
Abstract
Hyperalgesia is an exaggerated response to noxious stimuli produced by peripheral or central plasticity. Stress modifies nociception, and humans with post-traumatic stress disorder (PTSD) exhibit co-morbid chronic pain and amygdala dysregulation. Predator odor stress produces hyperalgesia in rodents. Systemic blockade of corticotropin-releasing factor (CRF) type 1 receptors (CRFR1s) reduces stress-induced thermal hyperalgesia. We hypothesized that CRF-CRFR1 signaling in central amygdala (CeA) mediates stress-induced hyperalgesia in rats with high stress reactivity. Adult male Wistar rats were exposed to predator odor stress in a conditioned place avoidance paradigm and indexed for high (Avoiders) and low (Non-Avoiders) avoidance of predator odor-paired context, or were unstressed Controls. Rats were tested for the latency to withdraw hindpaws from thermal stimuli (Hargreaves test). We used pharmacological, molecular, and immunohistochemical techniques to assess the role of CRF-CRFR1 signaling in CeA in stress-induced hyperalgesia. Avoiders exhibited higher CRF peptide levels in CeA that did not appear to be locally synthesized. Intra-CeA CRF infusion mimicked stress-induced hyperalgesia. Avoiders exhibited thermal hyperalgesia that was reversed by systemic or intra-CeA injection of a CRFR1 antagonist. Finally, intra-CeA infusion of tetrodotoxin produced thermal hyperalgesia in unstressed rats and blocked the anti-hyperalgesic effect of systemic CRFR1 antagonist in stressed rats. These data suggest that rats with high stress reactivity exhibit hyperalgesia that is mediated by CRF-CRFR1 signaling in CeA.
Similar content being viewed by others

Introduction
Hyperalgesia is an exaggerated and prolonged response to noxious stimuli that can be produced by plasticity at peripheral sites (eg, reduced threshold and/or amplified nociceptor response to noxious stimuli) and centrally in the spinal cord or brain. Central sensitization is a process whereby nociceptive neurons and circuits exhibit increased function in response to activity, inflammation, or injury through a variety of processes that include changes in receptor field size, increases in neuronal excitability, increases in synaptic efficiency/coupling, and changes in neuronal connectivity (Latremoliere and Woolf, 2009). Therefore, altered pain perception is not contingent on changes in sensitivity to, or even the presence of, a noxious stimulus.
The amygdala, and more specifically the central nucleus of the amygdala (CeA), is a convergence point for multiple pathways carrying nociceptive information into the central nervous system. Peptide-expressing nociceptors synapse on spinal cord lamina I neurons that project upward and synapse onto parabrachial nucleus neurons that project to amygdala (Jasmin et al, 1997). Peptide-deplete nociceptors synapse on spinal cord lamina II/V neurons, which send ascending projections directly to amygdala (Braz et al, 2005). The cortex also sends polymodal sensory information, including nociceptive information, to CeA via relays in basolateral amygdaloid complex (Shi and Davis, 1999). The convergence of nociceptive information in CeA is particularly interesting because CeA is the primary output nucleus of the amygdala, and sends projections to effector regions that initiate physiological and behavioral responses to external events, including the response to noxious stimuli (Gilpin et al, 2015).
CeA projections to periacqueductal gray (PAG) gate descending pathways that modulate nociceptive afferent activity in the dorsal horn of the spinal cord. Electrical stimulation of CeA in rodents produces analgesic effects that are blocked either by lidocaine ‘silencing’ of PAG or by blocking opioid receptors in PAG, suggesting that CeA-to-PAG projections are critical for the role of CeA signaling in modulating the nocifensive response (Oliveira and Prado, 2001). Therefore, under normal physiological conditions, CeA functions as a hub between incoming nociceptive information and nocifensive responses to noxious stimuli. As such, it is reasonable to hypothesize that dysregulation of CeA function modifies nociceptive processing and/or responses to noxious stimuli.
Traumatic stress disorders such as PTSD are highly co-morbid with chronic pain (Asmundson and Katz, 2009; Lew et al, 2009; Moeller-Bertram et al, 2012; Shipherd et al, 2007), and studies report lower experimental pain thresholds and acute stress-induced hyperalgesia in PTSD humans (Defrin et al, 2008; Orr et al, 2000); but see Asmundson and Katz (2009). Our laboratory uses a predator odor stress model in which rats are divided into groups that exhibit persistently high (Avoiders) or low (Non-Avoiders) avoidance of a predator odor-paired context; these animals are then compared with each other and with unstressed Controls for post-stress behavioral and neural changes. In our model, predator odor increases anxiety-like behavior and corticosterone in both Avoiders and Non-Avoiders (Whitaker and Gilpin, 2015), but animals exhibit measurable and persistent (at least 6 weeks) differences in their avoidance of a context paired with predator odor (Edwards et al, 2013). Because one of the major diagnostic criteria for PTSD in humans (DSM-5) is persistent avoidance of stimuli related to traumatic stress, we use avoidance to index animals for stress reactivity.
Animal models of chronic pain (eg, arthritis) increase the magnitude and duration of CeA neuronal responses to innocuous mechanical stimuli, independent of altered nociceptive input or plasticity in ascending pathways (Neugebauer and Li, 2003). The notion that strictly psychological factors (eg, stress) can produce central sensitization is a relatively newer concept. For example, psychosocial stress produces allodynia in both healthy humans and fibromyalgia patients (Crettaz et al, 2013). Rodents exposed to restraint stress, swim stress, single prolonged stress (restraint+swim), unpredictable sound stress, or social defeat stress exhibit mechanical and thermal hyperalgesia that may be mediated by a combination of brain, spinal, and nociceptor mechanisms (Dina et al, 2011; Li et al, 2014). That said, stress-induced analgesia is also a robust behavioral phenomenon, and there is a large body of basic science literature exploring the neurobiology of this effect (eg, see Millan, 2002). Whether stress produces analgesia or hyperalgesia likely depends on factors that include, but are not limited to, post-stress time course, number of stress exposures, type and modality of stress, modality of pain stimulus (heat vs cold or mechanical stimuli, etc.), gender, and age. Post-stress time course may be particularly important since most stress-induced analgesia experiments test nociception immediately after or during the stress, whereas here we test nociception several days post-stress.
Hypothalamic corticotropin-releasing factor (CRF) drives the neuroendocrine stress response, and CRF signaling through the CRF-1 receptor (CRFR1) in CeA coordinates the emotional response to stress (Heilig et al, 1994; Regev et al, 2012; Sakanaka et al, 1986). CRF modulates inhibitory and excitatory transmission in CeA, thereby gating the activity of CeA projection neurons to downstream effector regions (Gilpin et al, 2015; Fu and Neugebauer, 2008; Ji et al, 2013). With respect to nociception, both stress and intra-CeA corticosterone pellet implants produce somatic hypersensitivity by increasing CRF expression in CeA, and these effects are blocked by antisense CRF knockdown in CeA (Johnson et al, 2015). Previous work by our group showed that predator odor exposure increases thermal nociception and this effect is reversed by systemic blockade of CRFR1s (Roltsch et al, 2014).
