
Abstract
The brain and the immune system are not fully formed at birth, but rather continue to mature in response to the postnatal environment. The two-way interaction between the brain and the immune system makes it possible for childhood psychosocial stressors to affect immune system development, which in turn can affect brain development and its long-term functioning. Drawing from experimental animal models and observational human studies, we propose that the psychoneuroimmunology of early-life stress can offer an innovative framework to understand and treat psychopathology linked to childhood trauma. Early-life stress predicts later inflammation, and there are striking analogies between the neurobiological correlates of early-life stress and of inflammation. Furthermore, there are overlapping trans-diagnostic patterns of association of childhood trauma and inflammation with clinical outcomes. These findings suggest new strategies to remediate the effect of childhood trauma before the onset of clinical symptoms, such as anti-inflammatory interventions and potentiation of adaptive immunity. Similar strategies might be used to ameliorate the unfavorable treatment response described in psychiatric patients with a history of childhood trauma.
Similar content being viewed by others
Introduction
The brain and the immune system are not fully formed at birth. Rather, they both emerge from fetal life with a minimal set of functions that enable newborns’ adaptation to a limited number of expected stimuli. Through experience in early phases of postnatal development, the brain and the immune system progressively expand their repertoire presumably to maximize adaptation to stimuli that are specific to the individual's unique environment (Bateson et al, 2004; Danese and McEwen, 2012; Greenough et al, 1987). Although these phenomena of developmental plasticity have been best studied with regard to the basic brain functions, such as vision (Hensch, 2005; Hubel and Wiesel, 2005), they are increasingly recognized as key determinants of complex brain and immune functions (Danese and McEwen, 2012; Rook et al, 2015). Here we will review evidence suggesting that early experiences are also important to set up long-term patterns of interaction between the brain and the immune system. Several excellent reviews have described how early-life infection and immune stimulation can affect brain development and exert long-term influence on psychopathology (Bilbo and Schwarz, 2009; Knuesel et al, 2014; Patterson, 2009). In this review, we describe how early-life stress can influence the developing immune system and, thereby, risk of psychopathology (Box 1).
Childhood trauma and inflammation
Experimental Studies in Animal Models
Because the development of the immune system is not completed at birth, but rather continues throughout childhood, environmental stimulation in childhood years can have profound effects on the immune system (Box 1). Initial evidence for the effect of psychosocial childhood experiences on immune system functioning emerged more than half a century ago from experimental animal models. Robert Ader and colleagues found that rats handled before weaning showed slower development of a transplanted tumor (Ader and Friedman, 1965). Furthermore, handled rats had greater serum antibody titer in response to flagellin, a protein from bacterial flagella (Solomon et al, 1968).
These seminal findings sparked interest in the field of developmental psychoneuroimmunology (Ader, 1983; Coe, 1996; Danese, 2014). Several studies have subsequently investigated the association between early-life stress and later immune functioning in rodents and non-human primates (for detailed reviews, see Ganguly and Brenhouse, 2015; Hennessy et al, 2010a; Shanks and Lightman, 2001). Manipulation of stress exposure in early life (ie, across postnatal days 1–20 in rodents) has been undertaken through multiple experimental models previously linked to heterogeneous effects on mothers' caregiving behavior and stress reactivity, including neonatal handling, maternal separation, maternal deprivation, nursery rearing, early weaning, and dexamethasone treatment. For example, although neonatal handling is associated with the increased maternal licking and grooming upon reunion and decreased stress reactivity in rats, maternal separation is associated with reduced maternal caregiving upon reunion and increase stress reactivity. Measures of immune functioning have included very different markers ranging from unstimulated pro-inflammatory cytokines in the plasma, to antigen-induced immunoproliferation in the spleen, to expression levels of pro-inflammatory genes in the brain, and intestinal microflora. Unsurprisingly, this mixture of designs and measures has produced mixed results with regard to the links between early-life stress and immunity (Ganguly and Brenhouse, 2015; Hennessy et al, 2010a; Shanks and Lightman, 2001). A clearer picture arguably emerges when looking at studies of maternal separation. In non-human primates, maternal separation led to an increase in macrophage activity (Coe et al, 1988) and long-term upregulation in pro-inflammatory gene transcription in monocytes (Cole et al, 2012). In rodents, maternal separation led to an increase in core temperature (Hennessy et al, 2010b) and in pro-inflammatory cytokines in the plasma (Wieck et al, 2013). These studies suggest that early-life stress resulting from the disruption of mother and child interaction is associated with inflammatory processes in later life.
Experimental studies in animal models have also highlighted a link between early-life stress and markers of immune function in the central nervous system (CNS)—an apparently sexually dimorphic phenomenon with greater effect sizes in male animals (Ganguly and Brenhouse, 2015). In pups, at a time when brain development is taking place (Box 2), maternal separation was found to be associated with blunted expression of pro-inflammatory mediators (lipopolysaccharide-binding protein) in the hippocampus (Wei et al, 2012) and with reduced microglial cell number in midbrain areas (Chocyk et al, 2011). In contrast, in adult animals, early maternal separation was associated with greater synaptic levels of the receptor for the pro-inflammatory cytokine interleukin-1 (IL-1) (Viviani et al, 2014), greater number and motility of cortical microglial processes (Takatsuru et al, 2015), and greater microglia activation (Brenhouse and Thompson, 2015).
Of note, early-life stress also appears to be associated with greater reactivity to subsequent psychosocial and immune challenges. For example, rodents exposed to maternal separation showed a greater increase in core temperature upon a second separation (Hennessy et al, 2010b), greater cytokine expression in response to subsequent viral infection (Avitsur et al, 2006), and greater cortical microglial activation following subsequent exposure to chronic food-restriction stress (Brenhouse and Thompson, 2015).
Observational Studies in Humans
By manipulating the early environment and randomly assigning animals to experiences, experimental studies have been pivotal in informing causal inference regarding the links between early-life stress and inflammation. Furthermore, animal models enabled researchers to use invasive measures to investigate the links between early-life stress and neuroinflammation (ie, the inflammation of the CNS), an area of investigation that is only starting to be applied to clinical populations (Bloomfield et al, 2016; Setiawan et al, 2015). Despite these two crucial advantages, the use of proxy models of human experiences and of brain functioning or behavior have important limitations (Clancy et al, 2007; van der Worp et al, 2010). Of note, although experimental studies have stronger internal validity over observational studies, generalization of causal inference from the narrow context of the experiment to the life of humans requires demonstration (or assumption) that the animals and their physiology are representative of human physiology and experiences (ie, external or ecological validity). In other words, experimental animal models have shown that early-life stress can affect later immune functioning, but observational human studies are needed to test if childhood trauma does affect later immune functioning in ordinary people. Below, we review key papers that have investigated the association between traumatic experiences in childhood (generally defined as pre-pubertal exposure) and immune abnormalities.
We have investigated the association between childhood trauma and plasma inflammation biomarkers among 1037 members of the Dunedin Multidisciplinary Health and Development Study (Poulton et al, 2015). Study members have been followed since their birth in Dunedin, New Zealand, in 1972–1973 through repeated waves of assessment until adult life with very low attrition. This design enabled us to undertake a longitudinal-prospective assessment of maltreatment experiences in the first decade of life as study members grew up, including behavioral observations, parental reports, and retrospective reports by study members once they reached adulthood. Furthermore, at age 32 years we measured plasma inflammation biomarkers, namely C-reactive protein (CRP), fibrinogen, and white blood cell count. We found that cumulative exposure to childhood maltreatment was associated with significant and graded elevation in inflammation levels 20 years later (Danese et al, 2007). This association generalized to all three clinically relevant inflammation biomarkers available. The association was not explained by key potential confounders, such as low birth weight, disadvantaged socio-economic conditions of the family, and low IQ, or by potential mediators, such as adult stressors, poor adult health, unhealthy behaviors, or acute infections at the time of inflammation assessment. Two dozen independent studies have since tested this association, and qualitative and quantitative reviews found reliable evidence of small elevations in inflammation biomarkers in maltreated individuals (Baumeister et al, 2015; Coelho et al, 2014). Of note, elevated inflammation levels are observed not only after maltreatment by adults, but also after other common and severe childhood stressors, such as bullying by peers (Takizawa et al, 2015). Finally, childhood maltreatment may also be associated with impaired acquired immunity (Shirtcliff et al, 2009).
The association between maltreatment and inflammation is already detectable in childhood years. We tested this association in 170 members of the Environmental-Risk (E-Risk) Longitudinal Twin Study. Study members were born in England and Wales, and have been followed since their birth in 1994–1995 through repeated waves of assessment until adolescence. We again undertook a longitudinal-prospective assessment of maltreatment experiences during childhood years. Furthermore, at the age 12 years, we measured an inflammation biomarker in blood drops collected on filtered paper (CRP in dried blood spots). We found that inflammation levels were already elevated in maltreated children who developed depression by age 12 years compared to controls (Danese et al, 2011). Subsequent studies have replicated and expanded these initial findings. For example, high inflammation levels were found in a large cohort of children and adolescents exposed to adverse childhood experiences (Slopen et al, 2013) and among children with Child Protective Service-documented history of maltreatment and genetic liability to high inflammation levels (Cicchetti et al, 2015).
Childhood maltreatment also predicts greater reactivity to subsequent psychosocial challenges. Physiological response to acute psychosocial stressors can be tested through experimental procedures, such as socially evaluated public speaking and mental arithmetical calculation tasks (the Trier Social Stress Test, or TSST; Kirschbaum et al, 1993). This and similar procedures have been shown to induce an acute inflammatory response in humans (Steptoe et al, 2007a). Of note, patients with major depression and history of childhood maltreatment showed greater increase in IL-6 levels and greater NF-kappaB DNA binding (indicating genomic activation of inflammatory pathways) in response to TSST compared to controls (Pace et al, 2006). Similarly, adults with a history of childhood maltreatment had greater inflammatory response to the TSST compared to individuals with no maltreatment history (Carpenter et al, 2010). Furthermore, adults with a history of maltreatment showed elevated inflammation levels in the context of depression, daily stressors, or caregiving stress in later life compared to controls (Danese et al, 2008; Gouin et al, 2012; Kiecolt-Glaser et al, 2011).
