
Abstract
Increased anxiety is commonly observed in individuals who illicitly administer anabolic androgenic steroids (AAS). Behavioral effects of steroid abuse have become an increasing concern in adults and adolescents of both sexes. The dorsolateral bed nucleus of the stria terminalis (dlBnST) has a critical role in the expression of diffuse anxiety and is a key site of action for the anxiogenic neuromodulator, corticotropin releasing factor (CRF). Here we demonstrate that chronic, but not acute, exposure of female mice during adolescence to AAS augments anxiety-like behaviors; effects that were blocked by central infusion of the CRF receptor type 1 antagonist, antalarmin. AAS treatment selectively increased action potential (AP) firing in neurons of the central amygdala (CeA) that project to the dlBnST, increased the frequency of GABAA receptor-mediated spontaneous inhibitory postsynaptic currents (sIPSCs) in dlBnST target neurons, and decreased both c-FOS immunoreactivity (IR) and AP frequency in these postsynaptic cells. Acute application of antalarmin abrogated the enhancement of GABAergic inhibition induced by chronic AAS exposure whereas application of CRF to brain slices of naïve mice mimicked the actions of this treatment. These results, in concert with previous data demonstrating that chronic AAS treatment results in enhanced levels of CRF mRNA in the CeA and increased CRF-IR in the dlBnST neuropil, are consistent with a mechanism in which the enhanced anxiety elicited by chronic AAS exposure involves augmented inhibitory activity of CeA afferents to the dlBnST and CRF-dependent enhancement of GABAergic inhibition in this brain region.
Similar content being viewed by others

INTRODUCTION
Anabolic androgenic steroids (AAS) comprise a large class of synthetic androgens designed for therapeutic purposes, but whose present use is predominantly illicit self-administration of supratherapeutic doses for image and performance enhancement (for review, Basaria et al, 2001; Bahrke and Yesalis, 2004; Trenton and Currier, 2005). Elevated levels of anxiety have been reported in AAS users (Su et al, 1993; Cooper et al, 1996; Galligani et al, 1996; Hall and Chapman, 2005), and AAS-dependent anxiety may contribute to other noted behavioral effects of AAS, including increased impulsivity, hostility, and aggression (Annitto and Layman, 1980; Pope and Katz, 1988; Burnett and Kleiman, 1994; Cooper et al, 1996; Hall and Chapman, 2005; Pagonis et al, 2006). Consistent with reports in human subjects, chronic AAS exposure increases anxiety-like behavior in adult male rodents, as assessed on the elevated plus maze (EPM), in open field paradigms, and in risk-assessment behaviors (Rocha et al, 2007; Ambar and Chiavegatto, 2009). Although the public focus of steroid abuse and the preponderance of animal studies have been on adult males, estimated use among adolescents indicates that several hundred thousand current AAS users are girls and young women (Elliot et al, 2007; Johnston et al, 2009). The risks associated with AAS use in this population may be compounded by the high degree of hormone sensitivity present during adolescence (O'Connor and Cicero, 1993; Sato et al, 2008; Schulz et al, 2009) and by sex-specific differences in susceptibility to behavioral disorders, such as anxiety, which is more prevalent in women (Zender and Olshansky, 2009).
The extended amygdala is a corridor of interrelated forebrain structures including the central amygdala (CeA) and adjoining structures of the bed nucleus of the stria terminalis (BnST) that are fundamental to the expression of anxious states (for review, Alheid, 2003; Davis et al, 2010). The CeA provides the major source of GABAergic input to the BnST (Krettek and Price, 1978; Sun and Cassell, 1993; Schmued, 1994; Petrovich and Swanson, 1997; Veinante and Freund-Mercier, 1998; Pitkanen, 2000), and GABAergic transmission in circuits of the extended amygdala has been shown to be critical in the expression of anxious states (Möhler, 2012). Prior studies in rodents suggest that the CeA is paramount in the acquisition and expression of phasic fear to specific threats whereas the BnST is key in sustained fear/ diffuse anxiety (for review, Shekhar et al, 2005; Davis et al, 2010; although see Resstel et al, 2008). Changes to both specific threats and generalized anxiety are noted in steroid abusers, but the latter may be particularly important with respect to the expression of inappropriate reactions and increased negative perception of self and others (Pagonis et al, 2006).
The corticotropin releasing factor (CRF) family of peptides acts to integrate sensory, endocrine and autonomic information and formulate an appropriate response to stress (for review, Korosi and Baram, 2008). Elevated levels of central CRF are implicated in anxiety and stress disorders in humans. The BnST is believed to be the primary site of anxiogenic action of CRF, and in rodents CRF stimulates anxiety-like behaviors, as assessed by the acoustic startle response (ASR), the EPM, open field tests, conditioned place aversion, and social interaction tests (for review, Bale and Vale, 2004; Davis et al, 2010). The lateral portion of the CeA (CeAL), which provides strong GABAergic input to the dorsolateral BnST (dlBnST), is also the major extrahypothalamic site of CRF mRNA expression (Swanson et al, 1983; Day et al, 1999; Asan et al, 2005). Corticotropin-releasing factor receptor type 1 (CRF-R1) mRNA is expressed throughout the BnST (van Pett et al, 2000; Justice et al, 2008) and has been shown to mediate CRF-dependent enhancement of the ASR (Swerdlow et al, 1986, 1989; Liang et al, 1992; Risbrough et al, 2003, 2004; Risbrough and Geyer, 2005; Walker et al, 2009a, 2009b).
Prolonged exposure of adolescent female mice to three AAS commonly self-administered in the human population results in enhanced anxiety-like behaviors, as assessed by the ASR (Costine et al, 2010). CRF signaling is likely to have an important role in this elevated level of anxiety, as treatment is also associated with increased CRF mRNA and CRF-immunoreactivity (IR) in the somata of CeA neurons and CRF-IR in the neuropil of their target region, the dlBnST (Costine et al, 2010). CRF has been shown to augment inhibitory transmission mediated by GABAA receptors in other regions of the extended amygdala (Kash and Winder, 2006; Kirby et al, 2008; Nie et al, 2004, 2009). These studies suggest that AAS-dependent increases in anxiety may involve enhanced GABAergic inhibition to the dlBnST and that CRF signaling may have a significant role in both the enhancement of this inhibition and in the expression of AAS-induced anxiety. The aim of the current study was to test this hypothesis.
MATERIALS AND METHODS
Animal Care and Use
Female C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) were group-housed (4/cage) with food and water ad libitum in a temperature-controlled and 12 h light cycle facility with lights on starting at 0700 h. Care was taken to minimize the discomfort and the number of animals used, and all procedures were approved by the Dartmouth College Institutional Animal Care and Use Committee and conducted in accordance with guidelines from the National Institutes of Health. Adolescent female mice were injected i.p. at postnatal day (PN) 24 for 6 days per week for 4 weeks with equal concentrations of testosterone cypionate (Sigma; St Louis, MO), nandrolone decanoate (Sigma) and methandrostenolone (Steraloids; Newport, RI) dissolved in sesame oil at a final concentration of 7.5 mg/kg per day. Age-matched control subjects were administered the same volume (20–30 μl based on body weight) of sesame oil alone. Behavioral testing commenced at ∼PN55, and treatment with either AAS or oil was continued during testing. AAS treatment imposes an anestrous state characterized by diestras-like profiles (Blasberg and Clark, 1997; Penatti et al, 2009a, 2011), and control mice were assessed in diestras (Cooper et al, 1993). In some experiments, PN55 diestras female mice that had not received any injections (naïve) were used.
Behavioral Testing
ASR testing was performed using the MED-ASR-PRO1 apparatus (Med Associates; St Albans, VT) (Costine et al, 2010). The day following acclimation, ASR testing consisted of 5 min of acclimation followed by exposure to the startle stimulus of a 50-ms/100 dB burst of white noise (1 ms rise/fall; 10 s intra-trial interval) for 60 trials. In one experiment, 55-day old mice received single injections of oil on day 1 and day 3 of testing followed by a single injection of either the AAS mixture or oil prior to testing on day 5. The EPM (Columbus Instruments, Columbus, OH) was 45 cm high and arms were 30 cm long. Walls of the closed arms were 15.25 cm high. EPM testing was done according to standard procedures (Lister, 1987; Walf and Frye, 2007). The luminous intensity in the center of the maze and at both ends of the open arms was 3.8 cd, equivalent to the intensity of the home cage during non-testing periods. The mouse was placed in the center of the EPM facing an open arm, and the time spent in the open and closed arms, and the number of entries was scored (5 min). Testing occurred between 4 and 6 h after lights on, and the experimenter was blind to treatment conditions.
