
Abstract
Prepulse inhibition (PPI) deficits are among the most reproducible phenotypic markers found in schizophrenic patients. We recently reported that nisoxetine, a selective norepinephrine transporter (NET) inhibitor, reversed the PPI deficits that have been identified in dopamine transporter (DAT) knockout (KO) mice. However, the mechanisms underlying nisoxetine-induced PPI recovery in DAT KO mice were unclear in previous experiments. To clarify these mechanisms, PPI was tested after microinjections of nisoxetine into the medial prefrontal cortex (mPFc) or nucleus accumbens (NAc) in wildtype (WT) and DAT KO mice. c-Fos immunohistochemistry provided an indicator of neural activation. Multiple-fluorescent-labeling procedures and the retrograde tracer fluorogold were employed to identify nisoxetine-activated neurons and circuits. Systemic nisoxetine activated the mPFc, the NAc shell, the basolateral amygdala, and the subiculum. Infusions of nisoxetine into the mPFc reversed PPI deficits in DAT KO mice, but produced no changes in WT mice, while infusion of nisoxetine into the NAc had no effect on PPI in both WT and DAT KO mice. Experiments using multiple-fluorescent labeling/fluorogold revealed that nisoxetine activates presumed glutamatergic pyramidal cells that project from the mPFc to the NAc. Activated glutamatergic projections from the mPFc to the NAc appear to have substantial roles in the ability of a NET inhibitor to normalize PPI deficits in DAT KO. Thus, this data suggest that selective NET inhibitors such as nisoxetine might improve information processing deficits in schizophrenia via regulation of cortico-subcortical neuromodulation.
Similar content being viewed by others

INTRODUCTION
Cognitive dysfunction, including problems in attention, learning, memory, and executive functions, are core symptoms of schizophrenia that are strongly associated with functional impairments in daily life (Mueser and McGurk, 2004). Most antipsychotic medications display only modest, if any, enhancement of cognitive performance in schizophrenics (Tamminga, 2006; Goldberg et al, 2007; Keefe et al, 2007). Medications that reverse aspects of the cognitive dysfunction in schizophrenic patients are thus urgently needed.
Hyperdopaminergic tone and hypofunction of glutamatergic signaling have both been suggested to contribute to cognitive dysfunction in schizophrenics and to underlie behavioral and cognitive deficits in rodent models of schizophrenia (Jentsch and Roth, 1999; Iversen and Iversen, 2007). Dopamine transporter knockout (DAT KO) mice exhibit increased extracellular dopamine levels in the striatum and nucleus accumbens (NAc), but display normal medial prefrontal cortical (mPFc) dopamine levels (Shen et al, 2004). The DAT KO mice display deficits in prepulse inhibition (PPI), a model of sensorimotor gating (Ralph et al, 2001; Yamashita et al, 2006; Ishii et al, 2010). PPI is the suppression of the startle response that occurs when a startle-eliciting stimulus is preceded by a brief low-intensity stimulus (the prepulse). As this response can be reliably elicited in rodents and humans, this model for identifying sensorimotor gating deficits has served as one of the few validated animal models of a clinical feature of abnormalities in sensorimotor gating impairments in schizophrenics (although PPI deficits can also be found in some other psychiatric disorders) (Braff et al, 2001; Geyer, 2006; Swerdlow et al, 2006). PPI has neuroanatomical substrates that include the mPFc, basolateral amygdala (BLA), hippocampus, and NAc, and neurochemical substrates that include dopamine, glutamate, and GABA (Swerdlow et al, 2001; Schmajuk and Larrauri, 2005). We previously reported that systemic treatment with a selective norepinephrine reuptake inhibitor (NRI), nisoxetine, normalized PPI deficits observed in DAT KO mice (Yamashita et al, 2006). However, the brain circuits at which nisoxetine might act to mediate recovery of PPI in DAT KO mice have yet to be determined (Schroeter et al, 2000; Gehlert et al, 2008). We now report results that elucidate the neural circuits that underlie PPI recovery in DAT KO mice by mapping the changes in neurons that express c-Fos (Sumner et al, 2004) after systemic nisoxetine administration, in combination with nisoxetine microinjection, specific neural markers and fluorogold.
MATERIALS AND METHODS
Animals
DAT KO mice and wildtype (WT) littermates (Sora et al, 1998) were bred at the Animal Laboratory Institute of Tohoku University Graduate School of Medicine and maintained on a mixed C57BL/6J-129Sv genetic background by crossing between heterozygous mice. Mice were weaned at 4 weeks of age and housed socially (segregated by sex), in a temperature- (22.0–24.0 °C), humidity- (45–65%) and light-controlled room (light on 0800–2000 hours). Food and water were available ad libitum. Male and female mice from 8–20 weeks old of each genotype were used in each experiment in equal proportions. All experiments were performed in accordance with the Guidelines for Care of Laboratory Animals of Tohoku University Graduate School of Medicine and conformed to all Japanese federal rules and guidelines.
