
Abstract
Blockade of the μ-opioid receptor (MOR) by naltrexone reduces relapse risk in a subpopulation of alcohol-dependent patients. Previous positron-emission-tomography (PET) studies using the MOR ligand [11C]carfentanil have found increased MOR availability in abstinent alcoholics, which may reflect either increased MOR expression or lower endogenous ligand concentration. To differentiate between both effects, we investigated two cohorts of alcoholic subjects using either post-mortem or clinical PET analysis. Post-mortem brain tissue of alcohol-dependent subjects and controls (N=43/group) was quantitatively analyzed for MOR ([3H]DAMGO)-binding sites and OPRM1 mRNA in striatal regions. [11C]carfentanil PET was performed in detoxified, medication free alcohol-dependent patients (N=38), followed by a randomized controlled study of naltrexone versus placebo and follow-up for 1 year (clinical trial number: NCT00317031). Because the functional OPRM1 variant rs1799971:A>G affects the ligand binding, allele carrier status was considered in the analyses. MOR-binding sites were reduced by 23–51% in post-mortem striatal tissue of alcoholics. In the PET study, a significant interaction of OPRM1 genotype, binding potential (BPND) for [11C]carfentanil in the ventral striatum, and relapse risk was found. Particularly in G-allele carriers, lower striatal BPND was associated with a higher relapse risk. Interestingly, this effect was more pronounced in the naltrexone treatment group. Reduced MOR is interpreted as a neuroadaptation to an alcohol-induced release of endogenous ligands in patients with severe alcoholism. Low MOR availability may explain the ineffectiveness of naltrexone treatment in this subpopulation. Finally, low MOR-binding sites are proposed as a molecular marker for a negative disease course.
Similar content being viewed by others

INTRODUCTION
According to the World Health Organization, approximately two billion people drink alcohol, and many of those alcohol users become dependent. Alcoholism is defined as a pathological syndrome characterized by compulsive drug use, craving and relapses that can occur even after years of abstinence.
Alcohol affects many neurochemical systems. Of particular relevance is the endogenous opioid system, which has a critical role in alcohol reward and relapse (Spanagel, 2009). Alcohol releases endogenous opioids, such as β-endorphin, into the orbitofrontal cortex, ventral tegmental area, and nucleus accumbens (ventral striatum; VS), key sites of the brain reward system. This has been demonstrated by in vivo microdialysis in rats (Olive et al., 2001) and positron-emission-tomography (PET) with the radiolabeled μ-opioid receptor (MOR) agonist [11C]carfentanil before and immediately after alcohol consumption in heavy drinkers and control subjects (Mitchell et al., 2012). Given that the endogenous opioid system is a main modulator of dopaminergic activity within the reward system (Spanagel et al., 1992), one of the theories in the alcohol-field posits that alcohol releases endogenous opioids, thereby activating MORs within the nucleus accumbens and subsequently enhancing dopamine release (Herz, 1997). Together, these neurochemical events are critical for alcohol reward and the development of addictive behavior. Accordingly, pharmacological or genetic inactivation of MOR reduces alcohol reward, craving and relapse in laboratory animals and alcohol-dependent patients (Spanagel, 2009). In line with this hypothesis, PET findings with [11C]carfentanil revealed that the binding potential (BPND) of MOR in the VS of recently detoxified alcohol-dependent patients correlates with alcohol craving (Heinz et al., 2005), and elevated BPND of MOR is seen in alcohol-dependent patients compared with healthy controls (Heinz et al., 2005; Weerts et al., 2011). Carfentanil is a potent and selective MOR agonist, and also at tracer doses binds to high affinity G-coupled (membrane bound and in a high-affinity state) receptors (Frost et al., 1989; Titeler et al., 1989). However, one inherent problem in PET is that BPND cannot directly be translated into the total number of surface opioid receptors, as the radiotracer can be displaced by endogenous opioids. Hence, low endogenous opioid levels may also cause elevated MOR availability despite an unchanged number of MOR. Thus, the status of the MOR system in alcohol-dependent patients remains an open question in the field. This is important to determine, as naltrexone and nalmefene (both non-selective MOR antagonists) are the main pharmacological treatment options for relapse prevention (Litten et al., 2012;Mann et al., 2013a), likely via suppression of alcohol-induced neural activity in the VS (Dudek et al., 2016). Furthermore, MOR BPND may represent a biomarker for treatment response.
Numerous genetic studies have suggested that alcohol dependence and treatment response to naltrexone is associated with the G-allele of the non-synonymous single-nucleotide polymorphism (SNP) rs1799971 (Asn40Asp, A118G), which is located in the first exon of the MOR gene (OPRM1) (Bond et al., 1998; Heilig et al., 2011; Ray et al., 2012). A pharmacogenetic role of this variant is of particular interest among individuals of European and Asian ancestry, where its frequency is 15–30% and 40–50%, respectively (Gelernter et al., 1999). Neuroimaging studies have revealed increased dopamine response to alcohol and altered alcohol-cue reactivity in G-allele carriers (Ramchandani et al., 2011;Schacht et al., 2013), supporting a differential integration and relevance of MOR status in the brains of G-allele carriers. Correspondingly, in the treatment of relapse, G-allele carriers of OPRM1 respond better to naltrexone, resulting in reduced heavy drinking days and an increase in abstinent days (Oslin et al., 2003; Anton et al., 2008; Chamorro et al., 2012). Recent results indicate that G-allele carriers may also benefit more from nalmefene treatment (Bach et al., 2015; Bilbao et al., 2015).