Intravcerebroventricular (i.c.v.) Infusions
Infusions of the CRF-R1 antagonist, antalarmin (10 μg/kg), or saline (vehicle) were made into the lateral ventricle (Briscoe et al, 2000; Pelleymounter et al, 2000; Risbrough et al, 2004). Infusate (5.0 μl) was pressure-ejected (30 s) in isofluorane-anesthetized mice through a 27-gauge 2.0 mm guide cannula placed lateral to the midline of the skull halfway between bregma and lambda. The incision was then sealed with cyanoacrylate. Mice were ambulatory within 10 min and were placed in the ASR chamber after 5 additional minutes. An additional group of AAS- and oil-injected mice received a sham surgical procedure that included exposure to anesthesia, skull exposure (but no injection), and cyanoacrylate wound closure. Following testing and euthanasia, brains were dissected and fixed overnight in 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB). Coronal brain sections were Nissl-stained with 0.25% cresyl violet and cover-slipped following serial dehydration. Placement of the guide cannula was verified under 200 × magnification and classified as either ventricular or extra-ventricular. Sera were collected and assayed for corticosterone in singlet by radioimmunoassay at the University of Virginia Ligand Assay Core Laboratory. Detection limits for the assay were 6.7–739.0 ng/ml.
c-FOS Immunolabeling
Mice were ASR-tested as described above and compared to mice that were injected with AAS or oil, but not tested on the ASR. At 1 h and 45 min after placement of a mouse into the chamber (or time matched for not-tested groups), animals received an overdose of ketamine/xylazine (9 : 1) and 0.05 ml heparin (1000 U/ml; APP Pharmaceuticals, Schaumburg, IL) and were intracardially perfused with 4% PFA in PB. Following overnight fixation and cryoprotection at 4 °C, coronal free-floating sections were labeled for c-FOS (Martel and Baum, 2009). Sections were reacted with 10% H2O2 solution, washed three times for 10 min in PB and incubated in blocking buffer: 10% normal goat serum in PBT (PB containing 0.3% Triton X-100; Thermo Fisher) for 3 h at room temperature. The sections were reacted overnight in primary antibody (rabbit anti-c-FOS, Santa Cruz sc-52; 1: 1000), followed by a 90-min incubation with a biotinylated goat anti-rabbit antibody (Vector Labs, Burlingame, CA; 1 : 2000 in PBT) and visualized with ABC Vectastain/Nickel-enhanced 3,3′ diaminobenzidene (Vector). Sections were washed in PB three times for 15 min between primary and secondary antibodies, and before and after ABC Vectastain. Bilateral images from two sections were obtained for the ventrolateral BnST (vlBnST) and the dlBnST (also called the anterolateral portion of the BnSTALG; Dong and Swanson, 2005; Hammack et al, 2007) (Bregma 0.14 mm); the CeAL (Bregma −1.46 mm); the basolateral nucleus of the amygdala (BLA; Bregma −1.46 mm); and the auditory cortex (AuC; Bregma −2.06) (Franklin and Paxinos, 1997). The numbers of c-FOS-IR cells in each image were counted and averaged for the four images per mouse using ImageJ and with the investigator blind to the experimental conditions.
Physiological Recordings
Coronal brain slices (250–300 μm) were prepared as described (Penatti et al, 2010, 2011). Sections corresponding the dlBnST (Bregma 0.14 mm) or the CeA (Bregma −1.46 mm) were superfused in artificial cerebrospinal fluid (aCSF: 95% O2/5% CO2 saturated aCSF, containing in mM: 125 NaCl, 1.2 CaCl2, 10 glucose, 4 KCl, 1 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, and 1 ascorbic acid at pH 7.35) for 20 min (1–2 ml aCSF per min) at 22 or 35 °C. Spontaneous action potentials (APs) were recorded from neurons using the ‘loose-patch’ on-cell conformation with electrodes (3–5 MΩ) filled with aCSF. In some experiments, either 100 μM picrotoxin or 1 μM strychnine (Sigma) were added to the recording bath to block GABAA or glycine receptors, respectively. All physiological data was acquired and stored using a HEKA-EPC9 amplifier (HEKA Instruments, Bellmore, NY, USA) (Penatti et al, 2005, 2009a, 2009b, 2010, 2011).
Fluorescent latex microspheres conjugated to Alexa-488 (Retrobeads, Lumafluor) were stereotaxically pressure injected in the dlBnST (Lammel et al, 2011; Carrillo et al, 2011). A 4.4-mm guide cannula was inserted to target the dlBnST, and 300 nl of retrobeads were delivered over 1 min through a Hamilton syringe, with the needle left in place for an additional minute. 4–7 days after Retrobead infusion, APs were recorded from the ipsilateral CeA from both labeled and neighboring unlabeled cells. The site of the Retrobead injection was verified in the dlBnST following recording; only those animals showing correct injection of the Retrobeads were used for analysis.
GABAA receptor-mediated spontaneous inhibitory postsynaptic currents (sIPSCs) were recorded in the whole-cell configuration (Vhold=−70 mV) in aCSF containing 2.0 mM kynurenic acid to block excitatory ion channels. Pipettes were filled with (in mM): 153 CsCl, 1 MgCl2, 5 EGTA, and 10 HEPES, to which 2 MgATP was added just before each experiment. Biocytin (0.5% w/v) or Lucifer yellow (0.5% w/v) was added in some experiments to confirm the neuron's location within the BnST (Campbell et al, 2005). The identity of synaptic currents as GABAergic was confirmed in some experiments by antagonism by bicuculline (20 μM). To record miniature IPSCs (mIPSCs), 1 μM tetrodotoxin (TTX) was added to the kynurenic acid-containing aCSF. Data were acquired for >5 min and accepted for analysis only if the seal resistance (>1 GΩ), access resistance (<25 MΩ), and the holding current did not change more than ±∼10% during the recording. In some experiments, antalarmin (40 or 4 μM), CRF (100 nM) (Anaspec, Fremont, CA), or the mixture of the three AAS (1 or 10 μM in 0.01% DMSO) was added to the aCSF.
Frequency analysis of APs was derived from direct assessment of inter-spike intervals. Patterning was determined using autocorrelation analysis and classified as regular, irregular, or ‘bursty’ (Penatti et al, 2009a, 2009b, 2010, 2011). Autocorrelational profiles correlated with the coefficient of variation (CV) of AP firing: the bursty pattern with high CVs (⩾4), the irregular pattern with CVs between 0.5 and 2.5, and the regular pattern with CVs <0.5. Cells with fewer 10 APs over the 5 min of recordings were classified as quiet and not used in frequency comparisons. Averaged IPSCs were analyzed for peak current amplitude (Ipeak), frequency, and decay kinetics (biphasic and fitted with two time constants, τ1 and τ2) or with a single weighted time constant (τw) (Penatti et al, 2010, 2011).
Statistical Analysis
Values are means±SE of the mean. Significance was determined by one-way, two-way or repeated measures analysis of variance (ANOVA) followed by post hoc analyses using the Student's t-test with Bonferroni correction. The Kolmogorov-Smirnov test was used for cumulative probability distributions. Non-normally distributed data were log-transformed prior to statistical assessment. For all data, the α level was set at p<0.05.
RESULTS
Chronic, but not Acute, Exposure to AAS Significantly Increases Anxiety-like Behavior in Female Mice
Female C57BL/6J mice were treated throughout a period that spans adolescence (Laviola et al, 2003) with three AAS that are commonly self-administered by human subjects and at a dosage that reflects a high human abuse regime (for discussion, Costine et al, 2010). Our previous study demonstrated that this same regime promotes anxiety-like behaviors in C57BL/6J female mice, as assessed by the ASR (Costine et al, 2010); results that were confirmed in the current study (Figure 1a). Anxiogenic effects of AAS were also indicated by a significant (F1,10=22.05, p<0.001) decrease in the percent time spent in the open arms on the EPM (3.90±0.96 for AAS vs 10.30±0.96 for oil; n=6, each group), a behavioral test that, like the ASR, is an assessment of sustained fear and heightened anxiety (Davis et al, 2010). No significant (F1,10=0.16, p=0.70) differences were evident in the total number of arm entries (oil: 22.67±1.87; AAS: 23.67±1.67), indicating that animals were not stationary in the closed arms and that the increased time spent in the closed arms was not attributable to AAS-dependent differences in locomotion.

Acoustic startle response (ASR) testing in mice that had received oil- or anabolic androgenic steroids (AAS)-injections. (a) Average amplitudes on the first 20 individual trials to the 50 ms 100 dB burst of white noise were significantly greater on all trials in the intact AAS-injected animals vs oil-injected animals (n=8 each group). Both cohorts exhibit comparable habituation over the testing period. (b) Average startle amplitudes over the first 20 trials for oil-injected and AAS-treated animals that received no infusions or surgery, mock surgery, ventricular infusion of saline, or ventricular infusion of antalarmin. n values represent the number of animals per group/condition. **p<0.01; ***p<0.001.
AAS have been reported to promote short-term effects on anxiety-like behaviors (Ågren et al, 1999; Aikey et al, 2002), as well as behavioral actions that necessitate prolonged exposure. AAS can act as acute allosteric modulators of GABAA receptors (Henderson and Jorge, 2004; Henderson, 2007; Oberlander et al, 2012), and such acute action may give rise to immediate behavioral effects. To determine whether a single exposure to the AAS altered anxiety-like behaviors, female mice that had not been treated during adolescence were tested on the ASR and the EPM beginning on PN55 (the age when behavioral testing was initiated for chronically treated mice) after a single injection of AAS or oil. No significant differences were observed on either the ASR or the EPM tests between animals receiving an acute injection of AAS vs oil (ASR: F1,590=0.18, p=0.68; EPM: F1,10=2.23, p=0.17). On the EPM, the percentage of time spent in the open arm was significantly higher for mice receiving a single AAS injection vs those that were chronically treated (18.50±2.81 vs 3.90±0.96, respectively; p<0.001), and those receiving a single AAS injection were not different than the chronic oil-injected group (p=0.16). These results suggest that the anxiogenic effect of AAS require long-term exposure to the AAS.