Potential mechanisms through which childhood trauma could trigger inflammation are portrayed in Figure 1.

Potential mechanisms linking childhood trauma and inflammation. Panel (a) portrays potential mechanisms through which exposure to childhood trauma could elicit an acute inflammatory response. On the one hand, exposure to acute psychological stress can elicit an acute inflammatory response by stimulation of the sympathetic nervous system (Bierhaus et al, 2003; Steptoe et al, 2007b). On the other hand, psychological trauma may occur in the context of physical trauma. In this case, physical injury and pathogen infection can induce inflammation by triggering innate immunity (Medzhitov and Janeway, 2000). Short-term activation of the inflammatory response during sensitive periods in early life may affect the brain development and later microglia and neuroendocrine reactivity (Figure 2). Panel (b) portrays potential biological and behavioral mechanisms through which response to childhood trauma could lead to a chronic inflammatory state. First, early-life stress may be indirectly linked to inflammation because of primary neuroendocrine abnormalities in the hypothalamic-pituitary-adrenal (HPA) axis. Childhood trauma is associated with later hyperactive HPA axis functioning (Danese and McEwen, 2012; Gunnar and Quevedo, 2007; Heim and Nemeroff, 2001; Lupien et al, 2009), presumably because of primary abnormalities of the glucocorticoid receptor. Both preclinical and clinical studies have shown that early-life stress is associated with epigenetic changes, leading to insufficient glucocorticoid signaling, namely, impaired functioning of the glucocorticoid-receptor-mediated signaling (Weaver et al, 2004; Klengel et al, 2013). These changes could, in turn, induce resistance to the anti-inflammatory properties of cortisol and, thus, high inflammation levels (Heim et al, 2000; Miller et al, 2002; Raison and Miller, 2003). However, because of the bi-directional association between HPA axis functioning and inflammation, it is also possible that primary inflammatory abnormalities could stimulate HPA axis activity (Besedovsky et al, 1986) and induce glucocorticoid resistance (Barnes and Adcock, 2009). Second, it is possible that early-life stress can influence the composition of the gut microbiota (Bailey and Coe, 1999; O'Mahony et al, 2009). In turn, gut dysbiosis could influence brain function through stimulation of the vagus nerve and other metabolic effects (Cryan and Dinan, 2012). Third, early-life stress is associated with hormonal and brain abnormalities that could contribute to a ‘thrifty’ phenotype characterized by increased energy intake and storage, and/or reduced energy expenditure resulting in obesity (Danese and Tan, 2014; Danese et al, 2014). In turn, obesity is associated with high systemic inflammation through the production of pro-inflammatory cytokines by adipocytes (Gregor and Hotamisligil, 2011). Fourth, early-life stress has been linked to alcohol and substance abuse disorders and smoking (Dube et al, 2003; Anda et al, 1999), which can increase inflammation levels (Crews et al, 2006; Shiels et al, 2014). Fifth, early-life stress has been associated with decreased total sleep and disruption in sleep architecture in rodents (Feng et al, 2007; Mrdalj et al, 2013; Tiba et al, 2004) and humans (Gregory and Sadeh, 2016; Kajeepeta et al, 2015). In turn, clinical experiments have shown that sleep deprivation is associated with an increase in the expression of pro-inflammatory cytokines in humans (Irwin et al, 2006), and epidemiological studies have found that reduced sleep is associated with elevated levels of inflammation biomarkers (Miller et al, 2009). Finally, early-life stress is associated with later abnormalities in brain functioning and behavior (Danese and McCrory, 2015). In particular, persistent or recurrent distress and self-harming behaviors described in individuals with a history of childhood trauma could contribute to maintaining elevated inflammation levels (Cohen et al, 2012; Molina, 2005).
Inflammation and psychopathology
Experimental Studies in Animal Models and Humans
Evidence of an association between childhood trauma and inflammation raises the possibility that inflammation could help explain the pathophysiology of trauma-related psychopathology. Indeed, inflammation is increasingly seen as a key determinant of brain development and functioning, as discussed below.
Initial evidence of links between the immune system and psychopathology, and particularly affective disorders, came from the recognition that animals and humans with acute infectious diseases developed a stereotypical pattern of behaviors characterized by lethargy, despair, anorexia, and reduction in grooming (Hart, 1988). Consistent with the hypothesized inflammatory origins of this ‘sickness behavior’, systemic or central administration of bacterial cells’ components (eg, lipopolysaccharide, or LPS, also known as endotoxin) or pro-inflammatory cytokines (eg, interleukin-1beta, or IL-1beta, and tumor necrosis factor-α, or TNF-α) could trigger a similar cluster of symptoms (Dantzer et al, 2008). Finally, expression of sickness behavior in response to pro-inflammatory stimuli was buffered by administration of the anti-inflammatory cytokine IL-10 and exacerbated in IL-10-deficient mice (Dantzer et al, 2008).
Similarly, administration of pro-inflammatory compounds in humans can induce cardinal mood symptoms. Depression-free patients with hepatitis C or cancer who were treated with interferon-α (IFN-α) to boost their immune system developed high rates of depression within weeks—about half of patients met criteria for a diagnosis of major depression and nearly all developed neurovegetative symptoms, including anorexia, fatigue, psychomotor retardation, and disrupted sleep (Musselman et al, 2001). Qualitatively similar but transient symptoms can also be induced by acute administration of pro-inflammatory mediators, such as typhoid vaccination and LPS (Reichenberg et al, 2001).
Additional experimental support for the role of inflammation in the pathophysiology of affective disorders comes from the evidence of anti-depressant effects of anti-inflammatory medications. A recent meta-analysis of 10 trials with non-steroidal anti-inflammatory drugs and four trials with cytokines inhibitors found small-to-moderate anti-depressant effects of these interventions (Köhler et al, 2014). Results from the meta-analyses and individual clinical trials also highlight the complexity of depression. There are several pathophysiological mechanisms leading to depression, and inflammation may be a key mechanism in a subset of depressed patients, who will then benefit from anti-inflammatory medications (Raison et al, 2013). However, anti-inflammatory medications may be ineffective or even detrimental in other cases of depression, where inflammation is not a key pathophysiological mechanism (Raison et al, 2013).
Observational Studies in Humans
Building on this experimental evidence, epidemiological and clinical studies in humans over the past 20–30 years have highlighted the role of the immune system in the pathophysiology of several psychiatric conditions, including both affective and psychotic disorders.
Further to the findings on sickness behavior (Hart, 1988), initial evidence for the role of inflammation in depression came from the observation that experimental administration of pro-inflammatory cytokines produced a clinical response resembling major depression (the ‘macrophage theory’ of depression; Smith, 1991) and that depressed patients showed systemic immune activation (Maes et al, 1992). Meta-analyses of clinical and population studies showed that depressed individuals have a small elevation in levels of several inflammation biomarkers (Howren et al, 2009). Longitudinal studies found that the elevated inflammation levels observed in depressed individuals likely result from bi-directional associations between depression and inflammation over time (Matthews et al, 2010). Of note, elevation in inflammation levels is more obvious in depressed patients with recurrent course of illness (Ford and Erlinger, 2004) and those who are resistant to conventional treatment strategies (Strawbridge et al, 2015).
Patients with bipolar disorder also have elevated inflammation levels (Leboyer et al, 2012). Meta-analyses of clinical studies showed that, compared to controls, bipolar patients have small-to-moderate elevation in levels of pro-inflammatory cytokines (Modabbernia et al, 2013) and of CRP (Dargél et al, 2015). Systemic inflammation can be observed not only during symptomatic episodes, but also in euthymic phases (Dargél et al, 2015), suggesting that inflammation may be a latent vulnerability factor (or trait markers) rather than a (state) marker of disease severity. Indeed, it has been suggested that inflammation may contribute not only to the onset of bipolar disorder but also to its progression (Berk et al, 2011).
The contribution of immune abnormalities to the pathophysiology of schizophrenia has long been debated (Khandaker et al, 2015). Meta-analyses of clinical studies showed that patients with schizophrenia have moderate-to-large elevation in levels of pro-inflammatory cytokines (Miller et al, 2011) and of CRP (Fernandes et al, 2016). Again, several inflammation biomarkers examined appeared to be trait markers, because their levels were found to be elevated both in first-episode psychosis and in chronic psychotic disorders, and did not reduce following antipsychotic treatment (Fernandes et al, 2016; Miller et al, 2011). Furthermore, initial evidence suggests that elevated baseline levels of inflammation predict poor treatment response in first-episode psychosis patients (Mondelli et al, 2015).
Individuals who develop post-traumatic stress disorder (PTSD) also show elevated inflammation levels. Meta-analyses showed that PTSD patients have moderate-to-large elevation in several pro-inflammatory cytokines (Passos et al, 2015). Of note, these associations were not simply explained by comorbid depression (Passos et al, 2015). In addition, genetic (Michopoulos et al, 2015) and longitudinal (Eraly et al, 2014) studies suggest that inflammation is a pre-existing vulnerability factor for the development of PTSD in trauma-exposed individuals rather than simply a correlate of subjective distress, disease severity, or maladaptive coping strategies following PTSD onset.
Key points emerge from observational human studies on the relationships between inflammation and psychiatric disorders. First, because elevated inflammation levels were observed in several groups of patients, inflammation appears to have trans-diagnostic effects on the brain likely resulting from the effect of inflammation on basic brain processes. Because of these trans-diagnostic effects, the frequent comorbidity of psychiatric diagnoses, and the prevailing focus of published studies on individual psychiatric diagnoses, studies so far may have overestimated the unique contribution of inflammation to each individual diagnosis. Second, several meta-analyses found significant levels of between-study heterogeneity, suggesting that not all patients with the above psychiatric diagnoses have elevated inflammation. It is, therefore, important to identify correlates of elevated inflammation in psychiatric patients and target interventions accordingly (Danese et al, 2008, 2011; Raison et al, 2013). Third, inflammation is not simply related to incident cases of psychiatric disorders (ie, the risk of psychopathology). In contrast, inflammation is in many cases seen as a trait marker of vulnerability linked to unfavorable longitudinal course and poor treatment response. Therefore, understanding and addressing the origins of inflammation in psychiatric patients can have important treatment implications (Figure 1).
