
Abstract
Smoking is the largest preventable cause of death in the United States. Furthermore, a recent study found that <10% of quit attempts resulted in continuous abstinence for 1 year. With the introduction of pharmacotherapies like Chantix (varenicline), a selective α4β2 nicotinic partial agonist, successful quit attempts have significantly increased. Therefore, novel subtype-specific nicotinic drugs, such as sazetidine-A, present a rich area for investigation of therapeutic potential in smoking cessation. The present studies examine the anxiety-related behavioral and functional effects of the nicotinic partial agonists varenicline and sazetidine-A during withdrawal from chronic nicotine in mice. Our studies indicate that ventral hippocampal-specific infusions of sazetidine-A, but not varenicline, are efficacious in reducing nicotine withdrawal-related anxiety-like phenotypes in the novelty-induced hypophagia (NIH) paradigm. To further investigate functional differences between these partial agonists, we utilized voltage-sensitive dye imaging (VSDi) in ventral hippocampal slices to determine the effects of sazetidine-A and varenicline in animals chronically treated with saline, nicotine, or undergoing 24 h withdrawal. These studies demonstrate a functional dissociation of varenicline and sazetidine-A on hippocampal network activity, which is directly related to previous drug exposure. Furthermore, the effects of the nicotinic partial agonists in VSDi assays are significantly correlated with their behavioral effects in the NIH test. These findings highlight the importance of drug history in understanding the mechanisms through which nicotinic compounds may be aiding smoking cessation in individuals experiencing withdrawal-associated anxiety.
Similar content being viewed by others


INTRODUCTION
Tobacco addiction affects an estimated 1.2 billion people and about 80% of smokers attempting to quit fail (Polosa and Benowitz, 2011). Nicotine, the addictive compound in tobacco, exerts its biological effects through activation of the nicotinic acetylcholine receptor (nAChR) (for review, see Benowitz, 2009). Drugs targeting nAChRs are an area of intense investigation for smoking cessation pharmacotherapies. Varenicline, the most effective medication for smoking cessation currently available (Chantix; Pfizer), is a potent partial agonist for α4β2 nAChRs, as well as a weak partial agonist at α3β4 nAChRs and a full agonist at α7 nAChRs (Coe et al, 2005; Rollema et al, 2007). Sazetidine-A is a recently developed nicotinic compound with high affinity and selectivity for α4β2 nAChRs, but not α3β4 or α7 nAChRs (Xiao et al, 2006), that quickly and thoroughly desensitizes the receptor after brief partial agonism (Xiao et al, 2006; Zwart et al, 2008). However, though both varenicline and sazetidine-A are α4β2 partial agonists, their differing activity profile, as well as subtype specificity, may result in divergent behavioral effects.
While the rewarding properties of nicotine have been extensively studied (Corrigall et al, 1992, 1994; Grieder et al, 2012; Laviolette et al, 2008; Laviolette and van der Kooy, 2003), the neural substrates associated with withdrawal are not as well characterized. Activation of diverse brain regions has been correlated with cognitive withdrawal symptoms and craving (Azizian et al, 2009; Davis and Gould, 2009; McClernon and Gilbert, 2004). Although craving and cognitive impairments are reliable markers of nicotine withdrawal (Kenney and Gould, 2008; Levin et al, 2006; McClernon et al, 2004; Rezvani and Levin, 2001), anxiety has been shown to impact relapse rate as well (Dani and Harris, 2005; DiMatteo et al, 2000). Furthermore, nicotine withdrawal itself induces an increased anxiety state in both human (Dani et al, 2005; Hogle et al, 2010; Picciotto et al, 2002; Piper et al, 2011; Pomerleau et al, 2005) and animal models (Costall et al, 1989; Irvine et al, 2001; Jackson et al, 2008, 2009; Jonkman et al, 2005; Stoker et al, 2008). A recent functional imaging study in smokers correlated affect with aberrant activation in the hippocampus during smoking cue presentation (McClernon et al, 2008). In addition, a follow-up study observed that hippocampal volume could be correlated with successful quit attempts (Froeliger et al, 2010). Hippocampal volume can also be correlated with anxiety levels in patients with anxiety disorders (Bremner et al, 1995; Stein et al, 1997). Furthermore, studies in rodents have shown that lesions to the hippocampus impact anxiety behaviors, including hyponeophagia (Bannerman et al, 2003, 2004; McHugh et al, 2004, 2011; Kalisch et al, 2006). Therefore, the present study examined the effects of sazetidine-A and varenicline following chronic nicotine exposure and withdrawal in two rodent models of anxiety, the novelty-induced hypophagia (NIH) test and the marble-burying test. In addition, hippocampal network function associated with the effects of sazetidine-A and varenicline during nicotine withdrawal was investigated.
MATERIALS AND METHODS
Animals
Male 129SvEv;C57Bl/6J F1 hybrid mice (7–12 weeks of age; 25–35 g; Taconic) were pair-housed and maintained on a 12-h light/dark cycle with food and water available ad libitum in accordance with the University of Pennsylvania Animal Care and Use Committee. All experimental testing sessions were conducted between 0900 and 1500 h, with animals randomly assigned to treatment conditions and tested in counterbalanced order.
Drugs
Doses of nicotine tartrate (Sigma-Aldrich, St Louis, MO), varenicline tartrate (Pfizer, Groton, CT), and sazetidine-A HCl (RTI International, Research Triangle Park, NC) are reported as free base weight. For injection studies, all drugs were prepared immediately before use in 0.9% saline.
Osmotic Minipumps
Nicotine tartrate was dissolved in sterile 0.9% saline solution and infused through subcutaneous osmotic minipumps for 14 days (Model 2002; Alzet, Palo Alto, CA). Mice were anesthetized with an isoflurane/oxygen vapor mixture (1–3%), and osmotic minipumps were inserted subcutaneously using aseptic surgery techniques. Minipumps were placed parallel to the spine at shoulder level with the flow moderator directed away from the wound. The wound was closed with 7 mm stainless steel wound clips (Reflex; Cellpoint Scientific, Gaithersburg, MD).
Minipump treatment groups
In all experiments, animals were implanted with osmotic minipumps to deliver chronic administration of either nicotine (18 mg/kg/day) or saline. Following 2 weeks of chronic administration, mice were anesthetized with an isoflurane/oxygen vapor mixture (1–3%), an incision was made above the pump at shoulder level and the pump was either removed (to initiate spontaneous withdrawal from either nicotine or saline) or left in place (to serve as sham surgical controls in the nicotine and saline groups). The incision was then closed with 7 mm stainless steel wound clips. In all experiments, mice were tested in behavioral paradigms or killed for tissue collection at 24 h following surgery.
Cannula Implantation Surgery
Mice were trained in the NIH test before surgery. Mice were anesthetized with an isoflurane/oxygen vapor mixture (5% induction, 2–3% maintenance) and placed in a stereotaxic device (Kopf Instruments, Tujunga, CA). Bilateral stainless steel guide cannulae (Plastics One, Roanoke, VA) were inserted into the ventral hippocampus and fixed to the skull with dental cement. Dummy cannulae (Plastics One) were inserted into the guide cannulae to prevent clogging. Coordinates for the ventral hippocampus, which were used previously (Kenney et al, 2012), were determined using the atlas of Paxinos and Franklin (2001): −2.8 mm posterior from Bregma, ±3.0 mm mediolateral, and −4.0 mm ventral to skull surface. Metacam (1 mg/kg; Midwest Veterinary Supply, Burnsville, MN) was administered subcutaneously following surgery to minimize postoperative pain. All animals were housed with one littermate and allowed at least 10 days to recover before infusions and behavioral testing were initiated.
Infusion of Drugs
Varenicline tartrate and sazetidine-A HCl were prepared immediately before use in 0.9% saline. Mice were gently restrained and dummy cannulae removed and replaced with 22 gauge infusion cannulae attached to PE50 polyethylene tubing (Plastics One) and a 10-μl Hamilton syringe (Reno, NV). Drugs were infused at a rate of 0.50 μl/min and an injection volume of 0.50 μl per side using a microinfusion pump (KD Scientific, New Hope, PA) over the period of 1 min. Total bilateral infusion procedures lasted 5–7 min. Infusion cannulae were left in place for 1 min following infusion to allow drug to diffuse away from cannulae tip. Direct infusions, as opposed to i.c.v. or systemic infusions, have very good anatomical precision, as the spread of infusion using this procedure is ∼1 mm3 after 24–48 h (Davis et al, 2007; Lewis and Gould, 2007). Behavioral testing was carried out 10–15 min after the infusion procedure because previous data with various behavioral assays as well as locomotor studies indicated that this was the best time and the earliest time point to observe a behavioral effect without drug delivery impacting the anxiety measure (Turner et al, 2010). A 5-min time window was allotted to allow the animal time to recover from the infusion procedure and to move the animal to the behavioral testing area. Drug doses and infusion rates were based on prior studies (Hendrickson et al, 2010; Turner et al, 2010), and any animal exhibiting a clogged cannula was excluded from the data analysis. Following NIH testing, mice were killed and brains were immersion fixed in a 4% formalin solution (Fisher Scientific, Waltham, MA) before sectioning. The sections were stained with cresyl violet and cover slipped and cannula placements were determined using a light microscope.