Drug Administration
For immunohistochemical studies, nisoxetine hydrochloride (Sigma-Aldrich) was dissolved in saline (10 mg/kg), and administered intraperitoneally (i.p.) in a volume of 10 ml/kg. For intracerebral microinjection studies, nisoxetine (2 and 8 μg/side) was dissolved in Ringer's solution (Na+ 145 mM, K+ 3 mM, Ca2+ 1.26 mM, and Mg2+ 1 mM).
Immunohistochemical Analysis of c-Fos Expression
WT and DAT KO mice were used to identify the regions of the brain activated by nisoxetine. Mice were placed in a temperature- (23.0 °C) and light-controlled (light on 0800–2000 hours) chamber on the day before the experiment. On the following day, 2 h after nisoxetine administration (10 mg/kg, i.p.), mice were deeply anesthetized with nembutal and transcardially perfused with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde in 0.1 M phosphate buffer (PB). This interval (2 h) was determined from the following: our previous work reported that amelioration of PPI deficits in DAT KO mice was observed 30 min after systemic nisoxetine treatment (Yamashita et al, 2006), and the period of peak production for c-Fos protein was between 90 and 120 min after treatment (Bisler et al, 2002). Brains were removed and post-fixed in the same fixative, transferred to 15% sucrose in 0.1 M PB, and then immersed in 30% sucrose in 0.1 M PB for cryoprotection. Brains were sectioned on a freezing microtome and serial coronal sections (14 μm) were incubated in PBS with 10% normal horse serum and 0.5% Triton X-100, and then incubated overnight at 4 °C with rabbit anti-c-Fos polyclonal antibody (1:1000, sc-253, Santa Cruz Biotechnology). Sections were rinsed with PBS and incubated with goat Alexa 488-conjugated anti-rabbit IgG (1:300, A11034, Molecular Probes) at 4 °C for 1 h. Sections were transferred to slides, rinsed with PBS, and imaged using a fluorescence microscope (Leica DMRXA).
Medial prefrontal cortex (mPFc), NAc shell and core, BLA, and dorsal and ventral hippocampus (dHPC and vHPC) were located according to the coordinates of the Paxinos mouse brain atlas (Paxinos, 2001). Subjects were numbered so that the investigator was blind to the groups during analysis. The numbers of c-Fos-positive cells in each region were quantified manually within a 744 × 555 μm2 grid placed over each area from each of four sections using ImageJ. Statistical analyses used the SPSS statistical package (SPSS 11.5J for Windows, SPSS, Tokyo, Japan).
Surgery
WT and DAT KO mice were anesthetized using Avertin (20 mg/ml), injected i.p., at an initial dose of 0.1 ml/10 gm body weight, and bilaterally implanted with guide cannulae targeting the mPFc (anterior +1.8 mm, lateral +0.5 mm, ventral −1.7 mm from bregma) or NAc shell (anterior +1.2 mm, lateral +0.6 mm, ventral −4.7 mm from bregma) according to the atlas of Paxinos (Paxinos, 2001). Mice were allowed to recover for at least 5 days before PPI testing and/or fluorogold infusions were performed.
PPI Testing in DAT KO Mice
PPI testing was conducted using previously reported methods (Geyer and Dulawa, 2003). Mice were tested in startle chambers (SR-LAB, San Diego Instruments, San Diego, CA), consisting of a nonrestrictive Plexiglas cylinder mounted on a frame inside a lighted, ventilated, and sound-proof chamber (35 × 35 × 47.5 cm3). Movement was detected by a piezoelectric accelerometer. Electrical signals were digitized and stored by a computer. A total of 65 readings were recorded at 1 ms intervals beginning at the stimulus onset. Average amplitude over this time was used as the measure of startle. Experimental sessions consisted of a 5 min acclimatization period with 65 dB background noise followed by startle trials, which were of five different types: no stimulus (nostim), consisting only of background noise (65 dB); startle pulse alone, 40 ms duration at 120 dB (p120); and three prepulse + pulse conditions, 20 ms duration prepulse at 68 dB (pp3), 71 dB (pp6), or 77 dB (pp12), followed by a 40 ms duration startle stimulus at 120 dB after a 100 ms interstimulus interval. The duration of the interstimulus interval was measured between the onset of the prepulse and the onset of the pulse (onset to onset). Test sessions began and ended with 12 presentations of the p120 trial, followed by 10 presentations of the nostim trial, and pp3, pp6, and pp12 prepulse trials given in pseudorandom order, with an intertrial interval of 8–23 s (mean 15 s). PPI was calculated as the percentage of the startle magnitude at each prepulse intensity using the following equation: % PPI = 100−((startle response for prepulse + pulse (pp3, pp6, or pp12))/(startle response for pulse alone (p120)) × 100). In the microinjection study, nisoxetine solution (2 and 8 μg/side or vehicle (Ringer's solution) was microinjected bilaterally into freely moving mice using a microsyringe pump (0.25 μl/min for 2 min). After the microinjection, the cannulae were left in place for 1 min to allow diffusion of the drug. Mice were then placed into the startle chamber for PPI testing. After the microinjection study, the brains were removed, sectioned at 100 μm thickness using a vibratome, and stained with cresyl violet for localization of cannula placements.