How Does Inflammation Affect Risk for Psychopathology?
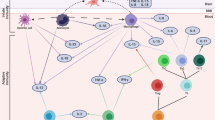
In light of the association between inflammation and psychopathology reported in animal models and humans, we describe below four broad mechanisms through which systemic immune activation can affect risk for psychopathology. Mechanisms include the effects of inflammation on concurrent brain function, the effects of early immune activation on brain development, the sensitization of immune brain cells to subsequent psychosocial stressors, and the cross-sensitization of the HPA axis response to subsequent psychosocial stressors (Figure 2). A better understanding of these mechanisms has largely emerged from the increasing appreciation that the traditional view of the brain as an ‘immune privileged’ organ—namely, that the brain is both immunologically inert and immunologically separated from the peripheral immune system—is incorrect (Galea et al, 2007).

Potential mechanisms linking childhood trauma-related inflammation to brain functioning and psychopathology. Mechanisms include the effects of inflammation on concurrent brain function, the effects of early immune activation on brain development, the sensitization of immune brain cells to subsequent psychosocial stressors, and the cross-sensitization of the HPA axis response to subsequent psychosocial stressors.
The brain is not immunologically inert. The activity of the brain immune system contributes to normal brain functioning (Yirmiya and Goshen, 2011). For example, microglia continuously scan the surrounding extracellular space for signals of cellular damage or infection and can respond by repairing micro-damages, eliminating cellular debris and dysfunctional synaptic structures, and supporting neurogenesis (Hanisch and Kettenmann, 2007). Auto-reactive T cells in the cerebrospinal fluid (CSF) monitor and respond to signals released from the brain and contribute to neurogenesis by producing the anti-inflammatory cytokine IL-4 (Kipnis et al, 2012). Low levels of pro-inflammatory cytokines produced locally or accumulated from the periphery (see below) are important to facilitate molecular mechanisms of brain plasticity (eg, long-term potentiation and depression; Yirmiya and Goshen, 2011). Despite the supportive roles of the immune system in normal brain physiology, systemic immune activation linked to inflammation or infections can cause significant brain damage. During systemic immune activation, ‘activated’ microglia (M1) respond through phagocytosis and/or by production of pro-inflammatory mediators. Although these mediators are functional in eliminating the triggering stimuli, they can also exert neurotoxic effects and suppress neurogenesis (Hanisch and Kettenmann, 2007). T cells can potentiate the immune response to pathogens and amplify the inflammatory response, ultimately increasing neurotoxicity (Kipnis et al, 2012). Finally, cytokines can have neurotoxic effects (eg, by stimulating the production of reactive oxygen species), reduce monoamine transmission, potentiate glutamate transmission (and excitotoxicity), and inhibit neurogenesis (Miller and Raison, 2015).
The brain is not immunologically separated from the peripheral immune system. In contrast, there are several pathways that ensure effective links between the peripheral immune system and the brain immune system (Dantzer et al, 2008). On the one hand, signals from peripheral cytokines can be translated into CNS signals in several ways. Peripheral cytokines can stimulate afferent fibers of the vagus nerve, which in turn can trigger cytokine production within the brain. Peripheral cytokines can also cross into the brain where the blood–brain barrier (BBB) is permeable or leaky (eg, area postrema) and can be carried through the BBB saturable, active transporters. Finally, peripheral cytokines can engage cell surface receptors on astrocytes and endothelial cells that form the BBB, which in turn can trigger cytokines production by these cells within the brain. On the other hand, a small number of peripheral immune cells—dendritic cells, leukocytes, and lymphocytes—may pass through the BBB, circumventricular organs, and the chorioid plexus. As a testament to the presence and functional role of these pathways, peripheral inflammation can induce neuroinflammation (Dantzer et al, 2008; Perry et al, 2003). For example, peripheral administration of LPS induces the expression of pro-inflammatory cytokines in the brain (Breder et al, 1994), the activation of the microglia (Monje et al, 2003), and the inhibition of adult neurogenesis (Monje et al, 2003).
Inflammation and Brain Function
Because the brain is neither immunologically inert nor immunologically separated from the peripheral immune system, peripheral/systemic immune activation exerts immediate effects on concurrent brain function, and in particular on domains (Research Domain Criteria; Insel, 2014) of cognition, positive valence, and negative valence. These effects have been investigated through experimental stimulation with of pro-inflammatory mediators, such as LPS, typhoid vaccine, and IFN-α and comprehensively described elsewhere (Miller and Raison, 2015; Schedlowski et al, 2014). For example, peripheral administration of LPS decreases verbal and nonverbal memory in humans (Reichenberg et al, 2001). Peripheral administration of different pro-inflammatory mediators reduces ventral striatum responses to reward (Capuron et al, 2012; Eisenberger et al, 2010). Peripheral administration of LPS potentiates amygdala activity in response to socially threatening stimuli (fear faces) (Inagaki et al, 2012), and typhoid vaccine reduces the connectivity of the subgenual anterior cingulate cortex (sACC) with the amygdala and increases the activity within the sACC during emotional face processing (Harrison et al, 2009; Box 2).
Early-Life Immune Activation and Brain Development
Early-life immune activation can influence brain development. Brain development does not end at birth, but rather continues into young adulthood and beyond (Box 2). The immune system modulates cellular events across different phases of the normal brain development (Boulanger, 2009; Deverman and Patterson, 2009), and here we give an overview of key immune influences on postnatal cellular events. With regard to synaptogenesis, the chemokine CXCL12 and its receptor CXCR4 regulate axonal elongation, branching, and pathfinding by modulating neuronal responses to axon guidance cues (eg, Slit-2, semaphorin 3A, and semaphorin 3C; Chalasani et al, 2003; Tran and Miller, 2003), and CXCR4 and CXCL12 mutant mice show disrupted axonal migration (Lieberam et al, 2005). With regard to synaptic refinement, the immune system modulates the strength of neuronal excitatory synapses to stabilize firing in the context of varying neuronal activity (ie, synaptic scaling). Microglia and astrocytes can sense the overall levels of neuronal activity and produce compensatory changes in synaptic strength by secreting TNF-α. Low synaptic activity induces secretion of TNF-α, which in turn promotes the surface expression of glutamate receptors in neurons increasing excitability and, thus, synaptic strength (Beattie et al, 2002; Stellwagen and Malenka, 2006). Furthermore, the surface expression of the major histocompatibility complex class-I is necessary for the regulation of both basal synaptic transmission and of acute synaptic plasticity (long-term potentiation and depression; Huh et al, 2000; Shatz, 2009). The immune system is also involved in activity-dependent mechanisms that regulate synaptic survival (ie, synaptic pruning). Inactive synapses express the complement proteins C1q and C3 (Stephan et al, 2012), and microglia recognize and phagocytize the complement-tagged synapses that are in this way eliminated (Paolicelli et al, 2011). With regard to myelination, microglia exert an important role in the survival and functioning of oligodendrocytes and, thus, myelin production. Stressed oligodendrocytes can recruit microglia through secretion of chemokines, cytokines, and other mediators. In turn, microglia can remove myelin debris and promote the differentiation of oligodendrocyte progenitor cell into mature myelinating oligodendrocytes (Peferoen et al, 2014). With regard to adult neurogenesis, this is inhibited by microglia activation induced through peripheral administration of LPS or radiation, effects that are minimized by systemic administration of anti-inflammatory drugs (Ekdahl et al, 2003; Monje et al, 2003). In addition, induction of adult neurogenesis by enriched environment was markedly impaired in immune-deficient mice and restored by T cells directed to a brain auto-antigen (Ziv et al, 2006).
Because of the important roles that the immune system has in regulating key cellular events during the brain development, postnatal early-life immune activation involving disruption of these fine-tuned processes could exert direct, long-term influences on brain and behavior. Consistent with this prediction, experimental studies of infection (eg, influenza virus and Escherichia coli) and systemic inflammation (eg, synthetic RNA poly(I:C) and LPS) during prenatal or neonatal periods found stable, long-term impairments in cognition, including learning, memory, and attention in rodents (Bilbo and Schwarz, 2009; Knuesel et al, 2014; Patterson, 2009) and non-human primates (Short et al, 2010). In line with these findings in animal models, elevated levels of systemic inflammation in children have been associated with an increased risk of developing depression and psychosis in young adulthood (Khandaker et al, 2014).
Early-Life Immune Activation and Glial Priming
In addition to its effects on concurrent brain function and on the brain development, early-life immune activation can bias microglia response to subsequent inflammatory stimuli (Perry and Holmes, 2014). By priming the microglia, early-life immune activation can thus influence later-life immune and behavioral responses to physical and psychological stressors. These priming effects are likely induced by the molecular composition of the CNS microenvironment. For example, the release of molecules expressed in the degenerating or injured brain, such as CSF-131 and C-C motif chemokine 2 (CCL2), may induce loss or downregulation of neuronal ligands that inhibit priming (Perry and Holmes, 2014). Furthermore, reduced sensitivity of microglia to the inhibitory effect of glucocorticoids may exert similar effects (Frank et al, 2013). Because of these priming signals, microglia undergo morphological changes and modifications of cell surface antigens, which in turn increase microglial sensitivity to subsequent inflammatory stimuli. Rodents with ‘primed’ microglia show heightened production of pro-inflammatory cytokines in the brain, neurotoxicity, and exaggerated fever and sickness behavior in response to subsequent inflammatory stimulation compared to rodents with ‘naive’ microglia (Perry and Holmes, 2014).