NIH Test
The NIH test is a well-validated model of anxiety-like behavior in mice (Balu et al, 2013; Bechtholt et al, 2007a, 2007b; Dulawa and Hen, 2005; Gamble-George et al, 2013; Goeldner et al, 2012; Gur et al, 2007; Merali et al, 2003; Onksen et al, 2012; Turner et al, 2010), which is sensitive to acute treatment with anxiolytic drugs, such as chlordiazepoxide (Merali et al, 2003; Turner et al, 2010), but not acute treatment with antidepressants (Gur et al, 2007; Santarelli et al, 2003). One week before training and for the duration of the experiment, mice were housed in groups of two. Training consisted of daily sessions in which mice were exposed to a highly palatable food (Reese’s peanut butter chips; Nestle, Glendale, CA (ingredients: partially defatted peanuts, sugar partially hydrogenated vegetable oil, corn syrup solids, dextrose, reduced minerals whey, salt vanillin, artificial flavor, soy lecithin)) in a clear plastic dish. Plastic dividers (dividing the standard mouse cage lengthwise) were placed inside each cage to separate the mice during the training and home cage testing periods. Mice were acclimated to the barriers for 1 h before placement of food. Food was placed in the cage for 15 min and latency to consume was measured. By the 12th day, a baseline latency to approach and consume the food was reached such that there was <20% variability between mice. Following training, mice were implanted with 14-day osmotic minipumps filled with nicotine (18 mg/kg/day) or 0.9% saline. Testing in the home cage (Home Day 1), novel environment, and home cage (Home Day 2) occurred on the last 3 days of minipump viability. On Home Day 1, all animals received a saline injection 10 min before presentation of food; following testing, minipumps were surgically removed for the withdrawal groups and sham surgeries were performed on the chronic nicotine group as well as saline animals. Twenty-four hours later on Novel Test Day, animals were acclimated for 1 h and injected 10 min before testing in the novel environment with sazetidine-A (0.01, 0.1, and 1.0 mg/kg), varenicline (0.01, 0.1, and 1.0 mg/kg), or saline. Mice were removed from the home cage and placed in an empty standard cage with no bedding. The cage was wiped with a cleanser (Pine Sol, 1 : 10 dilution) to emit a novel odor and placed in a white box with bright light illumination (2150 lux). Latency to consume was recorded. Mice were tested once again in the home environment on day 3 (Home Day 2), but were injected with either saline or drug. On both home test days, the amount consumed was recorded as grams peanut butter chips to ensure there were no appetitive treatment effects. Data with the 1.0 mg/kg dose of sazetidine-A were excluded due to profound reduction in locomotor activity.
Marble-Burying Test
The marble-burying test is a rodent model of anxiety that has high predictive value to detect anxiolytic-like activity of acutely administered drugs (Nicolas et al, 2006). Mice were implanted with 14-day osmotic minipumps filled with nicotine (18 mg/kg/day) or 0.9% saline. Following chronic treatment, animals were tested in the marble-burying test (Turner et al, 2010, 2011). After a period of acclimation (1 h), mice were injected i.p. with saline, sazetidine-A, or varenicline at the doses indicated. Ten minutes later, the mice were placed individually in small cages (26 × 20 × 14 cm), in which 20 marbles had been equally distributed on top of mouse bedding (5-cm deep), and a wire lid was placed on top of the cage. Mice were left undisturbed for 15 min, after which time the number of buried marbles (ie, those covered by bedding three-quarters or more) was counted by a blind observer. Again, data with the 1.0 mg/kg dose of sazetidine-A were excluded due to profound hypolocomotion.
Locomotor Activity
Locomotor activity was assessed in a white Plexiglas box (40 × 40 × 40 cm), which was wiped down with 70% ethanol between animals. To ensure consistent results, animals undergoing chronic treatment were acclimated to the open field environment for 15 min each day over a period of 3 days. On the fourth day, animals in all treatment groups were injected with either saline or the nicotinic drugs. Ten minutes later, they were placed in the locomotor activity box for 15 min. Locomotion was quantified using the TopScan suite (Clever Sys, Reston, VA).
Receptor Binding
Cortical and hippocampal tissues were homogenized in 50 mM Tris–HCl buffer, pH 7.4 at 24 °C, and centrifuged twice at 35 000 g for 10 min in fresh buffer. The membrane pellets were resuspended in fresh buffer and added to tubes containing [3H]Epibatidine (EB; Perkin-Elmer, Boston, MA) or [3H]2-FA-85380 (a kind gift from AstraZeneca, Wilmington, DE) with or without competing drugs. Incubations were performed in Tris buffer at pH 7.4 for 2 h at 24 °C with [3H]EB and [3H]2-FA-85380. Bound receptors were separated from free ligand by vacuum filtration over GF/C glass-fiber filters (Brandel, Gaithersburg, MD) that were prewet with 0.5% polyethyleneimine, and the filters were then counted in a liquid scintillation counter. Non-specific binding was determined in the presence of 300 μM nicotine, and specific binding was defined as the difference between total binding and non-specific binding.
Voltage-Sensitive Dye Imaging
Mice undergoing chronic treatment with saline, nicotine (18 mg/kg/day), or 24 h nicotine withdrawal (24 h WD) were decapitated following isoflurane anesthesia. The brain was removed and horizontal hippocampal slices (350 μm) were cut with an Integraslice 7550 PSDS (Campden Instruments, Lafayette, IN) in an ice-cold artificial cerebrospinal fluid (ACSF) containing an equiosmolar concentration of sucrose. ACSF consisted of 130 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 26 mM NaHCO3, 10 mM glucose, 1 mM MgCl2, 2 mM CaCl2 (pH 7.2–7.4 when saturated with 95% O2/5% CO2). Hippocampal slices (between −5.60 and −6.60 from Bregma) were then transferred to a static interface chamber (34 °C) for 30 min and kept at 22–25 °C thereafter, until transfer to the recording chamber. The osmolarity of all solutions was 305–315 mOsm.
Slices were stained with 0.125 mg/ml of the voltage-sensitive dye di-3-ANEPPDHQ (D36801, Invitrogen) in ACSF for 20 min, and imaged in an oxygenated interface chamber (34 °C) using an 80 × 80 CCD camera recording at a 2-kHz frame rate (NeuroCCD: RedShirtImaging, Decatur, GA). Epi-illumination was provided by a custom LED illuminator. Compared with the more commonly used photodiode array, the CCD chip well size (215 000 electrons) requires use of relatively low light intensities minimizing photodynamic damage. A 10 × objective lens (Olympus, Tokyo, Japan) imaged a 2.5 × 2.5 mm CA1 region (32 × 32 μm region per pixel). Schaffer collateral stimulation was administered with the electrode placed near the border of CA1 in the stratum radiatum (SR). Each trial consisted of 12 stimuli, 20 s apart.
Data Analyses
Voltage-sensitive dye imaging
While in some experiments multiple slices were tested from the same animal, these were treated as replicates and n refers to the total number of animals in each group. VSD data were analyzed in IGOR (Wavemetrics, Lake Oswego, OR) on 12 trial averages as previously described (Ang et al, 2005; Ortinski et al, 2010). Briefly, fluorescence changes were calculated as the percentage change in fluorescence divided by the resting fluorescence (dF/F). Fitted double exponentials were subtracted from this normalized dF/F to compensate for photobleaching. Local VSD signals were quantified from visually identified regions of interest (ROIs) in the SR of the CA1 in the hippocampus. Identical ROIs were used during analysis of within-slice parameters, while strict adherence to anatomical landmarks was used to ensure consistency of ROI areas across slices. To make comparisons between groups, we averaged the peak dF/F signal during an integration window corresponding to the peak of the response. To evaluate the extent of spatial activation, we measured the percent of area significantly activated in each ROI by summing the number of pixels that exhibited significant depolarization after stimulation (Δ0.04% dF/F, corresponding to responses⩾3 SD over noise levels). This ‘active’ pixel number was normalized to the total number of pixels in the ROI and the average number of pixels in a 10-ms window around the peak was calculated.