The purpose of these experiments was to test the overall hypothesis that animals with high stress reactivity exhibit sensitized CRF/CRFR1 signaling in CeA, which leads to exaggerated nociception, by testing: (1) the effect of stress on nociception, (2) the effect of stress on CRF peptide, CRF mRNA, CRFR1 mRNA, and CRF cell numbers in CeA, the effect of intra-CeA CRF on (3) conditioned place avoidance and (4) nociception in naive animals, (5) the effect of systemic CRF-CRFR1 antagonism on stress-induced hyperalgesia in Avoiders, and (6) the effect of CeA CRF-CRFR1 antagonism on stress-induced hyperalgesia in Avoiders. Our results confirm our predictions that (1) Avoiders exhibit post-stress thermal hyperalgesia, (2) Avoiders have more CRF in CeA, (3) intra-CeA CRF produces dose-dependent conditioned place avoidance, (4) intra-CeA CRF infusion produces thermal hyperalgesia, (5) systemic CRFR1 antagonism reverses stress-induced hyperalgesia in Avoiders in a manner that is contingent on CeA function, and (6) intra-CeA CRFR1 antagonism reverses stress-induced hyperalgesia in Avoiders.
Materials and Methods
Animals
Adult male Wistar rats (Charles River) weighing ~300 g at start of experiments were pair-housed in groups of two in a humidity- and temperature-controlled (22 °C) vivarium on a 12-h light/dark cycle. Rats were acclimated for 1 week before start of experiments. Behavioral tests occurred during the dark period. Animals had ad libitum access to food and water throughout experiments. All procedures were approved by the Institutional Animal Care and Use Committee of the Louisiana State University Health Sciences Center and were in accordance with the National Institute of Health guidelines.
Hargreaves Method
To assess thermal nociception, each hindpaw was stimulated using a halogen heat source from an IITC model 309 Hargreaves apparatus (IITC Life Sciences, Woodland Hills, CA). A 20-second cut-off was employed to prevent tissue damage in non-responsive subjects. On test days, rats were allowed 5 min to acclimate to the testing room, then 10 min to acclimate to the testing apparatus, which consisted of Plexiglas enclosures on glass floors suspended 30 cm from tabletop. On each test day, each hindpaw was tested twice in an alternating order (ie, left, right, 1 min wait, left, right), and these scores were averaged to yield a single score for analysis. This average hindpaw withdrawal latency was the thermal nociception score for each rat (ie, lower scores indicate hyperalgesia).
Predator Odor Conditioned Place Aversion
As previously described (Edwards et al, 2013), rats were assigned to control or stress groups. The stressed group was exposed to predator odor (bobcat urine) or no odor in contexts that differed on visual and tactile cues, in an unbiased procedure, while the controls were never exposed to predator odor. On day 1, rats were allowed 5 min to freely explore the apparatus. On day 2, rats were placed in one context with no odor for 15 min. On day 3, rats were placed in the other context with predator odor (no odor for Controls) for 15 min. On day 4, rats were allowed 5 min to freely explore the apparatus. Avoidance of predator odor-paired context was quantified as post-conditioning time minus pre-conditioning time in the predator odor-paired context. Stressed rats that exhibited >10 s decrease in time spent in predator odor-paired context were grouped as Avoiders; all other stressed rats were grouped as Non-Avoiders. Controls underwent the same procedure but were never exposed to predator odor.
Drug Cannula Implantation
Rats were anesthetized with isoflurane and mounted in a stereotaxic frame (Stoelting). An incision was made, small bilateral holes were drilled into the skull, and stainless steel guide cannulae (26 gauge) lowered such that tips were 1 mm dorsal to CeA at these coordinates: AP −2.3, ML +4.0, DV −7.5. Guide cannulae were secured to skull with metal screws and dental cement, the incision closed, and dummy cannulae inserted. Rats were monitored for 1 week or until they resumed normal activity.
Drug Microinfusions
Infusions were administered bilaterally at a rate of 0.2 μl/min over 2.5 min via injectors (33 ga, stainless steel) that extended 1 mm beyond the tip of the guide cannulae. Injection cannulae were left in guide cannulae for additional 1 min to allow for diffusion. Infusions were delivered via polyethylene tubing connected to a 10-μl Hamilton syringe. Animals underwent sham infusions on days preceding infusion to acclimate them to the procedure.
Statistical Analysis
Data were analyzed using paired t-test, one-way and two-way RM ANOVAs, and Pearson correlations, except where otherwise indicated. In Experiment 4, CRF cell counts failed the Shapiro–Wilk Normality Test; therefore, data were analyzed using Kruskal–Wallis ANOVA on Ranks. In Experiment 7, avoidance data were analyzed using trend analysis to test for dose-response drug effect. In the case of significant interaction and/or main effects, pairwise comparisons were probed with Student–Newman–Keuls (SNK) post hoc analysis.
Histological Verification of Intra-CeA Cannulae
Rats were deeply anesthetized with isoflurane and bilaterally injected with Evans Blue dye using the same rate and volume as drug injections (0.5 μl per side over 2.5 min). Brains were removed before being snap-frozen in isopentane (Fisher Scientific) on dry ice. Brains were stored at −80 °C, then coronally sectioned on a cryostat. Brain sections containing CeA (−1.2 mm to −3.2 mm relative to bregma) were mounted on slides and stained with cresyl violet before being cover slipped. Placement of the Evens Blue dye was visually estimated by a treatment-blind observer using a rat brain atlas (Paxinos and Watson, 2005). Rats that did not have bilaterally accurate cannulae were excluded from all data analysis.
Experiment 1: Hargreaves testing over time
Rats (N=8) were tested for baseline thermal nociception using the Hargreaves test, then periodically over 32 days to determine whether repeated testing altered thermal nociception.
Experiment 2: stress effects on thermal nociception
Rats (N=23) were tested for baseline thermal nociception using the Hargreaves test. Upon stabilization of hindpaw withdrawal latencies, rats were tested for conditioned place aversion (CPA) using predator odor and separated into Control (N=5), Non-Avoider (N=8), and Avoider (N=10) groups. Rats were tested for thermal nociception 48 h after predator odor exposure, then re-tested for avoidance of predator odor-paired chamber 8 days post stress.