Early-Life Immune Activation and HPA Axis ‘Programming’
In addition to sensitizing the immune system (glial priming), early-life immune activation can also cross-sensitize the neuroendocrine response to stress. Similar to psychosocial stress exposure, experimental administration of IL-1, and to a lesser extent IL-6 and TNF-α, can trigger HPA axis and central catecholamine activation (Besedovsky et al, 1986; Dunn, 2006). On the basis of this initial evidence, later studies have tested whether immune activation in early life—when HPA axis development may be more sensitive to environmental stimuli—can have long-term effects on HPA axis activation. Indeed, research in rodents has shown that immune stimulation with LPS in early-life-induced elevated unstimulated levels of corticotropin-releasing hormone, greater adrenocorticotrophic hormone, and corticosterone responses to restraint stress, decreased negative-feedback sensitivity to glucocorticoids, and reduction in glucocorticoid receptor density across the brain in adult animals (Shanks et al, 1995, 2000). Of note, these changes in HPA axis functioning are strikingly similar to the hallmark abnormalities described in affective disorders (Pariante and Miller, 2001). Therefore, the immune programming of the HPA axis system could contribute to explaining the effects of early-life immune activation on psychopathology.
Inflammatory links between childhood trauma and psychopathology
Findings reviewed above suggest that childhood trauma is linked to the later activation of the innate immune system, which in turn can affect risk of psychopathology. However, this provides only limited support for the mediating role of inflammation in the association between childhood trauma and psychopathology. Further support for the mediating role of inflammation comes from the critical examination of analogy, synergy, specificity, and reversibility.
Analogy
At the most basic level of critical examination, it is possible to identify several analogies between the long-term consequences of childhood trauma and of early-life immune activation (Danese and McEwen, 2012). For example, early-life immune activation is associated with impaired neurogenesis and cognition (Monje et al, 2003; Reichenberg et al, 2001), blunted reward sensitivity (Capuron et al, 2012; Eisenberger et al, 2010), and heightened threat reactivity (Harrison et al, 2009; Inagaki et al, 2012; see Inflammation and Brain Function). Similarly, childhood trauma is linked to impaired cognition (Hart and Rubia, 2012; Mirescu et al, 2004; Pechtel and Pizzagalli, 2011), blunted reward sensitivity (Matthews and Robbins, 2003; Mehta et al, 2010), and heightened threat reactivity (McCrory et al, 2013; Pollak and Kistler, 2002; Tottenham et al, 2011; van Harmelen et al, 2013). Furthermore, both childhood trauma (Varese et al, 2012; Widom, 1999; Widom et al, 2007) and early-life immune activation (see Inflammation and Psychopathology: Observational Human Studies) are associated with a large range of psychiatric disorders, including depression, bipolar disorder, schizophrenia, and PTSD. Finally, both factors appear to be associated not only with higher risk of incident disorders, but also with unfavorable longitudinal course of illness and treatment response (Agnew-Blais and Danese, 2016; Berk et al, 2011; Ford and Erlinger, 2004; Nanni et al, 2012; Strawbridge et al, 2015).
Synergy
Evidence that childhood trauma and inflammation have synergistic effects suggests that both factors are likely to be on the same causative pathways influencing brain and behavior. On the one hand, there is evidence of cross-sensitization between childhood trauma and inflammation. For example, compared to individuals without a history of childhood trauma, those with a history of childhood trauma have greater inflammatory response to subsequent stressors including experimental psychosocial stressors and daily stressors (Carpenter et al, 2010; Gouin et al, 2012; Kiecolt-Glaser et al, 2011; Pace et al, 2006). Similar findings have been described in rodents (Avitsur et al, 2006; Brenhouse and Thompson, 2015; Hennessy et al, 2010b). Furthermore, early-life immune activation has been linked to greater neuroendocrine activation in the context of subsequent stressors (Shanks et al, 1995, 2000), and higher baseline levels of inflammation have been associated with greater stress-induced responses in animal models (Hodes et al, 2014) and humans (Eraly et al, 2014; Michopoulos et al, 2015). On the other hand, there is initial evidence that genes coding for pro-inflammatory cytokines could moderate the effect of childhood trauma on brain function. For example, genetic variation in the IFN-γ gene could moderate the association between a history of childhood maltreatment and the amygdala response to angry and fearful faces (Redlich et al, 2015).
Specificity
Evidence that inflammation levels are elevated in psychiatric patients with history of childhood trauma, but not in patients without history of childhood trauma, suggests that inflammation may contribute to psychopathology after childhood trauma. For example, we have reported that members of the Dunedin Study who had current depression but no prospectively collected evidence of childhood maltreatment showed small and non-significant elevation in inflammation levels at age 32 years compared to controls (Danese et al, 2008). In contrast, depressed individuals with a history of maltreatment had moderate elevation in inflammation levels that reached statistical significance compared to controls (Danese et al, 2008). We have reported similar findings among 12-year-old members of the E-Risk Longitudinal Twin Study (Danese et al, 2011). Similarly, longitudinal analyses found that adolescents with history of early-life stressors had greater increases in both IL-6 and CRP, when becoming depressed than their counterparts without a history of early-life stressors (Miller and Cole, 2012). Of note, because elevated inflammation levels have been linked to unfavorable course of illness and treatment response in mood disorders (Ford and Erlinger, 2004; Strawbridge et al, 2015), the specificity of these biological effects could explain the link between childhood maltreatment and these clinical outcomes (Agnew-Blais and Danese, 2016; Nanni et al, 2012).
Reversibility
Evidence that administration of anti-inflammatory agents could prevent the expression of long-term consequences of childhood trauma suggests that inflammation may contribute to the pathophysiology of trauma-related psychopathology. For example, rodent pups separated from their mothers later showed more depressive-like symptoms and lower prefrontal cortex parvalbumin levels (a GABAergic marker) compared to unchallenged pups (Leussis et al, 2012), and these differences were reduced by the administration of an anti-inflammatory cyclo-oxygenase 2 (COX-2) inhibitor (Brenhouse and Andersen, 2011b) or anti-inflammatory IL-10 (Wieck et al, 2013). More indirectly, physical exercise and anti-depressant medications have anti-inflammatory effects (Gleeson et al, 2011; Hannestad et al, 2011) and they have both appeared to buffer the effects of early-life stress on brain and behavioral outcomes (Harrison and Baune, 2014).
Future research directions
The psychoneuroimmunology of early-life stress offers an innovative perspective to understand the effects of childhood trauma on psychopathology. We propose that psychological trauma in childhood could trigger biological responses qualitatively similar to physical trauma (Molina, 2005), including the activation of the innate immune system. In turn, early-life immune activation could affect brain function and development, and sensitize traumatized individuals to the effects of subsequent stressors. These ‘hidden wounds’ may help explain the long-term toll of childhood trauma on mental health. Although early-life stress can affect both risk for elevated inflammation and for psychopathology, the direct involvement of activated innate immunity in the etiology of psychiatric conditions associated with childhood trauma remains untested. Further research is needed to better understand the mediating role of inflammation and the implications of these findings for prevention and treatment of trauma-related psychopathology.
Mediation
Although the findings reviewed above suggest that inflammation might help explain the high risk of psychopathology in individuals with a history of childhood trauma (see Inflammatory Links Between Childhood Trauma And Psychopathology), none of them in isolation directly provides definitive evidence for the mediating role of inflammation (eg, anti-inflammatory medications may exert buffering effects through other more proximal mechanisms, eg, by reducing glucocorticoid-mediated neurotoxicity). Formal or direct test of this mediation hypothesis in humans (MacKinnon et al, 2007) requires new studies with peculiar design features, such as assessment of childhood trauma, inflammation biomarkers, and psychopathology in the same individuals (for comprehensive models); longitudinal-prospective assessment of childhood trauma and repeated measurement of inflammation and psychopathology (to rule out reverse causality); assessment of multiple inflammation biomarkers (to reduce measurement error); assessment of key potential confounders, including socio-economic status, childhood psychopathology, and genetic liability to inflammation (to minimize risk of omitted variables).
In addition to design issues, future studies will be needed to address more general questions on childhood trauma and psychopathology. For example, childhood trauma is a broad term encompassing various experiences ranging from child sexual abuse, to parental loss, and road traffic accidents. However, different types of trauma may have different correlates and consequences (Terr, 1991). Further understanding of the heterogeneity of experiences included in the definition of childhood trauma will help clarify the links between childhood trauma and inflammation. In addition, childhood trauma and inflammation are associated with elevated risk for several psychiatric diagnoses that often co-occur. As such, further research is needed to test whether there is any significant specificity in the links of childhood trauma and inflammation with psychopathology (Caspi et al, 2014; Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium, 2015).
Reversibility
Further research is needed to test the impact of immune mechanisms in stress resilience and, more practically, in remediating (or reversing) the biological effects of early-life stress before the onset of clinical symptoms. On the one hand, different lines of research suggest that anti-inflammatory strategies could prevent the onset of clinical outcomes. First, higher baseline levels of inflammation have been associated with greater stress-induced responses in animal models (Hodes et al, 2014) and humans (Eraly et al, 2014; Michopoulos et al, 2015). Second, administration of anti-inflammatory agents, such as COX-2 inhibitors or IL-10, can buffer the expression of biological and clinical sequelae of early-life stress in rodents, including the reduction in prefrontal cortex parvalbumin levels and the onset of depressive-like symptoms (Brenhouse and Andersen, 2011b; Wieck et al, 2013). Third, non-pharmacological strategies with anti-inflammatory properties, physical exercise, and anti-depressant medications, can buffer the effects of early-life stress on brain and behavioral outcomes in animal models (Harrison and Baune, 2014). Similarly, psychosocial interventions with anti-inflammatory properties, such as psychotherapy and mindful meditation (Black and Slavich, 2016; Koh et al, 2008; Pace et al, 2013; Thornton et al, 2009), might be beneficial in the aftermath of trauma exposure (Macmillan et al, 2009).