General data analysis
Statistical analyses were performed with GraphPad Prism 5.0 software package (GraphPad Software, San Diego, CA). Differences between groups were assessed using one- or two-way ANOVA followed by Bonferroni’s multiple comparison tests. To better assess significant correlations between in vivo behavior and CA1 reactivity, we used group data in the generation of plotted points for a Pearson correlation analysis with 95% CI. All data are expressed as mean±SEM.
RESULTS
Withdrawal from Chronic Nicotine Elicits an Anxiogenic Effect in the NIH Test
In humans, nicotine withdrawal is often characterized by an increase in anxiety. To model this pre-clinically, we utilized the NIH test, a well-validated model of anxiety-like behavior in mice (Balu et al, 2013; Bechtholt et al, 2007a, 2007b; Dulawa et al, 2005; Goeldner et al, 2012; Gur et al, 2007; Merali et al, 2003; Onksen et al, 2012; Turner et al, 2010). Animals chronically treated with nicotine show a reduced latency to feed in a novel environment relative to saline controls, indicating an anxiolytic response (Figure 1) (Turner et al, 2010). In contrast, those animals undergoing 24 h WD show an increased latency to feed relative to saline controls, indicative of an anxiogenic response. These effects were not attributable to changes in appetitive behavior as there are no differences in the latency to feed (P>0.05) or in the amount consumed (P>0.05) in the home environment on Home Day 1 or Home Day 2.

Behavioral effects of chronic nicotine and nicotine withdrawal. Chronic nicotine is anxiolytic while 24 h withdrawal (WD) from nicotine is anxiogenic in the NIH test. Mice were implanted with 14-day osmotic minipumps filled with saline or nicotine. On day 13, osmotic minipumps were extracted from ½ of the animals to elicit spontaneous withdrawal from either saline or nicotine. After chronic treatment, mice were tested in the NIH paradigm and the 24-h WD time point corresponds to novel day testing. Latency to approach and consume food is shown as seconds±SEM. Chronic treatment with nicotine significantly reduced the latency to consume food in a novel environment (***P<0.001). In contrast, spontaneous withdrawal from nicotine at the 24-h time point (24 h WD) significantly increased the latency to consume food in a novel environment, compared with both saline (***P<0.001) and chronic nicotine (###P<0.001) (n=6).
Chronic In Vivo Treatment Increases nAChRs in the Hippocampus and Cortex
A hallmark of chronic nicotine administration is the upregulation of nicotinic receptors in the brain (Marks et al, 1983; Schwartz and Kellar, 1983) and recent studies in humans have suggested a functional role of these receptors in relapse vulnerability (Cosgrove et al, 2009). Radioligand binding to nicotinic receptors is an effective way of evaluating upregulation. Epibatidine is a broad-spectrum nAChR ligand that binds with very high affinity (25–300 pM) to all known heteromeric nAChRs (Avalos et al, 2002; Houghtling et al, 1995; Xiao and Kellar, 2004). In contrast, [3H]2-FA-85380 binds selectively to β2-containing nAChRs (Brody et al, 2006; Deuther-Conrad et al, 2006; Vaupel et al, 2005). Therefore, examination of the nAChR binding sites labeled by these two ligands in the ventral hippocampus (which is an area important in modulating anxiety responses) and in the cortex (which possesses almost exclusively α4β2 nAChRs) helps to grossly identify those receptors possessing a β2 subunit in the upregulated pool of nAChRs. Following chronic administration of nicotine, [3H]epibatidine (1.5 nM) binding to both hippocampal and cortical homogenates was increased (Figure 2ai and ii), indicating significant upregulation of heteromeric nAChRs following in vivo treatment with nicotine. Furthermore, binding experiments using a saturating concentration of [3H]2-FA-85380 (1.8 nM) demonstrate that this upregulated pool of nAChRs in both the hippocampus and the cortex contains the β2 subunit (Figure 2bi and ii). In both the hippocampus and cortex, nAChR upregulation persisted despite 24 h cessation of nicotine treatment (Figure 2b), indicating that these increased numbers of receptors are present during withdrawal. Therefore, investigation of drugs specific for these receptor subtypes is relevant.

Chronic nicotine and 24 h nicotine WD effects on nicotinic receptor regulation in the hippocampus and cortex. Homogenate-binding experiments with a saturating concentration of (a) [3H]epibatidine ([3H]EB, 1.5 nM) or (b) [3H]2-FA-85380 (1.8 nM) were performed on hippocampal (i) and cortical (ii) homogenates. Chronic nicotine upregulated heteromeric nAChRs labeled by (a) [3H]EB and (b) [3H]2-FA-85380 in both the hippocampus (i) and the cortex (ii), which persisted following 24 h of nicotine withdrawal (**P<0.01; ***P<0.001) (n=3–7).
Effects of Sazetidine-A and Varenicline in the NIH Test and Marble-Burying Test During Nicotine Withdrawal
Aversive symptoms such as anxiety during smoking abstinence often prompt relapse and the upregulation of β2-containing nAChRs has been suggested to underlie both the aversive symptoms and relapse vulnerability (Cosgrove et al, 2009). Therefore, understanding the effects of potential smoking cessation aids targeting β2-containing nAChRs on nicotine withdrawal-induced anxiety is critical. To do this, we examined the effects of varenicline and sazetidine-A in animals undergoing 24 h WD from nicotine in both the NIH test and the marble-burying test. As shown previously, chronic nicotine has a significant anxiolytic effect while nicotine withdrawal produces a strong anxiogenic response in this test (Figure 3a and b). However, an acute i.p. administration of sazetidine-A (0.01 or 0.1 mg/kg) given to a mouse undergoing nicotine withdrawal 10 min before testing reversed the anxiogenic effects of 24 h WD (Figure 3a). In contrast, acute administration of varenicline at a range of doses (0.01, 0.1, or 1.0 mg/kg) did not ameliorate nicotine withdrawal behaviors in the NIH test (Figure 3b). To evaluate the effects of these drugs in another model of anxiety, we utilized the marble-burying test, which has been shown to be sensitive to both acute and chronic administration of nicotinic compounds in drug-naive animals (Anderson and Brunzell, 2012; Turner et al, 2010, 2011). In this task, we observe that treatment with sazetidine-A (0.1 mg/kg) as well as varenicline (1.0 mg/kg) significantly reduces the number of marbles buried in this task compared with animals undergoing 24 h WD (Figure 3c). Because altered locomotor activity can confound results both in the NIH test and in the marble-burying test, treatment effects on locomotor activity were evaluated. However, as shown in Figure 3d, no differences in locomotor activity were observed.

Effect of sazetidine-A and varenicline on nicotine withdrawal-induced behaviors in the NIH test and marble-burying test. Mice treated with saline, chronic nicotine, or 24 h withdrawal from chronic nicotine were tested in the NIH paradigm, the marble-burying test, or in the open field. (a, b) On Novel Test Day, mice received intraperitoneal injections of saline or drug 10 min before testing. The mean latency to approach and consume food is shown as seconds±SEM. Chronic nicotine reduced the latency to consume food in the novel environment relative to both saline (***P<0.001; *P<0.05) and 24 h WD (###P<0.001, #P<0.05). Nicotine withdrawal significantly increased the latency to approach and consume food in the novel environment compared with saline (***P<0.001; **P<0.01). No significant treatment effects were observed in the home environment (n=7–13). (a) Acute sazetidine-A treatment at the low dose (0.01 mg/kg) during 24 h WD significantly reduced latencies compared with 24 h WD (###P<0.001), while the 0.1 mg/kg sazetidine-A dose administered during nicotine withdrawal significantly reduced latency to feed relative to both saline treatment (***P<0.05) and 24 h WD (###P<0.001) (n=7–13). (b) In contrast, acute treatment with varenicline (0.1 mg/kg) during 24 h nicotine withdrawal remained significantly elevated from saline levels (*P<0.05) and no dose of varenicline resulted in significantly different values relative to 24 h WD (n=7–13). (c) Chronic treatment with nicotine resulted in significantly fewer marbles buried in the marble-burying test compared with saline (*P<0.05). Additionally, the chronic nicotine treatment group as well as administration of sazetidine-A (0.1 mg/kg) or varenicline (1.0 mg/kg) during 24 h WD resulted in significantly fewer marbles buried compared with 24 h WD (###P<0.001; ##P<0.01) (n=6–8). (d) There were no significant treatment effects on locomotor activity (n=7–8).