Experiment 3: CRF content in CeA
Radioimmunoassay (RIA) was used to measure total CRF peptide levels in CeA of rats exposed to predator odor stress. Rats (N=22) were tested for CPA using predator odor and separated into Control (N=6), Non-Avoider (N=4), and Avoider (N=12) groups. Twelve days post stress, rats were deeply anesthetized with isoflurane and decapitated. Brains were extracted, frozen in isopentane, cut into 500 μm sections, and CeA was ‘punched’ out with a 17-g needle (see Supplemental Material for location of micropunches). CeA CRF total protein content was determined by RIA in extracted samples. Frozen tissue punches from the CeA were homogenized in PBS containing EDTA, aprotinin, and 12.5% acetic acid. Lyophilized supernatant was stored at −80 °C until extraction. During the extraction procedure, lyophilized powder was acidified with 1% trifluoroacetic acid in water (buffer A) and centrifuged for 10 min at 10 000 r.p.m. and 4 °C. C-18 SEP-Columns (Waters Corporation, Milford, MA) were washed with 1 ml buffer B (60% acetonitrile+40% buffer A) followed by three washes with 3 ml buffer A. The acidified sample was loaded onto the pre-treated column and washed three times with buffer A. The peptides were eluted with 3 ml of buffer B and lyophilized until dry. The residue was stored at −80 °C until analysis. On the day of the assay, the residue was reconstituted using the buffer provided by a rat-specific RIA kit (Phoenix Pharmaceuticals, Burlingame, CA). Values were normalized to total protein measured in the reconstituted sample using a BCA protein assay kit (Thermo Scientific). The range of detection of the assay was 10–1280 pg/ml.
Experiment 4: intra-CeA CRF-induced conditioned place avoidance
Rats (N=38) in this experiment were all naive to predator odor (ie, no stress). Rats were acclimated to handling before surgery. Guide cannulae were bilaterally implanted 1 mm dorsal to CeA of rats. After 1 week of recovery, rats were acclimated to handling in the experimental rooms and sham injected for 3 days before testing. On test day 1, rats were allowed 5 min to freely explore the apparatus. Designation of vehicle vs. CRF paired chambers were assigned in an unbiased procedure. On days 2, 4, and 6, all rats received intra-CeA vehicle infusions immediately before being placed in one chamber. On days 3, 5, and 7, all rats received intra-CeA 0.5 μg infusions of one of four CRF doses (0, 0.05, 0.25, and 0.5 μg/side) immediately before being placed in the second chamber. On day 8, rats were allowed 5 min to freely explore both chambers and a CPA score was calculated. Avoidance of CRF-paired context was quantified as post-conditioning time minus pre-conditioning time in the CRF-paired context. Immediately after the final test session, rats were deeply anesthetized with isoflurane, injected with Evans Blue dye (0.5 μl), and decapitated. Brains were extracted, frozen in isopentane, cut into 50-μm sections, mounted on glass slides, stained with cresyl violet, cover slipped, and visually inspected for cannula placement. Rats that did not have bilaterally accurate intra-CeA cannulae placements were excluded from data analysis.
Experiment 5: intra-CeA CRF effects on thermal nociception
Rats (N=9) were tested for baseline thermal nociception using the Hargreaves test. Rats in this experiment were all naive to predator odor (ie, no stress). Upon stabilization of hindpaw withdrawal latencies, guide cannulae were bilaterally implanted 1 mm dorsal to CeA of rats. After 1 week of recovery, rats were re-tested for baseline thermal nociception. Rats were tested for the effects of intra-CeA CRF on thermal nociception. On day 1 of the infusion protocol, all rats received intra-CeA vehicle infusions before testing; on day 2, all rats received intra-CeA CRF (0.5 μg) infusions. Rats that did not have bilaterally accurate intra-CeA cannulae placements were excluded from data analysis.
Experiment 6: CRF mRNA and CRFR1 mRNA content in CeA
Rats (N=16) were exposed to predator odor stress, indexed for avoidance, and separated into unstressed Control (N=5), Non-Avoider (N=6), and Avoider (N=5) groups. Forty-eight hours post stress, rats were deeply anesthetized with isoflurane and decapitated. Brains were extracted, frozen in isopentane, cut into 500-μm sections, and CeA was ‘punched’ out with a 17-g needle. Quantitative real-time PCR (qRT-PCR) was used to measure CRF and CRF1R gene expression in CeA of rats 48 h post stress. Total RNA was extracted from brain tissue using an RNeasy Plus Universal Mini Kit (Qiagen, Valencia, CA). Total RNA was reverse transcribed using TaqMan Reverse Transcription Reagent kit (Life Technologies Corporation, Carlsbad, CA). The primer concentrations used were 500 nmol. The primer sequences (Integrated DNA Technologies, Coralville, IA) used in this study were as follows: CRF 5′-TGATCCGCATGGGTGAAGAATACTTCCTC-3′ (forward), 5′-CCCGATAATCTCCATCAGTTTCCTGTTGCTG-3′ (reverse), CRFR1 5′-TCCACTACATCTGAGACCATTCAGTACA-3′ (forward), 5′-CCTGCCACCGGCGCCACCTCTTCCGGA-3′ (reverse), housekeeping 36B4 5′-TTCCCACTGGCTGAAAAGGT-3′ (forward), 5′-CGCAGCCGCAAATGC-3′ (reverse). The RT2 SYBR Green Mastermixes (Qiagen) were used for real-time PCR. All reactions were performed on a CFX96 system (Bio-Rad Laboratories, Hercules, CA). RT-qPCR data were analyzed using the DDCT method. Target genes were compared with housekeeping gene 36B4 and normalized to control values.
Experiment 7: CRF cell counts in medial and lateral CeA (CeM and CeL)
Rats (N=31) underwent a CPA procedure using predator odor and were separated into Control (N=9), Non-Avoider (N=12), and Avoider (N=10) groups. Nine days post stress, rats were deeply anesthetized with isoflurane, injected with chloral hydrate (35%, 2 ml), and intracardially perfused with 4% paraformaldehyde/0.1 M borate buffer, pH 9.5. Brains were post-fixed in the same fixative for 4 h at 4 °C and submerged in the 20% sucrose/0.1 M phosphate buffer, pH 7.4 for 48–72 h before being snap-frozen in isopentane on dry ice. Brains were coronally sectioned at 35 μm on a freezing microtome, and sections were stored in cryoprotectant (50% 0.1 M phosphate-buffered saline, 30% ethylene glycol, 30% sucrose, and 1% polyvinyl pyrrolidone) at −20 °C until immunolabeling. Brain sections containing CeA (from −2.04 mm to −3.24 mm relative to bregma) were sorted and labeled by rabbit anti-h/rCRF antiserum (1 : 5000, generously provided by Dr Wylie Vale, Salk Institute) and biotin-conjugated goat anti-rabbit antiserum (1 : 200, Vector Laboratories) using a free-floating immunohistochemistry procedure. CRF labeling signal was developed with 3,3′-diaminobenzidine (DAB, Vector Laboratories), and sections were mounted on slides for microscopic analysis.
