
Abstract
Maize may be transformed very efficiently using Agrobacterium tumefaciens-mediated methods. The most critical factor in the transformation protocol is the co-cultivation of healthy immature embryos of the correct developmental stage with A. tumefaciens; the embryos should be collected only from vigorous plants grown in well-conditioned glasshouses. With the protocol described here, approximately 50% of immature embryos from the inbred line A188 and 15% from inbred lines A634, H99 and W117 will produce transformants. About half of the transformed plants are expected to carry one or two copies of the transgenes, which are inherited by the progeny in a mendelian fashion. More than 90% of transformants are expected to be normal in morphology. The protocol takes about 3 months from the start of co-cultivation to the planting of transformants into pots.
Similar content being viewed by others


Introduction
Transgenic maize plants were first obtained from protoplasts by an electroporation method1, but fertile plants have never been produced by this method. Other direct gene transfer methods, which did not require the prior culture of protoplasts, were then tried2,3,4,5, and microprojectile bombardment6 of cells in suspension cultures or immature embryos became quite popular in basic and applied studies. Efficiency of transformation by microprojectile bombardment has been higher than other direct methods, and quite a few fertile plants have been generated to date7. Microprojectile bombardment is also useful for the analysis of the transient expression of foreign genes in intact, fully developed tissues. However, high copy numbers and extensive rearrangement of the foreign DNA have frequently been found in plants transformed with direct gene transfer methods8,9.
For the last two decades, dicotyledonous plants have been transformed using the soil phytopathogen A. tumefaciens. A. tumefaciens is first transformed with the DNA construct of interest (T-DNA); this modified bacterial strain is then used to introduce the T-DNA into plants. A major advantage of Agrobacterium-mediated transformation is that a small number of copies (often one or two) of relatively large segments (can be larger than 10 kb) of T-DNA with defined ends are integrated into the plant genome with minimal rearrangement, resulting in transgenic plants of high quality. Initially, it was not clear if this technology could be extended to monocotyledonous plants, as they are not natural hosts of A. tumefaciens. However, a highly efficient method of transformation of rice by A. tumefaciens was reported10, followed by successful reports of the transformation of important cereals such as maize11, wheat12, barley13 and sorghum14 by A. tumefaciens. Key factors in these achievements include the optimization of types of plant material for infection with A. tumefaciens, choice of vectors, choice of strains of A. tumefaciens and optimization of tissue culture techniques. Transformation mediated by A. tumefaciens is now highly recommended for maize varieties with good tissue culture responses.
For the successful production of transgenic plants in any species, foreign genes must be delivered to undifferentiated, dedifferentiated or dedifferentiating cells that are actively dividing or about to divide and that are capable of regenerating plants. In maize, the material of choice is immature embryos, and all protocols mediated by particle bombardment or A. tumefaciens for efficient production of transgenic maize have solely employed immature embryos. Thus, the primary determinants of a successful transformation are the response of immature embryos in tissue culture, the types of cells that grow from immature embryos and subsequent characteristics in growth and regeneration. Unfortunately, many genotypes of maize, especially so-called elite varieties, are poor in these aspects, and thus only a limited number of genotypes have been efficiently transformed so far.
In the original protocol of Ishida et al.11, transgenic plants were obtained from between 5% and 30% of A. tumefaciens-infected immature inbred A188 embryos. The protocol was successfully employed in a number of studies in molecular biology and biotechnology15,16,17,18,19. Since then, the methods have been greatly improved, and a highly optimized protocol routinely used in our laboratory is presented in this article20,21. An overview and typical timeline of this optimized protocol is given in Figure 1. The modifications made to the protocol include pretreatment by heat and centrifuging addition of silver and copper ions to the co-cultivation medium and extension of the co-cultivation period from 3 to 7 days. The effects of heat, centrifuging and ions are evident21, but the mechanisms are not understood. In protocols described by other authors, a resting culture, which is a non-selective incubation of embryos on a medium that contains an antibiotic to kill bacteria, is performed after co-cultivation22. In our experience, the growth of transformed cells was better in the selection culture if the resting culture was performed; however, the same effect was produced by an extended period of co-cultivation. Thus, if the co-cultivation is extended, no resting culture is necessary. In general, the bar gene23, which confers resistance to the herbicide phosphinothricin, is a more effective selection agent than the hpt gene24, which confers resistance to the antibiotic hygromycin. Further, use of vectors that carry additional virB and virG genes from pTiBo542 (see refs. 25,26) gives much higher transformation frequencies. The function of virB is related to formation of a transmembrane channel between the bacterium and the plant cell and the function of virG is related to the activation of the other vir genes27.

It is assumed that 100 immature embryos are collected in Step 3 and that materials derived from the 100 embryos are handled in the subsequent steps by a single, skilled technician. Similarly, seeds from ten transgenic plants are examined in Steps 32–35.
Transgenic plants may be routinely obtained from more than 50% of the immature embryos from the A188 genotype, and the range of transformable genotypes has been extended to inbreds A634, H99 and W117 and hybrids between pairs of these varieties in our laboratory20,21. Hybrid genotype Hi-II (ref. 28) and inbreds Mo17, B104, B114 and Ky21 have been transformed by similar protocols in other laboratories22,29,30,31.