Here we examined MOR expression in post-mortem brain tissue of 43 deceased alcoholics and 43 controls by performing mRNA expression (qRT-PCR) and receptor ([3H]DAMGO)-binding studies in striatal areas, including the VS and nucleus caudatus (NC). In addition, we performed a separate prospective [11C]carfentanil PET study to determine whether the MOR BPND predicts the treatment response to naltrexone. Previous MOR PET studies have only assessed craving as an outcome parameter, but never the more relevant relapse behavior. Therefore, we focused on relapse risk as the primary outcome variable and, accordingly, did not include control subjects. Given the relevance of the OPRM1 A118G variant in alcohol dependence and the treatment response to non-selective MOR antagonists, we also studied the influence of this SNP on MOR expression and binding in post-mortem brain tissue, and on MOR availability in association with disease course and naltrexone response in our prospective PET study.
METHODS AND MATERIALS
Human post-mortem brain samples from 86 male Caucasians (43 alcoholics and 43 controls) were obtained from the NSW Tissue Resource Center (University of Sydney, Australia, see Table 1 and Supplementary Table S1). Two brain regions (VS, NC) were analyzed for MOR expression by quantitative RT-PCR and receptor autoradiography. Thirty-eight alcoholics were recruited for the [11C]carfentanil PET study at the Central Institute of Mental Health Mannheim and the University of Tübingen, Germany (Table 2) and treated according to a modified protocol of the PREDICT study (Mann et al., 2013b). All participants of the clinical study signed an informed consent statement that had been approved by the Ethics Committee of the Mannheim Medical Faculty of the University of Heidelberg. Both post-mortem and PET experiments are described in detail in Supplementary Material and Methods.
RESULTS
Post-Mortem MOR Expression Study: Binding Sites and Transcripts are Decreased in Striatal Brain Tissue of Alcoholics
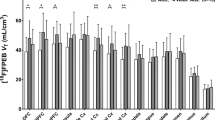
Averaged demographical data and tissue characteristics of 86 male Caucasian subjects used for post-mortem analysis are shown in Table 1. A detailed description of each subject is provided in Supplementary Table S1. There were no differences in age (range: 24–73 years), post-mortem interval (PMI) (controls: 29 h±2 h, alcoholics: 32 h±2 h) or tissue pH (~6.5) between alcohol-dependent patients (N=43) and controls (N=43). However, subjects differed by other potentially confounding factors that can affect MOR binding or expression, including blood alcohol level, nicotine use, psychotropic medications, and rs1799971 genotype. These factors were subsequently included into the analysis as covariates. First, the alcoholics were further divided into subjects with positive blood alcohol levels at death (N=16; here referred to as intoxicated) and those with negative blood alcohol levels (N=27; non-intoxicated, Table 1). A significant decrease of MOR-binding sites in NC and VS was found in both alcohol intoxicated and non-intoxicated alcohol-dependent subjects as compared with controls (NC: F(2,77)=13.83; p<0.001; VS: F (2,75)=12.6, p<0.001, Figure 1a). MOR-binding sites in NC were reduced by ~30% (p<0.001) in both ethanol non-intoxicated and intoxicated alcoholics (Figure 1a). In the VS-binding sites were decreased by 23% (p<0.001) in non-intoxicated alcoholics, this effect was even more pronounced in ethanol intoxicated alcoholics (51% reduction, p<0.001). There was a significantly difference between non-intoxicated and intoxicated alcoholics on MOR-binding sites (p<0.05).

Post-mortem study: μ-opioid receptor (MOR)-binding sites and OPRM1 mRNA expression in the nucleus caudatus and VS. Controls (white bar) are compared with non-intoxicated (dashed bar) and intoxicated (black bar) alcoholic subjects. (a and b) Data show protein and mRNA expression of μ-opioid receptor on post-mortem brain sections by means of saturated [3H]DAMGO receptor autoradiography (a) and qRT-PCR (b). Data are expressed as mean±SEM (receptor autoradiography: fmol/mg±SEM, n=14–40/group, OPRM1 mRNA: mean ddCT±SEM, n=12–35/group). Statistical analysis for both binding sites and mRNA was performed by analysis of covariance followed by Fisher’s LSD post hoc test, *p<0.05, **p<0.01, ***p<0.001 non-intoxicated/intoxicated alcoholics vs controls, #p<0.05 intoxicated alcoholics vs non-intoxicated alcoholics. (c) Schematic overview of the nucleus caudatus (NC) and ventral striatum (VS) is shown on a coronal human brain section. (d) Representative [3H]DAMGO receptor autoradiography on a frontal lobe section (upper panel) showing a specific pattern of MOR similar to Hiller and Fan (Hiller and Fan, 1996) and Mathieu-Kia et al. (2001). Non-specific signal was determined by [3H]DAMGO binding in presence of the MOR antagonist CTOP (lower panel).
Concordant with MOR binding, a significant decrease of OPRM1 mRNA was observed in NC (p<0.01) and VS (p<0.05) of non-intoxicated alcoholics as compared with controls (NC: F(2,58)=5.65, p=0.006; VS: F(2,59)=4.76, p=0.012; Figure 1b). Alcohol-intoxicated alcoholics showed significantly lower OPRM1 mRNA levels in the VS (p<0.05), and transcripts in the NC showed a trend towards decrease (p=0.058) as compared with controls. There was no difference between intoxicated and non-intoxicated alcoholics on OPRM1 mRNA levels in both regions.