Five coronal sections containing CeA (each separated by 175 μm) were collected for quantification of CRF-positive cells. For CeM and CeL, a treatment-blind experimenter counted cells expressing CRF peptide using a Nikon microscope. Cells containing labeling throughout the soma and neuronal processes with a clear border around the soma were considered CRF-immunolabeled and counted as described above. Adjusting the optical focus (z axis) of the microscope allowed for clearer visualization of soma depth to distinguish CRF-ir somata from ambiguous particles, and of individual cell borders when cells were clustered (Karanikas et al, 2013).
Experiment 8: systemic CRFR1 antagonism and intra-CeA tetrodotoxin effects on stress-induced changes in thermal nociception
Rats (N=14) were tested for baseline thermal nociception using the Hargreaves test. Upon stabilization of hindpaw withdrawal latencies, cannulae were surgically implanted 1 mm dorsal to CeA bilaterally and secured to skull. After 1 week of recovery, rats were re-tested for baseline thermal nociception, underwent a CPA procedure using predator odor and separated into Control (N=3), Non-Avoider (N=5), and Avoider (N=6) groups. On days 2–5 post stress, rats were tested for thermal nociception. Each test was preceded by systemic (i.p.) injection of the CRFR1 antagonist R121919 (10 mg/2 ml/kg) or vehicle (20% HBC, 2 ml/kg) 60 min pre-test, and intra-CeA infusion of TTX (10 ng/0.5 μl) or vehicle (aCSF, 0.5 μl) 40 min pre-test. On day 2 post stress, all rats received systemic vehicle and intra-CeA vehicle. On days 3 and 4 post stress, all rats received systemic R121919, then intra-CeA TTX or vehicle, in a counterbalanced order. On day 5 post stress, all rats received systemic vehicle and intra-CeA TTX. Rats that did not have bilaterally accurate intra-CeA cannulae placements were excluded from data analysis.
Experiment 9: intra-CeA CRFR1 antagonism effects on stress-induced changes in thermal nociception
Rats (N=26) were tested for baseline thermal nociception using the Hargreaves test. Upon stabilization of hindpaw withdrawal latencies, cannulae were surgically implanted 1 mm dorsal to CeA bilaterally and secured to skull. After 1 week of recovery, rats were re-tested for baseline thermal nociception, then underwent a CPA procedure using predator odor and separated into Control (N=7), Non-Avoider (N=11), and Avoider (N=8) groups. On days 2–5 post stress, rats were tested for thermal nociception. Each test was preceded by intra-CeA infusion of vehicle (20% HBC, 0.5 μl/side) or one of three R121919 doses (0.0625, 0.125, or 0.25 μg/0.5 μl/side) 5 min before the test. All rats received all doses in a counterbalanced order. Rats that did not have bilaterally accurate intra-CeA cannulae placements were excluded from data analysis.
Drugs
The CRFR1 antagonist R121919 (generously supplied by Neurocrine) was solubilized first in 1 M HCl (10% final volume), then diluted into 2-hydroxypropyl-β-cyclodextrin (HBC; Sigma-Aldrich, 20% wt/vol final concentration in distilled water) and back-titrated with NaOH to pH 4.5. Tetrodotoxin (TTX, Tocris Bioscience) and CRF (Sigma Aldrich) were solubilized in artificial cerebrospinal fluid (aCSF, Tocris Bioscience).
Results
Repeated Testing Does Not Alter Thermal Nociception
In Experiment 1, naive rats were tested for thermal nociception seven times over the course of 32 days. A one-way RM ANOVA indicated that there were no significant differences across test days, indicative of the stability of responding and a lack of tolerance/sensitization after repeated thermal stimulation (F[6, 55]=0.937, p=0.479; Figure 1a).

Repeated testing does not alter thermal nociception, and predator odor increases thermal nociception in avoiders. (a) Mean±SEM paw withdrawal latency for experimentally naive rats. Latency remained stable over 32 days, indicating that repeated testing does not lead to sensitization nor habituation. (b) Mean±SEM paw withdrawal latency of Control (open triangles, solid line), Non-Avoider (open circles, dotted line), and Avoider (solid circle, solid line) rats at baseline and 48 h after odor (Avoider and Non-Avoider) or air (Control) exposure. Scatter plots denote (c) individual rat baseline paw withdrawal latency before odor exposure vs avoidance of predator paired chamber 24 h after odor exposure, (d) avoidance of predator paired chamber 24 h after odor exposure vs paw withdrawal latency 48 h after odor exposure, (e) avoidance of predator paired chamber 24 h after odor exposure vs avoidance of predator odor paired chamber 8 days after odor exposure. *Denotes p<0.05 when compared with pre-odor baseline. #Denotes p<0.05 when compared with Control and Non-Avoider 48 h after odor exposure.
Predator Odor Produces Persistent Avoidance and Increases Thermal Nociception in Avoiders
In Experiment 2, rats were sorted into Controls, Non-Avoiders, and Avoiders, tested for thermal nociception 48 h after predator odor exposure, and re-tested for avoidance 8 days post stress. Forty-eight hours after predator odor exposure, Avoiders exhibited significantly lower nociceptive thresholds (ie, hyperalgesia) than Non-Avoiders and Controls. A two-way RM ANOVA yielded a significant interaction effect (F[2, 21]=4.124, p=0.031; Figure 1b). Post hoc analyses revealed that Avoiders exhibited significantly lower post-stress nociceptive thresholds relative to pre-odor baseline (p=0.006; Figure 1b), and also relative to post-stress Non-Avoiders and Controls (p=0.037 and p=0.023; Figure 1b). There was no difference in nociceptive thresholds between groups before stress, suggesting that higher thermal nociception in Avoiders 48 h post stress was elicited by stress (ie, not pre-existing). There was no significant correlation between pre-odor exposure hindpaw withdrawal latencies and 24-h post-stress avoidance (p>0.05; Figure 1c), but there was a significant correlation between 24-h post-stress avoidance of predator odor-paired chamber and 48-h post-stress hindpaw withdrawal latency (R2=0.445; p=0.003; Figure 1d), suggesting that avoidance by individual rats predicts post-stress thermal sensitivity. Consistent with previous work from our laboratory (Edwards et al, 2013), avoidance persisted over time, as evidenced by the significant correlation between avoidance of predator odor-paired chamber 24 h and 8 days post odor exposure (R2=0.626; p<0.001; Figure 1e).
Predator Odor Stress Increases Total CRF Peptide Content in CeA of Avoiders
In Experiment 3, rats were sorted into Controls, Non-Avoiders, and Avoiders, brains were extracted and frozen 12 days post stress, tissue was ‘punched’ to isolate CeA samples, and RIA was used to determine CeA CRF total protein content. There was a significant main effect of stress group on CRF peptide levels, as measured by RIA, in CeA 12 days post stress (F[2]=6.018, p=0.009; Figure 2a). Avoiders exhibited significantly higher CRF levels than unstressed Controls and Non-Avoiders (p=0.032 and p=0.010, respectively). A correlation between Avoidance and CRF levels yielded a weak negative correlation (R=−0.306) that did not reach statistical significance (p=0.250).