It should be noted that two types of embryogenic callus, type I and type II, may proliferate from the scutellum of immature embryos, depending on the genotypes of maize32. The type I callus is a relatively hard and compact embryogenic callus, whereas the type II callus is relatively soft and friable. A type I callus is usually obtained from inbreds A188, A634, H99 and W117 on the media described in this article. For genotypes such as Hi-II, from which the type II callus is frequently produced, modification of the media is recommended22,29,33. Protocols recommended for Hi-II and some other genotypes have been reported by other groups22,29,30,31. These protocols are similar to those presented in this article and differ essentially only in the composition of the media. While the macro components in all of the media in this article are based on the LS medium, those in the protocols of other groups are based mostly on the N6 medium. In addition, Frame et al.29 used media that contained cysteine for co-cultivation. Thus, especially when other genotypes are to be transformed, media based on N6 and media with cysteine should also be considered.
Elimination of selection marker genes from transgenic plants for commercialization is an important task because the presence of selection marker genes, which are unnecessary once transgenic plants are established, is of high public concern. A simple approach to remove selection markers is to perform A. tumefaciens-mediated co-transformation of plants with two T-DNA segments, one with a selection marker and the other with genes of interest, followed by segregation of marker-free progeny34. Because the frequency of co-transformants among initial transformants is never 100% and about a half of the co-transformants do not segregate marker-free progeny, this approach requires a high frequency of transformation, desirably more than 20% (independent transgenics/immature embryo). With the highly optimized protocol presented here, production of marker-free transgenics has become a realistic option in maize; selection-marker-free transformants may be obtained from about 50% of co-transformed plants when co-transformation vectors are employed.
It is clear that the protocol presented in this article still needs to be improved. Despite the high frequency of transformation in A188 and related genotypes, the majority of genotypes—including the so-called elite inbreds—are not expected to be transformed efficiently. In addition, there are many critical parameters in the protocol and the 'windows' of optimal conditions are very narrow; the assistance of an experienced tissue culture specialist may be necessary to establish and maintain efficient maize transformation programs.
Materials
Reagents
-
Maize seeds of inbreds A188, A634, H99 and W117 were supplied from the National Institute of Agrobiological Sciences of Japan (http://www.nias.affrc.go.jp/index_e.html)
-
A. tumefaciens strain carrying appropriate T-DNA-containing vectors (see Table 1)
Table 1 Typical frequency of transformation. -
10× LS major salts (see REAGENT SETUP)
-
100× FeEDTA (see REAGENT SETUP)
-
100× LS minor salts (see REAGENT SETUP)
-
100× modified LS vitamins (see REAGENT SETUP)
-
100 mg liter−1 2,4-dichlorophenoxy-acetic acid (2,4-D) (see REAGENT SETUP)
-
100 mg liter−1 Zeatin (see REAGENT SETUP)
-
100 mg liter−1 indole-3-butyric acid (IBA) (see REAGENT SETUP)
-
100 mg liter−1 6-benzylamino-purine (6BA) (see REAGENT SETUP)
-
100 mM acetosyringone (see REAGENT SETUP)
-
100 mM 5-bromo-4-chloro-3-indolyl β-D-glucuronide (X-gluc) (see REAGENT SETUP)
-
50 mM Na2HPO4 (see REAGENT SETUP)
-
50 mM NaH2PO4 (see REAGENT SETUP)
-
YP plates (see REAGENT SETUP)
-
LS-inf medium (see REAGENT SETUP)
-
LS-inf-AS medium (see REAGENT SETUP)
-
LS-AS medium (see REAGENT SETUP)
-
LSD1.5A medium (see REAGENT SETUP)
-
LSD1.5B medium (see REAGENT SETUP)
-
LSZ medium (see REAGENT SETUP)
-
LSF medium (see REAGENT SETUP)
-
ELA medium (see REAGENT SETUP)
-
Soil for pots (see REAGENT SETUP)
-
Buffer P, for β-glucuronidase (GUS) staining (see REAGENT SETUP and Box 1)
-
Buffer X, for GUS staining (see REAGENT SETUP and Box 1)
Equipment
-
Scalpel blade (no. 19, Futaba Co., Ltd)
-
Surgical tape made of unwoven fabric (no. 25, Nichiban Co., Ltd, http://www.nichiban.co.jp/)
-
Parafilm (Pechiney Plastic Packaging)
-
270 mm pot (270 mm in diameter and 270 mm in height) for maize cultivation
-
230 mm pot (230 mm in diameter and 190 mm in height) for maize cultivation
Reagent setup
-
10× LS major salts Dissolve 19.0 g KNO3, 16.5 g NH4NO3, 4.4 g CaCl2·2H2O, 3.7 g MgSO4·7H2O and 1.7 g KH2PO4 in 900 ml distilled water and make up the volume to 1,000 ml (see ref. 35). Store at 4 °C. The final concentrations of components in this solution are 188 mM KNO3, 206 mM NH4NO3, 30 mM CaCl2·2H2O, 15 mM MgSO4·7H2O and 12.5 mM KH2PO4.
-
100× FeEDTA Dissolve 2.78 g FeSO4·7H2O in 900 ml of hot distilled water and add 3.73 g ethylenediamine-N,N,N′,N′-tetraaceticacid, disodium salt (Na2EDTA). Cool and make up the volume to 1,000 ml. Store at 4 °C. The final concentrations of components in this solution are 10 mM FeSO4·7H2O and 10 mM Na2EDTA.