Hippocampal-Specific Injection of Sazetidine-A, But Not Varenicline, Rescues Anxiogenic Nicotine Withdrawal Effects in the NIH Test
The selective differential effects of sazetidine-A and varenicline in the NIH test suggested divergent effects on the neural substrates driving this behavioral response. The NIH test is an anxiety model that is sensitive to ventral hippocampal perturbation (Bannerman et al, 2002, 2003; McHugh et al, 2004), suggesting that the behavioral effects of sazetidine-A and varenicline may be mediated within this structure. To determine if the ventral hippocampus is a critical neural substrate underlying the behavioral effects of sazetidine and varenicline in the NIH, we injected these compounds directly into this structure during 24 h WD. As shown in Figure 4a, infusion of sazetidine-A at either the low (10 pM) or high (1000 pM) concentration resulted in significant reduction in the latency to feed during 24 h WD in the NIH test. In contrast, ventral hippocampal infusions of varenicline at either dose (10 or 1000 pM) had no significant effect on the latency to feed in the novel environment.

Hippocampal-specific infusions of sazetidine-A, but not varenicline, rescue anxiety-like nicotine withdrawal impairments. (a) Mice undergoing 24 h withdrawal from chronic nicotine were tested in the NIH paradigm. On Novel Test Day, mice received intrahippocampal infusions of saline or drug 10–15 min before testing. The mean latency to approach and consume food is shown as seconds±SEM. Hippocampal-specific treatment with sazetidine-A at either 10 or 1000 pM reduced the latency to consume food in the novel environment (*P<0.05; **P<0.01), compared with nicotine withdrawal. In contrast, intrahippocampal treatment with varenicline (10 or 1000 pM) during nicotine withdrawal had no effect. No significant treatment effects were observed in the home environment. (b) Schematic of bilateral infusion sites in the ventral hippocampus (n=6–7).
Alterations in Anxiety-like Behavioral Responses in the NIH are Paralleled by Functional Changes in the Hippocampal Network Activity
Because of the ventral hippocampal-specific effects of these drugs in the NIH test, we chose to evaluate their effects on ventral hippocampal network activity. However, to do this, we first investigated hippocampal functional changes following in vivo treatment with chronic nicotine and withdrawal, which may impact the efficacy of these drugs. To do this, evoked responses were recorded from ventral hippocampal slices taken from animals treated with saline, chronic nicotine (18 mg/kg/day), or 24 h WD.
Electrical stimulation of the Schaffer collateral pathway (Figure 5a and b) results in robust activation of the CA1, a region of the hippocampus sensitive to stress-induced synaptic remodeling (Dalla et al, 2009; Joels et al, 2004, 2008; McEwen, 2001; McEwen and Magarinos, 2001; Shors et al, 1997, 2001) and shown to be selectively affected in individuals with anxiety disorders (Cole et al, 2010). As shown in Figure 5ci and ii (+10ms), responses evoked in the CA1 by Schaffer collateral stimulation were significantly increased following chronic administration of nicotine relative to saline controls. In contrast, hippocampal slices from animals undergoing 24 h WD showed responses comparable to saline controls (Figure 5ci and iii, +10 ms). These increases with nicotine and decreases with 24 h WD are reflected both in the intensity of response (Figure 5d) and in the amount of area recruited following stimulation (Figure 5e). Interestingly, although there was an apparent loss of inhibition following chronic nicotine treatment which is evident 100 ms after the stimulation (Figure 5ci and ii, +100 ms), there was no effect of in vivo treatment on Tau (Figure 5f), a measurement reflecting inhibitory activity. This suggests that chronic administration of nicotine impacts predominantly the excitatory components governing network activity of the CA1, which return to control levels following 24 h of WD.

CA1 responsivity to Schaffer collateral stimulation is increased following in vivo chronic nicotine treatment. (a) Diagram of hippocampus circuitry illustrates the stimulus paradigm utilized in this study. A stimulating electrode was placed in the Schaffer collateral axons from CA3 pyramidal cells and a recording electrode was placed in the distal end of SR in CA1. The light gray line represents the pyramidal cell layer and the dotted black line delineates the path of the Schaffer collateral axons. The dark gray box depicts the area visualized in b. (b) Horizontal slices containing the hippocampus were visualized under a 10 × lens. The black triangle shows the stimulating electrode placement and the white triangle shows the placement of the recording electrode. Labels correspond to: SO—stratum oriens, SR—stratum radiatum, SLM—stratum lacunosum moleculare, CTX—cortex, DG—dentate gyrus. (c) Representative images of the average pixel changes for the duration indicated following stimulation demonstrates the peak excitatory (top panel, +10 ms) and inhibitory (bottom panel, +100 ms) responses in saline (i), nicotine (ii), or 24 h nicotine WD (iii) groups. Changes in membrane voltage are illustrated in red (excitation) or blue (inhibition). (d–f) Chronic administration of nicotine increased both the amplitude of peak excitation (d) and the area of peak excitation (e) in stratum radiatum (SR) compared with saline (*P<0.05; **P<0.01) and 24 h nicotine withdrawal (##P<0.01) conditions. No differences in tau (f) were observed in the SR between treatment groups. All data are presented as mean±SEM (n=6–14).
Previous Nicotine Exposure Differentially Impacts Effects of Sazetidine-A and Varenicline on CA1 Evoked Responses
To investigate underlying functional mechanisms associated with the behavioral response to sazetidine-A and varenicline during nicotine withdrawal, we examined the effects of these drugs in vitro on ventral hippocampal function following in vivo administration of nicotine and 24 h WD. Microdialysis studies in naïve animals have found low micromolar levels of endogenous acetylcholine in the hippocampus (Marrosu et al, 1995). In addition, deposition studies show that varenicline levels in brain following an acute administration remain in the low micromolar range for up to 6 h (Rollema et al, 2010). Based on these studies, we chose to utilize bath application of low micromolar concentrations of the partial agonists. As shown previously, hippocampal responsivity was increased following chronic nicotine administration and returned to saline levels after 24 h of nicotine cessation (Figure 6a, top row). In saline-treated animals (Figure 6a, first column), bath application of sazetidine-A (1 μM) slightly dampened baseline activity while varenicline (1 μM) mildly enhanced it. Conversely, in nicotine-treated animals (Figure 6a, middle column), sazetidine-A application had no effect while varenicline greatly attenuated the increased excitability resulting from chronic in vivo treatment with nicotine. During nicotine WD (Figure 6a, last column), bath application of sazetidine-A enhances the CA1 responsivity to near nicotine-treated levels, while varenicline application results in a small attenuation of the evoked response. Thus, the modulatory effects of sazetidine-A and varenicline in the hippocampus differ depending on the in vivo drug history (eg, saline, nicotine, or 24 h WD) (Figure 6b). Sazetidine-A effectively reduces CA1 excitability in a saline-treated animal, has no measurable effect in the nicotine-treated animal, and significantly enhances CA1 excitability in animals undergoing 24 h WD. In contrast, varenicline mildly enhances CA1 excitability in naïve animals, while attenuating responses in both the nicotine and 24 h WD groups.

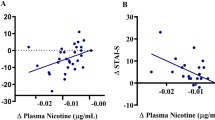
Effects of sazetidine-A and varenicline on CA1 excitatory responses are dependent upon in vivo drug treatment. (a) Representative images of the average pixel changes for the peak excitatory responses (+10 ms) in saline (i), nicotine (ii), or 24 h nicotine WD (iii) groups are shown at baseline (top row), during bath application of sazetidine-A (1 μM, middle row), or during bath application of varenicline (1 μM, bottom row). Excitatory changes in membrane voltage are illustrated in red. (b) Effects of in vitro application of sazetidine-A and varenicline on peak SR excitatory responses are shown as mean percent change from their respective in vivo drug treatment baseline. In vitro sazetidine-A (1 μM) treatment attenuates CA1 excitatory responses in saline animals, has no effect in chronic nicotine-treated animals, and enhances peak excitatory responses during 24 h withdrawal from chronic nicotine. In contrast, in vitro treatment with varenicline (1 μM) slightly enhances CA1 excitatory responses in saline animals, while significantly attenuating CA1 excitatory responses in both chronic nicotine and 24 h nicotine withdrawal conditions. Significantly different from respective saline in vivo condition: *P<0.05; **P<0.01; ***P<0.001. Significantly different from respective nicotine in vivo condition: #P<0.05. Data represent mean changes observed in 7–14 animals per group. (c) The amplitude of peak SR excitation significantly correlated with the latency to feed in the NIH paradigm on Novel Test Day (P<0.05). Group data for the in vivo treatment groups are represented in the correlational graphs. Dashed lines indicate the 95% CI (n=5–7 per group).