On the other hand, it is possible that resilience to stress might be boosted by harnessing the adaptive immune system (Lewitus and Schwartz, 2009). Consistent with previous research on stress-related potentiation of immune surveillance in the skin (Dhabhar and McEwen, 1997), more recent studies showed that stress can increase lymphocyte trafficking to the brain though expression of ICAM-1 by choroid plexus cells in rodents (Lewitus et al, 2008). Lymphocyte trafficking to the brain is likely protective in the face of stress. Indeed, mice who showed greater lymphocyte trafficking in response to predator odor developed lower anxiety levels and weaker acoustic startle response, and showed faster recovery to pre-stress hippocampal brain-derived neurotrophic factor (BDNF) levels (Cohen et al, 2006; Lewitus et al, 2008). These protective effects are thought to be due to modulation of the stress-induced microglia activation by invading T cells and, thus, to reduction in secondary neuronal damage (Moalem et al, 1999; Yoles et al, 2001). Lymphocyte trafficking to the brain also generates memory T cells reactive to CNS antigens (likely signaling cellular damage under stress), which build immune memory and thereby boost secondary adaptive immune response and resilience in the face of subsequent stressors (Cohen et al, 2006). Of note, inoculation (vaccination and immunization) with a CNS-derived peptide has been associated with lower anxiety levels, weaker acoustic startle response, and faster recovery to pre-stress BDNF levels in rodents exposed to predator odor (Lewitus et al, 2008). However, immunization approaches have not yet been studied in relation to early-life stress.
Stratified Medicine
Finally, it is possible that similar immuno-modulatory interventions could improve treatment of psychiatric patients with a history of childhood trauma. Stratified (or precision) medicine aims to identify subgroups of patients with distinct mechanisms of disease or particular responses to treatments, in order to develop treatments that are effective for these groups of patients (National Research Council (US) Committee on A Framework for Developing a New Taxonomy of Disease, 2011). It is possible that childhood trauma could identify one such subgroup of psychiatric patients.
Childhood trauma identifies a subgroup of psychiatric patients who respond poorly to treatment and—possibly because of this—have unfavorable course of illness. For example, depressed patients with a history of childhood maltreatment are at greater risk of showing poor treatment response and particularly are nearly twice as likely to show poor response to combined treatment (psychotherapy and pharmacotherapy; a regime often used to treat severe cases of depression) compared to non-maltreated-depressed patients (Nanni et al, 2012). In addition, depressed individuals with a history of childhood maltreatment are more than twice as likely to have unfavorable course of illness compared to depressed individuals without a history of childhood maltreatment (Nanni et al, 2012). Similarly, compared to non-maltreated counterparts, bipolar patients with a history of childhood maltreatment have a heightened risk of severe manic, depressive, and psychotic symptoms, heightened risk of comorbid PTSD, anxiety, substance or alcohol misuse disorders, early age of bipolar disorder onset, heightened risk of rapid cycling, greater number of manic and depressive episodes, and heightened risk of suicide attempts (Agnew-Blais and Danese, 2016).
Childhood trauma also identifies a subgroup of psychiatric patients with elevated inflammation levels. For example, depressed individuals with a history of childhood maltreatment showed higher inflammation biomarker levels compared to controls, whereas depressed individuals without a history of childhood maltreatment did not (Danese et al, 2008, 2011).
Because treatment-resistant depressed patients with elevated baseline inflammation levels may benefit from treatment with anti-inflammatory compounds, such TNF-α inhibitor, infliximab (Raison et al, 2013), the stratified biological findings in maltreated patients suggest potential treatment targets. Future research should test if immuno-modulatory interventions could improve treatment of psychiatric patients with a history of childhood trauma.
Funding and disclosure
Dr Lewis receives funding from the National Institute for Health Research (NIHR) Biomedical Research Centres and at South London and Maudsley NHS Foundation Trust, Guy's and St Thomas' NHS Foundation Trust and King's College London. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health. The authors declare no conflict of interest.
References
Ader R (1983). Developmental psychoneuroimmunology. Dev Psychobiol 16: 251–267.
Ader R, Friedman SB (1965). Differential early experiences and susceptibility to transplanted tumor in the rat. J Comp Physiol Psychol 59: 361–364.
Agnew-Blais J, Danese A (2016). Childhood maltreatment and unfavourable clinical outcomes in bipolar disorder: a systematic review and meta-analysis. Lancet Psychiatry 3: 342–349.
Anda RF, Croft JB, Felitti VJ, Nordenberg D, Giles WH, Williamson DF et al (1999). Adverse childhood experiences and smoking during adolescence and adulthood. JAMA 282: 1652–1658.
Avitsur R, Hunzeker J, Sheridan JF (2006). Role of early stress in the individual differences in host response to viral infection. Brain Behav Immun 20: 339–348.
Bailey MT, Coe CL (1999). Maternal separation disrupts the integrity of the intestinal microflora in infant rhesus monkeys. Dev Psychobiol 35: 146–155.
Balleine BW, Delgado MR, Hikosaka O (2007). The role of the dorsal striatum in reward and decision-making. J Neurosci 27: 8161–8165.
Barnes PJ, Adcock IM (2009). Glucocorticoid resistance in inflammatory diseases. Lancet 373: 1905–1917.
Bateson P, Barker D, Clutton-Brock T, Deb D, D'Udine B, Foley RA et al (2004). Developmental plasticity and human health. Nature 430: 419–421.
Baumeister D, Akhtar R, Ciufolini S, Pariante CM, Mondelli V (2015). Childhood trauma and adulthood inflammation: a meta-analysis of peripheral C-reactive protein, interleukin-6 and tumour necrosis factor-α. Mol Psychiatry 21: 642–649.
Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Zastrow Von M et al (2002). Control of synaptic strength by glial TNFalpha. Science 295: 2282–2285.
Berk M, Kapczinski F, Andreazza AC, Dean OM, Giorlando F, Maes M et al (2011). Pathways underlying neuroprogression in bipolar disorder: focus on inflammation, oxidative stress and neurotrophic factors. Neurosci Biobehav Rev 35: 804–817.
Besedovsky H, del Rey A, Sorkin E, Dinarello CA (1986). Immunoregulatory feedback between interleukin-1 and glucocorticoid hormones. Science 233: 652–654.
Bierhaus A, Wolf J, Andrassy M, Rohleder N, Humpert PM, Petrov D et al (2003). A mechanism converting psychosocial stress into mononuclear cell activation. Proc Natl Acad Sci 100: 1920–1925.
Bilbo SD, Schwarz JM (2009). Early-life programming of later-life brain and behavior: a critical role for the immune system. Front Behav Neurosci 3: 14.
Black DS, Slavich GM (2016). Mindfulness meditation and the immune system: a systematic review of randomized controlled trials. Ann NY Acad Sci 1373: 13–24.
Bloomfield PS, Selvaraj S, Veronese M, Rizzo G, Bertoldo A, Owen DR et al (2016). Microglial activity in people at ultra high risk of psychosis and in schizophrenia: an [11c]pbr28 pet brain imaging study. Am J Psychiatry 173: 44–52.
Boulanger LM (2009). Immune proteins in brain development and synaptic plasticity. Neuron 64: 93–109.
Breder CD, Hazuka C, Ghayur T, Klug C, Huginin M, Yasuda K et al (1994). Regional induction of tumor necrosis factor alpha expression in the mouse brain after systemic lipopolysaccharide administration. Proc Natl Acad Sci 91: 11393–11397.
Brenhouse HC, Andersen SL (2011a). Developmental trajectories during adolescence in males and females: a cross-species understanding of underlying brain changes. Neurosci Biobehav Rev 35: 1687–1703.
Brenhouse HC, Andersen SL (2011b). Nonsteroidal anti-inflammatory treatment prevents delayed effects of early life stress in rats. Biol Psychiatry 70: 434–440.
Brenhouse HC, Thompson V (2015). Maternal separation increases IBA-1 expression: a microglia activation marker in the prefrontal cortex of adolescent males following a second hit of stress (abstract). Soc Biol Psychiatry.
Capuron L, Pagnoni G, Drake DF, Woolwine BJ, Spivey JR, Crowe RJ et al (2012). Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon alfa administration. Arch Gen Psychiatry 69: 1044–1053.
Carpenter LL, Gawuga CE, Tyrka AR, Lee JK, Anderson GM, Price LH (2010). Association between plasma IL-6 response to acute stress and early-life adversity in healthy adults. Neuropsychopharmacology 35: 2617–2623.
Casey BJ, Jones RM, Hare TA (2008). The adolescent brain. Ann NY Acad Sci 1124: 111–126.
Caspi A, Houts RM, Belsky DW, Goldman-Mellor SJ, Harrington H, Israel S et al (2014). The p factor: one general psychopathology factor in the structure of psychiatric disorders? Clin Psychol Sci 2: 119–137.
Chalasani SH, Sabelko KA, Sunshine MJ, Littman DR, Raper JA (2003). A chemokine, SDF-1, reduces the effectiveness of multiple axonal repellents and is required for normal axon pathfinding. J Neurosci 23: 1360–1371.
Chocyk A, Dudys D, Przyborowska A, Majcher I, Maćkowiak M, Wędzony K (2011). Maternal separation affects the number, proliferation and apoptosis of glia cells in the substantia nigra and ventral tegmental area of juvenile rats. Neuroscience 173: 1–18.
Chugani HT, Phelps ME, Mazziotta JC (1987). Positron emission tomography study of human brain functional development. Ann Neurol 22: 487–497.
Cicchetti D, Handley ED, Rogosch FA (2015). Child maltreatment, inflammation, and internalizing symptoms: Investigating the roles of C-reactive protein, gene variation, and neuroendocrine regulation. Dev Psychopathol 27: 553–566.
Clancy B, Finlay BL, Darlington RB, Anand KJS (2007). Extrapolating brain development from experimental species to humans. Neurotoxicology 28: 931–937.
Coe C (1996). Developmental psychoneuroimmunology revisited. Brain Behav Immun 10: 185–187.
Coe CL, Rosenberg LT, Levine S (1988). Prolonged effect of psychological disturbance on macrophage chemiluminescence in the squirrel monkey. Brain Behav Immun 2: 151–160.
Coelho R, Viola TW, Walss-Bass C, Brietzke E, Grassi-Oliveira R (2014). Childhood maltreatment and inflammatory markers: a systematic review. Acta Psychiatr Scand 129: 180–192.
Cohen H, Ziv Y, Cardon M, Kaplan Z, Matar MA, Gidron Y et al (2006). Maladaptation to mental stress mitigated by the adaptive immune system via depletion of naturally occurring regulatory CD4+CD25+ cells. J Neurobiol 66: 552–563.