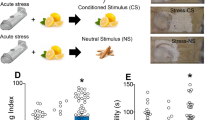
Predator odor increases CRF in CeA of Avoiders, and Intra-CeA CRF produces CPA and hyperalgesia. (a) Mean±SEM pg CRF/mg total protein in CeA of CeA of Control (white bar), Non-Avoider (gray bar), and Avoider (black bar) rats. *Denotes p<0.05 when compared with Control and Non-Avoider. (b) Mean±SEM change in time spent in the Intra-CeA CRF-paired chamber. *Denotes p<0.05 trend analysis of CRF dose. (c) Mean±SEM paw withdrawal latency in rats infused in CeA with vehicle or CRF (0.5 μg). *Denotes p<0.05 when compared with vehicle infusion.
Intra-CeA CRF Produces Conditioned Place Avoidance
In Experiment 4, stress-naive rats received three pairings of one of four CRF doses (0, 0.05, 0.25, 0.5 μg/0.5 μl/side) with one of two contexts in a CPA chamber, and three pairings of intra-CeA vehicle (saline, 0.5 μl/side) with the other context. A trend analysis revealed a dose-dependent avoidance of the context paired with Intra-CeA CRF infusions (p=0.017; Figure 2b).
Intra-CeA CRF Produces Thermal Hyperalgesia
In Experiment 5, stress-naive rats were tested for thermal nociception after receiving intra-CeA vehicle and again 24 h later after receiving intra-CeA CRF (0.5 μg/side) infusions. A paired t-test revealed that intra-CeA CRF infusion reduced nociceptive thresholds (ie, produced hyperalgesia) relative to vehicle in predator odor naive rats (p=0.009; Figure 2c).
Predator Odor Stress Does Not Increase CRF mRNA, CRFR1 mRNA, or CRF-Peptide Labeled Cell Counts in CeA
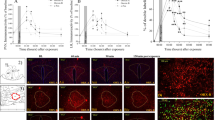
Experiments 6 and 7 measured CRF mRNA, CRFR1 mRNA, and CRF-immunoreactive cell body counts in CeA. CeA punches were taken from brains harvested 48 h (Experiment 6) or 8 days (Experiment 7) post stress. Separate one-way ANOVAs indicated no effect of stress history on CRF mRNA (F[2, 15]=3.805, p=0.44; Figure 3a) or CRFR1 mRNA (F[2, 15]=3.805, p=0.85; Figure 3b) in the CeA 48 h post stress. Across all groups, CRF-immunoreactive cell counts were much higher in lateral CeA than in medial CeA (Figure 3e–f). Because the results of CRF-positive cell counts were not normally distributed (Shapiro–Wilk Normality Test, lateral CeA: W=0.88, p<0.01; medial CeA: W=0.86, p<0.001), a Kruskal–Wallis Test on ranks was used, and revealed no effect on CRF-positive cell number in CeA of Avoiders 10 days post stress (CeL, H(2)=3.5, p=0.17; CeM, H(2)=1, p=0.60; Figure 3d–f). These data suggest that lasting increases in CRF peptide in the CeA of Avoiders may not be synthesized in the CeA.

Predator odor does not increase CRF mRNA, CRFR1 mRNA, or CRF-positive cell counts. (a) Mean±SEM fold change of CRF mRNA level in CeA of Control (white bar), Non-Avoider (gray bar), and Avoider (black bar) rats. (b) Mean±SEM fold change of CRFR1 mRNA level in CeA of Control (white bar), Non-Avoider (gray bar), and Avoider (black bar) rats. (c) Photomicrograph shows representative CRF labeling in CeA under × 5 objective. Scale bar, 200 μm. White box indicates area of inset figure. The inset shows the individual CRF-ir cells under × 100 objective. Scale bar, 20 μm. (d) Mean±SEM CRF cell count per CeA section in Control (white bar), Non-Avoider (gray bar), and Avoider (black bar) rats. (e) Mean±SEM CRF cell count per CeL section in Control (white bar), Non-Avoider (gray bar), and Avoider (black bar) rats. (f) Mean±SEM CRF cell count per CeM section in Control (white bar), Non-Avoider (gray bar), and Avoider (black bar) rats.
Intra-CeA TTX Blocks Anti-Hyperalgesic Effect of Systemic CRFR1 Antagonist in Avoiders
In Experiment 8, rats were sorted into Controls, Non-Avoiders, and Avoiders, then tested for thermal nociception 2–5 days post stress. All rats received all combinations of systemic vehicle/R121919+intra-CeA vehicle/TTX across 4 test days. A two-way ANOVA yielded a significant effect of drug treatment (F[3, 32]=11.487, p<0.001; Figure 4) and a group × drug interaction effect (F[6,32]=2.498, p=0.043). As in Experiment 2, Avoiders exhibited significantly lower nociceptive thresholds than Controls and Non-Avoiders (p=0.034 and p=0.032, respectively) 48 h post stress under vehicle (i.p.)+vehicle (CeA) conditions. Systemic R121919 normalized thermal nociception in Avoiders, as evidenced by a significant increase in hindpaw withdrawal latency relative to vehicle (p=0.011). Regardless of whether animals were injected systemically with R121919 or vehicle, intra-CeA TTX significantly reduced hindpaw withdrawal latencies (ie, produced hyperalgesia) in Avoiders, Non-Avoiders, and unstressed Controls relative to vehicle (p<0.05 in all cases). These data suggest that intact CeA neurotransmission is necessary for CRFR1 antagonist reversal of stress-induced thermal hyperalgesia in Avoiders, and that blockade of CeA action potentials is sufficient to produce thermal hyperalgesia.

Intra-CeA tetrodotoxin blocks anti-hyperalgesia effects of systemic R121919. Mean±SEM percent baseline paw withdrawal latency of Control (white bars), Non-Avoider (gray bars), and Avoider (black bars) rats with vehicle or R121919 systemic treatment and vehicle or tetrodotoxin infusion into CeA. *Denotes p<0.05 when compared with Control and Non-Avoider at the same treatment. #Denotes p<0.05 when compared with vehicle/vehicle treatment within a group. $Denotes p<0.05 when compared with R121919/vehicle treatment within a group.