There were 68% smokers among the alcoholic patients and 31% in the control group (Supplementary Table S1). Backward stepwise linear regression analysis was used to identify potential covariates. This procedure excluded smoking as a non-significant covariate for striatal MOR ([3H]-DAMGO)-binding and mRNA levels. In order to ensure the robustness of the results, we further conducted an additional regression analysis using smoking as a covariate. Following this analysis, smoking still appears to have no significant effect on binding and mRNA levels.
Given the proposed role of the OPRM1-A118G SNP, all subjects from the post-mortem collection were genotyped, but there are no significant effects on MOR expression at both mRNA and binding sites to report (Supplementary Tables S1 and S2).
Because our sample set included subjects that either committed suicide or were positive for some substances known to alter MOR binding (ie, benzodiazepines, opioids, cannabis) at time of death, we excluded those cases in a sensitivity analysis. A total of 30–38 controls and 21–31 alcoholics (N depending on brain region and group) with and without BAL were analyzed. Similar results were found as above, ie, lower MOR-binding sites and mRNA in alcoholics as compared with controls (Supplementary Fig. S1).
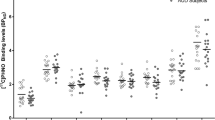
Thus, excluding the above-mentioned factors known to alter MOR expression did not change our finding of decreased MOR expression in heavy alcoholics. For a final validation of these findings, we tested in a more homogeneous subset consisting of age-matched alcoholics (N=14) and control (N=15) subjects (range: 34–73 years) with PMI <40 h (PMI controls: 21 h±2 h, PMI alcoholics: 25 h±2 h), brain pH ~6, no blood alcohol, no significant amounts of substances known to alter MOR binding (eg, opioids, benzodiazepines, concentration <0.1 mg/l) at death, and no suicides. These individuals are described in Supplementary Table S1, highlighted in gray as our ‘core samples’. Here, both MOR-binding sites and OPRM1 mRNA were further reduced in NC and VS of alcoholics (Supplementary Figure S2). In this ‘core sample set’ we also assessed MOR expression in two additional cortical regions, ie, Brodmann area 9 and anterior cingulate cortex (BA24), and found no differences at both the binding and mRNA levels (Supplementary Fig. S2), demonstrating that there is no general MOR reduction in the brains of alcoholics and that our findings are specific to striatal areas. In addition, we provide further evidence of the specificity of this effect by evaluating the expression of κ-opioid receptor (KOR) in our defined core sample set using the selective KOR agonist [3H]U69,593 for receptor autoradiography experiments. Thus, in the NC, KOR-binding sites were not changed in alcoholics as compared with controls, whereas in the VS the number of binding sites was numerically increased but did not reach statistical significance (Supplementary Fig. S3).
In conclusion, independent of the smoking status and other putative confounding factors, we found a strong reduction in [3H]DAMGO binding; ie, a strongly reduced number of MOR-binding sites in the NC and the VS of alcoholic patients—an effect that was augmented in brains from deceased alcoholic patients with positive blood alcohol levels.
Meta-Analysis on OPRM1 mRNA Expression and DAMGO Binding in Alcohol-Dependent Rats
To further corroborate the human post-mortem findings, we performed a meta-analysis on published rodent studies that included 233 rats to analyze striatal OPRM1 mRNA and MOR-binding sites during abstinence. In agreement with our human data, we found decreased mRNA levels and MOR-binding during the first 3 days of alcohol withdrawal and protracted abstinence (Supplementary Table S3).
[11C]Carfentanil PET study: Low Binding Potential of MOR Predicts Relapse
The PET study included 38 alcohol-dependent inpatients (Table 2). After 3 weeks of supervised abstinence, a [11C]carfentanil PET was performed in the absence of medication, followed by a randomized placebo-controlled naltrexone treatment for 90 days and a follow up for 1 year (N=20 naltrexone; N=18 placebo). Abstinence rates after 90 days and 1 year were not significantly different between treatment groups (90 days: naltrexone 40% vs placebo 55%; 1 year: naltrexone 30% vs placebo 17%; log rank test P=0.8, χ2=0.07, df=1). Kaplan–Meier estimates are provided in Supplementary Fig. S4, see also (Mann et al., 2013b). Three regions of interest (ROI) were defined: NC, putamen (P), and VS.
To investigate the relationship between BPND and relapse, we accounted for the fact that BPND cannot directly be compared between subjects with a potentially different MOR affinity (ie, between OPRM1 genotype AA and AG) and that MOR affinity for synaptic endorphins determines the extent to which BPND measured with PET is affected by opioidergic activity. Cox-regression analysis in all 38 patients, accounting for a possible BPND × genotype interaction (Table 3), showed a significant association between low BPND and higher relapse risk in all three ROI, with the strongest association in the VS (p=0.002). From clinical data, a relapse-prevention effect of naltrexone is especially indicated in G-allele carriers of the OPRM1 (Chamorro et al., 2012). We found a BPND × genotype interaction significant in the VS (p=0.010) and at the border of significance in the two other ROIs (P, p=0.052; caudate, p=0.085), indicating that the association between BPND and time to relapse is stronger in AG carriers than in AA carriers, with the highest relapse rate found in AG carriers with low BPND. In addition, controlling for age, gender, and smoking status did not change results (Table 3).