Cohen S, Janicki-Deverts D, Doyle WJ, Miller GE, Frank E, Rabin BS et al (2012). Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proc Natl Acad Sci USA 109: 5995–5999.
Cole SW, Conti G, Arevalo JMG, Ruggiero AM, Heckman JJ, Suomi SJ (2012). Transcriptional modulation of the developing immune system by early life social adversity. Proc Natl Acad Sci USA 109: 20578–20583.
Crews FT, Bechara R, Brown LA, Guidot DM, Mandrekar P, Oak S et al (2006). Cytokines and alcohol. Alcohol Clin Exp Res 30: 720–730.
Crone EA, Dahl RE (2012). Understanding adolescence as a period of social-affective engagement and goal flexibility. Nat Rev Neurosci 13: 636–650.
Cryan JF, Dinan TG (2012). Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci 13: 701–712.
Danese A (2014). Developmental psychoneuroimmunology: from bench to bedside. Brain Behav Immun 36: 27–28.
Danese A, Caspi A, Williams B, Ambler A, Sugden K, Mika J et al (2011). Biological embedding of stress through inflammation processes in childhood. Mol Psychiatry 16: 244–246.
Danese A, Dove R, Belsky DW, Henchy J, Williams B, Ambler A et al (2014). Leptin deficiency in maltreated children. Transl Psychiatry 4: e446.
Danese A, McCrory E (2015). Child maltreatment. In: Rutter's Child & Adolescent Psychiatry. Wiley Blackwell.
Danese A, McEwen BS (2012). Adverse childhood experiences, allostasis, allostatic load, and age-related disease. Physiol Behav 106: 29–39.
Danese A, Moffitt TE, Pariante CM, Ambler A, Poulton R, Caspi A (2008). Elevated inflammation levels in depressed adults with a history of childhood maltreatment. Arch Gen Psychiatry 65: 409–415.
Danese A, Pariante CM, Caspi A, Taylor A, Poulton R (2007). Childhood maltreatment predicts adult inflammation in a life-course study. Proc Natl Acad Sci USA 104: 1319–1324.
Danese A, Tan M (2014). Childhood maltreatment and obesity: systematic review and meta-analysis. Mol Psychiatry 19: 544–554.
Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW (2008). From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9: 46–56.
Dargél AA, Godin O, Kapczinski F, Kupfer DJ, Leboyer M (2015). C-reactive protein alterations in bipolar disorder: a meta-analysis. J Clin Psychiatry 76: 142–150.
Delves PJ, Roitt IM (2000a). The immune system. First of two parts. N Engl J Med 343: 37–49.
Delves PJ, Roitt IM (2000b). The immune system. Second of two parts. N Engl J Med 343: 108–117.
Deverman BE, Patterson PH (2009). Cytokines and CNS development. Neuron 64: 61–78.
Dhabhar FS, McEwen BS (1997). Acute stress enhances while chronic stress suppresses cell-mediated immunity in vivo: a potential role for leukocyte trafficking. Brain Behav Immun 11: 286–306.
Dowling DJ, Levy O (2014). Ontogeny of early life immunity. Trends Immunol 35: 299–310.
Dube SR, Felitti VJ, Dong M, Chapman DP, Giles WH, Anda RF (2003). Childhood abuse, neglect, and household dysfunction and the risk of illicit drug use: the adverse childhood experiences study. Pediatrics 111: 564–572.
Dunn AJ (2006). Cytokine activation of the HPA axis. Ann NY Acad Sci 917: 608–617.
Eisenberger NI, Berkman ET, Inagaki TK, Rameson LT, Mashal NM, Irwin MR (2010). Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry 68: 748–754.
Ekdahl CT, Claasen J-H, Bonde S, Kokaia Z, Lindvall O (2003). Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci 100: 13632–13637.
Eraly SA, Nievergelt CM, Maihofer AX, Barkauskas DA, Biswas N, Agorastos A et al (2014). Assessment of plasma C-reactive protein as a biomarker of posttraumatic stress disorder risk. JAMA Psychiatry 71: 423–431.
Ernst A, Frisén J (2015). Adult neurogenesis in humans- common and unique traits in mammals. PLoS Biol 13: e1002045.
Feng P, Vurbic D, Wu Z, Strohl KP (2007). Brain orexins and wake regulation in rats exposed to maternal deprivation. Brain Res 1154: 163–172.
Fernandes BS, Steiner J, Bernstein H-G, Dodd S, Pasco JA, Dean OM et al (2016). C-reactive protein is increased in schizophrenia but is not altered by antipsychotics: meta-analysis and implications. Mol Psychiatry 21: 554–564.
Flajnik MF, Du Pasquier L (2004). Evolution of innate and adaptive immunity: can we draw a line? Trends Immunol 25: 640–644.
Ford DE, Erlinger TP (2004). Depression and C-reactive protein in US adults: data from the Third National Health and Nutrition Examination Survey. Arch Intern Med 164: 1010–1014.
Frank MG, Watkins LR, Maier SF (2013). Stress-induced glucocorticoids as a neuroendocrine alarm signal of danger. Brain Behav Immun 33: 1–6.
Galea I, Bechmann I, Perry VH (2007). What is immune privilege (not)? Trends Immunol 28: 12–18.
Ganguly P, Brenhouse HC (2015). Broken or maladaptive? Altered trajectories in neuroinflammation and behavior after early life adversity. Dev Cogn Neurosci 11: 18–30.
Garner CC, Zhai RG, Gundelfinger ED, Ziv NE (2002). Molecular mechanisms of CNS synaptogenesis. Trends Neurosci 25: 243–250.
Giedd JN, Rapoport JL (2010). Structural MRI of pediatric brain development: what have we learned and where are we going? Neuron 67: 728–734.
Gleeson M, Bishop NC, Stensel DJ, Lindley MR, Mastana SS, Nimmo MA (2011). The anti-inflammatory effects of exercise: mechanisms and implications for the prevention and treatment of disease. Nat Rev Immunol 11: 607–615.
Gogtay N, Giedd JN, Lusk L, Hayashi KM, Greenstein D, Vaituzis AC et al (2004). Dynamic mapping of human cortical development during childhood through early adulthood. Proc Natl Acad Sci USA 101: 8174–8179.
Gouin J-P, Glaser R, Malarkey WB, Beversdorf D, Kiecolt-Glaser JK (2012). Childhood abuse and inflammatory responses to daily stressors. Ann Behav Med 44: 287–292.
Gould E (2007). How widespread is adult neurogenesis in mammals? Nat Rev Neurosci 8: 481–488.
Greenough WT, Black JE, Wallace CS (1987). Experience and brain development. Child Dev 58: 539–559.
Gregor MF, Hotamisligil GS (2011). Inflammatory mechanisms in obesity. Ann Rev Immunol 29: 415–445.
Gregory AM, Sadeh A (2016). Annual research review: sleep problems in childhood psychiatric disorders - a review of the latest science. J Child Psychol Psychiatry 57: 296–317.
Gunnar M, Quevedo K (2007). The neurobiology of stress and development. Ann Rev Psychology 58: 145–173.
Hanisch U-K, Kettenmann H (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 10: 1387–1394.
Hannestad J, DellaGioia N, Bloch M (2011). The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: a meta-analysis. Neuropsychopharmacology 36: 2452–2459.
Harrison EL, Baune BT (2014). Modulation of early stress-induced neurobiological changes: a review of behavioural and pharmacological interventions in animal models. Transl Psychiatry 4: e390.
Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Critchley HD (2009). Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol Psychiatry 66: 407–414.
Hart BL (1988). Biological basis of the behavior of sick animals. Neurosci Biobehav Rev 12: 123–137.
Hart H, Rubia K (2012). Neuroimaging of child abuse: a critical review. Front Hum Neurosci 6: 52.
Heim C, Ehlert U, Hellhammer DH (2000). The potential role of hypocortisolism in the pathophysiology of stress-related bodily disorders. Psychoneuroendocrinology 25: 1–35.
Heim C, Nemeroff CB (2001). The role of childhood trauma in the neurobiology of mood and anxiety disorders: preclinical and clinical studies. Biol Psychiatry 49: 1023–1039.
Hennessy MB, Deak T, Schiml-Webb PA (2010a). Early attachment-figure separation and increased risk for later depression: potential mediation by proinflammatory processes. Neurosci Biobehav Rev 34: 782–790.
Hennessy MB, Deak T, Schiml-Webb PA, Carlisle CW, O'Brien E (2010b). Maternal separation produces, and a second separation enhances, core temperature and passive behavioral responses in guinea pig pups. Physiol Behav 100: 305–310.
Hensch TK (2005). Critical period plasticity in local cortical circuits. Nat Rev Neurosci 6: 877–888.
Hodes GE, Pfau ML, Leboeuf M, Golden SA, Christoffel DJ, Bregman D et al (2014). Individual differences in the peripheral immune system promote resilience versus susceptibility to social stress. Proc Natl Acad Sci USA 111: 16136–16141.
Howren MB, Lamkin DM, Suls J (2009). Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med 71: 171–186.
Hubel DH, Wiesel TN Brain and visual perception. Oxford University Press: Oxford & New York; (2005).
Huh GS, Boulanger LM, Du H, Riquelme PA, Brotz TM, Shatz CJ (2000). Functional requirement for class I MHC in CNS development and plasticity. Science 290: 2155–2159.
Inagaki TK, Muscatell KA, Irwin MR, Cole SW, Eisenberger NI (2012). Inflammation selectively enhances amygdala activity to socially threatening images. Neuroimage 59: 3222–3226.
Insel TR (2014). The NIMH research domain criteria (RDoC) project: precision medicine for psychiatry. Am J Psychiatry 171: 395–397.
Irwin MR, Wang M, Campomayor CO, Collado-Hidalgo A, Cole S (2006). Sleep deprivation and activation of morning levels of cellular and genomic markers of inflammation. Arch Intern Med 166: 1756–1762.
Janeway CA, Medzhitov R (2002). Innate immune recognition. Ann Rev Immunol 20: 197–216.