Double Immunostaining for c-Fos and Neural Markers
Brain sections obtained from naïve WT (n=4; n=2 male and n=2 female) and DAT KO mice (n=4; n=2 male and n=2 female) were boiled in 10 mM citrate solution for 5 min. Sections were washed with PBS, incubated in PBS with 10% normal horse serum and 0.5 % Triton X-100, and then incubated overnight at 4 °C with rabbit anti-c-Fos polyclonal antibody (1:1000, sc-253, Santa Cruz Biotechnology), anti-GAD67 monoclonal antibody (1:500, MAB5406, Chemicon), anti-phosphate-activated glutaminase (PAG) monoclonal antibody (1:500, 5 mg/ml; a gift from Takeshi Kaneko, Kyoto University) or anti-tyrosine hydroxylase (TH) monoclonal antibody (1:1000, MAB318, Chemicon) as markers for activated neurons, GABAergic, glutamatergic or dopaminergic neurons, respectively. Sections were washed with PBS, incubated with biotinylated anti-mouse IgG (5 μg/ml, BA-2000, Vector Laboratories) at 4 °C for 2 h, incubated with goat Alexa 594-conjugated anti-rabbit IgG (1:300, A11012, Molecular Probes) and Alexa 488-conjugated streptavidin (1:300, S11223, Molecular Probes) at 4 °C for 1 h, rinsed with PBS and imaged using a fluorescence microscope (Keyence, BZ-9000). Quantitative analysis of c-Fos and PAG colocalization was calculated as the percentage of colocalized neurons: Colocalized neurons (%) = (Number of c-Fos-positive and PAG-positive cells)/(Number of c-Fos-positive cells) × 100.
Fluorogold Infusion and Triple Fluorescence Imaging
WT and DAT KO mice implanted with guide cannulae in the NAc, as described above, were used for fluorogold-retrograde labeling and immunostaining to detect multiple neuronal markers. Fluorogold (80014, Biotium) was dissolved in saline at a concentration of 4% and injected bilaterally (0.5 μl/side) into the NAc. Sections were prepared as described above, up to the secondary antibody step, but were then incubated with goat Alexa 555-conjugated anti-rabbit IgG (1:200, A21429, Molecular Probes) and Alexa 633-conjugated streptavidin (1:200, S21375, Molecular Probes) at 4 °C for 1 h, rinsed with PBS, and imaged by fluorescence microscopy (Keyence, BZ-9000). Quantitative analysis of colocalization of c-Fos, neuronal markers, and fluorogold was calculated as the percentage of colocalized neurons: Colocalized neurons (%) in the mPFc = (Number of triple-labeled cells for c-Fos, PAG, and fluorogold)/(Number of c-Fos-positive cells) × 100, colocalized neurons (%) in the VTA = (Number of triple-labeled cells for c-Fos, TH, and fluorogold)/(Number of c-Fos-positive cells) × 100.
Statistical Analysis
For c-Fos immunohistochemistry, cell counts were analyzed by analysis of variance (ANOVA), with brain region, genotype, and drug as between-subjects factors, followed by the Tukey HSD or Bonferroni post hoc comparisons. PPI data in microinjection studies were analyzed by ANOVA with drug treatment and genotype as between-subjects factors and prepulse intensities as a within-subjects factor followed by the Bonferroni post hoc comparisons. The alpha level <5% (p<0.05) was considered as significant difference. All data are presented as means ±SEM.
RESULTS
Effects of Nisoxetine on c-Fos Induction
c-Fos immunoreactivity was examined in WT and DAT KO mice to identify the brain regions that were activated by nisoxetine. Systemic nisoxetine (10 mg/kg), which normalized PPI defects in DAT KO mice (see below), increased the number of c-Fos-positive nuclei in several brain regions (Figure 1). ANOVA revealed that there was no significant main effect of genotype on the number of c-Fos-positive nuclei, there were significant main effect of drug (F(1, 404) = 91.286, p<0.001) and brain region (F(5, 404)=14.286, p<0.001), there was a significant drug × brain region interaction (F(5, 404)=11.726, p<0.001), and there was no significant brain region × drug × genotype interaction. Post hoc analysis revealed a significant induction of c-Fos by nisoxetine in the mPFc (F(1, 404)=70.003, p<0.001), NAc shell (F(1, 404)=32.114, p<0.001), BLA (F(1, 404)=48.684, p<0.001), and vHPC (F(1, 404)=6.009, p<0.05). By contrast, there were no changes in the number of c-Fos-positive nuclei in the NAc core and dHPC.

The effects of vehicle (V) or selective norepinephrine reuptake inhibitor, nisoxetine (N) 10 mg/kg, on the numbers of c-Fos-positive nuclei in WT (n=4–5; n=2–3 for male and n=2–3 for female per groups) and DAT KO mice (n=4–5; n=2–3 for male and n=2–3 for female per groups). Nisoxetine significantly increased the numbers of c-Fos-positive nuclei in the medial prefrontal cortex (mPFc), nucleus accumbens (NAc) shell, basolateral amygdala (BLA), and ventral hippocampus (vHPC) in both WT and DAT KO mice. Nisoxetine had no effect on c-Fos expression in the NAc core and dorsal hippocampus (dHPC). ***p<0.001, *p<0.05 compared with vehicle-treated mice.