This would support the view of a more functional relevant opioid system in G-allele carriers, as already demonstrated in rhesus macaques (Barr et al., 2008). For illustration we performed a Kaplan–Meier estimate of a median split of MOR BPND in both genotypes (Supplementary Fig. S5). The corresponding Log Rank Test revealed differences between the four groups (χ2=8.757; p=0.033). However, this analysis has to be interpreted with caution because of the low number of OPRM1 G-allele carriers (N=7).
To investigate if the observed association between BP in the VS and relapse risk can be attributed to either the naltrexone or placebo group, we exploratively repeated the analysis of interaction for each group separately. In the group of patients treated with naltrexone (N=20), significant interactions of BPND, OPRM1 genotype and relapse risk were observed in all three ROIs (VS: p=0.021; P: p=0.022; caudate: p=0.022). In the placebo group (N=18), no significant interaction was found. However, in the VS, a trend (p=0.052) emerged.
We further compared MOR BPND from the 38 alcohol-dependent patients with age-matched healthy controls (N=10) from a previous study (Heinz et al., 2005) using the same scanner and measurement protocol. A non-significant tendency for enhanced MOR BPND in alcoholics vs controls was found in the VS (p=0.089, Supplementary Fig. S6).
The same PET study (Heinz et al., 2005) found a positive correlation of BPND in the VS and alcohol craving measured with the Obsessive Compulsive Drinking Scale (OCDS). We were able to replicate this finding only for the OCDS subscale obsessive thoughts (R=0.36, p=0.025), but not for the total OCDS score. This suggests that patients with high BPND also show high OCDS scores for alcohol-associated obsessive thoughts.
DISCUSSION
Based on previous opioid receptor PET studies in alcoholism, it has been suggested that MORs are increased in the reward system of alcohol-dependent patients when compared with healthy controls (Heinz et al., 2005; Williams et al., 2009;Weerts et al., 2011). It has also been shown that elevated MOR BPND in the VS correlates with alcohol craving in abstinent patients (Heinz et al., 2005). These findings provide the rationale for the effectiveness of naltrexone and nalmefene in reducing alcohol reward, craving, and relapse in alcohol-dependent patients (Spanagel, 2009; Litten et al., 2012; Mann et al., 2013b). However, PET studies on MORs are difficult to interpret, as elevated binding potential of the radiotracer can result from high rates of receptor expression (either trait-like or adaptive) and/or low competition from endogenous opioid peptides following chronic alcohol exposure. The latter effect is particularly important, as [11C]carfentanil—the only MOR-selective PET ligand established to date—is an agonist, and therefore, owing to G-protein-mediated internalization, particularly sensitive to synaptic levels of the endogenous ligand (Zubieta et al., 2001).
In contrast to the generally held view, we show here—with our post-mortem brain sample—a clear and selective reduction of OPRM1 mRNA and MOR-binding sites in the brain reward system of severe alcoholics. Analysis of post-mortem brain tissue in psychiatric patients is often confounded by suicide, pre-mortem medication, heavy smoking, and co-abuse. However, after excluding such cases in a secondary analysis, our findings did not change, and in fact using the ‘core sample set’ the reduction of MOR was even more pronounced. Long-term alcohol abuse induces neurodegeneration, which could potentially lead to a general non-specific MOR reduction. To control for this possibility, we determined the MOR status in prefrontocortical brain regions, known to be highly sensitive to alcohol's neurotoxic effects (Zahr et al., 2011). We found no difference in MOR expression between alcoholics and controls in these regions. Furthermore, within striatal regions, KORs are not affected by disease state. Thus, the strong reduction of striatal MOR seems to be dependence-specific and most likely not related to brain pathology as a secondary effect of alcohol. Another critical issue is the stability of mRNA and protein during the post-mortem period. We addressed this by correlating levels of OPRM1 mRNA and MOR-binding sites with PMI, brain pH, and RIN in each group and found no significant effect. Nevertheless, the heterogeneous PMI (8.5–72 h) could present a potential confound. However, a recent rodent study has addressed the effect of PMI on protein expression (Li et al., 2003). The most marked effect on protein abundance was observed between 0 and 8 h PMI, thereafter protein levels were reasonably stable. Also, a previous study concluded that the levels of many proteins are stable in human post-mortem tissue (Stan et al., 2006), and ligand binding does not seem to be affected by PMI (Kornhuber et al., 1988). Based on these studies and our statistical analysis, we conclude that PMI has not confounded the MOR-binding results. Overall, we used samples of good quality, which were obtained from one of the best providers of post-mortem human specimens, the New South Wales Tissue Resource Centre, University of Sydney, Australia, from which samples have been already successfully used in numerous studies (Sheedy et al., 2008; Meinhardt et al., 2013;Hirth et al., 2016).
Other important information is the time of abstinence of non-intoxicated alcoholics. Unfortunately, information about abstinence times is not available for most cases from the brain bank. However, we addressed this in our recent study by Hirth et al. (2016) using identical samples for convergent analysis of the dopamine system during alcohol abstinence in both humans and rats. Based on time-dependent changes on dopamine receptor/transporter during abstinence, the human post-mortem findings appeared most closely related to those seen with 21 days of abstinence in rodents. Thus, we assume that most non-intoxicated subjects may have died in protracted abstinence, ie, beyond the first weeks of abstinence.
Our finding of reduced MOR expression in the brain of alcoholics is consistent with published rodent data, as shown by the meta-analysis. In further support of our data, reduced naloxone binding in the dentate gyrus of Cloninger type I alcoholics has been reported by Laukkanen et al. (2015).