Intra-CeA CRFR1 Antagonist Blocks Hyperalgesia in Avoiders
In Experiment 9, rats were sorted into Controls, Non-Avoiders, and Avoiders, then tested for thermal nociception 2−5 days post stress. All rats received all four doses of Intra-CeA R121919 (0, 0.0625, 0.125, or 0.25 μg/0.5 μl/side) across 4 test days. A two-way RM ANOVA yielded a significant effect of stress group (F[2, 103]=12.51, p<0.001) and a stress group × drug treatment interaction effect (F[6, 51]=6.90, p<0.001; Figure 5). Post hoc analyses revealed that Avoiders exhibited significantly lower nociceptive thresholds than Controls and Non-Avoiders under vehicle and low dose R121919 conditions (p<0.001 in all cases), but not after infusion with middle and high R121919 doses. Furthermore, within Avoiders, the high R121919 dose increased nociceptive thresholds relative to all three other drug treatment conditions (p<0.05 for all conditions). Within Avoiders, the middle R121919 dose also increased nociceptive thresholds relative to vehicle and low dose conditions (p<0.01 for both). These data suggest that CRFR1 antagonism in CeA is sufficient to reverse stress-induced thermal hyperalgesia in Avoiders.

Inhibition of CRFR1s in CeA blocks predator odor-induced increases in thermal nociception in avoiders. Mean±SEM paw withdrawal latency of Control (white bars), Non-Avoider (gray bars), and Avoider (black bars) rats infused in CeA with vehicle or one of three R121919 doses (0.0625, 0.125, and 0.25 μg). *Denotes p<0.05 when compared with Controls and Non-Avoider rats. #Denotes p<0.05 when compared with all other drug doses within Avoider rats.
Discussion
Using a predator odor conditioning model that allows for the identification of rats that exhibit high (Avoiders) or low (Non-Avoiders) stress reactivity, we show here that Avoiders exhibit thermal hyperalgesia that is predicted by post-stress avoidance but is not present before the stress event. We also show that Avoiders have higher total CRF peptide content in CeA following predator odor stress relative to Non-Avoiders or unstressed Controls, and that intra-CeA CRF infusion mimics stress-induced thermal hyperalgesia. We also report that systemic antagonism of CRFR1 reverses stress-induced thermal hyperalgesia in Avoiders without affecting thermal nociception in Non-Avoiders or unstressed Controls, and that this effect is contingent on intact CeA neurotransmission. Furthermore, inactivation of CeA with TTX produced hyperalgesia in all stress groups, regardless of R121919 treatment. Intra-CeA TTX may have produced a floor (ie, maximal) hyperalgesic effect on hindpaw withdrawal latency. This floor effect may explain the lack of a stress and/or R121919 effects in animals treated with intra-CeA TTX. We also found that intra-CeA antagonism of CRFR1 reverses stress-induced thermal hyperalgesia in Avoiders without affecting thermal nociception in Non-Avoiders or unstressed Controls. These results implicate CRF-CRFR1 signaling in CeA as a potential mediator of hyperalgesia in humans diagnosed with PTSD.
The CeA is uniquely situated to mediate the cross-talk between pain and negative affective states. The CeA sends descending GABAergic projections to effector regions that mediate physiological and behavioral responses to noxious stimuli (Neugebauer et al, 2004; Bourgeais et al, 2001). Our current understanding of CeA circuitry suggests that increased activity of GABAergic projections to downstream effector regions would reduce physiological and behavioral responses to stress, whereas lower activity of these neurons would disinhibit neuronal firing in effector regions and thereby facilitate physiological and behavioral stress responses.
CRF signaling through CRFR1 increases CeA GABAergic transmission by facilitating pre-synaptic GABA release (Bourgeais et al, 2001), and also facilitates NMDA receptor-mediated EPSCs in laterocapuslar CeA (Fu and Neugebauer, 2008; Ji et al, 2013). Therefore, our observation that intra-CeA CRF injection produces hyperalgesia may be due to CRF-CRFR1 modulation of inhibitory and/or excitatory transmission. We also report that inactivation of CeA with TTX produces hyperalgesia in control rats, and abolishes the ability of a systemic CRFR1 antagonist to reverse stress-induced hyperalgesia. We interpret this effect to be attributable to blockade of action potentials in CeA projection neurons (along with upstream CeA interneurons), and this result may suggest that behavioral effects of CeA CRF are attributable to CRF-induced increases in GABAergic transmission, rather than CRF-induced decreases in glutamatergic transmission. The direction of our effect is not consistent with a past report that bilateral CeA lesion with ibotenic acid reverses footshock-induced hyperalgesia, although that study measured tail-flick during footshock exposure (Crown et al, 2000). The effects of CeA manipulations on nociception are likely dictated by whether these manipulations affect interneurons or projection neurons or both, and also by the extent and laterality of the lesion. For example, one prior study showed that unilateral optogenetic activation of right CeA promotes nociception in a mouse model of chronic bladder pain (Crock et al, 2012), whereas other work shows that bilateral CeA lesion blocks anti-nociception (ie, promotes nociception; Werka, 1994; Werka and Marek, 1990). In humans with PTSD, neuroimaging studies report higher amygdala activity at rest (Semple et al, 2000), and hyper-reactivity of the amygdala to trauma-related stimuli (Dickie et al, 2008; Morey et al, 2009; Rauch et al, 2000), although those studies lacked the resolution to isolate the CeA.
PAG projections to rostral ventromedial medulla (RVM) initiate descending modulation of incoming pain signals via RVM projections to spinal cord (Lau and Vaughan, 2014). This PAG-RVM-spinal pathway mediates the analgesic effects of opioids and cannabinoids, as well as stress-induced analgesia. Here, we show that stress-induced hyperalgesia is modulated by CRF-CRFR1 signaling in CeA, a region that projects heavily to PAG (Gray and Magnuson, 1992). Collectively, our data and prior work by other laboratories suggest that stress bi-directionally modulates nociception, and CeA projections to PAG are a strong candidate pathway for this bi-directional modulation (Butler and Finn, 2009).
Our results suggest that post-stress increases in CRF-CRFR1 signaling in CeA mediate thermal hyperalgesia in Avoider animals. Because CRF is produced in high quantities in CeA (McDonald, 1989; Justice et al, 2008) and is also imported from other regions (Uryu et al, 1992), it is not known whether local or distal CRF sources (or both) are the critical modulator of CeA projection neurons. Stress can trigger descending inhibition to induce anti-nociception, in order to enhance performance under conditions of actual or impending tissue damage (Millan, 2002). Our current data show that Avoiders exhibit higher total CRF peptide content in CeA, but not increases in CRF mRNA, CRFR1 mRNA, and CRF-positive cell counts in CeA, suggesting that stress may increase CRF release into CeA from distal afferents. However, unchanged mRNA levels do not necessarily rule out a role for locally synthesized CRF, as stress may alter mRNA stability. Another alternative explanation is that Avoiders may exhibit higher CRF content per neuron. Future studies will use anatomical and genetic strategies to identify the CeA CRF source (local or distal) that is altered by predator odor stress and important for hyperalgesia.