Effects of Nisoxetine Microinjection on PPI
There were no effects of surgery on PPI in WT or DAT KO mice; neither PPI nor startle response differed between preoperative and postoperative assessments (data not shown). After intracerebral vehicle infusions into either the mPFC or NAc, PPI deficits were observed in DAT KO mice compared with WT mice (Figures 2, 3a and b). ANOVA revealed a significant main effect of genotype (F(1, 61)=13.0, p<0.01), drug (F(2, 61)=3.49), and prepulse intensity (F(2, 122)=107.3, p<0.001) on PPI in mice with mPFC cannula. Intracerebral injection of nisoxetine into the mPFC reversed these deficits that were confirmed by a significant genotype × drug interaction in the ANOVA (F(1, 61)=12.7, p<0.01). Post hoc comparisons revealed that vehicle-treated DAT KO mice displayed PPI that was significantly reduced when compared with vehicle-treated WT mice (F(1, 61)=30.8, p<0.001). Nisoxetine 8 μg/side in the mPFc reversed the PPI deficits displayed by DAT KO mice compared with vehicle-treated DAT KO mice (F(2, 61)=9.72, p<0.001) (Figure 3a). Nisoxetine in the mPFC was without effect in WT mice. In contrast to the effects of nisoxetine in the mPFC, nisoxetine in the NAc did not significantly alter PPI in either WT or DAT KO mice (Figure 3b). ANOVA revealed a significant main effect of genotype (F(1, 47)=14.791, p<0.001), but there was no significant genotype × drug interaction.

Schematic representation of the injection sites, mapped on drawings of mouse brain coronal sections (adapted from Paxinos, 2001). Each black circle in (a) medial prefrontal cortex (mPFc) and (b) nucleus accumbens (NAc) represents the approximate injection cannula placement.

The effects of vehicle (VEH) or nisoxetine (NSX) microinjection on PPI in WT (n=11–16 for medial prefrontal cortex (mPFc) infusion; n=6–8 for male and n=5–8 for female per groups; n=12 for nucleus accumbens (NAc) infusion; n=6 for male and n=6 for female per groups) and DAT KO mice (n=12–16 for mPFc infusion; n=6–8 for male and n=6–8 for female per groups; n=11–12 for NAc infusion; n=5–6 for male and n=6 for female per groups). After administration of VEH into the mPFc (a) or NAc (b), DAT KO mice displayed significantly reduced PPI compared with WT VEH. mPFc infusions of nisoxetine (8 μg/side, NSX8) significantly increased PPI in DAT KO mice without affecting PPI in WT mice, while NAc infusions of nisoxetine (8 μg/side, NSX8) had no effect on PPI. % PPI values represent mean ± SEM. ***p<0.001 compared with WT VEH; ###p<0.01 compared with KO VEH.
The magnitude of the acoustic startle response was not affected by genotype or drug in either injection group (Table 1). In the ANOVA, there was no significant effect of genotype or drug, nor was there a significant genotype × drug interaction.
Double Immunostaining for c-Fos and Neuron Markers
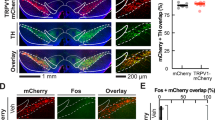
To investigate the types of neurons activated by nisoxetine treatment, double immunostaining was employed for c-Fos and neuronal markers for glutamatergic (PAG) and GABAergic (GAD67) neurons. Systemic treatment with the selective norepinephrine transporter (NET) blocker nisoxetine activated PAG-positive neurons in the mPFc (Figure 4a–h). Table 2 shows that nearly all (WT mice: 94.2% and DAT KO mice: 87.4%) of the nisoxetine-induced c-Fos-positive cells in the mPFc express PAG. In addition to the glutamatergic neurons in these areas, nisoxetine activated GABAergic neurons in the NAc shell in both WT and DAT KO mice (Figure 4i–l).

Representative micrographs of c-Fos (a1–l1, magenta), PAG (a2–d2, green), and GAD67 (e2–l2, green) immunoreactivity and overlay (a3–l3, merge) in WT (n=4; n=2 for male and n=2 for female) and DAT KO mice (n=4; n=2 for male and n=2 for female) treated with vehicle and nisoxetine (10 mg/kg, i.p.). Detailed quantitative data are shown in Table 2. PAG: glutamatergic neuronal marker, NAc: nucleus accumbens. Scale bars, 100 μm.
Activated Neural Circuits by Nisoxetine Treatment
Retrograde labeling produced by fluorogold injection in the NAc was observed in neurons in the mPFc (Figure 5b and f) and VTA (Figure 5j and n) in both WT and DAT KO mice. Triple fluorescence immunostaining revealed the majority of nisoxetine-induced c-Fos expression in the mPFc was labeled for both PAG and fluorogold (WT mice: 84.0% and DAT KO mice: 90.9%; Figure 5d and h and Table 3). By contrast, no c-Fos expression was observed in both TH- and fluorogold-positive cells in the VTA (WT mice: 0% and DAT KO mice: 0%; Figure 5l and p).