Antagonism of the CRF-R1 Abrogated AAS-Dependent Enhancement of the ASR
Sustained fear and heightened anxiety-like behaviors are believed to arise from changes in neural circuitry of the extended amygdala. In particular, CRF-containing afferents from the CeAL that project to the dlBnST are believed to be key mediators in the expression of diffuse anxiety and sustained fear. CRF, acting primarily through CRF-R1, augments the ASR, and the BnST is the critical site for CRF-dependent enhancement of the ASR (Swerdlow et al, 1986, 1989; Liang et al, 1992; Risbrough et al, 2003, 2004; Risbrough and Geyer, 2005; Walker et al, 2009a, 2009b). To test whether CRF-R1-mediated signaling is also required for AAS-induced increases in anxiety-like behaviors, chronically treated female mice were given i.c.v. infusions of the CRF-R1 antagonist, antalarmin (Briscoe et al, 2000; Pelleymounter et al, 2000), or saline prior to ASR testing. Antagonism of CRF-R1 has been shown to block the ASR (Risbrough et al, 2003), and antalarmin, a non-peptide antagonist of the CRF-R1 receptor (Webster et al, 1996) reduces anxiety-like behavior on the EPM (Zorrilla et al, 2002) without affecting the stress response to restraint (Wong et al, 1999; however, see Deak et al, 1998). Both AAS- and oil-injected mice habituated to the 100 dB white noise burst during the ASR testing at similar rates (F11,418=10.48, p<0.01). The effects of antalarmin were, therefore, assessed for the first 20 trials of the ASR. As expected, AAS-injected groups exhibited significantly higher ASR amplitudes than oil-injected groups, following sham surgery procedures (F1,4=4.58, p<0.05; Figure 1b) or i.c.v. infusion of saline (F1,6=24.83, p<0.001) (Figure 1b). However, the effect of the AAS treatment was abrogated by infusion of antalarmin into the lateral ventricle (F1,8 =0.40, p=0.70) (Figure 1b). Significant AAS-induced potentiation of the ASR was evident in animals where infusions of antalarmin were made off-target in either the corpus callosum or the cerebellum (F1,13=16.69, p<0.0001). Although these data do not localize the actions of antalarmin solely to the dlBnST, they do suggest that the ability of the AAS to augment anxiety-like behaviors requires signaling mediated by CRF-R1 in brain regions in the extended amygdala, including the dlBnST, that are likely to be the predominant targets of antalarmin during this i.c.v. infusion procedure.
An alternative explanation for the anxiogenic actions of AAS is that they could arise from changes in the activation of the HPA axis, as CRF is an important central regulator of the stress response via its actions in the paraventricular nucleus (PVN) of the hypothalamus (Baram et al, 1997; Gallagher et al, 2008; for review, Gray and Bingaman, 1996; Herman et al, 1996). Our previous studies argue against this mechanism as, although AAS treatment enhanced CRF expression in the extended amygdala, exposure to these steroids did not alter CRF expression in the median eminence or the PVN and did not increase basal levels of serum corticosterone in animals not subjected to behavioral testing (Costine et al, 2010). Interestingly, central treatment of adult male rats with the androgen, dihydrotestosterone, also failed to augment CRF expression in the PVN (Bingham et al, 2011). Here, we extended this analysis by assessing corticosterone levels following i.c.v. infusions and ASR testing.
Corticosterone levels in animals subjected to sham surgery procedures (no infusion), saline infusions, or extra-ventricular administration of antalarmin and subsequently tested on the ASR were significantly higher in the AAS-injected mice than in oil-injected mice (sham surgery: F1,4=4.16, p<0.05; saline: F1,7=5.65, p<0.001; extra-ventricular antalarmin: F1,14=2.99, p<0.01). Mice that had received i.c.v. antalarmin showed no differences in corticosterone levels between AAS- and oil-injected groups (F1,10=0.40, p=0.70), consistent with the lack of differences in ASR amplitudes between these two conditions. As previously reported (Costine et al, 2010), corticosterone levels were indistinguishable between AAS- and oil-injected mice that had received no infusions or testing (F1,7=0.41, p=0.69). Moreover, mice who had not been subjected to ASR testing had significantly lower levels of corticosterone than any group of behaviorally tested mice (p<0.0001), irrespective of the permutations of manipulations associated with infusions to which they had been subjected. Taken together with the prior results (Costine et al, 2010), these data suggest that although the basal state of the HPA axis is not altered by AAS exposure, AAS-treatment results in an enhanced response to a stressor, in this case the startle noise, that is CRF-R1-mediated and results in elevated peripheral levels of corticosterone.
Neuronal Excitability is Increased in the CeAL by AAS Exposure
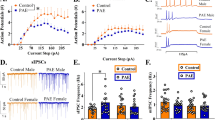
CRF action on neural activity in the BnST is believed to be critical for the expression of diffuse anxiety (for review, Davis et al, 1997; Shekhar et al, 2005; Davis et al, 2010). Our previous work demonstrated that AAS treatment increased the levels of CRF mRNA and somal-associated CRF-IR in the CeAL and enhanced CRF-IR in the neuropil of the dlBnST (Costine et al, 2010). The CeA provides a robust projection to the dlBnST from neurons that express both CRF and the classical neurotransmitter, GABA (Sun et al, 1991; Sun and Cassell, 1993; Arluison et al, 1994; Cassell et al, 1999). We hypothesized that AAS treatment would increase the activity of these CeAL projection neurons, and thus also promote an increase in CRF release and CRF-R1-medaited signaling in the dlBnST. On-cell recordings of neurons in the rostral extent of the CeAL support this assertion. The frequency of APs was found to be significantly higher (F1,69=8.40, p<0.005; 35 °C) in CeAL neurons from AAS-treated than oil-injected animals (Figures 2a–c). The percentage of bursty cells in this region was greater in AAS-treated animals (31.6% in oil-injected vs 42.9 % AAS-treated), but the difference was not significant.

(a) Representative on-cell recordings from the lateral part of the central amygdala (CeAL) from oil- and anabolic androgenic steroids (AAS)-injected mice in recordings made at 35 °C. (b) Action potential (AP) frequency; *p<0.05. (c) Cumulative probability distributions of AP frequencies. Inset: Kolmogorov-Smirnov ‘D’ values.
Immunoreactivity for the immediate early gene, c-FOS (Hoffman et al, 1993; Sharp et al, 1993; Clayton, 2000) can provide an assessment of concurrent neural activity in inter-related brain regions involved in the expression of fear and anxiety (Scicli et al, 2004; Veening et al, 2009; Skórzewska et al, 2008). Surprisingly, whereas electrophysiological recordings demonstrated a significant increase in AP frequency in CeAL neurons with AAS treatment, c-FOS-IR did not reveal a significant effect of treatment in this area (Figure 3). Such a discrepancy between these two approaches might arise if the AAS promote an increase in electrical activity that occurs within a select subset of CeAL neurons, so that the differences in activity in this more restricted population are lost in the broader assessment of c-FOS expression. To test whether or not this restricted population may correspond to the neurons within the CeAL that project to the dlBnST, fluorescent latex microspheres (Retrobeads) were stereotaxically injected into the dlBnST, and recordings were then made from retrogradely labeled neurons in the CeAL(Figure 4a). Labeled neurons localized in the CeAL were significantly more active in the AAS-treated mice than either non-labeled neurons in AAS-injected mice or either population of neurons in the oil-injected mice (F1,39=12.98, p<0.001) (Figure 4b). These data are consistent with the hypothesis that AAS selectively enhance the activity of CeA neurons that project to the dlBnST, and are likely to provide both GABAergic inhibition and CRF input in this region. Interestingly, AAS treatment also promoted a significant shift in the distribution of bursty vs non-bursty cells in the CeAL projection neurons (Figure 4c). The mechanism underlying this shift is not yet known, but it suggests that the AAS impose changes that may alter the network interactions in this key region either through direct effects on the electrical excitability of these neurons or on the synaptic inputs to them.

(a) Representative photomicrographs of the dorsolateral bed nucleus of the stria terminalis (dlBnST) (as indicated by the triangular lines) demonstrating c-FOS-immunoreactivity. Scale bar=50 μm. (b) Mean number of c-FOS-immunoreactive cells in regions of the extended amygdala and the hypothalamus that have a role in the expression of anxiety and stress as well as the auditory cortex (AuC) (n=5 for oil/untested; n=8 for oil/acoustic startle response (ASR); n=5 for AAS/untested; n =7 for anabolic androgenic steroids (AAS)/ASR). *Two-way analysis of variance (ANOVA) indicated a significant effect of treatment (p<0.027) with the number significantly lower for AAS-treated than oil-injected animals not subjected to testing (untested; p<0.01); for untested vs ASR-tested animals in the AAS-group (p<0.04) and for untested/AAS-treated vs oil/ASR (p<0.01). ^ Two-way ANOVA indicated a significant effect of testing (p<0.035).

(a) Representative photomicrographs of the Retrobead injection site in the dorsolateral bed nucleus of the stria terminalis (dlBnST) (left), the transverse mesh wire is holding down the coronal slice, and of retrogradely labeled neurons in the lateral part of the central amygdala (CeAL) (right) as captured by the DAGE-MTI VE-1000 camera on the recording set-up. (b) Average data indicating the mean action potential (AP) frequency in labeled (gray bars) and unlabeled (black bars) neurons from oil-injected and anabolic androgenic steroids (AAS)-injected mice. The ‘b’ indicates that the value for labeled neurons in AAS-treated mice was significantly different from the other three states (‘a’); n values represent the number of cells. (c) The distribution of action potential (AP) patterning amongst labeled (L) and unlabeled (U) neurons from AAS- and oil-injected mice. *The distribution between labeled and unlabeled neurons in the AAS-injected mice was significantly different (p<0.001).
Neuronal Excitability is Decreased in the dlBnST by AAS Exposure
The BnST is the key site of action of CRF in eliciting diffuse anxiety (Davis et al, 1997; Shekhar et al, 2005; Davis et al, 2010). To determine whether the AAS-dependent increases in anxiety-like behavior is correlated with significant changes in neuronal activity within the dlBnST, on-cell recordings of AP activity were made from neurons within this region. These recordings revealed that the frequency of APs in dlBnST neurons was significantly lower in AAS-injected than in oil-injected animals (F1,32=16.70, p<0.0003) (Figures 5a–c). Autocorrelational analysis of firing patterns revealed that irregularly firing cells accounted for the majority of recordings under both treatment conditions, and although the percentage of irregularly firing cells was lower in AAS-treated than oil-injected animals (35.3% vs 52.9%, with a concomitant increase in the percentage of cells with regular firing patterns), the difference was not significant.

(a) Representative on-cell recordings of action potentials (AP) from the dorsolateral bed nucleus of the stria terminalis (dlBnST) from oil- and anabolic androgenic steroids (AAS)-injected mice in recordings made at 35 °C. (b) AP frequency in recordings from dlBnST neurons at 22 and 35 °C; *p<0.05. (c) Cumulative probability distributions of AP frequencies at 22 and 35 °C. Inset: Kolmogorov-Smirnov ‘D’ values. (d) Distribution of AP frequencies (22 °C).