Interestingly, in our post-mortem samples, G-allele carriers (20% of the subjects) did not show an altered number of MOR-binding sites in comparison with the AA genotype. This is in agreement with a recent study from Ramchandani et al. (2011) using a mouse model in which a mutation was introduced in a position corresponding to the human A118G marker. However, our findings seem contradictory with other publications showing decreased MOR expression (in vitro cell culture study by Kroslak et al. (2007); human autopsy brain tissue analyses of MOR mRNA and protein, N=76 AA, N=8 AG and N=3 GG OPRM1 genotype by Zhang et al., (2005)) and a reduced binding potential (human PET study with N=11 AG versus N=29 AA carrier of OPRM1 by Reimold et al. (2010); human PET study of 25 alcohol-dependent patients after 5 days of abstinence (19 AA and 6 AG carrier) and 28 healthy controls (20 AA and 8 AG carrier) by Weerts et al. (2013)) in G-allele carriers. Several factors may account for this inconsistency. First, our small sample size (controls: AA=31/AG=4, alcoholics: AA=17/AG=13) prevented a detection of genetic effects. Second, in these cited autopsy studies, tissue samples were not of striatal origin. And third, the allelic effects of OPRM1 A118G on ligand binding can be masked by receptor reserve (Robinson et al., 2015), which is difficult to assess in post-mortem tissue.
We further show from our prospective PET study that striatal BPND for [11C]carfentanil is predictive of the risk of relapse. This means that low striatal BPND is associated with an increased relapse risk, and vice versa. In our group of patients, relapse rates were not different between the naltrexone and placebo group, so naltrexone failed to show a beneficial effect, likely owing to small sample sizes of N=20 naltrexone versus N=18 placebo. However, in the naltrexone treated subgroup, high BPND in all three ROIs still predicts low relapse risk, whereas in the placebo subgroup this association fails significance. This may suggest a stronger association of MOR status and relapse risk if remaining MORs are blocked by naltrexone in contrast to not blocking MORs. Furthermore, high BPND was associated with high alcohol craving (OCDS subscale of obsessive thoughts). If we merge the latter two findings, high BPND is associated with high craving and, if treated with naltrexone, with a diminished relapse risk. This suggests that relapse-prevention effects of naltrexone are mediated by reducing craving, and from the perspective of individualized medicine, that alcoholics experiencing high craving may have a high number of striatal opioid receptors and therefore may especially benefit from naltrexone. Together, the strongly reduced MOR status in the reward system of alcoholics and the increased relapse risk associated with low MOR BPND suggest that low MORs are accompanied by a state of anhedonia, a well-known risk factor of for relapse (Heilig et al., 2010). Thus, patients having low MOR BPND after detoxification may be more likely to relapse and may not benefit from naltrexone treatment. In other words, sufficient MOR BPND may be a precondition for naltrexone to reveal its relapse-prevention effect. These results should be interpreted with caution, because the number of participants is low for complex interaction analyses of BPND, relapse, and OPRM1 genotype. However, our PET results are consistent with our post-mortem data and can substantially add to the puzzle of dynamic alterations of the opioid system in alcoholism.
According to our combined post-mortem brain and in vivo PET data, we now propose the following dynamical alterations of the MOR and opioid peptide status in the course of the addiction cycle (Figure 2). Acute alcohol consumption results in the release of endogenous opioids (Mitchell et al., 2012), mediating feelings of pleasure or ‘high’. Repeated alcohol consumption results in an increased frequency and strength of opioidergic neurotransmission. As an adaptive mechanism, MOR expression is reduced, resulting in a subsequent reduction in MOR-binding sites. This decreased surface density of MORs may contribute to tolerance to the rewarding effects of alcohol, driving further enhanced alcohol consumption. In alcohol withdrawal and early abstinence, the alcohol-induced release of endogenous opioids is suddenly stopped, whereas MOR-binding sites are strongly diminished. These molecular events may contribute to the anhedonic state in early abstinence, characterized by dysphoria, increased anxiety, and depressiveness (Heilig et al., 2010). During protracted abstinence, the responsiveness of the opioidergic system is still diminished and must adapt to less-intense natural rewards in comparison with alcohol in order to recover. During recovery, natural rewards, such as from social interactions, may progressively regain the potential to stimulate the release of endogenous opioids. During this recovery phase, reduced endogenous opioids may constitute the neurochemical stimulus to increase and progressively normalize the surface density of MORs. Our post-mortem finding of a stronger reduction of MOR expression in those alcoholics drinking until death support an allostatic view of the MOR system and opioid-mediated reward, which is in line with published papers indicating a progressive loss of ventro-striatal-mediated alcohol reward as detected by functional magnetic resonance imaging (Gilman et al., 2008; Gilman et al., 2012; Spagnolo et al., 2014).

Molecular model of alcohol-mediated dynamic alterations in the striatal opioid system during the addiction. Initial and repeated alcohol consumption results in the release of endogenous opioids by progressively enhancing the frequency and strength of opioidergic neurotransmission. This neurochemical event is accompanied by reward and hedonia. As an adaptive mechanism, MOR expression is reduced and a subsequent reduction in MOR-binding sites ensures maintained homeostasis. In alcohol withdrawal and early abstinence, alcohol-induced release of endogenous opioids is suddenly stopped, whereas MOR expression is strongly diminished. These molecular events may contribute to anhedonia in early abstinence. During protracted abstinence, the responsiveness of the opioidergic system is still diminished and must adapt to less intense natural rewards in comparison to alcohol in order to recover. [11C]carfentanil PET assesses MOR availability, which depends on the absolute number of MOR-binding sites and endogenous opioid levels. Therefore, PET data can only be correctly be interpreted if additional data on either one of these measures is available. The binding potential (BPND, solid black arrows) is the specific to non-specific equilibrium partition coefficient that is determined by the concentration of the endogenous opioids and the absolute number of MOR-binding sites. Saturated [3H]DAMGO autoradiographies in post-mortem tissue are measures of number of available MOR-binding sites (BS), which are reduced in alcoholics at different times of abstinence in relation to controls.
