were never there: misassigned natural products and the role of chemical synthesis in modern structure elucidation. Angew. Chem. Int. Ed. 44, 1012–1044 (2005).
Pretsch, E., Bühlmann, P. & Affolter, C. Structure Determination of Organic Compounds: Tables of Spectral Data 3rd edn. (Springer, 2000).
Abraham, R. & Mobli, M. Modelling 1H NMR Spectra of Organic Compounds: Theory, Applications and NMR Prediction Software 1st edn. (John Wiley & Sons, 2008).
Wiitala, K.W., Al-Rashid, Z.F., Dvornikovs, V., Hoye, T.R. & Cramer, C.T. Evaluation of various DFT protocols for computing 1H and 13C chemical shifts to distinguish stereoisomers: diastereomeric 2-, 3-, and 4-methylcyclohexanols as a test set. J. Phys. Org. Chem. 20, 345–354 (2007).
Wiitala, K.W., Hoye, T.R. & Cramer, C.J. Hybrid density functional methods empirically optimized for the computation of 13C and 1H chemical shifts in chloroform solution. J. Chem. Theory Comput. 2, 1085–1092 (2006).
MacroModel, version 10.0. http://www.schrodinger.com/citations/41/11/1/ (Schrödinger, New York, 2013).
Chang, G., Guida, W.C. & Still, W.C. An internal coordinate Monte Carlo method for searching conformational space. J. Am. Chem. Soc. 111, 4379–4386 (1989).
Smith, S.G. & Goodman, J.M. Assigning stereochemistry to single diastereomers by GIAO NMR calculation: The DP4 probability. J. Am. Chem. Soc. 132, 12946–12959 (2010).
Zhao, Y. & Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06 functionals and twelve other functionals. Theor. Chem. Acc. 120, 215–241 (2008).
Zhao, Y. & Truhlar, D.G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 41, 157–167 (2008).
London, F. Théorie quantique des courants interatomiques dans les cominaisons aromatiques. J. Phys. Radium 8, 397–409 (1937).
Cramer, C.J. Chapter 9 (charge distribution and spectroscopic properties) in Essentials of Computational Chemistry 2nd edn. (John Wiley & Sons, 2004).
Rablen, P.R., Pearlman, S.A. & Finkbiner, J. A comparison of density functional methods for the estimation of proton chemical shifts with chemical accuracy. J. Phys. Chem. A 103, 7357–7363 (1999).
Tomasi, J., Mennucci, B. & Cances, E. The IEF version of the PCM solvation method: an overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. (Theochem) 464, 211–226 (1999).
Barone, V., Cossi, M. & Tomasi, J. A new definition of cavities for the computation of solvation free energies by the polarizable continuum model. J. Chem. Phys. 107, 3210–3221 (1997).
Bondi, A. van der Waals volumes and radii. J. Phys. Chem. 68, 441–451 (1964).
Smith, S.G. & Goodman, J.M. Assigning the stereochemistry of pairs of diastereomers using GIAO NMR shift calculation. J. Org. Chem. 74, 4597–4607 (2009).
Sarotti, A.M. Successful combination of computationally inexpensive GIAO 13C NMR calculations and artificial neural network pattern recognition: a new strategy for simple and rapid detection of structural misassignments. Org. Biomol. Chem. 11, 4847–4859 (2013).
Wiitala, K.W., Cramer, C.J. & Hoye, T.R. Comparison of various density functional methods for distinguishing stereoisomers based on computed 1H or 13C NMR chemical shifts using diastereomeric penam ß-lactams as a test set. Magn. Reson. Chem. 45, 819–829 (2007).
Brown, S.G., Jansma, M.J. & Hoye, T.R. Case study of empirical and computational chemical shift analyses: reassignment of the relative configuration of phomopsichalasin to that of diaporthichalasin. J. Nat. Prod. 75, 1326–1331 (2012).
Saielli, G., Nicolaou, K.C., Ortiz, A., Zhang, H. & Bagno, A. Addressing the stereochemistry of complex organic molecules by density functional theory-NMR: vannusal B in retrospective. J. Am. Chem. Soc. 133, 6072–6077 (2011).
Rychnovsky, S.D. Predicting NMR spectra by computational methods: structure revision of hexacyclinol. Org. Lett. 8, 2895–2898 (2006).
Lodewyk, M.W. & Tantillo, D.J. Prediction of the structure of nobilisitine a using computed NMR chemical shifts. J. Nat. Prod. 74, 1339–1343 (2011).
Maestro, version 9.4 http://www.schrodinger.com/citations/41/12/1/. (Schrödinger, New York, 2013).
Macrae, C.F. et al. Mercury CSD 2.0–new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 41, 466–470 (2008).
Frisch, M.J. et al. Gaussian 09, Revision A. http://www.gaussian.com/g_tech/g_ur/m_citation.htm (Gaussian, 2009).
Dennington, R., Keith, T. & Millam, J. GaussView, Version 5. http://www.gaussian.com/g_tech/gv5ref/gv5citation.htm. (Semichem, 2009).
Marell, D.J., Emond, S.J., Kulshrestha, A. & Hoye, T.R. Analysis of seven-membered lactones by computational NMR methods: proton NMR chemical shift data are more discriminating than carbon. J. Org. Chem. 79, 753–758 (2014).
Solomons, T.W.G. & Fryhle, C.B. Organic Chemistry 9th edn. (John Wiley & Sons, 2007).