Consistent with the electrophysiological assessment in the dlBnST, two-way ANOVA of c-FOS-IR revealed a significant effect of treatment in this region (F1,21=5.37, p<0.030). Post-hoc analysis indicated that the number of c-FOS-IR cells was significantly lower in AAS-treated vs oil-injected animals not subjected to behavioral testing (untested; p<0.01), and also lower (p<0.04) for untested mice than those subjected to the startle stimulus in the AAS-treated group (Figures 3a and b). No significant effects of either testing or treatment were observed in c-FOS expression in other regions of the extended amygdala (the BLA and the vlBnST), although, as expected for an auditory stimulus, startle testing elicited a significant increase in c-FOS in the AuC (F1,21=5.26, p<0.034) (Figure 3b). These electrophysiological and immunocytochemical data suggested that AAS exposure might impose a lower basal level of activity in dlBnST neurons, and thus accentuate the response to the startle itself. Such an action of the AAS may arise if there is an enhanced inhibition of dlBnST neurons in the AAS-treated mice.
AAS Exposure Augments GABAA Receptor-Mediated Inhibition in the dlBnST
We next used whole-cell recording to determine whether the lower level of activity in the dlBnST of AAS-treated mice arose from increased GABAergic inhibition in these neurons. To allow prolonged recordings of GABAA receptor-mediated sIPSCs in the dlBnST, these experiments were performed at 22 °C. As a prelude to these whole-cell recordings, we first determined in on-cell recordings that the AAS-dependent decrease in AP frequency in dlBnST neurons was still significant at this lower temperature, and indeed it was (Figures 5a–c). As with recordings made at 35 °C, the mean AP frequency was significantly lower in AAS-treated vs oil-injected animals for recordings made from the dlBnST at 22 °C (F1,60=4.88, p<0.031) (Figures 5a–c). We next assessed whether blockade of GABAA receptor-mediated transmission abrogated the effects of AAS treatment on AP frequency in the dlBnST by recording from oil-injected and AAS-treated animals in the presence of the non-competitive GABAA receptor antagonist, picrotoxin (100 μM). In the presence of picrotoxin, the AP frequencies were not different between the two groups (oil: 2.19±0.69 Hz; AAS: 2.89±1.08 Hz; F1,49=0.51, p=0.48). Addition of the glycine receptor antagonist strychnine had no effect on AP frequency (data not shown), further implicating GABAA receptors as the mediators of AAS-dependent enhanced inhibition in the dlBnST.
Whole-cell recordings revealed that the frequency of GABAA receptor-mediated sIPSCs was significantly (F1,77=24.13, p<0.0001) higher in recordings from AAS-injected mice than from oil-injected mice (Figures 6a and b). AAS treatment had no effect on sIPSC amplitudes or the time constants of synaptic decay (Figure 5c; legend). Analysis of mIPSCs recorded in the presence of 1 μM TTX revealed no significant differences in the frequency, amplitude, or decay kinetics of these currents (frequency: 1.82±0.27 Hz vs 1.97±0.32 Hz; Ipeak: 27.2±4.0 pA vs 26.6±3.9 pA; τw: 27.7±3.6 ms vs 35.2±3.5 ms in dlBnST neurons from oil-injected vs AAS-treated animals, respectively). These data suggest that AAS treatment augments AP-dependent presynaptic release of GABA onto dlBnST neurons, but does not alter postsynaptic sensitivity to GABA.

(a) Representative whole-cell recordings of GABAA receptor-mediated spontaneous inhibitory postsynaptic currents (sIPSCs) in dorsolateral bed nucleus of the stria terminalis (dlBnST) neurons from oil- and anabolic androgenic steroids (AAS)-injected mice. (b) sIPSC frequencies from individual dlBnST neurons from oil-injected (black circles) and AAS-treated (white circles) mice. Histograms display the same data as means with error bars (SEM); n values represent the number of cells. *p<0.0001. (c) Averaged sIPSCs from dlBnST neurons under the two treatment conditions. No differences between oil-injected and AAS-treated mice in Ipeak (36.8±2.5 vs 42.0±3.3 pA) or decay kinetics (τ1: 18.1±1.1 vs 21.2±1.3 ms), (τ2: 76.3±10.0 vs 86.0±11.5) or (τw: 33.2±2.9 vs 37.1±2.3 ms) were evident. Fitted line to data reflects τw.
We have shown that AAS treatment augments AP firing selectively in CeAL neurons that project to the dlBnST and separately that AAS augments GABAA receptor-mediated responses in dlBnST cells, together suggesting that the enhanced GABAergic drive arises from these CeA afferents. A recognized caveat to this interpretation, however, is that in the slice configuration used here, the cell bodies of the CeA neurons would not have been retained in the BnST slice. CeA neurons may continue to generate spontaneous APs even in the absence of cell bodies if the sites of AP initiation are distal to the somata (see Peng et al, 2007; Sheffield et al, 2011). Conversely, AAS may augment GABA release from the population of intrinsic interneurons within the BnST. If AAS enhanced the activity of interneurons within the dlBnST itself, we might expect to see a bimodal distribution of AP frequencies: local interneurons whose activity was enhanced by AAS treatment and target dlBnST neurons who received the corresponding increase in inhibitory input. Assessment of the distribution of AP frequencies in dlBnST neurons did not reveal such an effect. At both recording temperatures (Figure 5d; 22 °C and 35 °C; data not shown), the effect of AAS was to shift the distribution to lower frequencies by the loss of cells with the highest firing rates. Moreover, there was no correlation between AP patterning and firing frequency at either temperature. Thus, the enhanced GABAergic drive to dlBnST neurons is likely to either reflect an increase of release from the desomatized CeA afferents and/or afferents arising from other regions of the extended amygdala, including neighboring subregions of the BnST.
The Ability of AAS to Enhance GABAergic Inhibition in the dlBnST is Mediated via a CRF-R1-Dependent Mechanism
AAS treatment augments CRF expression (Costine et al, 2010) and activity in CeAL projection neurons to the dlBnST; neurons that provide not only GABA, but also CRF signaling to the dlBnST (Sun et al, 1991; Sun and Cassell, 1993; Arluison et al, 1994; Cassell et al, 1999). Moreover, our data demonstrate that AAS enhance CRF expression in the CeAL and that the increase in anxiety-like behaviors observed in ASR testing in AAS-treated mice is abrogated by infusion of the CRF-R1 antagonist, antalarmin (Costine et al, 2010; present study). Because CRF has been shown to increase the frequency of GABAA receptor-mediated sIPSCs in other forebrain regions (Kirby et al, 2008; Nie et al, 2004, 2009), we hypothesized that enhanced CRF release from CeAL afferents into the dlBnST might also mediate, via a CRF-R1-dependent mechanism, the AAS-dependent increase in sIPSCs observed in dlBnST target neurons. To test this hypothesis, antalarmin was added to the bath during recordings from dlBnST neurons in acutely isolated brains slices of oil-injected or AAS-treated animals. As previously observed, the frequency of sIPSCs was significantly higher in dlBnST neurons from the AAS-treated than oil-treated groups prior to addition of antalarmin (F1,35=15.08, p<0.001). However, in the presence of 40 μM antalarmin, no significant differences (F1,19=0.019, p=0.89) were evident in sIPSC frequency between oil-injected and AAS-treated mice (Figures 7a and b). Moreover, the effect of antalarmin was found to be due to a significant decrease in sIPSC frequency only in recordings from the AAS-injected group (p<0.001). The effects of antalarmin were reversible upon washout, and addition of this antagonist did not alter either the peak current or the decay kinetics of the sIPSCs (data not shown). Acute application of 4 μM antalarmin had an intermediate effect, lowering sIPSC frequency in AAS-treated animals to a value partway between that observed in control solutions and that observed in the presence of 40 μM antalarmin (Figure 7c). As with the 40 μM concentration, 4 μM antalarmin had no effect on the frequency of sIPSCs in dlBnST neurons of oil-injected mice (Figure 7c).

Representative whole-cell recordings of GABAA receptor-mediated sIPSCs from dorsolateral bed nucleus of the stria terminalis (dlBnST) neurons of (a) oil- or (b) anabolic androgenic steroids (AAS)-injected mice made in artificial cerebrospinal fluid (aCSF) alone (control, top), following the addition of 40 μM antalarmin (middle) or in the presence of 20 μM bicuculline (bottom). (c) Average spontaneous inhibitory postsynaptic currents (sIPSC) frequency in the dlBnST in aCSF alone (control) or in the presence of 4 or 40 μM antalarmin. *Frequency for AAS (control) value significantly (p<0.05) different from oil-injected (control); n values represent the number of cells.