PET studies during early abstinence have revealed increased BPND compared with healthy controls (Heinz et al., 2005; Weerts et al., 2011), which is, on a trend level, in line with an exploratory comparison of our PET sample with 10 healthy controls from a previous study (Heinz et al., 2005). In the light of reduced MOR binding in our post-mortem sample, we conclude that increased PET-BPND reflects reduced competition from endogenous opioids rather than increased receptor density (Figure 2).
Although our study provides new insights into the dynamic alterations of the MOR and opioid peptide status in the course of the addiction cycle, it fails to clarify the mode of action of MOR antagonists. Given diminished MORs during withdrawal and protracted abstinence, a pharmacological blockade would be expected to worsen the condition and eventually precipitate relapse. Thus, there must be other, still unknown mechanisms in place—for example, augmented signaling from enhanced G-protein-MOR coupling (Bjork et al., 2013) or other molecular downstream effects—that explain the mode of action of opioid receptor antagonist treatment in alcoholism.
In conclusion, combined post-mortem brain and PET analysis allows the characterization of receptor status—the number of membrane surface receptors—in a disease condition and thus provides more certainty in the interpretation of PET results. This approach has enabled us to provide a new model of the dynamics of the endogenous opioid system and MOR activity in the addiction cycle. We show strongly diminished MORs in alcohol-dependent patients. Furthermore, diminished MOR BPND may potentially be used as a biomarker in alcoholics to predict early relapse and low-treatment response to naltrexone.
FUNDING AND DISCLOSURE
The authors declare no conflict of interest.
References
Anton RF, Oroszi G, O'malley S, Couper D, Swift R, Pettinati H et al (2008). An evaluation of mu-opioid receptor (OPRM1) as a predictor of naltrexone response in the treatment of alcohol dependence: results from the Combined Pharmacotherapies and Behavioral Interventions for Alcohol Dependence (COMBINE) study. Arch Gen Psychiatry 65: 135–144.
Bach P, Vollsta Dt-Klein S, Kirsch M, Hoffmann S, Jorde A, Frank J et al (2015). Increased mesolimbic cue-reactivity in carriers of the mu-opioid-receptor gene OPRM1 A118G polymorphism predicts drinking outcome: a functional imaging study in alcohol dependent subjects. Eur Neuropsychopharmacol 25: 1128–1135.
Barr CS, Schwandt ML, Lindell SG, Higley JD, Maestripieri D, Goldman D et al (2008). Variation at the mu-opioid receptor gene (OPRM1) influences attachment behavior in infant primates. Proc Natl Acad Sci USA 105: 5277–5281.
Bilbao A, Robinson JE, Heilig M, Malanga CJ, Spanagel R, Sommer WH, Thorsell A (2015). A pharmacogenetic determinant of mu-opioid receptor antagonist effects on alcohol reward and consumption: evidence from humanized mice. Biol Psychiatry 77: 850–858.
Bjork K, Tronci V, Thorsell A, Tanda G, Hirth N, Heilig M et al (2013). beta-Arrestin 2 knockout mice exhibit sensitized dopamine release and increased reward in response to a low dose of alcohol. Psychopharmacology (Berl) 230: 439–449.
Bond C, Laforge KS, Tian M, Melia D, Zhang S, Borg L et al (1998). Single-nucleotide polymorphism in the human mu opioid receptor gene alters beta-endorphin binding and activity: possible implications for opiate addiction. Proc Natl Acad Sci USA 95: 9608–9613.
Chamorro AJ, Marcos M, Miron-Canelo JA, Pastor I, Gonzalez-Sarmiento R, Laso FJ (2012). Association of micro-opioid receptor (OPRM1) gene polymorphism with response to naltrexone in alcohol dependence: a systematic review and meta-analysis. Addict Biol 17: 505–512.
Dudek M, Canals S, Sommer WH, Hyytia P (2016). Modulation of nucleus accumbens connectivity by alcohol drinking and naltrexone in alcohol-preferring rats: a manganese-enhanced magnetic resonance imaging study. Eur Neuropsychopharmacol 26: 445–455.
Frost JJ, Douglass KH, Mayberg HS, Dannals RF, Links JM, Wilson AA et al (1989). Multicompartmental analysis of [11C]-carfentanil binding to opiate receptors in humans measured by positron emission tomography. J Cereb Blood Flow Metab 9: 398–409.
Gelernter J, Kranzler H, Cubells J (1999). Genetics of two mu opioid receptor gene (OPRM1) exon I polymorphisms: population studies, and allele frequencies in alcohol- and drug-dependent subjects. Mol Psychiatry 4: 476–483.
Gilman JM, Ramchandani VA, Crouss T, Hommer DW (2012). Subjective and neural responses to intravenous alcohol in young adults with light and heavy drinking patterns. Neuropsychopharmacology 37: 467–477.