Correlation of Ventral Hippocampal Function with Anxiety-like Behavior in the NIH Test
To determine if there is an association between hippocampal function and nicotine withdrawal-related anxiety behaviors, we correlated CA1 excitability in the hippocampus with the NIH behavioral responses in all treatment groups. We found that heightened CA1 evoked responses in the hippocampus significantly correlate with reduced anxiety in the NIH test (Figure 6c). These data conclude that increased hippocampal excitability accompanies both anxiolytic effects of chronic nicotine (Figures 1a and 2) and anxiolytic effects of sazetidine-A in 24 h WD animals (Figure 2). Conversely, reduced hippocampal excitability accompanies anxiogenic effects observed in 24 h WD animals (Figures 1a and 2) and following treatment with varenicline during 24 h WD (Figure 2).
DISCUSSION
Gray and McNaughton (1996, 2000) have proposed the hippocampus as the neurobiological correlate of trait anxiety in the brain and theorized that those systems responsible for anxious behaviors during conflict are mediated by the hippocampus. Recently, this has been demonstrated experimentally in human subjects in fMRI connectivity studies showing that the hippocampus controls emotional responses in the amygdala and other brain regions (Hahn et al, 2010). Furthermore, there is a large body of rodent literature demonstrating that lesions of the ventral hippocampus specifically, but not the dorsal hippocampus or amygdala, impact anxiety behaviors in conflict and hyponeophagia paradigms like the NIH test (Bannerman et al, 2002, 2003, 2004; McHugh et al, 2004, 2011). This hippocampal control of emotional responses could be accomplished via endogenous cholinergic drive within the hippocampus working synergistically with GABAergic inputs from the septum to affect hippocampal circuitry, thus directly inhibiting anxiety responses (Degroot and Treit, 2003, 2004).
Nicotine withdrawal results in an increase in anxiety symptoms (for review, see Dani et al, 2005, Picciotto et al, 2008, and Picciotto et al, 2002), which can be dissociated from fear responses during nicotine withdrawal (Hogle et al, 2010). Clinically, studies in smoking populations have found that hippocampal activation due to smoking cue presentation is associated with anxiety symptoms in abstinent smokers (McClernon et al, 2008). Furthermore, hippocampal volume has been shown to predict smoking cessation treatment outcomes (Froeliger et al, 2010). Though clinical studies have described associations between nicotine withdrawal-induced anxiety, hippocampal function, and smoking cessation treatment outcomes, the present studies are the first concerted effort to (1) determine the anxiolytic efficacy of these drugs in nicotine-dependent animals and (2) evaluate the effect of these drugs on hippocampal network function following chronic treatment with nicotine and withdrawal.
Our results show that the NIH test is sensitive to the effects of chronic nicotine and 24 h nicotine withdrawal. In concordance with clinical findings in abstinent smokers (McClernon et al, 2008), these alterations in anxiety-related behaviors are paralleled by a nicotine-induced change in ventral hippocampal activation; specifically, chronic nicotine increases evoked responses from the CA1 of the hippocampus following Schaffer collateral stimulation (Figures 5 and 6). The first report of increased excitability as a consequence of in vivo nicotine administration was recently described in the hippocampus using single-cell and extracellular field recordings; this study found that increased excitability in CA1 pyramidal neurons persists even after prolonged withdrawal (24 h–9 months) (Penton et al, 2011). We extend these single-cell findings to demonstrate the alterations in hippocampal network function following chronic nicotine treatment and withdrawal, specifically demonstrating an increase in network-level neuronal activity following chronic in vivo nicotine administration (Figure 5). Recent voltage-sensitive dye imaging (VSDi) studies have demonstrated the importance of cholinergic signaling in the integration of excitatory inputs, specifically via α4β2 nAChRs (Bell et al, 2011; McQuiston, 2010). Continuous modulation of these inputs, as in the case with chronic nicotine exposure, may lead to the long-lasting CA1 hyperexcitability, which we observe following chronic nicotine treatment and withdrawal. This hyperexcitability may underlie many of the nicotine withdrawal symptoms (Kenney et al, 2008).
In addition to the functional changes observed following chronic in vivo nicotine and nicotine withdrawal, we also show that the α4β2 selective nicotinic compounds sazetidine-A and varenicline exhibit very different effects during nicotine withdrawal. Previous studies show that both sazetidine-A and varenicline ameliorate cognitive withdrawal symptoms (Raybuck et al, 2008; Rezvani et al, 2011) and reduce nicotine self-administration (Levin et al, 2010; O'Connor et al, 2010; Rezvani et al, 2010). Furthermore, both varenicline and sazetidine-A have shown antidepressant efficacy in the forced swim test (Caldarone et al, 2011; Kozikowski et al, 2009; Mineur et al, 2011; Rollema et al, 2009; Turner et al, 2010). In contrast, in conflict anxiety paradigms like the NIH test, we have previously shown that chronic sazetidine-A has anxiolytic effects, while varenicline does not (Turner et al, 2010). However, these previous studies examined the effects of sazetidine-A and varenicline in naïve animals, in not nicotine-dependent animals. The present studies examine the effects of these drugs following 24 h withdrawal from chronic nicotine treatment. During nicotine withdrawal, we find that acute administration of sazetidine-A, whether administered i.p. or specifically infused into the ventral hippocampus, successfully rescued nicotine-withdrawal deficits in the NIH test (Figures 3a and 4). In contrast, neither i.p. injections nor hippocampal infusions of varenicline had any effect on nicotine-withdrawal deficits in the NIH test (Figures 3b and 4). While nicotine withdrawal may potentially impair cognitive processing related to novelty, it is unlikely that this impacts our interpretation of the NIH test. First, our previous work has shown that while nicotine withdrawal at the same dose significantly impairs hippocampal-dependent contextual learning in C57/Bl6 mice, the 129SvEv;C57Bl/6J F1 hybrid mice utilized in these studies do not exhibit learning impairments (Wilkinson et al, 2013). Second, these studies specifically target alterations in the ventral hippocampus, which in contrast to the dorsal hippocampus, is thought to contribute to anxiety-related phenotypes and not spatial memory, per se. For example, lesions to the ventral hippocampus affect anxiety but not spatial learning in rodents (Bannerman et al, 2002, 2003, 2004; McHugh et al, 2004, 2011). And third, we have previously shown that while there is a selective deficit in spatial object recognition during nicotine withdrawal, there was no effect on novel object recognition (Kenney et al, 2011), suggesting that the cognitive impairments following nicotine withdrawal are not novelty related. Because alterations in motor ability following chronic treatment with nicotine and/or 24 h WD would seriously impede proper interpretation of our NIH test results, we evaluated the effects of our treatments in an open field analysis. None of these effects were due to differences in locomotion (Figure 3d). We also find that the dissociable effects of varenicline and sazetidine-A are also paradigm specific. For example, in the marble-burying test, we observe that acute treatment with either sazetidine-A (0.1 mg/kg) or varenicline (1.0 mg/kg) during 24 h WD is anxiolytic, which is similar to our previous observations with acute or chronic treatment of sazetidine-A and varenicline in naïve animals (Turner et al, 2010, 2011). However, the precise neural substrates underlying the marble-burying task are unknown.
In addition, the dissociable effects of varenicline and sazetidine-A were also evident in vitro, where application of sazetidine-A potentiated evoked responses in the hippocampus, while application of varenicline did not. Current studies indicate a network level balance between activation and desensitization is important in eliciting the behavioral effects of nicotinic compounds (for review, see Picciotto et al, 2008). In vitro studies show that low concentrations of sazetidine-A profoundly desensitize α4β2 nAChRs for prolonged periods (Xiao et al, 2006; Zwart et al, 2008). This is in contrast to varenicline, where recent evidence suggests that the therapeutic window for varenicline corresponds to the point at which there is enough unbound agonist to elicit combined desensitization and receptor activation (Rollema et al, 2010). Therefore, these results may be due to differential effects of varenicline and sazetidine-A on nAChR activation and desensitization (Caldarone et al, 2011; Rollema et al, 2010; Xiao et al, 2006; Zwart et al, 2008).
Nicotine dependence impacts 46 million American adults (CDC, 2007). Current treatment options, whether nicotine replacement therapy, bupropion, or varenicline, have a best treatment outcome of <20% of participants maintaining abstinence for 12 months (Gonzales et al, 2006). Furthermore, while prolonged varenicline treatment has been shown to significantly decrease relapse rates during the period of administration (Lee et al, 2008), >50% of the test subjects relapsed at 1 year post quit irrespective of the treatment group (Tonstad et al, 2006). This phenomenon of increased relapse rate following cessation of varenicline treatment may be related to varenicline’s ability to induce nAChR upregulation (Turner et al, 2011). Of interest, sazetidine-A does not upregulate nAChRs (Hussmann et al, 2012). Though the consequence of this upregulation is not fully understood, a recent SPECT study has suggested that nicotinic receptor availability is directly related to increased relapse rate (Cosgrove et al, 2009). However, studies examining the effects of smoking cessation drugs are often conducted in naïve animals and it is therefore unknown whether or not the upregulated pool of nAChRs is critical for therapeutic efficacy of smoking cessation aids, perhaps making the findings less representative of clinical outcomes. These studies, which directly assess two candidate pharmacotherapeutics during 24 h withdrawal from chronic nicotine, demonstrate that the effects of both sazetidine-A and varenicline are critically dependent upon previous nicotine administration. Thus, these combined studies provide a paradigm for using currently available drugs to broadly probe nAChR plasticity. In doing so, we provide a novel behaviorally validated outcome measure for future drug and therapy development that target the still recalcitrant problem of successful smoking cessation.