AAS given to mice during the treatment period are likely washed out or substantially diluted during slice preparation. To assess whether direct exposure to CRF (100 nM) altered GABAergic inhibition in the dlBnST and whether the action of CRF was different if AAS were present, recordings were made from dlBnST neurons recordings were made from dlBnST neurons either in aCSF alone or with co-application of AAS from slices of age-matched naïve mice that had not received injections of any type. Real-time PCR analysis indicates that BnST neurons express high levels of GABAA receptor subunits, including α1, α2, γ2, and γ1 (data not shown), that we have shown form receptors that are sensitive to acute positive allosteric modulation by the AAS (Yang et al, 2002, 2005; Clark et al, 2004). Thus, not surprisingly, we found that acute exposure to the AAS mixture at a concentration (1 μM) that approximates the concentration measured in the CSF of adult volunteers administered AAS on a moderate, short-term basis (Daly et al, 2001), had a modest, but significant effect on the peak amplitude of sIPSCs in dlBnST neurons (22.2±1.4 pA in aCSF; 26.3±1.8 pA in the presence of 1 μM AAS;n=13) (Figure 8a). Acute AAS application had no significant effect on sIPSC decay kinetics or frequency. Acute exposure of brain slices to 100 nM CRF significantly increased the frequency of sIPSCs in dlBnST neurons in recordings made in aCSF alone (47%; p<0.016) and in neurons in which CRF was applied following exposure to 1 μM AAS (39%; p<0.023) (Figures 8b and c). CRF was without effect on either the peak amplitudes of the sIPSCs or the decay kinetics of sIPSCs. A similar, albeit somewhat smaller, increase in sIPSC frequency (21%) was also observed in slices exposed to 10 μM AAS; a concentration that may be present in the CNS in individuals who administer high doses for a prolonged period of time. The percentage of neurons that showed an increase in sIPSC frequency when exposed to CRF that was >10% was comparable in both recording conditions (70% for saline alone; 69% for saline+1 μM AAS). As with sIPSCs, CRF had no effect on the amplitude or frequency of mIPSCs recorded in the presence of 1 μM TTX. In contrast to sIPSCs, CRF had no significant effect on mIPSC frequency (0.91±0.1 Hz in saline alone vs 0.99±0.15 in saline+CRF, n=10 cells each condition). These results suggest that the actions of CRF in the dlBnST are presynaptic and require AP-dependent release.

(a) Representative recordings illustrating effects of acute application of corticotropin releasing factor (CRF) on GABAA receptor-mediated spontaneous inhibitory postsynaptic currents (sIPSCs) from dorsolateral bed nucleus of the stria terminalis (dlBnST) neurons of naïve mice in recordings made in saline alone or saline supplemented with 1 μM anabolic androgenic steroids (AAS). (b) Data points indicate the change in sIPSC frequency in individual dlBnST neurons exposed to CRF alone (100 nM) or CRF following exposure to 1 μM AAS. (c) Averaged data indicating the percent increase (±SEM) in sIPSC frequency elicited by acute exposure to CRF for dlBnST neurons in saline alone or saline supplemented by 1 μM AAS. *p<0.016 (saline); p<0.023 (1 μM AAS). n values represent the number of cells.
DISCUSSION
The prevalence of illicit self-administration of supratherapeutic doses of AAS has been estimated to be approximately 0.5% of adolescent girls and 2% of adolescent boys (Bahrke et al, 2000; Miller et al, 2005; Johnston et al, 2009). In both adults and adolescents, AAS use has been associated with changes in behavior, including anxiety, irritability, poor impulse control, and mood fluctuations, in addition to their actions on physique and performance (Annitto and Layman, 1980; Pope and Katz, 1988; Burnett and Kleiman, 1994; Cooper et al, 1996; Franke and Berendonk, 1997; Rashid, 2000; Pagonis et al, 2006). Interestingly, human subjects have reported both transient euphoria and hypomania early in the course of AAS administration (Pope and Katz, 1994; Thiblin and Petersson, 2005; Basaria, 2010), and higher levels of anxiety with prolonged, chronic use (Cooper et al, 1996; Galligani et al, 1996; Hall and Chapman, 2005; Pagonis et al, 2006). Consistent with the multiplicity of effects on anxiety observed in humans who self-administer these drugs, treatment of rodents with AAS has also been reported to elicit anxiolytic/diminished fear responses (Bitran et al, 1993; Ågren et al, 1999; Barreto-Estrada et al, 2004; Steensland et al, 2005; Frye et al, 2008b; Kouvelas et al, 2008), anxiogenic/enhanced fear responses (Rocha et al, 2007; Agis-Balboa et al, 2009; Ambar and Chiavegatto, 2009; Costine et al, 2010), or minimal observable effects on anxiety-like behaviors (Rojas-Ortiz et al, 2005). The plethora of effects on anxiety-like behaviors is likely to reflect not only the temporal chronology in the exposure to AAS (transient vs prolonged exposure), but also a complex matrix of interactions that include, age, sex, the genetic and the social background of the subjects, and the interface of these factors with the types of AAS taken and the paradigms of their administration (Clark and Henderson, 2003; Clark et al, 2006; Pinna et al, 2008).
Here, we report that chronic exposure of C57BL/6J female mice during adolescence to a mixture of three commonly abused AAS at a dose that reflects a high human abuse concentration augments anxiety-like behaviors. These results are consistent with our prior study in female mice (Costine et al, 2010). In contrast, a single acute injection of this mixture of three AAS promoted neither anxiolytic nor anxiogenic effects, suggesting that effects on anxiety of this combination of AAS in these mice require long-term, likely genomic actions. The increase in anxiety in mice chronically treated with AAS was paralleled by a concomitant increase in the expression of CRF and CRF-dependent changes in neural signaling within the extended amygdala, as both the behavioral and the physiological effects of AAS treatment were shown to require signaling through the CRF-R1 (Costine et al, 2010; current study). Our findings provide a mechanistic link between a large body of literature highlighting the importance of CRF signaling in the BnST to the production of diffuse anxiety/sustained fear (for review, Reul and Holsboer, 2002; Holsboer and Ising, 2008; Davis et al, 2010) and the demonstrated anxiogenic actions of prolonged AAS use (Cooper et al, 1996; Galligani et al, 1996; Hall and Chapman, 2005; Pagonis et al, 2006; Rocha et al, 2007; Kanayama et al, 2008; Agis-Balboa et al, 2009; Ambar and Chiavegatto, 2009; Costine et al, 2010). Specifically, our data demonstrate that CRF signaling is required for the AAS-dependent increase in anxiety-like behaviors and for changes in neural circuits of the extended amygdala that underlie their generation.
Although we cannot rule out the possibility that AAS-dependent changes in CRF expression act to alter synaptic inputs onto neurons in the amygdala itself, previous studies argue against this being the critical site with respect to the AAS-elicited increase in diffuse anxiety and the enhanced startle response. First, our previous study (Costine et al, 2010) demonstrated that CRF mRNA is enhanced in CeA neurons, as is somal-associated CRF-IR. However, the overall IR in the CeA is not affected by AAS treatment. These data suggest that the AAS increase CRF expression within CeA neurons, but not in neurons that provide the afferent CRF innervation to the CeA neurons themselves. Second, a substantial body of work from Davis and colleagues supports the postulate that although the CeA is an integral part of the startle circuitry and that afferent disinhibition of CeA neurons is involved in the startle response, the action of CRF in enhancing the startle is localized to the BnST (Liang et al, 1992; Davis et al, 1997; Walker et al, 2009a, 2009b; for review Davis et al, 2010). Finally, we note that AAS treatment enhances generalized anxiety as measured in the ASR, but not fear-potentiated startle (Costine et al, 2010), again, suggesting that the BnST is the critical site of AAS actions.
Our data also suggest that CRF-R1-dependent changes in GABAA receptor-mediated transmission between the CeAL and its major projection site, the dlBnST, is a critical intermediary in the AAS-dependent expression of anxiety. Specifically, AAS treatment was shown to promote increased AP frequency selectively in those CeAL neurons that project to the dlBnST and to increase presynaptic release of GABA in this region. We note that while AAS do promote this selective increase in firing of CeA projection neurons, CeA somata are unlikely to be present in the slice preparations used for dlBnST recordings. Although we do not know the site of AP initiation in the CeA afferents, and initiation sites quite distal from cell somata have been described in other preparations (Peng et al, 2007; Sheffield et al, 2011), CRF effects on local GABAergic interneurons must also be considered as the source of the increase in GABAergic sIPSCs recorded in the dlBnST. Irrespective of the source, the enhanced GABAergic tone in the dlBnST is, in turn, associated with a lower level of neural activity in this region, as indicated by both c-FOS expression and the frequency of spontaneous APs. The role of GABA in imposing AAS-dependent effects in the dlBnST was further reinforced by experiments demonstrating that the effects of the AAS on AP activity in the dlBnST could be abrogated by the GABAA receptor antagonist, picrotoxin, and that CRF-R1 antagonism could negate the AAS-induced increase in sIPSC frequency in dlBnST neurons. This proposed mechanism (Figure 9), by which AAS act to enhance transmission from GABA- and CRF-containing CeAL afferents, resulting in enhanced GABAergic inhibition imposed on target dlBnST neurons, is consistent with several separate, but interrelated, findings from previous studies. First, CRF-positive terminals originating in the CeA co-express GABA (Veinante et al, 1997), and CRF has been shown to augment GABAergic transmission via CRF-R1-mediated signaling in other brain regions (Nie et al, 2004, 2009; Kash and Winder, 2006; Bajo et al, 2008; Kirby et al, 2008; Roberto et al, 2010). Second, it has been well established that the GABAA receptor is one of the key molecular targets for drugs that alter the expression of anxiety (for review, Reddy, 2010; Möhler, 2012). Third, steroid-dependent changes in GABAA receptor-mediated transmission significantly modify anxiety-like behaviors (Shen et al, 2007; Frye et al, 2008a,b; Smith et al, 2009; Maguire and Mody, 2009).

Putative model for anabolic androgenic steroids (AAS) actions in the extended amygdala: Chronic AAS exposure acting through classical androgen receptors (1) increases CRF expression (2) and increases action potential firing/bursting of GABAergic/CRF projection neurons from the lateral central amygdala (CeAL) (3) that promotes both a direct increase in GABA release and increased release of CRF. CRF acting through the CRF type 1 receptor (CRF R1) (4) on CeAL or other bed nucleus of the stria terminalis (BnST) terminals further enhances the release of GABA (5). The increased release of GABA is reflected in an increased frequency of sIPSCs (6), hyperpolarization (7), and decreased action potential activity in postsynaptic dorsolateral BnST (dlBnST) neurons (8). The diminished output from the dlBnST gives rise to increased anxiety as measured by the acoustic startle response and on the elevated plus maze, potentially through disinhibition (double red line) of the CeA itself through reciprocal connections (9).