-
100× LS minor salts Dissolve 2.23 g MnSO4·5H2O, 1.06 g ZnSO4·7H2O, 620 mg H3BO3, 83 mg KI, 25.0 mg Na2MoO4·2H2O, 2.5 mg CuSO4·5H2O and 2.5 mg CoCl2·6H2O in 900 ml of distilled water and make up the volume to 1,000 ml. Store at 4 °C. The final concentrations of components in this solution are 9.3 mM MnSO4·5H2O, 3.7 mM ZnSO4·7H2O, 10 mM H3BO3, 0.5 mM KI, 0.1 mM Na2MoO4·2H2O, 0.01 mM CuSO4·5H2O and 0.01 mM CoCl2·6H2O.
-
100× modified LS vitamins Dissolve 10 g myoinositol, 100 mg thiamine hydrochloride, 50 mg pyridoxine hydrochloride and 50 mg nicotinic acid in 900 ml of distilled water and make up the volume to 1,000 ml. Store at 4 °C. The final concentrations of components in this solution are 55.5 mM myoinositol, 0.30 mM thiamine hydrochloride, 0.24 mM pyridoxine hydrochloride and 0.41 mM nicotinic acid.
-
100 mg liter−1 2,4-D Add 1 N NaOH dropwise to 100 mg 2,4-D until completely dissolved. Make up to 1,000 ml with distilled water. Store at 4 °C. The final concentration of 2,4-D in this solution is 0.45 mM.
-
100 mg liter−1 zeatin Add 1 N NaOH dropwise to 100 mg zeatin until completely dissolved. Make up to 1,000 ml with distilled water. Store at 4 °C. The final concentration of zeatin in this solution is 0.46 mM.
-
100 mg liter−1 IBA Add 1 N NaOH dropwise to 100 mg IBA until completely dissolved. Make up to 1,000 ml with distilled water. Store at 4 °C. The final concentration of IBA in this solution is 0.49 mM.
-
100 mg liter−1 6BA Add 1 N NaOH dropwise to 100 mg 6BA until completely dissolved. Make up to 1,000 ml with distilled water. Store at 4 °C. The final concentration of 6BA in this solution is 0.44 mM.
-
100 mM acetosyringone Dissolve 392.4 mg acetosyringone in 10 ml of dimethyl sulfoxide and dilute with 10 ml distilled water. Filter-sterilize and store in the dark at 4 °C.
-
100 mM X-gluc Dissolve 52 mg X-gluc (Sigma B6650) in 1 ml of ethylene glycol monomethyl ether (Sigma E5378). Store in the dark at −20 °C.
Caution
Ethylene glycol monomethyl ether is toxic. Wear suitable protective clothing, gloves and eye/face protection.
-
50 mM Na2HPO4 Dissolve 17.91 g Na2HPO4·12H2O in 900 ml of distilled water and make up to 1,000 ml.
-
50 mM NaH2PO4 Dissolve 7.8 g NaH2PO4·2H2O in 900 ml of distilled water and make up to 1,000 ml.
-
YP plate (for A. tumefaciens) Dissolve 5 g yeast extract, 10 g peptone and 5 g sodium chloride in 900 ml of distilled water and adjust pH to 6.8 with NaOH. Make up to 1,000 ml and add 15 g agar (Difco). Autoclave at 121 °C for 15 min. Cool the medium to 50 °C, add appropriate antibiotics, which depend on the type of plasmid(s) in the strain, and pour 20 ml aliquots into Petri dishes (90 × 15 mm).
-
LS-inf medium (for preparation of immature embryos) Add 100 ml of 10× LS major salts, 10 ml of 100× FeEDTA, 10 ml of 100× LS minor salts, 10 ml of 100× modified LS vitamins and 15 ml of 100 mg liter−1 2,4-D (final concentration is 1.5 mg liter−1) to 700 ml of distilled water. Dissolve 68.46 g sucrose, 36.04 g glucose and 1.0 g Casamino acids in the mixture and make up the volume to 1,000 ml. Adjust pH to 5.2 and sterilize with a 0.22 μm cellulose-acetate filter.
-
LS-inf-AS medium (for infection) Add 1 μl of 100 mM acetosyringone to 1 ml of LS-inf medium.
-
LS-AS medium (for co-cultivation) Add 100 ml of 10× LS major salts, 10 ml of 100× FeEDTA, 10 ml of 100× LS minor salts, 10 ml of 100× modified LS vitamins, 15 ml of 100 mg liter−1 2,4-D (final concentration is 1.5 mg liter−1) and 0.05 ml of 100 mM CuSO4 to 700 ml of distilled water. Dissolve 20 g sucrose, 10 g glucose, 0.7 g proline and 0.5 g MES in the mixture and make up the volume to 1,000 ml. Adjust pH to 5.8 and add 8 g agarose. Autoclave and cool to 50 °C, and add 1 ml of 100 mM acetosyringone and 0.05 ml of 100 mM AgNO3 and pour 30 ml aliquots into Petri dishes (90 × 20 mm). Store in the dark at room temperature (20–25 °C).
-
LSD1.5A medium (for first selection of transformed cells) Add 100 ml of 10× LS major salts, 10 ml of 100× FeEDTA, 10 ml of 100× LS minor salts, 10 ml of 100× modified LS vitamins and 15 ml of 100 mg liter−1 2,4-D (final concentration is 1.5 mg liter−1) to 700 ml of distilled water. Dissolve 20 g sucrose, 0.7 g proline and 0.5 g MES in the mixture and make up the volume to 1,000 ml. Adjust pH to 5.8 and add 8 g agar (Sigma, A6013-500G). Autoclave at 121 °C for 15 min. Cool to 50 °C and add 1 ml of 250 g liter−1 carbenicillin (final concentration is 250 mg liter−1), 0.4 ml of 250 g liter−1 cefotaxime (final concentration is 100 mg liter−1), 0.1 ml of 100 mM AgNO3 and either 0.25 ml of 20 g liter−1 phosphinothricin (Riedel-de Haën AG) (final concentration is 5 mg liter−1) for bar selection or 0.3 ml of 50 g liter−1 hygromycin (Calbiochem) (final concentration is 15 mg liter−1) for hpt selection. Pour 30 ml aliquots into Petri dishes (90 × 20 mm) and store in the dark at room temperature.