Triple fluorescence micrographs for c-Fos (blue; a, e, i, and m), PAG (magenta: c and g), TH (magenta; k and o), and fluorogold (green; b, f, j, and n) for WT and DAT KO mice treated with nisoxetine (10 mg/kg, i.p.) after fluorogold injection into the NAc. PAG (phosphate-activated glutaminase): glutamatergic neuronal marker and TH (tyrosine hydroxylase): dopaminergic marker. Scale bars, 100 μm.
DISCUSSION
The present study found that (1) selective NET blockade by microinjection of nisoxetine into the mPFc, but not the NAc, ameliorated PPI deficits in DAT KO mice and (2) systemic nisoxetine activated glutamatergic pyramidal cells projecting to the NAc. This confirms the observation of nisoxetine-induced normalization of PPI deficits in DAT KO mice (Yamashita et al, 2006) and suggests that it is produced, at least in part, through enhancement of glutamatergic neurotransmission in the prefrontal-accumbal pathway. In addition, nisoxetine might also activate other glutamatergic projections to the NAc, including those from the BLA and vHPC (Grace et al, 2007).
In support of this conclusion, c-Fos activation produced by systemic treatment with nisoxetine was observed in the mPFc, NAc shell, BLA, and vHPC in both WT and DAT KO mice, all areas known to be important for the regulation of PPI. In most brain areas assessed there was no basal expression of c-Fos, consistent with previous observations (Sumner et al, 2004). Systemic nisoxetine produced substantial increases in c-Fos expression in all of these structures as well as the NAc shell and BLA, which would appear to identify a particular neural circuit activated by nisoxetine, which includes the NAc shell (Hasue and Shammah-Lagnado, 2002) and glutamatergic projections to the NAc shell from the mPFc, BLA, and vHPC (Brog et al, 1993; Grace et al, 2007). This neuroanatomical circuitry that includes mPFc, BLA, vHPC, and NAc has also been identified as an important portion of the circuitry that regulates PPI (Swerdlow et al, 2001; Schmajuk and Larrauri, 2005).
As the next step in identifying the critical site of action of nisoxetine that normalizes PPI in DAT KO mice, the consequences of local intracerebral administration of nisoxetine on PPI was examined. Based on prior data and c-Fos mapping results, the mPFc and NAc were selected for local application of nisoxetine. In addition to the c-Fos study presented here, a number of studies have suggested that the mPFC and NAc are important anatomical sites regulating PPI (Swerdlow et al, 2001; Schmajuk and Larrauri, 2005). The mPFC, in particular, might be relevant to the effects of nisoxetine as mPFc dopamine regulates PPI, and NET is the primary mediator of frontocortical dopamine uptake (Carboni et al, 1990; Yamamoto and Novotney, 1998; Moron et al, 2002). That NET effects upon mPFC dopamine, and not norepinephrine, levels might be most relevant to mPFc regulation of PPI is suggested by the ability of dopamine D1 and D2 antagonists in the PFc to reduce PPI (Ellenbroek et al, 1996; Shoemaker et al, 2005). Furthermore, systemic nisoxetine enhances mPFc dopamine levels in both WT and DAT KO mice (Arime et al, 2006), and atomoxetine increases dopamine in the mPFc, but not the NAc (Bymaster et al, 2002). Thus, specific NET blockade by local administration of nisoxetine into the mPFc would be expected to selectively enhance dopamine neurotransmission in the mPFC, making this action of nisoxetine a strong candidate to mediate normalization of PPI in DAT KO mice. Indeed, the present study found that selective NET blockade in the mPFc, but not the NAc, ameliorated PPI deficits in DAT KO mice, confirming that this structure is an important site of action for this effect. That the effects of NET blockade are mediated by mPFC dopamine is also consistent with a recent imaging study that identified altered prefrontal circuitry in DAT KO mice indicative of hypofrontality (Zhang et al, 2010). This does not rule out contributions from other structures, and given that c-Fos activation was observed in both the BLA and vHPC, which also have glutamatergic projections to the NAc, these other structures might also contribute to this effect. Previous studies reported that blockade of NMDA receptors in the BLA reduces PPI in rats, that this impairment is reversed by the dopamine D2 receptor antagonist haloperidol (Fendt et al, 2000), and that enhanced NMDA receptor activity in the vHPC also reduces PPI (Wan et al, 1996a). The involvement of amygdalo-accumbens or subiculo-accumbens glutamatergic neurotransmission via NMDA receptors in PPI regulation suggests that NRIs might ameliorate PPI deficits via activation of these subcortical glutamatergic pathways in addition to the prefrontal-accumbens pathway. However, it is known that there are species differences in the regulation of PPI during systemic administration of dopamine D1 and D2 agonist (Ralph and Caine, 2005). Therefore, it remains unclear which type of receptors in the mPFc and hippocampus have a role in the amelioration of PPI deficits in DAT KO mice.