Although our data are consistent with previous studies demonstrating that CRF enhances neurotransmitter release from GABAergic neurons in regions of the extended amygdala (Kirby et al, 2008; Nie et al, 2004, 2009), our results do diverge from these previous studies in that we found that CRF in the dlBnST enhanced the frequency of sIPSCs, but not mIPSCs. A growing body of literature suggests that the processes leading to mIPSCs, sIPSCs, and evoked ISPCs are not always coordinately regulated (Sara et al, 2005; Wasser et al, 2007; Wasser and Kavalali, 2009; Fredj and Burrone, 2009; Chung et al, 2010), and region-specific effects of CRF may exist between the CeA and the dlBnST. In addition, we note that CRF has been shown to enhance evoked IPSCs in the vlBnST through a postsynaptic mechanism (Kash and Winder, 2006). Taken together, these data raise the possibility that this neuropeptide may have separate synapse-specific mechanisms of augmenting GABAergic inhibition within the different regions of the extended amygdala.
Prior studies have suggested that activity within the BnST as a whole is correlated with enhanced anxiety (for review, Shekhar et al, 2005; Hammack et al, 2009; Davis et al, 2010). Thus, at first blush increased GABAergic inhibition to the dlBnST may seem paradoxical with this proposed role. However, it must be considered that the BnST is a complex and heterogeneous structure in which the neurons make abundant and often reciprocal connections with other structures, as well as with a rich network of local inhibitory circuits (Dong and Swanson, 2005). The majority of GABAergic neurons in the BnST are localized to the dlBnST (Sun and Cassell, 1993). Thus, inhibition of these dlBnST GABAergic neurons may, through local circuit interactions, result in disinhibition of BnST efferents. The CeA not only sends a dense projection to the dlBnST, but receives an equivalently robust one from this region as well (Dong et al, 2001; Dong and Swanson, 2005). This reciprocal innervation may promote disinhibition in the CeA, which has been suggested to be an important component of the startle response (Davis et al, 1997). Conversely, the AAS may alter glutamatergic excitatory synaptic drive onto CeA neurons either directly or indirectly, or may affect synaptic inputs to these cells by altering the expression of key peptides, such as oxytocin, Neuropeptide Y, arginine vasopressin, and PACAP that have important effects on the expression of anxiety. The AAS are also known to significantly alter the expression of receptors for serotonin in other brain regions (Grimes and Melloni, 2005; Ricci et al, 2006; Schwartzer et al, 2009). As regulation of 5-HT transmission is the cornerstone of current treatments for anxiety, and changes in serotonergic modulation of transmission within the BnST have been proposed to have an important role in the etiology of pathological anxiety (for review, Hammack et al, 2009), steroid-dependent changes in serotonergic signaling within the extended amygdala and its targets may also have an important role in AAS-dependent increases in anxiety.
Androgens are also known to have widespread and significant effects on the expression of voltage-gated channels that regulate electrical excitability (for review, Penatti and Henderson, 2009). Preliminary microarray data from our lab suggests that AAS treatment may alter the expression of several different potassium channel genes (including GIRK3, Kv1.3, Kv3.4, and HCN4) that are known to regulate repetitive firing behavior. Interestingly, some of these channel genes are selectively expressed in the populations of neurons that exhibit distinctly different firing patterns as assessed in neighboring subregions of the BnST (Egli and Winder, 2003; Hammack et al, 2007; Hazra et al, 2011). These findings raise the possibility that AAS may also affect the output of the BnST by altering the intrinsic membrane properties of these neurons, although care must be made in extrapolating from studies that have been made in adult male rats to properties of BnST neurons in adolescent female mice, especially as the BnST displays a marked sexual dimorphism (Polston et al, 2004; Forger et al, 2004).
The signaling mechanisms by which AAS increase CRF in the extended amygdala have yet to be determined. Costine et al (2010) demonstrated that AAS treatment results in increases in CRF mRNA in the CeA and CRF peptide in the BnST. Although previous studies have underscored the importance of the androgen receptor in mediating the chronic effects of some of the AAS used in this study in both males and females (Penatti et al, 2009a, 2009b) and in regulating the HPA axis response to stress in males (Handa et al, 1994), estrogens have also been shown to alter CRF expression in vivo and in vitro (Paulmyer-Lacroix et al, 1996; Lunga and Herbert, 2004; Lalmansingh and Uht, 2010; for review, Kageyama and Suda, 2009), raising the possibility that estrogen receptors may also have a pivotal role in AAS-dependent changes in CRF in the extended amygdala. The demonstrated ability of androgens to interact directly with estrogen receptor β to affect the stress response (Handa et al, 2009) further highlights the complexity in the potential hormone pathways that may mediate the effects of the AAS. In addition, it is possible that AAS may regulate the levels of CRF within the extended amygdala by altering the expression or binding interactions of the CRF-binding protein (Potter et al, 1992; Westphal and Seasholtz, 2006). Finally, data demonstrating that down-stream coupling of CRF receptors with different G proteins varies with strain (Blank et al, 2003), and that there are sex-specific differences in CRF receptor trafficking (Bangasser et al, 2010) highlight that variables such as sex and genetic background are likely to be significant parameters in determining the effects of AAS on the neural circuits that underlie the expression of anxiety.
An interesting facet to emerge from this study is the concept that AAS may impose their effects on the production of anxious behaviors by lowering the basal level of activity in the dlBnST, as was indicated by both c-FOS and AP frequency. This would be expected to extend the dynamic range of the circuits in this system that regulate the expression of behaviors to an anxiety-producing stimulus. In the hippocampus, it has been proposed that stress associated with a novel environment may impair learning and memory retrieval by saturating the mechanisms that generate glutamate receptor-mediated long-term potentiation (LTP) such that they are limited in their ability to subsequently respond to other stimuli that would normally promote learning (for review, Diamond et al, 2005; Kim et al, 2006; Howland and Wang, 2008; Collingridge et al, 2010). Consistent with this model, CRF has been shown to prevent LTP in the hippocampus (Rebaudo et al, 2001). It is provocative to consider a mirror mechanism by which AAS-dependent increases in CRF at the synapse may ‘de-saturate’ transmission between the CeA and dlBnST by enhancing GABAergic inhibition, extending the response range in this critical circuit in the extended amygdala and thus allowing the more pronounced expression of sustained fear/anxiety to stressful stimuli that was observed in these animals. It will be of great interest to ascertain whether or not chronic AAS exposure also alters glutamate receptor-mediated synaptic plasticity at the CeA to dlBnST synapse as changes in anxiety-like behaviors in animals exposed to social isolation conditions and to ethanol result in blunted LTP in the dlBnST (Conrad and Winder, 2011).
Our results also suggest important and intriguing parallels in the effects of the AAS and the actions of ethanol. Human studies have documented covariability in use of AAS and alcohol (Brower et al, 1991; Perry et al, 2005; Dodge and Hoagland, 2011), and data from animal models suggest a convergence in behavioral and neurophysiological effects of these two classes of drugs on CRF-dependent modulation of GABAergic transmission. In rodents, increased anxiety and augmented ASR are associated with ethanol withdrawal (Rassnick et al, 1992), and the anxiety elicited by ethanol withdrawal is associated with increased levels of CRF release in both the CeA (Merlo Pich et al, 1995) and the BnST (Olive et al, 2002). Furthermore, the increase in CRF with withdrawal is counteracted by subsequent ethanol intake (Olive et al, 2002). Both CRF mRNA and the sensitivity of GABA release to CRF modulation are increased in ethanol-dependent rats (Roberto et al, 2004, 2010; Sommer et al, 2008), and CRF/CRF-R1-dependent changes in transmission mediated by GABAA receptors have been implicated as having a major role in anxiety associated with ethanol dependence and in ethanol consumption (for discussion, see Roberto et al, 2010). The BnST has been identified as the critical region in CRF-dependent reinstatement of drug-seeking behavior (Erb and Stewart, 1999; Erb et al, 2001), and acute injections of ethanol have been shown to elicit increases in c-FOS expression in the lateral BnST in GABAergic neurons (Leriche et al, 2008). Finally, injections of GABAA receptor antagonists into the BnST reduce ethanol responding in rats (Hyytiä and Koob, 1995). These studies, as well as preliminary data from our laboratory indicating enhanced ethanol consumption and preference on the two-bottle test in AAS-treated mice (M. Onakomaiya, unpublished data) strongly support the assumption that there is a common neural basis for the anxiety associated with ethanol withdrawal and with AAS use. This convergence further suggests that this shared neural mechanism may underlie the covariance in AAS and alcohol use. Future experiments designed to test the interactions of AAS and ethanol on CRF-dependent modulation of GABAergic transmission in the extended amygdala will provide important information to alert AAS users, especially adolescents, that they could be at an increased risk for alcohol abuse.
References
Agis-Balboa RC, Pibiri F, Nelson M, Pinna G (2009). Enhanced fear responses in mice treated with anabolic androgenic steroids. Neuroreport 20 (6): 617–621.
Ågren G, Thiblin I, Tirassa P, Lundeberg T, Stenfors C (1999). Behavioural anxiolytic effects of low-dose anabolic androgenic steroid treatment in rats. Physiol Behav 66: 503–509.
Aikey JL, Nyby JG, Anmuth DM, James PJ (2002). Testosterone rapidly reduces anxiety in male house mice (Mus musculus). Horm Behav 42: 448–460.
Alheid GF (2003). Extended amygdala and basal forebrain. Ann NY Acad Sci 985: 185–205.
Ambar G, Chiavegatto S (2009). Anabolic-androgenic steroid treatment induces behavioral disinhibition and downregulation of serotonin receptor messenger RNA in the prefrontal cortex and amygdala of male mice. Genes Brain Behav 8: 161–173.
Annitto WJ, Layman WA (1980). Anabolic steroids and acute schizophrenic episode. J Clin Psychiatry 41 (4): 143–144.