-
LSD1.5B medium (for second and third selection of transformed cells) This is identical to LSD1.5A medium except for the amount of selective agent. Add 0.5 ml of 20 g liter−1 phosphinothricin (final concentration is 10 mg liter−1) instead of 0.25 ml or 0.6 ml of 50 g liter−1 hygromycin (final concentration is 30 mg liter−1) instead of 0.3 ml in this case.
-
LSZ medium (for regeneration of transformed plants) Add 100 ml of 10× LS major salts, 10 ml of 100× FeEDTA, 10 ml of 100× LS minor salts, 10 ml of 100× modified LS vitamins, 50 ml of 100 mg liter−1 zeatin (final concentration is 5 mg liter−1) and 0.1 ml of 100 mM CuSO4 to 700 ml of distilled water. Dissolve 20 g sucrose and 0.5 g MES in the mixture and make up the volume to 1,000 ml. Adjust pH to 5.8 and add 8 g agar (Sigma, A6013-500G). Autoclave at 121 °C for 15 min. Cool to 50 °C, and add 1 ml of 250 g liter−1 carbenicillin (final concentration is 250 mg liter−1), 0.4 ml of 250 g liter−1 cefotaxime (final concentration is 100 mg liter−1) and either 0.25 ml of 20 g liter−1 phosphinothricin (final concentration is 5 mg liter−1) for bar selection or 0.6 ml of 50 g liter−1 hygromycin (final concentration is 30 mg liter−1) for hpt selection. Pour 30 ml aliquots into Petri dishes (90 × 20 mm) and store in the dark at room temperature.
-
LSF medium (for rooting of transformed plants) Add 100 ml of 10× LS major salts, 10 ml of 100× FeEDTA, 10 ml of 100× LS minor salts, 10 ml of 100× modified LS vitamins and 2 ml of 100 mg liter−1 IBA (final concentration in 0.2 mg liter−1 to 700 ml of distilled water. Dissolve 15 g sucrose and 0.5 g MES in the mixture and make up the volume to 1,000 ml and adjust pH to 5.8. Add 3 g gellan gum and warm to 90 °C to dissolve. Pour 10 ml aliquots to glass test tubes (25 mm in diameter × 100 mm in height). Cover the tubes with polypropylene caps and autoclave at 121 °C for 15 min. Store at room temperature.
-
ELA medium (for detached leaf analysis) Add 100 ml of 10× LS major salts, 10 ml of 100× FeEDTA, 10 ml of 100× LS minor salts and 5 ml of 100 mg liter−1 6BA (final concentration is 0.5 mg liter−1) to 700 ml of distilled water. Dissolve 0.5 g MES in the mixture and make up the volume to 1,000 ml. Adjust pH to 5.8 and add 8 g agar (Sigma, A6013-500G). Autoclave at 121 °C for 15 min. Cool to 50 °C and add either 0.1 ml of Basta (Bayer Crop Science) for analysis of expression of bar gene or 2 ml of 50 mg ml−1 (final concentration is 100 mg liter−1) hygromycin for analysis of expression of hpt gene. Pour 30 ml aliquots into Petri dishes (90 × 20 mm) and store in the dark at room temperature.
-
Soil for pots A soil mixture for horticultural use that is commercially available and well drained is usually good. Adjust the major nutrients to 0.4 g N per liter, 0.4 g P per liter and 0.4 g K per liter by adding a commercial fertilizer.
-
Buffer P Add 50 mM Na2HPO4 (about 500 ml) to 1,000 ml of 50 mM NaH2PO4 until the pH reaches 6.8. Sterilize using a 0.22 μm cellulose-acetate filter and store at room temperature. Mix 9.9 ml of this buffer and 0.1 ml of Triton X-100 before use.
-
Buffer X Mix 8 ml of buffer P, 0.1 ml of 100 mM X-gluc and 2 ml methanol just before use.
Procedure
Preparation of immature embryos
Timing 35 min for handling, 90 days for growing plants
-
1
Grow maize plants in individual 270 mm pots in a greenhouse. Maintain daytime temperature between 30 and 35 °C and night time temperature between 20 and 25 °C. Ideally, the light intensity should be stronger than 60,000 lx and the photoperiod should be more than 12 h.
Critical Step
The quality of the immature embryos is one of the most import factors for achieving highly efficient maize transformation. Good embryos are obtained only from vigorous plants grown in a well-conditioned greenhouse. Air-conditioning and supplemental lights are needed to ensure a supply of good embryos year-round. Usually, more than 150 kernels can be collected from a single ear of A188. Production of a much lower number of kernels on a single cob implies that the growth conditions are not optimal. If transformation efficiency is poor, the greenhouse conditions should be optimized before investigating other aspects of the protocol, such as types of vectors and strains, and media compositions.
-
2
Between 8 and 15 days after pollination (DAP), harvest an ear that contains immature embryos at the right developmental stage (see Fig. 2).