Although it would appear likely that nisoxetine effects upon mPFc dopamine function likely contribute to these effects, the present experiments cannot determine whether c-Fos activation, and the consequent effects of these activated mPFc glutamatergic neurons, is in fact mediated primarily by increased dopamine or norepinephrine neurotransmission. Systemic administration of the dopamine D1/D2 agonist apomorphine and local infusion of norepinephrine into the mPFc both increase mPFc c-Fos expression (Stone et al, 1991; Deutch and Duman, 1996). Clozapine and olanzapine have been shown to induce c-Fos expression in the mPFc via noradrenergic mechanisms (Ohashi et al, 2000), and the selective NET blocker atomoxetine also increases mPFc c-Fos immunoreactivity (Bymaster et al, 2002). Our immunohistochemical double-labeling results for c-Fos and the glutamatergic cell marker PAG identified co-labeling in pyramidal glutamatergic mPFc neurons in ∼90% of c-Fos-positive cells expressing PAG in both WT and DAT KO mice, indicating that these mPFc neurons were indeed activated by nisoxetine (Table 2). Furthermore, triple fluorescence studies for c-Fos, specific neuronal markers, and the retrograde tracer fluorogold indicated that nisoxetine activates mPFc glutamatergic neurons that project to the NAc shell, which receives mPFc projections (Wright and Groenewegen, 1995). Furthermore, dopamine terminals form synapses on these mPFc pyramidal cells (Carr et al, 1999). Taken together, these data suggest that glutamatergic inputs from the mPFc to the NAc shell region are modulated by effects of nisoxetine on catecholaminergic inputs to the mPFc, and modulation of this pathway is responsible, at least in part, for the normalization of PPI by nisoxetine in DAT KO mice.
The present results also demonstrated that systemic nisoxetine activates GABAergic neurons in the NAc shell, based on immunohistochemical double-labeling for c-Fos and GAD67, a marker of GABAergic neurons. In the NAc, GABAergic medium spiny neurons constitute ∼90% of all neurons (Meredith, 1999). These neurons receive excitatory inputs from the mPFc, BLA, and ventral subiculum (Brog et al, 1993; Grace et al, 2007), as well as dopamine innervation from the VTA (Margolis et al, 2006), and PPI is regulated by both of these projections (Wan and Swerdlow, 1996b; Swerdlow et al, 2001). Systemic nisoxetine activated the mPFC, BLA, and ventral subiculum. However, there were no nisoxetine-induced increases in the number of c-Fos-positive cells projecting from the VTA to NAc. Thus, although nisoxetine might inhibit hyperdopaminergic neurotransmission indirectly, there was no evidence for direct actions on dopaminergic neurons. Therefore, it would seem that NAc GABAergic neurons that showed nisoxetine-induced increases in c-Fos expression were secondarily activated by excitatory glutamatergic inputs from other regions, such as the mPFc. These results suggest that nisoxetine enhances glutamatergic signaling in the NAc shell and that subsequent GABAergic activation might also be involved in the normalization of PPI in DAT KO mice.
The behavioral, pharmacological, and anatomical data presented here indicate that the NET inhibitor nisoxetine normalizes PPI deficits in hyperdopaminergic mouse model, and that the mechanism underlying these effects most likely involves activation of glutamatergic neurons that includes, but may not be limited to, those projecting from the mPFc to the NAc. Schizophrenia is associated with alterations in the anatomy and function of several cortical and subcortical brain areas, including those that have been shown to be most relevant to the effects of nisoxetine in the present studies, and are thought to underlie the cognitive impairments observed in schizophrenic patients (Lewis and Lieberman, 2000; Marek et al, 2010). PPI is not considered to be a cognitive measure per se but such abnormalities in preattentive information processing might be predictive of cognitive deficits (Geyer, 2006). Our results suggest that selective NRIs, such as nisoxetine, might improve information processing deficits in schizophrenia via regulation of this malfunctioning cortico-subcortical and mesolimbic circuitry.
References
Arime Y, Yamashita M, Fukushima S, Shen H-W, Hagino Y, Hall FS et al (2006). Norepinephrine transporter blockade reversed the prepulse inhibition deficits in dopamine transporter knockout mice. The Society for Neuroscience 36th Annual Meeting, Atlanta, USA.
Bisler S, Schleicher A, Gass P, Stehle JH, Zilles K, Staiger JF (2002). Expression of c-Fos, ICER, Krox-24 and JunB in the whisker-to-barrel pathway of rats: time course of induction upon whisker stimulation by tactile exploration of an enriched environment. J Chem Neuroanat 23: 187–198.
Braff DL, Geyer MA, Swerdlow NR (2001). Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) 156: 234–258.
Brog JS, Salyapongse A, Deutch AY, Zahm DS (1993). The patterns of afferent innervation of the core and shell in the ″accumbens″ part of the rat ventral striatum: immunohistochemical detection of retrogradely transported fluoro-gold. J Comp Neurol 338: 255–278.
Bymaster FP, Katner JS, Nelson DL, Hemrick-Luecke SK, Threlkeld PG, Heiligenstein JH et al (2002). Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology 27: 699–711.