Gilman JM, Ramchandani VA, Davis MB, Bjork JM, Hommer DW (2008). Why we like to drink: a functional magnetic resonance imaging study of the rewarding and anxiolytic effects of alcohol. J Neurosci 28: 4583–4591.
Heilig M, Egli M, Crabbe JC, Becker HC (2010). Acute withdrawal, protracted abstinence and negative affect in alcoholism: are they linked? Addict Biol 15: 169–184.
Heilig M, Goldman D, Berrettini W, O'brien CP (2011). Pharmacogenetic approaches to the treatment of alcohol addiction. Nat Rev Neurosci 12: 670–684.
Heinz A, Reimold M, Wrase J, Hermann D, Croissant B, Mundle G (2005). Correlation of stable elevations in striatal mu-opioid receptor availability in detoxified alcoholic patients with alcohol craving: a positron emission tomography study using carbon 11-labeled carfentanil. Arch Gen Psychiatry 62: 57–64.
Herz A (1997). Endogenous opioid systems and alcohol addiction. Psychopharmacology (Berl) 129: 99–111.
Hiller JM, Fan LQ (1996). Laminar distribution of the multiple opioid receptors in the human cerebral cortex. Neurochem Res 21: 1333–1345.
Hirth N, Meinhardt MW, Noori HR, Salgado H, Torres-Ramirez O, Uhrig S et al (2016). Convergent evidence from alcohol-dependent humans and rats for a hyperdopaminergic state in protracted abstinence. Proc Natl Acad Sci USA 113: 3024–3029.
Kornhuber J, Retz W, Riederer P, Heinsen H, Fritze J (1988). Effect of antemortem and postmortem factors on [3H]glutamate binding in the human brain. Neurosci Lett 93: 312–317.
Kroslak T, Laforge KS, Gianotti RJ, Ho A, Nielsen DA, Kreek MJ (2007). The single nucleotide polymorphism A118G alters functional properties of the human mu opioid receptor. J Neurochem 103: 77–87.
Laukkanen V, Karkkainen O, Kautiainen H, Tiihonen J, Storvik M (2015). Decreased [3H]naloxone binding in the dentate gyrus of cloninger type 1 anxiety-prone alcoholics: a postmortem whole-hemisphere autoradiography study. Alcohol Clin Exp Res 39: 1352–1359.
Li J, Gould TD, Yuan P, Manji HK, Chen G (2003). Post-mortem interval effects on the phosphorylation of signaling proteins. Neuropsychopharmacology 28: 1017–1025.
Litten RZ, Egli M, Heilig M, Cui C, Fertig JB, Ryan ML et al (2012). Medications development to treat alcohol dependence: a vision for the next decade. Addict Biol 17: 513–527.
Mann K, Bladstrom A, Torup L, Gual A, Van Den Brink W (2013a). Extending the treatment options in alcohol dependence: a randomized controlled study of as-needed nalmefene. Biol Psychiatry 73: 706–713.
Mann K, Lemenager T, Hoffmann S, Reinhard I, Hermann D, Batra A et al (2013b). Results of a double-blind, placebo-controlled pharmacotherapy trial in alcoholism conducted in Germany and comparison with the US COMBINE study. Addict Biol 18: 937–946.
Mathieu-Kia AM, Fan LQ, Kreek MJ, Simon EJ, Hiller JM (2001). Mu-, delta- and kappa-opioid receptor populations are differentially altered in distinct areas of postmortem brains of Alzheimer's disease patients. Brain Res 893: 121–134.
Meinhardt MW, Hansson AC, Perreau-Lenz S, Bauder-Wenz C, Stahlin O, Heilig M et al (2013). Rescue of infralimbic mGluR2 deficit restores control over drug-seeking behavior in alcohol dependence. J Neurosci 33: 2794–2806.
Mitchell JM, O'neil JP, Janabi M, Marks SM, Jagust WJ, Fields HL (2012). Alcohol consumption induces endogenous opioid release in the human orbitofrontal cortex and nucleus accumbens. Sci Transl Med. 4 116ra116.
Olive MF, Koenig HN, Nannini MA, Hodge CW (2001). Stimulation of endorphin neurotransmission in the nucleus accumbens by ethanol, cocaine, and amphetamine. J Neurosci 21: RC184.
Oslin DW, Berrettini W, Kranzler HR, Pettinati H, Gelernter J, Volpicelli JR et al (2003). A functional polymorphism of the mu-opioid receptor gene is associated with naltrexone response in alcohol-dependent patients. Neuropsychopharmacology 28: 1546–1552.
Ramchandani VA, Umhau J, Pavon FJ, Ruiz-Velasco V, Margas W, Sun H et al (2011). A genetic determinant of the striatal dopamine response to alcohol in men. Mol Psychiatry 16: 809–817.
Ray LA, Barr CS, Blendy JA, Oslin D, Goldman D, Anton RF (2012). The role of the Asn40Asp polymorphism of the mu opioid receptor gene (OPRM1) on alcoholism etiology and treatment: a critical review. Alcohol Clin Exp Res 36: 385–394.
Reimold MH, Reischl G, Smolka G, Batra M, Rietschel A, Kiefer M et al (2010). Reduced mu-opiate receptor availability in a genetic variant of the mu-opiate receptor (A118G): a [11C]carfentanil PET study. J Nucl Med 51: 49.
Robinson JE, Vardy E, Diberto JF, Chefer VI, White KL, Fish EW et al (2015). receptor reserve moderates mesolimbic responses to opioids in a humanized mouse model of the OPRM1 A118G polymorphism. Neuropsychopharmacology 40: 2614–2622.