Figure 2: Size of immature embryo. 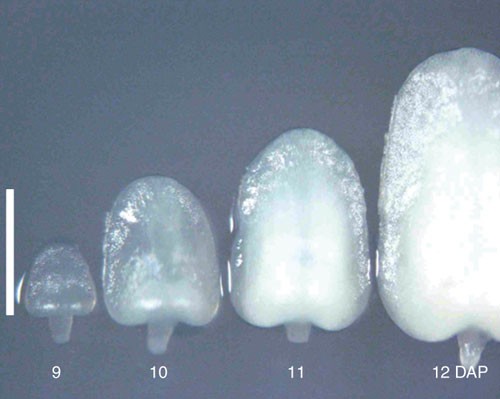
Immature embryos of 9–12 DAP were collected from maize inbred line A188 on June 2004 in Iwata, Shizuoka, Japan. Immature embryo of 10 DAP is 1.2 mm in length and suitable for transformation in this case. Scale bar, 1 mm.
Critical Step
The use of immature embryos at the right developmental stage is a critical factor, and the size of the embryos is a very good indicator of the stage. Immature embryos that are between 1.0 and 1.2 mm in length along the axis are optimal for transformation. Time (DAP) required for embryos to reach the best stage differs depending on the genotypes and the season. So carefully examine the sizes of embryos and determine the time of collection. A typical pattern of the growth of embryos is shown in Figure 2. For example, the time of collection for A188 was 8 DAP in August, 10 DAP in June and 15 DAP in January at our facility. As long as the sizes of embryos are in the above range, the frequency of transformation is reproducible year-round.
-
3
Husk the ear and detach kernels from the cob by cutting the base of the kernel with a scalpel (Fig. 3a,b). Insert a scalpel into the detached kernels (Fig. 3c) and remove the immature embryos (Fig. 3d). If plants are grown in a greenhouse free from disease and pest, cobs in husks are clean and immature embryos can be removed without surface sterilization of the cobs.
Figure 3: Isolation of immature embryo. 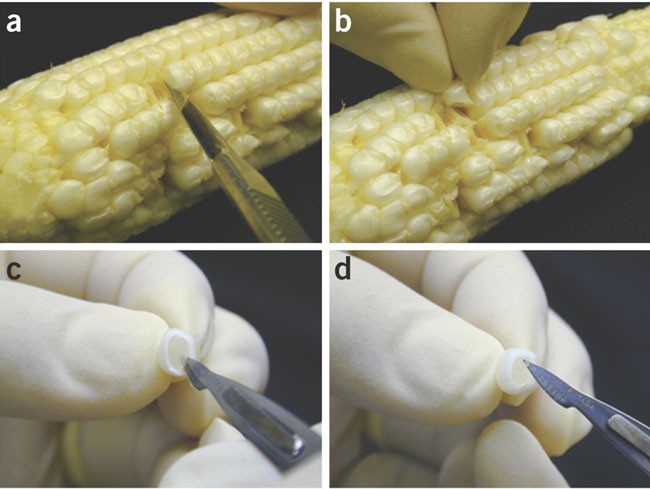
(a) Cut a kernel; (b) take out the kernel; (c) insert a scalpel into the kernel; and (d) lift out an immature embryo.
-
4
Immerse the embryos in 2.0 ml of LS-inf medium in a 2.0 ml microcentrifuge tube at room temperature until the remaining embryos have been collected. Finish the collection of immature embryos within 1 h. More than 200 immature embryos can be collected in 1 h by a single, skilled technician.
-
5
Vortex the microcentrifuge tube at 2,700 r.p.m. at room temperature for 5 s and remove the medium.
-
6
Add 2.0 ml of LS-inf medium and vortex as in Step 5.


Pre-treatment with heat and centrifuging
Timing 30 min
-
7
Incubate the embryos in the microcentrifuge tube in a water bath at 46 °C for 3 min.
Critical Step
The optimal conditions for this heat treatment will differ depending on genotype. The conditions described here work well for genotypes A188, H99 and A634. Since immature embryos of W117 were more sensitive to heat than these genotypes, lower temperature and/or shorter treatment may be suitable for successful transformation in W117. Optimization will be necessary to find the best conditions for other strains and can be assessed by co-culturing with a strain of A. tumefaciens carrying an intron-GUS gene (Steps 11–13) and GUS staining at Step 18 (see Box 1). In addition, it is also important to assess callus induction from heat-treated immature embryos.
-
8
Cool the microcentrifuge tube on ice for 1 min.
-
9
Remove the medium and add 2.0 ml of LS-inf medium.
-
10
Centrifuge the microcentrifuge tube with a fixed-angle rotor with a maximum radius of 83 mm at 20,000g at 4 °C for 10 min.
Critical Step
The optimal centrifugation conditions will differ depending on genotype. The conditions described here work well for genotypes A188, A634, W117 and H99. Optimization will be necessary to find the best conditions for other strains and can be assessed by co-culturing with a strain of A. tumefaciens carrying an intron-GUS gene (Steps 11–13) and GUS staining at Step 18 (see Box 1).
Preparation of inoculum
Timing 5 min for handling, 2 days for cultivation
-
11
Culture A. tumefaciens strain on a YP plate that contains appropriate antibiotics in the dark at 28 °C for 2 days.
-
12
Collect the bacteria with a loop and suspend in 1.0 ml of LS-inf-AS medium at a density of 1 × 109 colony-forming units per ml (OD = 1.0 at 660 nm). Inoculum should be prepared fresh. Growth of Agrobacterium in liquid culture before transformation is not necessary.