Arluison M, Brochier G, Vankova M, Leviel V, Villalobos J, Tramu G (1994). Demonstration of peptidergic afferents to the bed nucleus of the stria terminalis using local injections of colchicine. A combined immunohistochemical and retrograde tracing study. Brain Res Bull 34 (4): 319–337.
Asan E, Yilmazer-Hanke DM, Eliava M, Hantsch M, Lesch KP, Schmitt A (2005). The corticotrophin-releasing factor (CRF)-system and monoaminergic afferents in the central amygdala: investigations in different mouse strains and comparison in the rat. Neuroscience 131: 953–967.
Bahrke MS, Yesalis CE (2004). Abuse of anabolic androgenic steroids and related substances in sport and exercise. Curr Opin Pharmacol 4 (6): 614–620.
Bahrke MS, Yesalis CE, Kopstein AN, Stephens JA (2000). Risk factors associated with anabolic-androgenic steroid use among adolescents. Sports Med 29 (6): 397–405.
Bajo M, Cruz MT, Siggins GR, Messing R, Roberto M (2008). Protein kinase C epsilon mediation of CRF- and ethanol-induced GABA release in central amygdala. Proc Natl Acad Sci USA 105: 8410–8415.
Bale TL, Vale WW (2004). CRF and CRF receptors: Role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol 44: 525–557.
Bangasser DA, Curtis A, Reyes BA, Bethea TT, Parastatidis I, Ischiropoulos H et al. (2010). Sex differences in corticotropin-releasing factor receptor signaling and trafficking: potential role in female vulnerability to stress-related psychopathology. Mol Psychiatry 15 (9): 896–904.
Baram TZ, Chalmers DT, Chen C, Koutsoukos Y, De Souza EB (1997). The CRF1 receptor mediates the excitatory actions of corticotropin releasing factor (CRF) in the developing rat brain: in vivo evidence using a novel, selective, non-peptide CRF receptor antagonist. Brain Res 770: 89–95.
Barreto-Estrada JL, Barreto J, Fortis-Santiago Y, Rivera-Ramos I, Fortis-Santiago A, Jorge JC (2004). Modulation of affect after chronic exposure to the anabolic steroid 17α-methyltestosterone in adult mice. Behav Neurosci 118: 1071–1079.
Basaria S (2010). Androgen abuse in athletes: detection and consequences. J Clin Endocrinol Metab 95 (4): 1533–1543.
Basaria S, Wahlstrom JT, Dobs AS (2001). Clinical review 138: Anabolic-androgenic steroid therapy in the treatment of chronic diseases. J Clin Endocrinol Metab 86: 5108–5117.
Bingham B, Myung C, Innala L, Gray M, Anonuevo A, Viau V (2011). Androgen receptors in the posterior bed nucleus of the stria terminalis increase neuropeptide expression and the stress-induced activation of the paraventricular nucleus of the hypothalamus. Neuropsychopharmacology 36: 1433–1443.
Bitran D, Kellogg CK, Hilvers RJ (1993). Treatment with an anabolic-androgenic steroid affects anxiety-related behavior and alters the sensitivity of cortical GABAA receptors in the rat. Horm Behav 27: 568–583.
Blank T, Nijholt I, Grammatopoulos DK, Randeva HS, Hillhouse EW, Spiess J (2003). Corticotropin-releasing factor receptors couple to multiple G-proteins to activate diverse intracellular signaling pathways in mouse hippocampus: role in neuronal excitability and associative learning. J Neurosci 23 (2): 700–707.
Blasberg ME, Clark AS (1997). Anabolic-androgenic steroid effects on sexual receptivity in ovariectomized rats. Horm Behav 32: 201–208.
Briscoe RJ, Cabrera CL, Baird TJ, Rice KC, Woods JH (2000). Antalarmin blockade of corticotropin releasing hormone-induced hypertension in rats. Brain Res 881 (2): 204–207.
Brower KJ, Blow FC, Young JP, Hill EM (1991). Symptoms and correlates of anabolic-androgenic steroid dependence. Br J Addict 86 (6): 759–768.
Burnett KF, Kleiman ME (1994). Psychological characteristics of adolescent steroid users. Adolescence 29 (113): 81–89.
Campbell RE, Han SK, Herbison AE (2005). Biocytin filling of adult gonadotropin-releasing hormone neurons in situ reveals extensive, spiny, dendritic processes. Endocrinology 146 (3): 1163–1169.
Carrillo M, Ricci LA, Melloni RH (2011). Glutamate and the aggression neural circuit in adolescent anabolic steroid-treated Syrian hamsters (Mesocricetus auratus). Behav Neurosci 125 (5): 753–763.
Cassell MD, Freedman LJ, Shi C (1999). The intrinsic organization of the central extended amygdala. Ann N Y Acad Sci 877: 217–241.
Chung C, Barylko B, Leitz J, Liu X, Kavalali ET (2010). Acute dynamin inhibition dissects synaptic vesicle recycling pathways that drive spontaneous and evoked neurotransmission. J Neurosci 30 (4): 1363–1376.
Clark AS, Costine BA, Jones BL, Kelton-Rehkopf MC, Meerts SH, Nutbrown-Greene LL et al. (2006). Sex- and age-specific effects of anabolic androgenic steroids on reproductive behaviors and on GABAergic transmission in neuroendocrine control regions. Brain Res 1126: 122–138.
Clark AS, Henderson LP (2003). Behavioral and physiological responses to anabolic-androgenic steroids. Neurosci Biobehav Rev 27 (5): 413–436.
Clark AS, Jones BL, Yang P, Henderson LP (2004). Anabolic androgenic steroids and the brain: novel actions at the GABAA receptor and on GABAA receptor mediated-behaviors. In: Smith, SS (ed). Neurosteroid Effects in the Central Nervous System: The Role of the GABAA Receptor. CRC Press: LLC pp 119–141.
Clayton DF (2000). The genomic action potential. Neurobiol Learn Mem 74 (3): 185–216.
Collingridge GL, Peineau S, Howland JG, Wang YT (2010). Long-term depression in the CNS. Nat Neurosci Revs 11: 459–473.
Conrad KL, Winder DG (2011). Altered anxiety-like behavior and long-term potentiation in the bed nucleus of the stria terminalis in adult mice exposed to chronic social isolation, unpredictable stress, and ethanol beginning in adolescence. Alcohol 45 (6): 585–593.
Cooper CJ, Noakes TD, Dunne T, Lambert MI, Rochford K (1996). A high prevalence of abnormal personality traits in chronic users of anabolic-androgenic steroids. Br J Sports Med 30 (3): 246–250.
Cooper RL, Goldman JM, Vandenbergh JG (1993). Monitoring of the Estrous Cycle in the Laboratory Rodent by Vaginal Lavage. In: Heindel JJ, Chapin RE (eds). Methods in Toxicology. Academic Press: New York, NY, pp 45–55.
Costine BA, Oberlander JG, Davis MC, Penatti CAA, Porter DM, Leaton RN et al. (2010). Chronic anabolic androgenic steroid exposure alters corticotropin releasing factor expression and anxiety-like behaviors in the female mouse. Psychoneuroendocrinology 35 (10): 1473–1485.
Daly RC, Su TP, Schmidt PJ, Pickar D, Murphy DL, Rubinow DR (2001). Cerebrospinal fluid and behavioral changes after methyltestosterone administration: preliminary findings. Arch Gen Psychiatry 58: 172–177.
Davis M, Walker DL, Lee Y (1997). Amygdala and bed nucleus of the stria terminalis: differtial roles in fear and anxiety measured with the acoustic startle reflex. Philos Trans R Soc Lond B Biol Sci 352 (1362): 1675–1687.
Davis M, Walker DL, Miles L, Grillon C (2010). Phasic vs sustained fear in rats and humans: role of the extended amygdala in fear vs anxiety. Neuropsychopharmacology 35: 105–135.
Day HEW, Curran EJ, Watson SJ, Akil H (1999). Distinct neurochemical populations in the rat central nucleus of the amygdala and bed nucleus of the stria terminalis: evidence for their selective activation by interleukin-1β. J Comp Neurol 413: 113–128.
Deak T, Meriwether JL, Fleshner M, Spencer RL, Abouhamze A, Moldawer LL et al. (1998). Evidence that brief stress may induce the acute phase response in rats. Am J Physiol 273 (6 Pt 2): R1998–R2004.
Diamond DM, Park CR, Campbell AM, Woodson JC (2005). Competitive interactions between endogenous LTD and LTP in the hippocampus underlie the storage of emotional memories and stress-induced amnesia. Hippocampus 15: 1006–1025.
Dodge T, Hoagland MF (2011). The use of anabolic androgenic steroids and polypharmacy: A review of the literature. Drug Alcohol Depend 114: 100–109.
Dong HW, Petrovich GD, Swanson LW (2001). Topography of projections from amygdala to bed nuclei of the stria terminalis. Brain Res Revs 38: 192–246.
Dong HW, Swanson LW (2005). Organization of axonal projections from the anterolateral area of the bed nucleus of the stria terminalis. J Comp Neurol 468: 277–298.
Egli RE, Winder DG (2003). Dopamine enhances fast excitatory synaptic transmission in the extended amygdala by a CRF-R1-dependent process. J Neurophysiol 90: 405–414.
Elliot DL, Cheong JW, Moe EL, Goldberg L (2007). Cross-sectional study of female students reporting anabolic steroid use. Arch Pediatr Adolesc Med 161: 572–577.
Erb S, Salmaso N, Rodaros D, Stewart J (2001). A role for the CRF-containing pathway from central nucleus of the amygdala to bed nucleus of the stria terminalis in the stress-induced reinstatement of cocaine seeking in rats. Psychopharmacology 158: 360–365.
Erb S, Stewart J (1999). A role for the bed nucleus of the stria terminalis, but not the amygdala, in the effects of corticotropin-releasing factor on stress-induced reinstatement of cocaine seeking. J Neurosci 19 (RC35): 106.