References
Anderson SM, Brunzell DH (2012). Low dose nicotine and antagonism of beta2 subunit containing nicotinic acetylcholine receptors have similar effects on affective behavior in mice. PLoS One 7: e48665.
Ang CW, Carlson GC, Coulter DA (2005). Hippocampal CA1 circuitry dynamically gates direct cortical inputs preferentially at theta frequencies. J Neurosci 25: 9567–9580.
Avalos M, Parker MJ, Maddox FN, Carroll FI, Luetje CW (2002). Effects of pyridine ring substitutions on affinity, efficacy, and subtype selectivity of neuronal nicotinic receptor agonist epibatidine. J Pharmacol Exp Ther 302: 1246–1252.
Azizian A, Monterosso J, O'Neill J, London ED (2009). Magnetic resonance imaging studies of cigarette smoking. Handb Exp Pharmacol 113–143.
Balu DT, Turner JR, Brookshire BR, Hill-Smith TE, Blendy JA, Lucki I (2013). Brain monoamines and antidepressant-like responses in MRL/MpJ versus C57BL/6J mice. Neuropharmacology 67: 503–510.
Bannerman DM, Deacon RM, Offen S, Friswell J, Grubb M, Rawlins JN (2002). Double dissociation of function within the hippocampus: spatial memory and hyponeophagia. Behav Neurosci 116: 884–901.
Bannerman DM, Grubb M, Deacon RM, Yee BK, Feldon J, Rawlins JN (2003). Ventral hippocampal lesions affect anxiety but not spatial learning. Behav Brain Res 139: 197–213.
Bannerman DM, Rawlins JN, McHugh SB, Deacon RM, Yee BK, Bast T et al (2004). Regional dissociations within the hippocampus—memory and anxiety. Neurosci Biobehav Rev 28: 273–283.
Bechtholt AJ, Hill TE, Lucki I (2007a). Anxiolytic effect of serotonin depletion in the novelty-induced hypophagia test. Psychopharmacology (Berl) 190: 531–540.
Bechtholt AJ, Valentino RJ, Lucki I (2007b). Overlapping and distinct brain regions associated with the anxiolytic effects of chlordiazepoxide and chronic fluoxetine. Neuropsychopharmacology 33: 2117–2130.
Bell KA, Shim H, Chen CK, McQuiston AR (2011). Nicotinic excitatory postsynaptic potentials in hippocampal CA1 interneurons are predominantly mediated by nicotinic receptors that contain alpha4 and beta2 subunits. Neuropharmacology 61: 1379–1388.
Benowitz NL (2009). Pharmacology of nicotine: addiction, smoking-induced disease, and therapeutics. Annu Rev Pharmacol Toxicol 49: 57–71.
Bremner JD, Randall P, Scott TM, Bronen RA, Seibyl JP, Southwick SM et al (1995). MRI-based measurement of hippocampal volume in patients with combat-related posttraumatic stress disorder. Am J Psychiatry 152: 973–981.
Brody AL, Mandelkern MA, London ED, Olmstead RE, Farahi J, Scheibal D et al (2006). Cigarette smoking saturates brain alpha 4 beta 2 nicotinic acetylcholine receptors. Arch Gen Psychiatry 63: 907–915.
Caldarone BJ, Wang D, Paterson NE, Manzano M, Fedolak A, Cavino K et al (2011). Dissociation between duration of action in the forced swim test in mice and nicotinic acetylcholine receptor occupancy with sazetidine, varenicline, and 5-I-A85380. Psychopharmacology (Berl) 217: 199–210.
CDC (2007). Cigarette smoking among adults—United States, 2006. Morbid Mortal Wkly Rep 56: 1157–1161.
Coe JW, Brooks PR, Vetelino MG, Wirtz MC, Arnold EP, Huang J et al (2005). Varenicline: an alpha4beta2 nicotinic receptor partial agonist for smoking cessation. J Med Chem 48: 3474–3477.
Cole J, Toga AW, Hojatkashani C, Thompson P, Costafreda SG, Cleare AJ et al (2010). Subregional hippocampal deformations in major depressive disorder. J Affect Disord 126: 272–277.
Corrigall WA, Coen KM, Adamson KL (1994). Self-administered nicotine activates the mesolimbic dopamine system through the ventral tegmental area. Brain Res 653: 278–284.
Corrigall WA, Franklin KB, Coen KM, Clarke PB (1992). The mesolimbic dopaminergic system is implicated in the reinforcing effects of nicotine. Psychopharmacology (Berl) 107: 285–289.
Cosgrove KP, Batis J, Bois F, Maciejewski PK, Esterlis I, Kloczynski T et al (2009). beta2-Nicotinic acetylcholine receptor availability during acute and prolonged abstinence from tobacco smoking. Arch Gen Psychiatry 66: 666–676.
Costall B, Kelly ME, Naylor RJ, Onaivi ES (1989). The actions of nicotine and cocaine in a mouse model of anxiety. Pharmacol Biochem Behav 33: 197–203.
Dalla C, Whetstone AS, Hodes GE, Shors TJ (2009). Stressful experience has opposite effects on dendritic spines in the hippocampus of cycling versus masculinized females. Neurosci Lett 449: 52–56.
Dani JA, Harris RA (2005). Nicotine addiction and comorbidity with alcohol abuse and mental illness. Nat Neurosci 8: 1465–1470.
Davis JA, Gould TJ (2009). Hippocampal nAChRs mediate nicotine withdrawal-related learning deficits. Eur Neuropsychopharmacol 19: 551–561.
Davis JA, Kenney JW, Gould TJ (2007). Hippocampal alpha4beta2 nicotinic acetylcholine receptor involvement in the enhancing effect of acute nicotine on contextual fear conditioning. J Neurosci 27: 10870–10877.
Degroot A, Treit D (2003). Septal GABAergic and hippocampal cholinergic systems interact in the modulation of anxiety. Neuroscience 117: 493–501.
Degroot A, Treit D (2004). Anxiety is functionally segregated within the septo-hippocampal system. Brain Res 1001: 60–71.
Deuther-Conrad W, Wevers A, Becker G, Schildan A, Patt M, Sabri O et al (2006). Autoradiography of 2-[18F]F-A-85380 on nicotinic acetylcholine receptors in the porcine brain in vitro. Synapse (New York, NY) 59: 201–210.
DiMatteo MR, Lepper HS, Croghan TW (2000). Depression is a risk factor for noncompliance with medical treatment: meta-analysis of the effects of anxiety and depression on patient adherence. Arch Intern Med 160: 2101–2107.
Dulawa SC, Hen R (2005). Recent advances in animal models of chronic antidepressant effects: the novelty-induced hypophagia test. Neurosci Biobehav Rev 29: 771–783.
Froeliger B, Kozink RV, Rose JE, Behm FM, Salley AN, McClernon FJ (2010). Hippocampal and striatal gray matter volume are associated with a smoking cessation treatment outcome: results of an exploratory voxel-based morphometric analysis. Psychopharmacology (Berl) 210: 577–583.
Gamble-George JC, Conger JR, Hartley ND, Gupta P, Sumislawski JJ, Patel S (2013). Dissociable effects of CB1 receptor blockade on anxiety-like and consummatory behaviors in the novelty-induced hypophagia test in mice. Psychopharmacology (Berl).
Gatt JM, Nemeroff CB, Dobson-Stone C, Paul RH, Bryant RA, Schofield PR et al (2009). Interactions between BDNF Val66Met polymorphism and early life stress predict brain and arousal pathways to syndromal depression and anxiety. Mol Psychiatry 14: 681–695.
Goeldner C, Spooren W, Wichmann J, Prinssen EP (2012). Further characterization of the prototypical nociceptin/orphanin FQ peptide receptor agonist Ro 64-6198 in rodent models of conflict anxiety and despair. Psychopharmacology (Berl) 222: 203–214.
Gonzales D, Rennard SI, Nides M, Oncken C, Azoulay S, Billing CB et al (2006). Varenicline, an alpha4beta2 nicotinic acetylcholine receptor partial agonist, vs sustained-release bupropion and placebo for smoking cessation: a randomized controlled trial. JAMA 296: 47–55.