Inoculation and co-cultivation
Timing 30 min for handling, 7 days for cultivation
-
13
Remove the medium from the microcentrifuge tube in Step 10 and add 1.0 ml of bacterial suspension from Step 12.
-
14
Vortex the microcentrifuge tube at 2,700 r.p.m. for 30 s.
-
15
Incubate for 5 min at room temperature.
-
16
Transfer the suspension of the embryos and bacteria to an empty Petri dish (60 × 15 mm).
-
17
Remove and discard 0.7 ml of the liquid from the suspension.
-
18
Transfer the embryos onto fresh LS-AS solid medium with the scutellum face up and seal the Petri dishes with Parafilm. Up to 200 embryos may be placed on a single plate. If the procedure is being optimized using strains that contain the intron-gus gene, transient expression of GUS can be analyzed at this point, as detailed in Box 1.
-
19
Incubate in the dark at 25 °C for 7 days; this is the co-cultivation step.
Selection of transformed calli
Timing 140 min for handling, 52 days for cultivation
-
20
Transfer the embryos to LSD1.5A medium and seal the Petri dishes with surgical tape. Up to 25 embryos may be placed on a single plate.
Critical Step
Do not rinse the embryos. Rinsing with an antibiotic solution tends to result in poor growth of cells. The surgical tape, which allows aeration, is much better for the growth of plant cells than air-tight tapes.
-
21
Incubate in the dark at 25 °C for 10 days; this is the first selection.
-
22
Transfer the embryos to LSD1.5B medium, and seal the Petri dishes with surgical tape. Up to 25 embryos may be placed on a single plate.
-
23
Incubate in the dark at 25 °C for 21 days; this is the second selection.
-
24
Cut type I calli proliferated from the scutellum into pieces of between 3 and 5 mm in diameter under a stereoscopic microscope (Fig. 4), transfer to LSD1.5B medium and seal the Petri dishes with the surgical tape. Up to 25 pieces may be placed on a single plate.
Figure 4: Guide to cutting type I callus for third selection and regeneration step. 
Proliferated type I callus is cut with a scalpel as indicated by yellow dotted lines. The size of the cut fragment is 3–5 mm at Step 24 or 2–3 mm at Step 26. Scale bar, 5 mm.
-
25
Incubate in the dark at 25 °C for 21 days; this is the third selection. Proliferated type I calli are transgenic.

Regeneration of transformed plants
Timing 240 min for handling, 28 days for cultivation, 100 days for growing plants
-
26
Cut the further proliferated type I calli from Step 25 into pieces of between 2 and 3 mm in diameter under a stereoscopic microscope, transfer to LSZ medium and seal the Petri dishes with Parafilm. Up to 25 pieces may be placed on a single plate.
-
27
Incubate under continuous illumination (5,000 lx) at 25 °C for 14 days.
-
28
Transfer a regenerated shoot to a tube of LSF medium, and cover with a polypropylene cap.
-
29
Incubate under continuous illumination (5,000 lx) at 25 °C for 14 days.
-
30
Transfer each plant to a 230 mm pot containing appropriately supplemented soil (see REAGENT SETUP).
-
31
Grow transgenic plants in a greenhouse as detailed in Step 1 for 3–4 months and harvest progeny seeds. Plants in the following generations may be grown as detailed in Step 1.
Examination of progeny for expression of a selection marker gene
Timing 10 days for growing plants, 30 min for handling, 2–4 days for incubation, 10 min for examination
-
32
This examination is performed by the modified method of Wang and Waterhouse36. Sow individual progeny seeds of transgenic plants in soil in 40 mm × 40 mm plastic pots and grow as detailed in Step 1. It is preferable that more than 30 seeds are sown and examined.
-
33
Excise a leaf segment of 10 mm in length from a 10-day-old seedling, insert 3 mm of the tip of the segment in ELA medium, and seal the Petri dish with Parafilm.
-
34
Incubate the plates under constant illumination (5,000 lx) at 25 °C.
-
35
Examine the segment for changes in the color 2, 3 and 4 days after the start of incubation. A resistant segment, which expresses the transgene, stays green whereas a sensitive segment (non-transgenic) turns yellow (Fig. 5).
Figure 5: Assay of segregation of transgene in detached leaves. 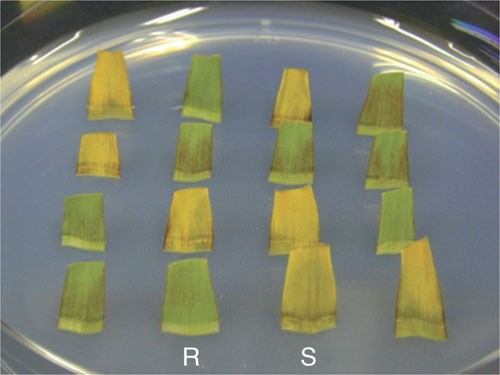
Leaves are excised from T1 plants of inbred line A188 transformed with LBA4404 (pSB131). Detached leaves are inserted in the medium containing 0.1% (w/v) Basta. The plate is incubated under continuous light at 25 °C for 3 days. Green leaves are resistant to Basta (R) and therefore transgenic, whereas yellow leaves are sensitive to Basta (S) and non-transgenic.