Kajeepeta S, Gelaye B, Jackson CL, Williams MA (2015). Adverse childhood experiences are associated with adult sleep disorders: a systematic review. Sleep Med 16: 320–330.
Khandaker GM, Cousins L, Deakin J, Lennox BR, Yolken R, Jones PB (2015). Inflammation and immunity in schizophrenia: implications for pathophysiology and treatment. Lancet Psychiatry 2: 258–270.
Khandaker GM, Pearson RM, Zammit S, Lewis G, Jones PB (2014). Association of serum interleukin 6 and C-Reactive protein in childhood with depression and psychosis in young adult life. JAMA Psychiatry 71: 1121–1128.
Kiecolt-Glaser JK, Gouin J-P, Weng N-P, Malarkey WB, Beversdorf DQ, Glaser R (2011). Childhood adversity heightens the impact of later-life caregiving stress on telomere length and inflammation. Psychosom Med 73: 16–22.
Kimbrell DA, Beutler B (2001). The evolution and genetics of innate immunity. Nat Rev Genet 2: 256–267.
Kipnis J, Gadani S, Derecki NC (2012). Pro-cognitive properties of T cells. Nat Rev Immunol 12: 663–669.
Kirschbaum C, Pirke KM, Hellhammer DH (1993). The “Trier Social Stress Test—”a tool for investigating psychobiological stress responses in a laboratory setting. Neuropsychobiology 28: 76–81.
Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM et al (2013). Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci 16: 33–41.
Knuesel I, Chicha L, Britschgi M, Schobel SA, Bodmer M, Hellings JA et al (2014). Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol 10: 643–660.
Koh KB, Lee Y, Beyn KM, Chu SH, Kim DM (2008). Counter-stress effects of relaxation on proinflammatory and anti-inflammatory cytokines. Brain Behav Immun 22: 1130–1137.
Köhler O, Benros ME, Nordentoft M, Farkouh ME, Iyengar RL, Mors O et al (2014). Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: a systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry 71: 1381–1391.
Leboyer M, Soreca I, Scott J, Frye M, Henry C, Tamouza R et al (2012). Can bipolar disorder be viewed as a multi-system inflammatory disease? J Affect Disord 141: 1–10.
Leussis MP, Freund N, Brenhouse HC, Thompson BS, Andersen SL (2012). Depressive-like behavior in adolescents after maternal separation: sex differences, controllability, and GABA. Dev Neurosci 34: 210–217.
Levitt P (2003). Structural and functional maturation of the developing primate brain. J Pediatr 143: 35–45.
Lewitus GM, Cohen H, Schwartz M (2008). Reducing post-traumatic anxiety by immunization. Brain Behav Immun 22: 1108–1114.
Lewitus GM, Schwartz M (2009). Behavioral immunization: immunity to self-antigens contributes to psychological stress resilience. Mol Psychiatry 14: 532–536.
Lieberam I, Agalliu D, Nagasawa T, Ericson J, Jessell TM (2005). A Cxcl12-CXCR4 chemokine signaling pathway defines the initial trajectory of mammalian motor axons. Neuron 47: 667–679.
Luo L, O'Leary DDM (2005). Axon retraction and degeneration in development and disease. Annu Rev Neurosci 28: 127–156.
Lupien SJ, McEwen BS, Gunnar MR, Heim C (2009). Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci 10: 434–445.
MacKinnon DP, Fairchild AJ, Fritz MS (2007). Mediation analysis. Ann Rev Psychol 58: 593–614.
Macmillan HL, Wathen CN, Barlow J, Fergusson DM, Leventhal JM, Taussig HN (2009). Interventions to prevent child maltreatment and associated impairment. Lancet 373: 250–266.
Maes M, Lambrechts J, Bosmans E, Jacobs J, Suy E, Vandervorst C et al (1992). Evidence for a systemic immune activation during depression: results of leukocyte enumeration by flow cytometry in conjunction with monoclonal antibody staining. Psychol Med 22: 45–53.
Matthews K, Robbins TW (2003). Early experience as a determinant of adult behavioural responses to reward: the effects of repeated maternal separation in the rat. Neurosci Biobehav Rev 27: 45–55.
Matthews KA, Schott LL, Bromberger JT, Cyranowski JM, Everson-Rose SA, Sowers M (2010). Are there bi-directional associations between depressive symptoms and C-reactive protein in mid-life women? Brain Behav Immun 24: 96–101.
McCrory EJ, De Brito SA, Kelly PA, Bird G, Sebastian CL, Mechelli A et al (2013). Amygdala activation in maltreated children during pre-attentive emotional processing. Br J Psychiatry 202: 269–276.
McDade TW (2012). Early environments and the ecology of inflammation. Proc Natl Acad Sci USA 109 (Suppl 2): 17281–17288.
Medzhitov R, Janeway C (2000). Innate immunity. N Engl J Med 343: 338–344.
Mehta MA, Gore-Langton E, Golembo N, Colvert E, Williams SCR, Sonuga-Barke E (2010). Hyporesponsive reward anticipation in the basal ganglia following severe institutional deprivation early in life. J Cogn Neurosci 22: 2316–2325.
Michopoulos V, Rothbaum AO, Jovanovic T, Almli LM, Bradley B, Rothbaum BO et al (2015). Association of CRP genetic variation and CRP level with elevated PTSD symptoms and physiological responses in a civilian population with high levels of trauma. Am J Psychiatry 172: 353–362.
Miller AH, Raison CL (2015). The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol 16: 22–34.
Miller BJ, Buckley P, Seabolt W, Mellor A, Kirkpatrick B (2011). Meta-analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry 70: 663–671.
Miller GE, Cohen S, Ritchey AK (2002). Chronic psychological stress and the regulation of pro-inflammatory cytokines: a glucocorticoid-resistance model. Health Psychol 21: 531–541.
Miller GE, Cole SW (2012). Clustering of depression and inflammation in adolescents previously exposed to childhood adversity. Biol Psychiatry 72: 34–40.
Miller MA, Kandala N-B, Kivimaki M, Kumari M, Brunner EJ, Lowe GDO et al (2009). Gender differences in the cross-sectional relationships between sleep duration and markers of inflammation: Whitehall II study. Sleep 32: 857–864.
Ming G-L, Song H (2011). Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70: 687–702.
Mirescu C, Peters JD, Gould E (2004). Early life experience alters response of adult neurogenesis to stress. Nat Neurosci 7: 841–846.
Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M (1999). Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med 5: 49–55.
Modabbernia A, Taslimi S, Brietzke E, Ashrafi M (2013). Cytokine alterations in bipolar disorder: a meta-analysis of 30 studies. Biol Psychiatry 74: 15–25.
Molina PE (2005). Neurobiology of the stress response: contribution of the sympathetic nervous system to the neuroimmune axis in traumatic injury. Shock 24: 3–10.
Mondelli V, Ciufolini S, Belvederi Murri M, Bonaccorso S, Di Forti M, Giordano A et al (2015). Cortisol and inflammatory biomarkers predict poor treatment response in first episode psychosis. Schizophr Bull 41: 1162–1170.
Monje ML, Toda H, Palmer TD (2003). Inflammatory blockade restores adult hippocampal neurogenesis. Science 302: 1760–1765.
Mrdalj J, Pallesen S, Milde AM, Jellestad FK, Murison R, Ursin R et al (2013). Early and later life stress alter brain activity and sleep in rats. PLoS ONE 8: e69923.
Musselman DL, Lawson DH, Gumnick JF, Manatunga AK, Penna S, Goodkin RS et al (2001). Paroxetine for the prevention of depression induced by high-dose interferon alfa. N Engl J Med 344: 961–966.
Nanni V, Uher R, Danese A (2012). Childhood maltreatment predicts unfavorable course of illness and treatment outcome in depression: a meta-analysis. Am J Psychiatry 169: 141–151.
National Research Council (US) (2011) Committee on A Framework for Developing a New Taxonomy of Disease. National Academies Press (US): Washington (DC).
Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium (2015). Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci 18: 199–209.
O'Mahony SM, Marchesi JR, Scully P, Codling C, Ceolho A-M, EMM Quigley et al (2009). Early life stress alters behavior, immunity, and microbiota in rats: implications for irritable bowel syndrome and psychiatric illnesses. Biol Psychiatry 65: 263–267.
Pace TWW, Mletzko TC, Alagbe O, Musselman DL, Nemeroff CB, Miller AH et al (2006). Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am J Psychiatry 163: 1630–1633.
Pace TWW, Negi LT, Dodson-Lavelle B, Ozawa-de Silva B, Reddy SD, Cole SP et al (2013). Engagement with Cognitively-Based Compassion Training is associated with reduced salivary C-reactive protein from before to after training in foster care program adolescents. Psychoneuroendocrinology 38: 294–299.
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P et al (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333: 1456–1458.
Pariante CM, Miller AH (2001). Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry 49: 391–404.
Passos IC, Vasconcelos-Moreno MP, Costa LG, Kunz M, Brietzke E, Quevedo J et al (2015). Inflammatory markers in post-traumatic stress disorder: a systematic review, meta-analysis, and meta-regression. Lancet Psychiatry 2: 1002–1012.
Patterson PH (2009). Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav Brain Res 204: 313–321.
Pechtel P, Pizzagalli DA (2011). Effects of early life stress on cognitive and affective function: an integrated review of human literature. Psychopharmacology 214: 55–70.
Peferoen L, Kipp M, van der Valk P, van Noort JM, Amor S (2014). Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology 141: 302–313.
Perry VH, Holmes C (2014). Microglial priming in neurodegenerative disease. Nat Rev Neurol 10: 217–224.
Perry VH, Newman TA, Cunningham C (2003). The impact of systemic infection on the progression of neurodegenerative disease. Nat Rev Neurosci 4: 103–112.
Petanjek Z, Judaš M, Šimic G, Rasin MR, Uylings HBM, Rakic P et al (2011). Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc Natl Acad Sci USA 108: 13281–13286.
Pollak SD, Kistler DJ (2002). Early experience is associated with the development of categorical representations for facial expressions of emotion. Proc Natl Acad Sci 99: 9072–9076.