Gray JA, McNaughton N (1996). The neuropsychology of anxiety: reprise. Nebr Symp Motiv 43: 61–134.
Gray JA, McNaughton N (2000) The Neuropsychology of Anxiety: An Enquiry into the Functions of the Septo-Hippocampal System 2nd edn Oxford University Press: Oxford, New York. xvi, 424 p..
Grieder TE, George O, Tan H, George SR, Le Foll B, Laviolette SR et al (2012). Phasic D1 and tonic D2 dopamine receptor signaling double dissociate the motivational effects of acute nicotine and chronic nicotine withdrawal. Proc Natl Acad Sci USA 109: 3101–3106.
Gur TL, Conti AC, Holden J, Bechtholt AJ, Hill TE, Lucki I et al (2007). cAMP response element-binding protein deficiency allows for increased neurogenesis and a rapid onset of antidepressant response. J Neurosci 27: 7860–7868.
Hahn T, Dresler T, Plichta MM, Ehlis AC, Ernst LH, Markulin F et al (2010). Functional amygdala-hippocampus connectivity during anticipation of aversive events is associated with Gray's trait "sensitivity to punishment". Biol Psychiatry 68: 459–464.
Hendrickson LM, Zhao-Shea R, Pang X, Gardner PD, Tapper AR (2010). Activation of alpha4* nAChRs is necessary and sufficient for varenicline-induced reduction of alcohol consumption. J Neurosci 30: 10169–10176.
Hogle JM, Kaye JT, Curtin JJ (2010). Nicotine withdrawal increases threat-induced anxiety but not fear: neuroadaptation in human addiction. Biol Psychiatry 68: 719–725.
Houghtling RA, Davila-Garcia MI, Kellar KJ (1995). Characterization of (+/−)(−)[3H]epibatidine binding to nicotinic cholinergic receptors in rat and human brain. Mol Pharmacol 48: 280–287.
Hussmann GP, Turner JR, Lomazzo E, Venkatesh R, Cousins V, Xiao Y et al (2012). Chronic sazetidine-A at behaviorally active doses does not increase nicotinic cholinergic receptors in rodent brain. J Pharmacol Exp Ther 343: 441–450.
Irvine EE, Cheeta S, File SE (2001). Tolerance to nicotine's effects in the elevated plus-maze and increased anxiety during withdrawal. Pharmacol Biochem Behav 68: 319–325.
Jackson KJ, Martin BR, Changeux JP, Damaj MI (2008). Differential role of nicotinic acetylcholine receptor subunits in physical and affective nicotine withdrawal signs. J Pharmacol Exp Ther 325: 302–312.
Jackson KJ, McIntosh JM, Brunzell DH, Sanjakdar SS, Damaj MI (2009). The role of alpha6-containing nicotinic acetylcholine receptors in nicotine reward and withdrawal. J Pharmacol Exp Ther 331: 547–554.
Joels M, Karst H, Alfarez D, Heine VM, Qin Y, van Riel E et al (2004). Effects of chronic stress on structure and cell function in rat hippocampus and hypothalamus. Stress 7: 221–231.
Joels M, Krugers H, Karst H (2008). Stress-induced changes in hippocampal function. Prog Brain Res 167: 3–15.
Jonkman S, Henry B, Semenova S, Markou A (2005). Mild anxiogenic effects of nicotine withdrawal in mice. Eur J Pharmacol 516: 40–45.
Kalisch R, Schubert M, Jacob W, Kessler MS, Hemauer R, Wigger A et al (2006). Anxiety and hippocampus volume in the rat. Neuropsychopharmacology 31: 925–932.
Kenney JW, Adoff MD, Wilkinson DS, Gould TJ (2011). The effects of acute, chronic, and withdrawal from chronic nicotine on novel and spatial object recognition in male C57BL/6J mice. Psychopharmacology (Berl) 217: 353–365.
Kenney JW, Gould TJ (2008). Modulation of hippocampus-dependent learning and synaptic plasticity by nicotine. Mol Neurobiol 38: 101–121.
Kenney JW, Raybuck JD, Gould TJ (2012). Nicotinic receptors in the dorsal and ventral hippocampus differentially modulate contextual fear conditioning. Hippocampus 22: 1681–1690.
Kozikowski AP, Eaton JB, Bajjuri KM, Chellappan SK, Chen Y, Karadi S et al (2009). Chemistry and pharmacology of nicotinic ligands based on 6-[5-(azetidin-2-ylmethoxy)pyridin-3-yl]hex-5-yn-1-ol (AMOP-H-OH) for possible use in depression. ChemMedChem 4: 1279–1291.
Laviolette SR, Lauzon NM, Bishop SF, Sun N, Tan H (2008). Dopamine signaling through D1-like versus D2-like receptors in the nucleus accumbens core versus shell differentially modulates nicotine reward sensitivity. J Neurosci 28: 8025–8033.
Laviolette SR, van der Kooy D (2003). Blockade of mesolimbic dopamine transmission dramatically increases sensitivity to the rewarding effects of nicotine in the ventral tegmental area. Mol Psychiatry 8: 50–59 9.
Lee JH, Jones PG, Bybee K, O'Keefe JH (2008). A longer course of varenicline therapy improves smoking cessation rates. Prev Cardiol 11: 210–214.
Levin ED, McClernon FJ, Rezvani AH (2006). Nicotinic effects on cognitive function: behavioral characterization, pharmacological specification, and anatomic localization. Psychopharmacology (Berl) 184: 523–539.
Levin ED, Rezvani AH, Xiao Y, Slade S, Cauley M, Wells C et al (2010). Sazetidine-A, a selective alpha4beta2 nicotinic receptor desensitizing agent and partial agonist, reduces nicotine self-administration in rats. J Pharmacol Exp Ther 332: 933–939.
Lewis MC, Gould TJ (2007). Reversible inactivation of the entorhinal cortex disrupts the establishment and expression of latent inhibition of cued fear conditioning in C57BL/6 mice. Hippocampus 17: 462–470.
Marks MJ, Burch JB, Collins AC (1983). Effects of chronic nicotine infusion on tolerance development and nicotinic receptors. J Pharmacol Exp Ther 226: 817–825.
Marrosu F, Portas C, Mascia MS, Casu MA, Fa M, Giagheddu M et al (1995). Microdialysis measurement of cortical and hippocampal acetylcholine release during sleep-wake cycle in freely moving cats. Brain Res 671: 329–332.
McClernon FJ, Gilbert DG (2004). Human functional neuroimaging in nicotine and tobacco research: basics, background, and beyond. Nicotine Tob Res 6: 941–959.
McClernon FJ, Kozink RV, Rose JE (2008). Individual differences in nicotine dependence, withdrawal symptoms, and sex predict transient fMRI-BOLD responses to smoking cues. Neuropsychopharmacology 33: 2148–2157.
McEwen BS (2001). Plasticity of the hippocampus: adaptation to chronic stress and allostatic load. Ann NY Acad Sci 933: 265–277.
McEwen BS, Magarinos AM (2001). Stress and hippocampal plasticity: implications for the pathophysiology of affective disorders. Hum Psychopharmacol 16 (S1): S7–S19.
McHugh SB, Deacon RM, Rawlins JN, Bannerman DM (2004). Amygdala and ventral hippocampus contribute differentially to mechanisms of fear and anxiety. Behav Neurosci 118: 63–78.
McHugh SB, Fillenz M, Lowry JP, Rawlins JN, Bannerman DM (2011). Brain tissue oxygen amperometry in behaving rats demonstrates functional dissociation of dorsal and ventral hippocampus during spatial processing and anxiety. Eur J Neurosci 33: 322–337.
McQuiston AR (2010). Cholinergic modulation of excitatory synaptic input integration in hippocampal CA1. J Physiol 588 (Pt 19): 3727–3742.
Merali Z, Levac C, Anisman H (2003). Validation of a simple, ethologically relevant paradigm for assessing anxiety in mice. Biol Psychiatry 54: 552–565.
Mineur YS, Einstein EB, Seymour PA, Coe JW, O'Neill BT, Rollema H et al (2011). alpha4beta2 nicotinic acetylcholine receptor partial agonists with low intrinsic efficacy have antidepressant-like properties. Behav Pharmacol 22: 291–299.
Nicolas L, Kolb Y, Prinssen E (2006). A combined marble burying-locomotor activity test in mice: a practical screening test with sensitivity to different classes of anxiolytics and antidepressants. Eur J Pharmacol 547: 106–115.