Troubleshooting
If transformation is unsuccessful and no transformants are obtained, ensure that the composition of all solutions and media is as indicated and perform all of the tissue culture steps without A. tumefaciens. Calli must grow vigorously from the immature embryos, and plants must be readily regenerated from the callus. Otherwise, ask an experienced maize breeder to examine the appearance of plants, temperature and light conditions of the greenhouse, soil conditions, watering frequency, etc. Take measures recommended by the expert. Check again if the sizes of the immature embryos are in the right range. If callus growth and plant regeneration without infection is fine, use an A. tumefaciens strain that carries the intron-gus gene to perform transient expression experiments (see Box 1). Typical results of this experiment are shown in Figure 6. If GUS expression is too low or too high, no transformants will be obtained. Adjust the concentration of A. tumefaciens, temperature and duration of heat treatment and centrifugal force and duration of centrifuging so that the expression is at the right level.

Upper row: expression is too weak and not suitable for high-frequency transformation. Middle row: good expression and suitable for high-frequency transformation. Lower row: expression is too strong and not suitable for high-frequency transformation. Immature embryos of inbred line A188 were treated with X-gluc 3 days after inoculation. Scale bar, 1 mm.
Timing
A typical timeline for the optimized protocol is given in Figure 1. It takes 87 days from the start of co-cultivation (Step 19) to the planting of transformants into soil in pots (Step 30).
Anticipated results
Typical average transformation frequencies for a number of maize genotypes are shown in Table 1 and raw data of transformation frequencies from a series of actual experiments are shown in Table 2. The optimized protocol has improved the frequency of transformation several fold over the original protocol for both selective markers, the bar gene and the hpt gene. In addition, some of the genotypes that could not be transformed using the original protocol have been successfully transformed using the optimized protocol (see Table 1).
To date, more than 90% of the maize plants transformed by A. tumefaciens have been normal in morphology and fertile11,20,21. About half of them had one or two copies of the transgenes. The other half had more than three copies of the transgene, but a majority of them had less than six copies11. Clear mendelian inheritance of the transgenes was observed in most of the cases11.
Co-transformation vectors34 have been tested in maize37,38. About half of the transformants carried the nonselective T-DNA, and selection marker-free transformants with solely the nonselective T-DNA were obtained from about half of these co-transformants. Thus, marker-free transformants may be segregated from a quarter of the transformants. With the highly optimized protocol, co-transformation processes may be performed very efficiently.
References
Rhodes, C.A., Pierce, D.A., Mettler, I.J., Mascarenhas, D. & Detmer, J.J. Genetically transformed maize plants from protoplasts. Science 240, 204–207 (1988).
Gordon-Kamm, W. et al. Transformation of maize cells and regeneration of fertile transgenic plants. Plant Cell 2, 603–618 (1990).
Fromm, M.E. et al. Inheritance and expression of chimeric genes in the progeny of transgenic maize plants. Biotechnology 8, 833–839 (1990).
D'Halluin, K., Bonne, K., Bossut, M., De Beuckeleer, M. & Leemans, J. Transgenic maize plants by tissue electroporation. Plant Cell 4, 1495–1505 (1992).
Frame, B.R. et al. Production of fertile transgenic maize plants by silicon carbide whisker-mediated transformation. Plant J. 6, 941–948 (1994).
Koziel, M.G. et al. Field performance of elite transgenic maize plants expressing an insecticidal protein derived from Bacillus thuringiensis. Biotechnology 11, 194–200 (1993).
Armstrong, C. The first decade of maize transformation: a review and future perspective. Maydica 44, 101–109 (1999).
Register, J.C. III et al. Structure and function of selectable and non-selectable transgenes in maize after introduction by particle bombardment. Plant Mol. Biol. 25, 951–961 (1994).
Shou, H., Frame, B.A., Whitham, S.A. & Wang, K. Assessment of transgenic maize events produced by particle bombardment or Agrobacterium-mediated transformation. Mol. Breed. 13, 201–208 (2004).
Hiei, Y., Ohta, S., Komari, T. & Kumashiro, T. Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J. 6, 271–282 (1994).
Ishida, Y. et al. High efficiency transformation of maize (Zea mays L.) mediated by Agrobacterium tumefaciens. Nat. Biotechnol. 14, 745–750 (1996).
Cheng, M. et al. Genetic transformation of wheat mediated by Agrobacterium tumefaciens. Plant Physiol. 115, 971–980 (1997).
Tingay, S. et al. Agrobacterium tumefaciens-mediated barley transformation. Plant J. 11, 1369–1376 (1997).
Zhao, Z.-y. et al. Agrobacterium-mediated sorghum transformation. Plant Mol. Biol. 44, 789–798 (2000).
Negrotto, D., Jolley, M., Beer, S., Wenck, A.R. & Hansen, G. The use of phosphomannose-isomerase as a selectable marker to recover transgenic maize plants (Zea mays L.) via Agrobacterium transformation. Plant Cell Rep. 19, 798–803 (2000).
Nomura, M. et al. The evolution of C4 plants: acquisition of cis-regulatory sequences in the promoter of C4-type pyruvate, orthophosphate dikinase gene. Plant J. 22, 211–221 (2000).
Ohta, S., Ishida, Y. & Usami, S. Expression of cold-tolerant pyruvate, orthophosphate dikinase cDNA, and heterotetramer formation in transgenic maize plants. Transgenic Res. 13, 475–485 (2004).
Ohta, S., Ishida, Y. & Usami, S. High-level expression of cold-tolerant pyruvate, orthophosphate dikinase from a genomic clone with site-directed mutations in transgenic maize. Mol. Breed. 18, 29–38 (2006).