Interestingly, the CeA receives CRF afferents from various brain regions (Uryu et al, 1992; Swanson et al, 1983) that modulate pain sensitivity (Dafny et al, 1996) and/or in which neurotransmission is altered by painful stimuli (eg, lateral hypothalamus, dorsal raphe, and bed nucleus of stria terminalis). We also report that the CeL contains many more CRF-positive cells than the CeM, but stress did not affect CRF-positive cell counts in these two sub-nuclei CRFR1 antagonism inhibits neuronal excitability in the laterocapsular CeA (CeLC), and reduces pain-related behavior in rats with arthritic pain (Fu and Neugebauer, 2008). Facilitation of NMDA receptor-mediated excitatory transmission in CeLC is blocked by CRFR1 antagonism but not by CRFR2 antagonism, and also by a PKA but not by a PKC inhibitor (Ji et al, 2013), although another study shows that PKCɛ inhibitor blocks CRF-induced GABA release (Bajo et al, 2008).
Increases in CRF-CRFR1 signaling may reflect increases in CRF peptide and/or increases in CRFR1 sensitivity. Here, we present data to support both of these effects, as Avoiders had higher CRF peptide content in CeA and exhibited higher behavioral sensitivity to CRFR1 antagonism. The observed increase in CeA CRF appears to be behaviorally relevant as intra-CeA CRF infusions produce conditioned place avoidance and thermal hyperalgesia in otherwise naive rats. Neither systemically administered CRFR1 antagonist nor intra-CeA CRFR1 antagonist affected thermal nociception in Non-Avoiders or unstressed Controls, which suggests that stress increases tonic CRF-CRFR1 signaling specifically in Avoiders. Our observation that Avoiders exhibit increased sensitivity to a CRFR1 antagonist is consistent with previous reports that animals with arthritic pain exhibit increased sensitivity to CRFR1 antagonists, and that CRFR1 antagonism reduces pain-related behaviors in animals with chronic pain (Fu and Neugebauer, 2008). In summary, we conclude that traumatic stress produces hyperalgesia in some but not in all animals and humans, and that this effect is mediated by increases in CRF-CRFR1 signaling in CeA.
Funding and Disclosure
This work was supported by NIH grants AA018400, AA023305, and AA021013. The authors declare no conflict of interest.
References
Asmundson GJ, Katz J (2009). Understanding the co-occurrence of anxiety disorders and chronic pain: state-of-the-art. Depress Anxiety 26: 888–901.
Bajo M, Cruz MT, Siggins GR, Messing R, Roberto M (2008). Protein kinase C epsilon mediation of CRF- and ethanol-induced GABA release in central amygdala. Proc Natl Acad Sci USA 105: 8410–8415.
Bale TL, Vale WW (2004). CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol 44: 525–557.
Bourgeais L, Gauriau C, Bernard JF (2001). Projections from the nociceptive area of the central nucleus of the amygdala to the forebrain: a PHA-L study in the rat. Eur J Neurosci 14: 229–255.
Braz JM, Nassar MA, Wood JN, Basbaum AI (2005). Parallel ‘pain’ pathways arise from subpopulations of primary afferent nociceptor. Neuron 47: 787–793.
Butler RK, Finn DP (2009). Stress-induced analgesia. Prog Neurobiol 88: 184–202.
Crettaz B, Marziniak M, Willeke P, Young P, Hellhammer D, Stumpf A et al (2013). Stress-induced allodynia—evidence of increased pain sensitivity in healthy humans and patients with chronic pain after experimentally induced psychosocial stress. PLoS One 8: e69460.
Crock LW, Kolber BJ, Morgan CD, Sadler KE, Vogt SK, Bruchas MR et al (2012). Central amygdala metabotropic glutamate receptor 5 in the modulation of visceral pain. J Neurosci 32: 14217–14226.
Crown ED, King TE, Meagher MW, Grau JW (2000). Shock-induced hyperalgesia: III. Role of the bed nucleus of the stria terminalis and amygdaloid nuclei. Behav Neurosci 114: 561–573.
Dafny N, Dong WQ, Prieto-Gomez C, Reyes-Vazquez C, Stanford J, Qiao JT (1996). Lateral hypothalamus: site involved in pain modulation. Neuroscience 70: 449–460.
Defrin R, Ginzburg K, Solomon Z, Polad E, Bloch M, Govezensky M et al (2008). Quantitative testing of pain perception in subjects with PTSD—implications for the mechanism of the coexistence between PTSD and chronic pain. Pain 138: 450–459.
Dickie EW, Brunet A, Akerib V, Armony JL (2008). An fMRI investigation of memory encoding in PTSD: influence of symptom severity. Neuropsychologia 46: 1522–1531.
Dina OA, Levine JD, Green PG (2011). Enhanced cytokine-induced mechanical hyperalgesia in skeletal muscle produced by a novel mechanism in rats exposed to unpredictable sound stress. Eur J Pain 15: 796–800.
Edwards S, Baynes BB, Carmichael CY, Zamora-Martinez ER, Barrus M, Koob GF et al (2013). Traumatic stress reactivity promotes excessive alcohol drinking and alters the balance of prefrontal cortex-amygdala activity. Transl Psychiatry 3: e296.
Fu Y, Neugebauer V (2008). Differential mechanisms of CRF1 and CRF2 receptor functions in the amygdala in pain-related synaptic facilitation and behavior. J Neurosci 28: 3861–3876.
Gilpin NW, Herman MA, Roberto M (2015). The central amygdala as an integrative hub for anxiety and alcohol use disorders. Biol Psychiatry 77: 859–869.
Gray TS, Magnuson DJ (1992). Peptide immunoreactive neurons in the amygdala and the bed nucleus of the stria terminalis project to the midbrain central gray in the rat. Peptides 13: 451–460.
Heilig M, Koob GF, Ekman R, Britton KT (1994). Corticotropin-releasing factor and neuropeptide Y: role in emotional integration. Trends Neurosci 17: 80–85.
Huang MM, Overstreet DH, Knapp DJ, Angel R, Wills TA, Navarro M et al (2010). Corticotropin-releasing factor (CRF) sensitization of ethanol withdrawal-induced anxiety-like behavior is brain site specific and mediated by CRF-1 receptors: relation to stress-induced sensitization. J Pharmacol Exp Ther 332: 298–307.
Jasmin L, Burkey AR, Card JP, Basbaum AI (1997). Transneuronal labeling of a nociceptive pathway, the spino-(trigemino-)parabrachio-amygdaloid, in the rat. J Neurosci 17: 3751–3765.
Ji G, Fu Y, Adwanikar H, Neugebauer V (2013). Non-pain-related CRF1 activation in the amygdala facilitates synaptic transmission and pain responses. Mol Pain 9: 1–16.
Johnson AC, Tran L, Greenwood-Van Meerveld B (2015). Knockdown of corticotropin-releasing factor in the central amygdala reverses persistent viscerosomatic hyperalgesia. Transl Psychiatry 5: e517.