O'Connor EC, Parker D, Rollema H, Mead AN (2010). The alpha4beta2 nicotinic acetylcholine-receptor partial agonist varenicline inhibits both nicotine self-administration following repeated dosing and reinstatement of nicotine seeking in rats. Psychopharmacology (Berl) 208: 365–376.
Onksen JL, Briand LA, Galante RJ, Pack AI, Blendy JA (2012). Running-induced anxiety is dependent on increases in hippocampal neurogenesis. Genes Brain Behav 11: 529–538.
Ortinski PI, Dong J, Mungenast A, Yue C, Takano H, Watson DJ et al (2010). Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci 13: 584–591.
Paxinos G, Franklin KBJ (2001) The Mouse Brain in Stereotaxic Coordinates 2nd edn Academic Press: San Diego, CA.
Penton RE, Quick MW, Lester RA (2011). Short- and long-lasting consequences of in vivo nicotine treatment on hippocampal excitability. J Neurosci 31: 2584–2594.
Picciotto MR, Addy NA, Mineur YS, Brunzell DH (2008). It is not "either/or": Activation and desensitization of nicotinic acetylcholine receptors both contribute to behaviors related to nicotine addiction and mood. Prog Neurobiol 84: 329–342.
Picciotto MR, Brunzell DH, Caldarone BJ (2002). Effect of nicotine and nicotinic receptors on anxiety and depression. Neuroreport 13: 1097–1106.
Piper ME, Cook JW, Schlam TR, Jorenby DE, Baker TB (2011). Anxiety diagnoses in smokers seeking cessation treatment: relations with tobacco dependence, withdrawal, outcome and response to treatment. Addiction 106: 418–427.
Polosa R, Benowitz NL (2011). Treatment of nicotine addiction: present therapeutic options and pipeline developments. Trends Pharmacol Sci 32: 281–289.
Pomerleau OF, Pomerleau CS, Mehringer AM, Snedecor SM, Ninowski R, Sen A (2005). Nicotine dependence, depression, and gender: characterizing phenotypes based on withdrawal discomfort, response to smoking, and ability to abstain. Nicotine Tob Res 7: 91–102.
Raybuck J, Portugal G, Lerman C, Gould T (2008). Varenicline ameliorates nicotine withdrawal-induced learning deficits in C57BL/6 mice. Behav Neurosci 122: 1166–1171.
Rezvani AH, Cauley M, Sexton H, Xiao Y, Brown ML, Paige MA et al (2011). Sazetidine-A, a selective alpha4beta2 nicotinic acetylcholine receptor ligand: effects on dizocilpine and scopolamine-induced attentional impairments in female Sprague-Dawley rats. Psychopharmacology (Berl) 215: 621–630.
Rezvani AH, Levin ED (2001). Cognitive effects of nicotine. Biol Psychiatry 49: 258–267.
Rezvani AH, Slade S, Wells C, Petro A, Lumeng L, Li TK et al (2010). Effects of sazetidine-A, a selective alpha4beta2 nicotinic acetylcholine receptor desensitizing agent on alcohol and nicotine self-administration in selectively bred alcohol-preferring (P) rats. Psychopharmacology (Berl) 211: 161–174.
Rollema H, Chambers LK, Coe JW, Glowa J, Hurst RS, Lebel LA et al (2007). Pharmacological profile of the alpha4beta2 nicotinic acetylcholine receptor partial agonist varenicline, an effective smoking cessation aid. Neuropharmacology 52: 985–994.
Rollema H, Guanowsky V, Mineur YS, Shrikhande A, Coe JW, Seymour PA et al (2009). Varenicline has antidepressant-like activity in the forced swim test and augments sertraline's effect. Eur J Pharmacol 605: 114–116.
Rollema H, Shrikhande A, Ward KM, Tingley FD 3rd, Coe JW, O'Neill BT et al (2010). Pre-clinical properties of the alpha4beta2 nicotinic acetylcholine receptor partial agonists varenicline, cytisine and dianicline translate to clinical efficacy for nicotine dependence. Br J Pharmacol 160: 334–345.
Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S et al (2003). Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science (New York, NY) 301: 805–809.
Schwartz R, Kellar K (1983). Nicotinic cholinergic receptor binding sites in the brain: regulation in vivo. Science (New York, NY) 220: 214–216.
Shors TJ, Chua C, Falduto J (2001). Sex differences and opposite effects of stress on dendritic spine density in the male versus female hippocampus. J Neurosci 21: 6292–6297.
Shors TJ, Gallegos RA, Breindl A (1997). Transient and persistent consequences of acute stress on long-term potentiation (LTP), synaptic efficacy, theta rhythms and bursts in area CA1 of the hippocampus. Synapse (New York, NY) 26: 209–217.
Stein MB, Koverola C, Hanna C, Torchia MG, McClarty B (1997). Hippocampal volume in women victimized by childhood sexual abuse. Psychol Med 27: 951–959.
Stoker AK, Semenova S, Markou A (2008). Affective and somatic aspects of spontaneous and precipitated nicotine withdrawal in C57BL/6J and BALB/cByJ mice. Neuropharmacology 54: 1223–1232.
Tonstad S, Tonnesen P, Hajek P, Williams KE, Billing CB, Reeves KR (2006). Effect of maintenance therapy with varenicline on smoking cessation: a randomized controlled trial. JAMA 296: 64–71.
Turner JR, Castellano LM, Blendy JA (2010). Nicotinic partial agonists varenicline and sazetidine-a have differential effects on affective behavior. J Pharmacol Exp Ther 334: 665–672.
Turner JR, Castellano LM, Blendy JA (2011). Parallel anxiolytic-like effects and upregulation of neuronal nicotinic acetylcholine receptors following chronic nicotine and varenicline. Nicotine Tob Res 13: 41–46.
Vaupel DB, Tella SR, Huso DL, Wagner VO 3rd, Mukhin AG, Chefer SI et al (2005). Pharmacological and toxicological evaluation of 2-fluoro-3-(2(S)-azetidinylmethoxy)pyridine (2-F-A-85380), a ligand for imaging cerebral nicotinic acetylcholine receptors with positron emission tomography. J Pharmacol Exp Ther 312: 355–365.
Wilkinson DS, Turner JR, Blendy JA, Gould TJ (2013). Genetic background influences the effects of withdrawal from chronic nicotine on learning and high-affinity nicotinic acetylcholine receptor binding in the dorsal and ventral hippocampus. Psychopharmacology (Berl) 225: 201–208.
Xiao Y, Fan H, Musachio JL, Wei ZL, Chellappan SK, Kozikowski AP et al (2006). Sazetidine-A, a novel ligand that desensitizes alpha4beta2 nicotinic acetylcholine receptors without activating them. Mol Pharmacol 70: 1454–1460.
Xiao Y, Kellar KJ (2004). The comparative pharmacology and up-regulation of rat neuronal nicotinic receptor subtype binding sites stably expressed in transfected mammalian cells. J Pharmacol Exp Ther 310: 98–107.
Zwart R, Carbone A, Moroni M, Bermudez E, Mogg A, Folly E et al (2008). Sazetidine-A is a potent and selective agonist at native and recombinant alpha4beta2 nicotinic acetylcholine receptors. Mol Pharmacol 73: 1838–1843.
Acknowledgements
We thank Drs Ken Kellar and Yingxian Xiao for helpful discussions of data and for providing sazetidine-A for these studies. We also thank Dr Hans Rollema (Pfizer) for providing a sample of varenicline and AstraZeneca (Wilmington, DE) for their kind give of [3H]2-FA-85380. This work was supported by the National Institutes of Health National Cancer Institute (P50-CA143187) and the National Institutes of Health National Institute on Drug Abuse (1-F32-DA026236, 1-K99-DA032681, DA024787-01A1S1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Turner, J., Wilkinson, D., Poole, R. et al. Divergent Functional Effects of Sazetidine-A and Varenicline During Nicotine Withdrawal. Neuropsychopharmacol 38, 2035–2047 (2013). https://doi.org/10.1038/npp.2013.105
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2013.105
Keywords
This article is cited by
-
Effects of nicotinic acetylcholine receptor-activating alkaloids on anxiety-like behavior in zebrafish
Journal of Natural Medicines (2021)
-
Differential effects of α4β2 nicotinic receptor antagonists and partial-agonists on contextual fear extinction in male C57BL/6 mice
Psychopharmacology (2018)
-
Distinct Roles of CREB Within the Ventral and Dorsal Hippocampus in Mediating Nicotine Withdrawal Phenotypes
Neuropsychopharmacology (2017)
-
Contributions of β2 subunit-containing nAChRs to chronic nicotine-induced alterations in cognitive flexibility in mice
Psychopharmacology (2015)
-
Evidence from mouse and man for a role of neuregulin 3 in nicotine dependence
Molecular Psychiatry (2014)