Poulton R, Moffitt TE, Silva PA (2015). The Dunedin Multidisciplinary Health and Development Study: overview of the first 40 years, with an eye to the future. Soc Psychiatry Psychiatr Epidemiol 50: 679–693.
Raison CL, Miller AH (2003). When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry 160: 1554–1565.
Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF et al (2013). A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry 70: 31–41.
Raznahan A, Shaw PW, Lerch JP, Clasen LS, Greenstein D, Berman R et al (2014). Longitudinal four-dimensional mapping of subcortical anatomy in human development. Proc Natl Acad Sci USA 111: 1592–1597.
Redlich R, Stacey D, Opel N, Grotegerd D, Dohm K, Kugel H et al (2015). Evidence of an IFN-γ by early life stress interaction in the regulation of amygdala reactivity to emotional stimuli. Psychoneuroendocrinology 62: 166–173.
Reichenberg A, Yirmiya R, Schuld A, Kraus T, Haack M, Morag A et al (2001). Cytokine-associated emotional and cognitive disturbances in humans. Arch Gen Psychiatry 58: 445–452.
Rook GA, Stanford JL (1998). Give us this day our daily germs. Immunol Today 19: 113–116.
Rook GAW, Lowry CA, Raison CL (2015). Hygiene and other early childhood influences on the subsequent function of the immune system. Brain Res 1617: 47–62.
Schedlowski M, Engler H, Grigoleit J-S (2014). Endotoxin-induced experimental systemic inflammation in humans: a model to disentangle immune-to-brain communication. Brain Behav Immun 35: 1–8.
Setiawan E, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Rajkowska G et al (2015). Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry 72: 268–275.
Shanks N, Larocque S, Meaney MJ (1995). Neonatal endotoxin exposure alters the development of the hypothalamic-pituitary-adrenal axis: early illness and later responsivity to stress. J Neurosci 15: 376–384.
Shanks N, Lightman SL (2001). The maternal-neonatal neuro-immune interface: are there long-term implications for inflammatory or stress-related disease? J Clin Invest 108: 1567–1573.
Shanks N, Windle RJ, Perks PA, Harbuz MS, Jessop DS, Ingram CD et al (2000). Early-life exposure to endotoxin alters hypothalamic-pituitary-adrenal function and predisposition to inflammation. Proc Natl Acad Sci 97: 5645–5650.
Shatz CJ (2009). MHC class I: an unexpected role in neuronal plasticity. Neuron 64: 40–45.
Shaw P, Eckstrand K, Sharp W, Blumenthal J, Lerch JP, Greenstein D et al (2007). Attention-deficit/hyperactivity disorder is characterized by a delay in cortical maturation. Proc Natl Acad Sci USA 104: 19649–19654.
Shaw P, Greenstein D, Lerch J, Clasen L, Lenroot R, Gogtay N et al (2006). Intellectual ability and cortical development in children and adolescents. Nature 440: 676–679.
Sherman DL, Brophy PJ (2005). Mechanisms of axon ensheathment and myelin growth. Nat Rev Neurosci 6: 683–690.
Shiels MS, Katki HA, Freedman ND, Purdue MP, Wentzensen N, Trabert B et al (2014). Cigarette smoking and variations in systemic immune and inflammation markers. J Natl Cancer Inst 106: dju294–dju294.
Shirtcliff EA, Coe CL, Pollak SD (2009). Early childhood stress is associated with elevated antibody levels to herpes simplex virus type 1. Proc Natl Acad Sci USA 106: 2963–2967.
Short SJ, Lubach GR, Karasin AI, Olsen CW, Styner M, Knickmeyer RC et al (2010). Maternal influenza infection during pregnancy impacts postnatal brain development in the rhesus monkey. Biol Psychiatry 67: 965–973.
Slopen N, Kubzansky LD, McLaughlin KA, Koenen KC (2013). Childhood adversity and inflammatory processes in youth: a prospective study. Psychoneuroendocrinology 38: 188–200.
Smith RS (1991). The macrophage theory of depression. Med Hypotheses 35: 298–306.
Solomon GF, Levine S, Kraft JK (1968). Early experience and immunity. Nature 220: 821–822.
Stellwagen D, Malenka RC (2006). Synaptic scaling mediated by glial TNF-alpha. Nature 440: 1054–1059.
Stephan AH, Barres BA, Stevens B (2012). The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci 35: 369–389.
Steptoe A, Hamer M, Chida Y (2007a). The effects of acute psychological stress on circulating inflammatory factors in humans: a review and meta-analysis. Brain Behav Immun 21: 901–912.
Steptoe A, Hamer M, Chida Y (2007b). The effects of acute psychological stress on circulating inflammatory factors in humans: a review and meta-analysis. Brain Behav Immun 21: 901–912.
Strachan DP (2000). Family size, infection and atopy: the first decade of the "hygiene hypothesis". Thorax 55 (Suppl 1): S2–S10.
Strawbridge R, Arnone D, Danese A, Papadopoulos A, Herane Vives A, Cleare AJ (2015). Inflammation and clinical response to treatment in depression: a meta-analysis. Eur Neuropsychopharmacol 25: 1532–1543.
Takatsuru Y, Nabekura J, Ishikawa T, Kohsaka S-I, Koibuchi N (2015). Early-life stress increases the motility of microglia in adulthood. J Physiol Sci 65: 187–194.
Takizawa R, Danese A, Maughan B, Arseneault L (2015). Bullying victimization in childhood predicts inflammation and obesity at mid-life: a five-decade birth cohort study. Psychol Med 45: 2705–2715.
Tau GZ, Peterson BS (2010). Normal development of brain circuits. Neuropsychopharmacology 35: 147–168.
Terr LC (1991). Childhood traumas: an outline and overview. Am J Psychiatry 148: 10–20.
Thornton LM, Andersen BL, Schuler TA, Carson WE (2009). A psychological intervention reduces inflammatory markers by alleviating depressive symptoms: secondary analysis of a randomized controlled trial. Psychosom Med 71: 715–724.
Tiba PA, Tufik S, Suchecki D (2004). Effects of maternal separation on baseline sleep and cold stress-induced sleep rebound in adult Wistar rats. Sleep 27: 1146–1153.
Tottenham N, Hare TA, Millner A, Gilhooly T, Zevin JD, Casey BJ (2011). Elevated amygdala response to faces following early deprivation. Dev Sci 14: 190–204.
Tottenham N, Sheridan MA (2009). A review of adversity, the amygdala and the hippocampus: a consideration of developmental timing. Front Hum Neurosci 3: 68.
Tran PB, Miller RJ (2003). Chemokine receptors: signposts to brain development and disease. Nat Rev Neurosci 4: 444–455.
Turrigiano GG (2008). The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135: 422–435.
Uematsu A, Matsui M, Tanaka C, Takahashi T, Noguchi K, Suzuki M et al (2012). Developmental trajectories of amygdala and hippocampus from infancy to early adulthood in healthy individuals. PLoS ONE 7: e46970.
van der Worp HB, Howells DW, Sena ES, Porritt MJ, Rewell S, O'Collins V et al (2010). Can animal models of disease reliably inform human studies? PLoS Med 7: e1000245.
van Harmelen A-L, van Tol M-J, Demenescu LR, van der Wee NJA, Veltman DJ, Aleman A et al (2013). Enhanced amygdala reactivity to emotional faces in adults reporting childhood emotional maltreatment. Soc Cogn Affect Neurosci 8: 362–369.
Varese F, Smeets F, Drukker M, Lieverse R, Lataster T, Viechtbauer W et al (2012). Childhood adversities increase the risk of psychosis: a meta-analysis of patient-control, prospective- and cross-sectional cohort studies. Schizophr Bull 38: 661–671.
Viviani B, Boraso M, Valero M, Gardoni F, Marco EM, Llorente R et al (2014). Early maternal deprivation immunologically primes hippocampal synapses by redistributing interleukin-1 receptor type I in a sex dependent manner. Brain Behav Immun 35: 135–143.
Weaver ICG, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR et al (2004). Epigenetic programming by maternal behavior. Nat Neurosci 7: 847–854.
Wei L, Simen A, Mane S, Kaffman A (2012). Early life stress inhibits expression of a novel innate immune pathway in the developing hippocampus. Neuropsychopharmacology 37: 567–580.
Widom CS (1999). Posttraumatic stress disorder in abused and neglected children grown up. Am J Psychiatry 156: 1223–1229.
Widom CS, DuMont K, Czaja SJ (2007). A prospective investigation of major depressive disorder and comorbidity in abused and neglected children grown up. Arch Gen Psychiatry 64: 49–56.
Wieck A, Andersen SL, Brenhouse HC (2013). Evidence for a neuroinflammatory mechanism in delayed effects of early life adversity in rats: relationship to cortical NMDA receptor expression. Brain Behav Immun 28: 218–226.
Ygberg S, Nilsson A (2012). The developing immune system - from foetus to toddler. Acta Paediatr 101: 120–127.
Yirmiya R, Goshen I (2011). Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun 25: 181–213.
Yoles E, Hauben E, Palgi O, Agranov E, Gothilf A, Cohen A et al (2001). Protective autoimmunity is a physiological response to CNS trauma. J Neurosci 21: 3740–3748.
Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N et al (2006). Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci 9: 268–275.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article

Cite this article
Danese, A., J Lewis, S. Psychoneuroimmunology of Early-Life Stress: The Hidden Wounds of Childhood Trauma?. Neuropsychopharmacol 42, 99–114 (2017). https://doi.org/10.1038/npp.2016.198
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2016.198
This article is cited by
-
Childhood maltreatment and health in the UK Biobank: triangulation of outcome-wide and polygenic risk score analyses
BMC Medicine (2024)
-
Brain temperature and free water increases after mild COVID-19 infection
Scientific Reports (2024)
-
SKA2 regulated hyperactive secretory autophagy drives neuroinflammation-induced neurodegeneration
Nature Communications (2024)
-
Proteome analysis of monocytes implicates altered mitochondrial biology in adults reporting adverse childhood experiences
Translational Psychiatry (2023)
-
Points of divergence on a bumpy road: early development of brain and immune threat processing systems following postnatal adversity
Molecular Psychiatry (2023)