Justice NJ, Yuan ZF, Sawchenko PE, Vale W (2008). Type 1 corticotropin-releasing factor receptor expression reported in BAC transgenic mice: implications for reconciling ligand-receptor mismatch in the central corticotropin-releasing factor system. J Comp Neurol 511: 479–496.
Karanikas CA, Lu Y-L, Richardson HN (2013). Adolescent drinking targets corticotropin-releasing factor peptide-labeled cells in the central amygdala of male and female rats. Neuroscience 249: 98–105.
Latremoliere A, Woolf CJ (2009). Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain 10: 895–926.
Lau BK, Vaughan CW (2014). Descending modulation of pain: the GABA disinhibition hypothesis of analgesia. Curr Opin Neurobiol 29: 159–164.
Lew HL, Otis JD, Tun C, Kerns RD, Clark ME, Cifu DX (2009). Prevalence of chronic pain, posttraumatic stress disorder, and persistent postconcussive symptoms in OIF/OEF veterans: polytrauma clinical triad. J Rehabil Res Dev 46: 697–702.
Li C, Yang Y, Liu S, Fang H, Zhang Y, Furmanski O et al (2014). Stress induces pain transition by potentiation of AMPA receptor phosphorylation. J Neurosci 34: 13737–13746.
McDonald AJ (1989). Coexistence of somatostatin with neuropeptide Y, but not with cholecystokinin or vasoactive intestinal peptide, in neurons of the rat amygdala. Brain Res 500: 37–45.
Millan MJ (2002). Descending control of pain. Prog Neurobiol 66: 355–474.
Moeller-Bertram T, Keltner J, Strigo IA (2012). Pain and post traumatic stress disorder - review of clinical and experimental evidence. Neuropharmacology 62: 586–597.
Morey RA, Dolcos F, Petty CM, Cooper DA, Hayes JP, LaBar KS et al (2009). The role of trauma-related distractors on neural systems for working memory and emotion processing in posttraumatic stress disorder. J Psychiatr Res 43: 809–817.
Neugebauer V, Li W (2003). Differential sensitization of amygdala neurons to afferent inputs in a model of arthritic pain. J Neurophysiol 89: 716–727.
Neugebauer V, Li W, Bird GC, Han JS (2004). The amygdala and persistent pain. Neuroscientist 10: 221–234.
Oliveira MA, Prado WA (2001). Role of PAG in the antinociception evoked from the medial or central amygdala in rats. Brain Res Bull 54: 55–63.
Orr SP, Metzger LJ, Lasko NB, Macklin ML, Peri T, Pitman RK (2000). De novo conditioning in trauma-exposed individuals with and without posttraumatic stress disorder. J Abnorm Psychol 109: 290–298.
Paxinos G, Watson C (2005) The Rat Brain in Stereotaxic Coordinates. Elsevier Academic Press: San Diego, CA, USA.
Rauch SL, Whalen PJ, Shin LM, McInerney SC, Macklin ML, Lasko NB et al (2000). Exaggerated amygdala response to masked facial stimuli in posttraumatic stress disorder: a functional MRI study. Biol Psychiatry 47: 769–776.
Regev L, Tsoory M, Gil S, Chen A (2012). Site-specific genetic manipulation of amygdala corticotropin-releasing factor reveals its imperative role in mediating behavioral response to challenge. Biol Psychiatry 71: 317–326.
Roltsch EA, Baynes BB, Mayeux JP, Whitaker AM, Baiamonte BA, Gilpin NW (2014). Predator odor stress alters corticotropin-releasing factor-1 receptor (CRF1R)-dependent behaviors in rats. Neuropharmacology 79: 83–89.
Sakanaka M, Shibasaki T, Lederis K (1986). Distribution and efferent projections of corticotropin-releasing factor-like immunoreactivity in the rat amygdaloid complex. Brain Res 382: 213–238.
Semple WE, Goyer PF, McCormick R, Donovan B, Muzic RF Jr, Rugle L et al (2000). Higher brain blood flow at amygdala and lower frontal cortex blood flow in PTSD patients with comorbid cocaine and alcohol abuse compared with normals. Psychiatry 63: 65–74.
Shi C, Davis M (1999). Pain pathways involved in fear conditioning measured with fear-potentiated startle: lesion studies. J Neurosci 19: 420–430.
Shipherd JC, Keyes M, Jovanovic T, Ready DJ, Baltzell D, Worley V et al (2007). Veterans seeking treatment for posttraumatic stress disorder: what about comorbid chronic pain? J Rehabil Res Dev 44: 153–166.
Swanson LW, Sawchenko PE, Rivier J, Vale WW (1983). Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology 36: 165–186.
Werka T (1994). Post-stress analgesia in rats with partial amygdala lesions. Acta Neurobiol Exp 54: 127–132.
Werka T, Marek P (1990). Post-stress analgesia after lesions to the central nucleus of the amygdala in rats. Acta Neurobiol Exp (Wars) 50: 13–22.
Whitaker AM, Gilpin NW (2015). Blunted hypothalamo-pituitary adrenal axis response to predator odor predicts high stress reactivity. Physiol Behav 147: 16–22.
Uryu K, Okumura T, Shibasaki T, Sakanaka M (1992). Fine structure and possible origins of nerve fibers with corticotropin-releasing factor-like immunoreactivity in the rat central amygdaloid nucleus. Brain Res 577: 175–179.
Acknowledgements
We thank Dimitri Grigoriadis of Neurocrine for generous donation of R121919 and Dr Wylie Vale of the Salk Institute for his generous donation of the CRF antisera. We thank Marina Shirokova and Dimitra Gomes for their technical assistance with these studies. Dr Gilpin is a consultant for Glauser Life Sciences.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Supplementary information
Rights and permissions
About this article

Cite this article
Itoga, C., Roltsch Hellard, E., Whitaker, A. et al. Traumatic Stress Promotes Hyperalgesia via Corticotropin-Releasing Factor-1 Receptor (CRFR1) Signaling in Central Amygdala. Neuropsychopharmacol 41, 2463–2472 (2016). https://doi.org/10.1038/npp.2016.44
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2016.44
This article is cited by
-
Traumatic Stress-Enhanced Alcohol Drinking: Sex Differences and Animal Model Perspectives
Current Addiction Reports (2024)
-
Examining sex differences in responses to footshock stress and the role of the metabotropic glutamate receptor 5: an [18F]FPEB and positron emission tomography study in rats
Neuropsychopharmacology (2023)
-
Effects of Glucocorticoid Hormones on Pain Sensitivity: Involvement of Glucocorticoid and Mineralocorticoid Receptors
Neuroscience and Behavioral Physiology (2023)
-
Rodent models of post-traumatic stress disorder: behavioral assessment
Translational Psychiatry (2020)