Schacht JP, Anton RF, Voronin KE, Randall PK, Li X, Henderson S et al (2013). Interacting effects of naltrexone and OPRM1 and DAT1 variation on the neural response to alcohol cues. Neuropsychopharmacology 38: 414–422.
Sheedy D, Garrick T, Dedova I, Hunt C, Miller R, Sundqvist N et al (2008). An Australian Brain Bank: a critical investment with a high return!. Cell Tissue Bank 9: 205–216.
Spagnolo PA, Ramchandani VA, Schwandt ML, Zhang L, Blaine SK, Usala JM et al (2014). Effects of naltrexone on neural and subjective response to alcohol in treatment-seeking alcohol-dependent patients. Alcohol Clin Exp Res 38: 3024–3032.
Spanagel R (2009). Alcoholism: a systems approach from molecular physiology to addictive behavior. Physiol Rev 89: 649–705.
Spanagel R, Durstewitz D, Hansson A, Heinz A, Kiefer F, Kohr G et al (2013). A systems medicine research approach for studying alcohol addiction. Addict Biol 18: 883–896.
Spanagel R, Herz A, Shippenberg TS (1992). Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc Natl Acad Sci USA 89: 2046–2050.
Stan AD, Ghose S, Gao XM, Roberts RC, Lewis-Amezcua K, Hatanpaa KJ et al (2006). Human postmortem tissue: what quality markers matter? Brain Res 1123: 1–11.
Titeler M, Lyon RA, Kuhar MJ, Frost JF, Dannals RF, Leonhardt S et al (1989). Mu opiate receptors are selectively labelled by [3H]carfentanil in human and rat brain. Eur J Pharmacol 167: 221–228.
Weerts EM, Mccaul ME, Kuwabara H, Yang X, Xu X, Dannals RF et al (2013). Influence of OPRM1 Asn40Asp variant (A118G) on [11C]carfentanil binding potential: preliminary findings in human subjects. Int J Neuropsychopharmacol 16: 47–53.
Weerts EM, Wand GS, Kuwabara H, Munro CA, Dannals RF, Hilton J et al (2011). Positron emission tomography imaging of mu- and delta-opioid receptor binding in alcohol-dependent and healthy control subjects. Alcohol Clin Exp Res 35: 2162–2173.
Williams TM, Davies SJ, Taylor LG, Daglish MR, Hammers A, Brooks DJ (2009). Brain opioid receptor binding in early abstinence from alcohol dependence and relationship to craving: an [11C]diprenorphine PET study. Eur Neuropsychopharmacol 19: 740–748.
Zahr NM, Kaufman KL, Harper CG (2011). Clinical and pathological features of alcohol-related brain damage. Nat Rev Neurol 7: 284–294.
Zhang Y, Wang D, Johnson AD, Papp AC, Sadee W (2005). Allelic expression imbalance of human mu opioid receptor (OPRM1) caused by variant A118G. J Biol Chem 280: 32618–32624.
Zubieta JK, Smith YR, Bueller JA, Xu Y, Kilbourn MR, Jewett DM et al (2001). Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science 293: 311–315.
Acknowledgements
We are very grateful for obtaining the brain samples from the Sydney Brain Bank, especially for Clive Harper, Donna Sheedy and Jillian J Kril. We thank E Röbel for technical support, Christine Jennen-Steinmetz for supporting statistical analysis, Valentina Vengeliene for providing some figures and Rick E Bernardi for English editing. Patient recruitment and phenotyping were largely performed by Sabine Zimmer-Butz, Rosie Krämer, and Tagrid Leménager. ACH, RS, WHS were responsible for study design of the post-mortem brain study and procured study funding. NH performed experiments in post-mortem brains. MR, DH, KM, FK, AB were responsible for study design of the PET study and procured study funding. MR and GR performed PET experiments. MR was responsible for PET measurements, MR and SH did statistical analysis. NH, ACH, WHS analyzed post-mortem data. HRN performed the meta-analysis. RS, ACH, DH, NH, WHS, CLF, KM wrote the manuscript. Our work is supported by the Bundesministerium für Bildung und Forschung (01EB0110; NGFN Plus, FKZ: 01GS08152; e:Med program, FKZ: 01ZX1311A 4, (Spanagel et al., 2013)) and the Ministerium für Wissenschaft, Forschung und Kunst (MWK) in Baden-Württemberg, Deutsche Forschungsgemeinschaft (DFG HA6102/1-1, DFG center grant (SFB1134), the European Union’s Horizon 2020 program (668863-SyBil-AA).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Supplementary information
PowerPoint slides
Rights and permissions
About this article

Cite this article
Hermann, D., Hirth, N., Reimold, M. et al. Low μ-Opioid Receptor Status in Alcohol Dependence Identified by Combined Positron Emission Tomography and Post-Mortem Brain Analysis. Neuropsychopharmacol 42, 606–614 (2017). https://doi.org/10.1038/npp.2016.145
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2016.145
This article is cited by
-
Hazardous drinking and alcohol use disorders
Nature Reviews Disease Primers (2022)
-
Risk for opioid misuse in chronic pain patients is associated with endogenous opioid system dysregulation
Translational Psychiatry (2022)
-
Blunted endogenous opioid release following an oral dexamphetamine challenge in abstinent alcohol-dependent individuals
Molecular Psychiatry (2020)