Carboni E, Tanda GL, Frau R, Di Chiara G (1990). Blockade of the noradrenaline carrier increases extracellular dopamine concentrations in the prefrontal cortex: evidence that dopamine is taken up in vivo by noradrenergic terminals. J Neurochem 55: 1067–1070.
Carr DB, O′Donnell P, Card JP, Sesack SR (1999). Dopamine terminals in the rat prefrontal cortex synapse on pyramidal cells that project to the nucleus accumbens. J Neurosci 19: 11049–11060.
Deutch AY, Duman RS (1996). The effects of antipsychotic drugs on Fos protein expression in the prefrontal cortex: cellular localization and pharmacological characterization. Neuroscience 70: 377–389.
Ellenbroek BA, Budde S, Cools AR (1996). Prepulse inhibition and latent inhibition: the role of dopamine in the medial prefrontal cortex. Neuroscience 75: 535–542.
Fendt M, Schwienbacher I, Koch M (2000). Amygdaloid N-methyl-D-aspartate and gamma-aminobutyric acid(A) receptors regulate sensorimotor gating in a dopamine-dependent way in rats. Neuroscience 98: 55–60.
Gehlert DR, Thompson LK, Hemrick-Luecke SK, Shaw J (2008). Monoaminergic compensation in the neuropeptide Y deficient mouse brain. Neuropeptides 42: 367–375.
Geyer MA (2006). Are cross-species measures of sensorimotor gating useful for the discovery of procognitive cotreatments for schizophrenia? Dialogues Clin Neurosci 8: 9–16.
Geyer MA, Dulawa SC (2003). Assessment of murine startle reactivity, prepulse inhibition, and habituation. Curr Protoc Neurosci Chapter 8, Unit 8 17.
Goldberg TE, Goldman RS, Burdick KE, Malhotra AK, Lencz T, Patel RC et al (2007). Cognitive improvement after treatment with second-generation antipsychotic medications in first-episode schizophrenia: is it a practice effect? Arch Gen Psychiatry 64: 1115–1122.
Grace AA, Floresco SB, Goto Y, Lodge DJ (2007). Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends Neurosci 30: 220–227.
Hasue RH, Shammah-Lagnado SJ (2002). Origin of the dopaminergic innervation of the central extended amygdala and accumbens shell: a combined retrograde tracing and immunohistochemical study in the rat. J Comp Neurol 454: 15–33.
Ishii D, Matsuzawa D, Kanahara N, Matsuda S, Sutoh C, Ohtsuka H et al (2010). Effects of aripiprazole on MK-801-induced prepulse inhibition deficits and mitogen-activated protein kinase signal transduction pathway. Neurosci Lett 471: 53–57.
Iversen SD, Iversen LL (2007). Dopamine: 50 years in perspective. Trends Neurosci 30: 188–193.
Jentsch JD, Roth RH (1999). The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 20: 201–225.
Keefe RS, Bilder RM, Davis SM, Harvey PD, Palmer BW, Gold JM et al (2007). Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE trial. Arch Gen Psychiatry 64: 633–647.
Lewis DA, Lieberman JA (2000). Catching up on schizophrenia: natural history and neurobiology. Neuron 28: 325–334.
Marek GJ, Behl B, Bespalov AY, Gross G, Lee Y, Schoemaker H (2010). Glutamatergic (N-methyl-D-aspartate receptor) hypofrontality in schizophrenia: too little juice or a miswired brain? Mol Pharmacol 77: 317–326.
Margolis EB, Lock H, Chefer VI, Shippenberg TS, Hjelmstad GO, Fields HL (2006). Kappa opioids selectively control dopaminergic neurons projecting to the prefrontal cortex. Proc Natl Acad Sci USA 103: 2938–2942.
Meredith GE (1999). The synaptic framework for chemical signaling in nucleus accumbens. Ann N Y Acad Sci 877: 140–156.
Moron JA, Brockington A, Wise RA, Rocha BA, Hope BT (2002). Dopamine uptake through the norepinephrine transporter in brain regions with low levels of the dopamine transporter: evidence from knock-out mouse lines. J Neurosci 22: 389–395.
Mueser KT, McGurk SR (2004). Schizophrenia. Lancet 363: 2063–2072.
Ohashi K, Hamamura T, Lee Y, Fujiwara Y, Suzuki H, Kuroda S (2000). Clozapine- and olanzapine-induced Fos expression in the rat medial prefrontal cortex is mediated by beta-adrenoceptors. Neuropsychopharmacology 23: 162–169.
Paxinos GFK (2001). The mouse brain in stereotaxic coordinates 2nd edn. Academic Press: San Diego.
Ralph RJ, Caine SB (2005). Dopamine D1 and D2 agonist effects on prepulse inhibition and locomotion: comparison of Sprague-Dawley rats to Swiss-Webster, 129X1/SvJ, C57BL/6J, and DBA/2J mice. J Pharmacol Exp Ther 312: 733–741.
Ralph RJ, Paulus MP, Fumagalli F, Caron MG, Geyer MA (2001). Prepulse inhibition deficits and perseverative motor patterns in dopamine transporter knock-out mice: differential effects of D1 and D2 receptor antagonists. J Neurosci 21: 305–313.