Taniguchi, M. et al. The promoter for the maize C4 pyruvate, orthophosphate dikinase gene directs cell- and tissue-specific transcription in transgenic maize plants. Plant Cell Physiol. 41, 42–48 (2000).
Ishida, Y., Saito, H., Hiei, Y. & Komari, T. Improved protocol for transformation of maize (Zea mays L.) mediated by Agrobacterium tumefaciens. Plant Biotechnol. 20, 57–66 (2003).
Hiei, Y., Ishida, Y., Kasaoka, K. & Komari, T. Improved frequency of transformation in rice and maize by treatment of immature embryos with centrifugation and heat prior to infection with Agrobacterium tumefaciens. Plant Cell Tissue Organ Cult. 87, 233–243 (2006).
Zhao, Z.-y. et al. High throughput genetic transformation mediated by Agrobacterium tumefaciens in maize. Mol. Breed. 8, 323–333 (2001).
De Block, M. et al. Engineering herbicide resistance in plants by expressing of a detoxifying enzyme. EMBO J. 6, 2513–2518 (1987).
van den Elzen, P.J.M., Townsend, J., Lee, K.Y. & Bedbrook, J.R. A chimaeric hygromycin resistance gene as a selectable marker in plant cells. Plant Mol. Biol. 5, 299–302 (1985).
Hood, E.E. et al. Restriction endonuclease map of pTiBo542, a potential Ti-plasmid vector for genetic engineering of plants. Biotechnology 2, 702–709 (1984).
Komari, T., Halperin, W. & Nester, E.W. Physical and functional map of supervirulent Agrobacterium tumefaciens tumor-inducing plasmid pTiBo542. J. Bacteriol. 166, 88–94 (1986).
Sheng, J. & Citovsky, V. Agrobacterium-plant cell DNA transport: have virulence proteins, will travel. Plant Cell 8, 1699–1710 (1996).
Armstrong, C.L., Green, C.E. & Phillips, R.L. Development and availability of germplasm with high type II culture formation response. Maize Genet. Coop. News Lett. 65, 92–93 (1991).
Frame, B.R. et al. Agrobacterium tumefaciens-mediated transformation of maize embryos using a standard binary vector system. Plant Physiol. 129, 13–22 (2002).
Huang, X. & Wei, Z. Successful Agrobacterium-mediated genetic transformation of maize elite inbred lines. Plant Cell Tissue Organ Cult. 83, 187–200 (2005).
Frame, B.R. et al. Improved Agrobacterium-mediated transformation of three maize inbred lines using MS salts. Plant Cell Rep. 25, 1024–1034 (2006).
Bajaj, Y.P.S. in Biotechnology in Agriculture and Forestry (ed. Bajaj, Y.P.S.) 3–23 (Springer-Verlag, Berlin, Heidelberg, 1994).
Frame, B.R., Paque, T. & Wang, K. in Methods in Molecular Biology (ed. Wang, K.) 185–199 (Humana Press Inc., Totowa, NJ, 2006).
Komari, T., Hiei, Y., Saito, Y., Murai, N. & Kumashiro, T. Vectors carrying two separate T-DNAs for co-transformation of higher plants mediated by Agrobacterium tumefaciens and segregation of transformants free from selection markers. Plant J. 10, 165–174 (1996).
Linsmaier, E. & Skoog, F. Organic growth factor requirements of tobacco tissue culture. Physiol. Plant 18, 100–127 (1965).
Wang, M.-B. & Waterhouse, P.M. A rapid and simple method of assaying plants transformed with hygromycin and PPT resistance genes. Plant Mol. Biol. Rep. 15, 209–215 (1997).
Miller, M. et al. High efficiency transgene segregation in co-transformation maize plants using an Agrobacterium tumefaciens 2 T-DNA binary system. Transgenic Res. 11, 381–396 (2002).
Ishida, Y. et al. Improved co-transformation of maize with vectors carrying two separate T-DNAs mediated by Agrobacterium tumefaciens. Plant Biotechnol. 21, 57–63 (2004).
Hiei, Y. & Komari, T. Improved protocols for transformation of indica rice mediated by Agrobacterium tumefaciens. Plant Cell Tissue Organ Cult. 85, 271–283 (2006).
Ohta, S., Mita, S., Hattori, T. & Nakamura, K. Construction and expression in tobacco of a β-glucuronidase (GUS) reporter gene containing an intron within the coding sequence. Plant Cell Physiol. 31, 805–813 (1990).
Jefferson, R.A. Assaying chimeric genes in plants: the GUS gene fusion system. Plant Mol. Biol. Rep. 5, 387–405 (1987).
Acknowledgements
We thank Ms. E. Usami and Ms. M. Noguchi for skillful assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Ishida, Y., Hiei, Y. & Komari, T. Agrobacterium-mediated transformation of maize. Nat Protoc 2, 1614–1621 (2007). https://doi.org/10.1038/nprot.2007.241
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2007.241
This article is cited by
-
Transcriptome-wide identification and characterization of genes exhibit allele-specific imprinting in maize embryo and endosperm
BMC Plant Biology (2023)
-
A transcription factor ZmGLK36 confers broad resistance to maize rough dwarf disease in cereal crops
Nature Plants (2023)
-
Efficient Agrobacterium tumefaciens-mediated genetic transformation of Aloe vera
Plant Cell, Tissue and Organ Culture (PCTOC) (2023)
-
Modern plant biotechnology as a strategy in addressing climate change and attaining food security
Agriculture & Food Security (2022)
-
Genome-wide dissection of changes in maize root system architecture during modern breeding
Nature Plants (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