Schlegel, B., Härtl, A., Dahse, H.-M., Gollmick, F.A. & Gräfe, U. Hexacyclinol, a new antiproliferative metabolite of Panus rudis HKI 0254. J. Antibiotics 55, 814–817 (2002).
La Clair, J.J. Total syntheses of hexacyclinol, 5-epi-hexacyclinol, and desoxohexacyclinol unveil an antimalarial prodrug motif. Angew. Chem. Int. Ed. 45, 2769–2773 (2006).
Porco, J.A. Jr., Su, S., Lei, X., Bardhan, S. & Rychnovsky, S.D. Total synthesis and structure assignment of (+)-hexacyclinol. Angew. Chem. Int. Ed. 45, 5790–5792 (2006).
Saielli, G. & Bagno, A. Can two molecules have the same NMR spectrum? Hexacyclinol revisited. Org. Lett. 11, 1409–1412 (2009).
Guella, G., Dini, F. & Pietra, F. Metabolites with a novel C30 backbone from marine ciliates. Angew. Chem. Int. Ed. 38, 1134–1136 (1999).
Guella, G. et al. Hemivannusal and prevannusadials–new sesquiterpenoids from the marine ciliate protist Euplotes vannus: the putative biogenetic precursors to dimeric terpenoid vannusals. Eur. J. Org. Chem. 2007, 5226–5234 (2007).
Nicolaou, K.C., Ortiz, A., Zhang, H. & Guella, G. Total synthesis and structural revision of vannusals A and B: synthesis of the true structures of vannusals A and B. J. Am. Chem. Soc. 132, 7153–7176 (2010).
Nicolaou, K.C., Ortiz, A., Zhang, H. & Guella, G. Total synthesis and structural revision of vannusals A and B: synthesis of the true structures of vannusals A and B. J. Am. Chem. Soc. 132, 7153–7176 (2010).
Nicolaou, K.C., Ortiz, A., Zhang, H. & Guella, G. Total synthesis and structural revision of vannusals A and B: synthesis of the true structures of vannusals A and B. J. Am. Chem. Soc. 132, 7153–7176 (2010).
Nicolaou, K.C., Ortiz, A., Zhang, H. & Guella, G. Total synthesis and structural revision of vannusals A and B: synthesis of the true structures of vannusals A and B. J. Am. Chem. Soc. 132, 7153–7176 (2010).
Acknowledgements
We thank T.A. Bedell and N.P. Labello for their contributions to the scripting efforts. We thank C.J. Cramer, A.N. Garr, A.M. Harned, S.S. Humble, K.A. Kalstabakken, J.C. Lo, D.J. Marell, K.W. Wiitala and B.P. Woods for helpful input, feedback and comments at various stages of the protocol development and manuscript preparation. This work was carried out in part using software and hardware resources made available through the University of Minnesota Supercomputing Institute (MSI). The research was supported by a grant awarded by the US National Science Foundation (NSF, CHE-0911696). P.H.W. was supported by an NSF Graduate Research Fellowship.
Author information
Authors and Affiliations
Contributions
P.H.W. and M.J.J. contributed equally to establishing and optimizing the protocol, and all authors participated in writing and editing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Data 1
Full list of chemical shift values for the trans- and cis-diastereomers of 3-methylcyclohexanol (PDF 4857 kb)
Supplementary Data 2
Four Python Scripts A-D (names in parentheses below) that are helpful in automating aspects of the procedure that are otherwise laborious and tedious. These are used to: A) Write a Gaussian input file for each structure (local minimum) obtained from Operation I. (write-g09-inputs.py and variants thereof that use different functional and basis set combinations). B) Check for redundant conformers or any that has an imaginary frequency. (duplicate_conf_and_imag_freq-check.py). C) Extract the NMR shielding tensor for a DFT-computed reference compound of interest. (get-ref-shifts.py). D) Perform the Boltzmann analysis to generate the weighted-averaged, referenced chemical shifts for each proton and carbon in the candidate structure. (nmr-data_compilation.py) (ZIP 275 kb)
Supplementary Data 3
Files for cis-3-methylcyclohexanol resulting from the Maestro, MacroModel, and Gaussian jobs (including “.chk” and “.out”) (ZIP 37624 kb)
Supplementary Data 4
Files for trans -3-methylcyclohexanol resulting from the Maestro, MacroModel, and Gaussian jobs (including “.chk” and “.out”) (ZIP 37749 kb)
Rights and permissions
About this article
Cite this article
Willoughby, P., Jansma, M. & Hoye, T. A guide to small-molecule structure assignment through computation of (1H and 13C) NMR chemical shifts. Nat Protoc 9, 643–660 (2014). https://doi.org/10.1038/nprot.2014.042
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2014.042
This article is cited by
-
Ilm-NMR-P31: an open-access 31P nuclear magnetic resonance database and data-driven prediction of 31P NMR shifts
Journal of Cheminformatics (2023)
-
An initial investigation of accuracy required for the identification of small molecules in complex samples using quantum chemical calculated NMR chemical shifts
Journal of Cheminformatics (2022)
-
Lignans and sesquiterpenoids from the stems of Schisandra bicolor var. tuberculata
Natural Products and Bioprospecting (2022)
-
From Relative to Absolute Stereochemistry of Secondary Metabolites: Applications in Plant Chemistry
Revista Brasileira de Farmacognosia (2022)
-
Addendum: A guide to small-molecule structure assignment through computation of (¹H and ¹³C) NMR chemical shifts
Nature Protocols (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