Schmajuk NA, Larrauri JA (2005). Neural network model of prepulse inhibition. Behav Neurosci 119: 1546–1562.
Schroeter S, Apparsundaram S, Wiley RG, Miner LH, Sesack SR, Blakely RD (2000). Immunolocalization of the cocaine- and antidepressant-sensitive l-norepinephrine transporter. J Comp Neurol 420: 211–232.
Shen HW, Hagino Y, Kobayashi H, Shinohara-Tanaka K, Ikeda K, Yamamoto H et al (2004). Regional differences in extracellular dopamine and serotonin assessed by in vivo microdialysis in mice lacking dopamine and/or serotonin transporters. Neuropsychopharmacology 29: 1790–1799.
Shoemaker JM, Saint Marie RL, Bongiovanni MJ, Neary AC, Tochen LS, Swerdlow NR (2005). Prefrontal D1 and ventral hippocampal N-methyl-D-aspartate regulation of startle gating in rats. Neuroscience 135: 385–394.
Sora I, Wichems C, Takahashi N, Li XF, Zeng Z, Revay R et al (1998). Cocaine reward models: conditioned place preference can be established in dopamine- and in serotonin-transporter knockout mice. Proc Natl Acad Sci USA 95: 7699–7704.
Stone EA, Zhang Y, John SM, Bing G (1991). c-Fos response to administration of catecholamines into brain by microdialysis. Neurosci Lett 133: 33–35.
Sumner BE, Cruise LA, Slattery DA, Hill DR, Shahid M, Henry B (2004). Testing the validity of c-fos expression profiling to aid the therapeutic classification of psychoactive drugs. Psychopharmacology (Berl) 171: 306–321.
Swerdlow NR, Geyer MA, Braff DL (2001). Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacology (Berl) 156: 194–215.
Swerdlow NR, Light GA, Cadenhead KS, Sprock J, Hsieh MH, Braff DL (2006). Startle gating deficits in a large cohort of patients with schizophrenia: relationship to medications, symptoms, neurocognition, and level of function. Arch Gen Psychiatry 63: 1325–1335.
Tamminga CA (2006). The neurobiology of cognition in schizophrenia. J Clin Psychiatry 67 (Suppl 9): 9–13 discussion 36–42.
Wan FJ, Caine SB, Swerdlow NR (1996a). The ventral subiculum modulation of prepulse inhibition is not mediated via dopamine D2 or nucleus accumbens non-NMDA glutamate receptor activity. Eur J Pharmacol 314: 9–18.
Wan FJ, Swerdlow NR (1996b). Sensorimotor gating in rats is regulated by different dopamine-glutamate interactions in the nucleus accumbens core and shell subregions. Brain Res 722: 168–176.
Wright CI, Groenewegen HJ (1995). Patterns of convergence and segregation in the medial nucleus accumbens of the rat: relationships of prefrontal cortical, midline thalamic, and basal amygdaloid afferents. J Comp Neurol 361: 383–403.
Yamamoto BK, Novotney S (1998). Regulation of extracellular dopamine by the norepinephrine transporter. J Neurochem 71: 274–280.
Yamashita M, Fukushima S, Shen HW, Hall FS, Uhl GR, Numachi Y et al (2006). Norepinephrine transporter blockade can normalize the prepulse inhibition deficits found in dopamine transporter knockout mice. Neuropsychopharmacology 31: 2132–2139.
Zhang X, Bearer EL, Boulat B, Hall FS, Uhl GR, Jacobs RE (2010). Altered neurocircuitry in the dopamine transporter knockout mouse brain. PLoS One 5: e11506.
Acknowledgements
We thank Dr Takeshi Kaneko for the gift of PAG monoclonal antibody, Nozomi Okayasu for technical assistance, and Dr Taku Sato and Dr Shiho Miyazawa for assistance with statistical analysis. This work was supported in part by Scientific Research on Priority Areas-System study on higher-order brain functions and Research on Pathomechanisms of Brain Disorders, Core Research for Evolutional Science and Technology (CREST) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, Global COE Program (Basic and Translational Research Center for Global Brain Science), MEXT, Japan, and through funding from the Intramural Research Program of the National Institute on Drug Abuse, NIH/DHHS, USA (GRU and FSH).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Arime, Y., Kasahara, Y., Hall, F. et al. Cortico-Subcortical Neuromodulation Involved in the Amelioration of Prepulse Inhibition Deficits in Dopamine Transporter Knockout Mice. Neuropsychopharmacol 37, 2522–2530 (2012). https://doi.org/10.1038/npp.2012.114
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2012.114
Keywords
This article is cited by
-
AgRP neurons control structure and function of the medial prefrontal cortex
Molecular Psychiatry (2022)
-
Reduced Dopamine Transporter Functioning Induces High-Reward Risk-Preference Consistent with Bipolar Disorder
Neuropsychopharmacology (2014)
-
Impaired cliff avoidance reaction in dopamine transporter knockout mice
Psychopharmacology (2013)
-
Transgenic mouse models for ADHD
Cell and Tissue Research (2013)