Forger NG, Rosen GJ, Waters EM, Jacob D, Simerly RB, de Vries GJ (2004). Deletion of Bax eliminates sex differences in the mouse forebrain. Proc Natl Acad Sci USA 101 (37): 13666–13671.
Franke WW, Berendonk B (1997). Hormonal doping and androgenization of athletes: a secret program of the German Democratic Republic government. Clin Chem 43: 1262–1279.
Franklin KJB, Paxinos G (1997). The Mouse Brain in Stereotaxic Coordinates. Academic Press: San Diego, CA.
Fredj NB, Burrone J (2009). A resting pool of vesicles is responsible for spontaneous vesicle fusion at the synapse. Nat Neurosci 12 (6): 751–758.
Frye CA, Edinger KL, Sumida K (2008a). Androgen administration to aged male mice increases anti-anxiety behavior and enhances cognitive performance. Neuropsychopharmacology 33: 1049–1061.
Frye CA, Koonce CJ, Edinger KL, Osborne DM, Walf AA (2008b). Androgens with activity at estrogen receptor beta have anxiolytic and cognitive-enhancing effects in male rats and mice. Horm Behav 54 (5): 726–734.
Gallagher JP, Orozco-Cabal LF, Liu J, Shinnick-Gallagher P (2008). Synaptic physiology of central CRH system. Eur J Pharmacol 583: 215–225.
Galligani N, Renck A, Hansen S (1996). Personality profile of men using anabolic androgenic steroids. Horm Behav 30 (2): 170–175.
Gray TS, Bingaman EW (1996). The amygdala: corticotropin-releasing factor, steroids, and stress. Crit Rev Neurobiol 10 (2): 155–168.
Grimes JM, Melloni Jr RH (2005). Serotonin-1B receptor activity and expression modulate the aggression-stimulating effects of adolescent anabolic steroid exposure in hamsters. Behav Neurosci 119 (5): 1184–1194.
Hall RC, Chapman MJ (2005). Psychiatric complications of anabolic steroid abuse. Psychosomatics 46 (4): 285–290.
Hammack SE, Guo JD, Hazra R, Dabrowska J, Myers KM, Rainnie DG (2009). The response of neurons in the bed nucleus of the stria terminalis to serotonin: implications for anxiety. Prog Neuropsychopharmacol Biol Pyschiatry 33 (8): 1309–1320.
Hammack SE, Irakli M, Ainnie DG (2007). Differential expression of intrinsic membrane currents in defined cell types of the anterolateral bed nucleus of the stria terminalis. J Neurophysiol 98: 638–656.
Handa RJ, Nunley KM, Lorens SA, Louie JP, McGivern RF, Bollnow MR (1994). Androgen regulation of adrenocorticotropin and corticosterone secretion in the male rat following novelty and foot shock stressors. Physiol Behav 55 (1): 117–124.
Handa RJ, Weiser MJ, Zuloaga DG (2009). A role for the androgen metabolite, 5α-androstane-3β,17β-diol in modulating oestrogen receptor β-mediated regulation of hormonal stress reactivity. Neuroendocrinology 21: 351–358.
Hazra R, Guo JD, Ryan SJ, Jasnow AM, Dabrowska J, Rainnie DG (2011). A transcriptome analysis of type I-III neurons in the bed nucleus of the stria terminalis. Molec Cell Neurosci 46: 699–709.
Henderson LP (2007). Steroid modulation of GABAA receptor-mediated transmission in the hypothalamus: Effects on reproductive function. Neuropharmacology 52: 1439–1453.
Henderson LP, Jorge JC (2004). Steroid modulation of GABAA receptors: CNS roles in reproduction, dysfunction and drug abuse. In: Maue RA (ed). Advances in Molecular and Cell Biology. Molecular Insights into Ion Channel Biology in Health and Disease: Elsevier, vol. 32 pp 217–249.
Herman JP, Prewitt CM, Cullinan WE (1996). Neuronal circuit regulation of the hypothalamo-pituitary-adrenocortical stresss axis. Crit Rev Neurobiol 10: 371–394.
Hoffman GE, Smith MS, Verbalis JG (1993). c-Fos and related immediate early gene products as markers of activity in neuroendocrine systems. Front Neuroendocrinol 14 (3): 173–213.
Holsboer F, Ising M (2008). Central CRH system in depression and anxiety--evidence from clinical studies with CRH1 receptor antagonists. Eur J Pharmacol 583: 350–357.
Howland JG, Wang YT (2008). Synaptic plasticity in learning and memory: stress effects in the hippocampus. Prog Brain Res 169: 145–158.
Hyytiä P, Koob GF (1995). GABAA receptor antagonism in the extended amygdala decreases ethanol self-administration in rats. Eur J Pharmacol 283: 151–159.
Johnston LD, O'Malley PM, Bachman JG, Schulenberg JE (2009). Monitoring the Future National Survey Results on Drug Use, 1975-2008. Volume I: Secondary School Students (NIH Publication No. 09-7402). National Institute on Drug Abuse: Bethesda, MD.
Justice NJ, Yuan ZF, Sawchenko PE, Vale W (2008). Type 1 corticotropin-releasing factor receptor expression reported in BAC transgenic mice: Implications for reconciling ligand-receptor mismatch in the central corticotropin-releasing factor system. J Comp Neurol 511: 479–496.
Kageyama K, Suda T (2009). Regulatory mechanisms underlying corticotropin-releasing factor gene expression in the hypothalamus. Endocrine J 56 (3): 335–344.
Kanayama G, Hudson JI, Pope HG (2008). Long-term psychiatric and medical consequences of anabolic-androgenic steroid abuse: a looming public health concern? Drug Alcohol Depend 98: 1–12.
Kash TL, Winder DG (2006). Neuropeptide Y and corticotropin-releasing factor bi-directionally modulate inhibitory synaptic transmission in the bed nucleus of the stria terminalis. Neuropharmacology 51: 1013–1022.
Kim JJ, Song EY, Kosten TA (2006). Stress effects in the hippocampus: synaptic plasticity and memory. Stress 9 (1): 1–11.
Kirby LG, Freeman-Daniels E, Lemos JC, Nunan JD, Lamy C, Akanwa A et al. (2008). Corticotropin-releasing factor increases GABA synaptic activity and induces inward current in 5-Hydroxytryptamine dorsal raphe neurons. J Neurosci 28 (48): 12927–12937.
Korosi A, Baram TZ (2008). The central corticotropin releasing factor system during development and adulthood. Eur J Pharmacol 583: 204–214.
Kouvelas D, Pourzitaki C, Papazisis G, Dagklis T, Dimou K, Kraus MM (2008). Nandrolone abuse decreases anxiety and impairs memory in rats via central androgenic receptors. Int J Neuropsychopharmacol 11: 925–934.
Krettek JE, Price JL (1978). Amygdaloid projections to subcortical structures within the basal forebrain and brainstem in the rat and cat. J Comp Neurol 178: 225–254.
Lalmansingh AS, Uht RM (2010). Estradiol regulates corticotropin-releasing hormone gene (crh) expression in a rapid and phasic manner that parallels estrogen receptor-α and -β recruitment to a 3′,5′-cyclic adenosine 5′-monophosphate regulatory region of the proximal crh promoter. Endocrinology 149 (1): 346–357.
Lammel S, Ion DI, Roeper J, Malenka RC (2011). Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron 70 (5): 855–862.
Laviola G, Macri S, Morley-Fletcher S, Adriani W (2003). Risk-taking behavior in adolescent mice: psychobiological determinants and early epigenetic influence. Neurosci Biobehav Revs 27: 19–31.
Leriche M, Méndez M, Zimmer L, Bérod A (2008). Acute ethanol induces Fos in GABAergic and non-GABAergic forebrain neurons: a double-labeling study in the medial prefrontal cortex and extended amygdala. Neuroscience 153: 259–267.
Liang KC, Melia KR, Miserendino MJD, Falls WA, Campeau S, Davis M (1992). Corticotropin-releasing factor: long-lasting facilitation of the acoustic startle reflex. J Neurosci 12: 2302–2312.
Lister RG (1987). The use of a plus-maze to measure anxiety in the mouse. Psychopharmacology (Berl) 92 (2): 180–185.
Lunga P, Herbert J (2004). 17β-oestradiol modulates glucocorticoid, neural and behavioural adaptations to repeated restrain stress in female rats. J Neuroendocrinol 16 (9): 776–785.
Maguire J, Mody I (2009). Steroid hormone fluctuations and GABAAR plasticity. Psychoneuroendocrinology 34S: S84–S90.
Martel KL, Baum MJ (2009). Adult testosterone treatment but not surgical disruption of vomeronasal function augments male-typical sexual behavior in female mice. J Neurosci 29 (24): 7658–7666.
Merlo Pich E, Lorang M, Yeganeh M, Rodriguez de Fonesca F, Raber J, Koob GF (1995). Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and ethanol withdrawal as measured by microdialysis. J Neurosci 15 (8): 5439–5447.
Miller KE, Hoffman JH, Barnes GM, Sabo D, Melnick MJ, Farrell MP (2005). Adolescent anabolic steroid use, gender, physical activity, and other problem behaviors*. Subst Use Misuse 40 (11): 1637–1657.
Möhler H (2012). The GABA system in anxiety and depression and its therapeutic potential. Neuropharmacology 62: 42–53.
Nie Z, Schweitzer P, Roberts AJ, Madamba SG, Moore SD, Siggins GR (2004). Ethanol augments GABAergic transmission in the central amygdala via CRF1 receptors. Science B 303 (5663): 1512–1514.
Nie Z, Zorilla EP, Madamba SG, Rice KC, Roberto M, Siggins GR (2009). Presynaptic CRF1 receptors mediate the ethanol enhancement of GABAergic transmission in the mouse central amygdala. Sci World J 9: 68–85.
Oberlander JG, Porter DM, Penatti CA, Henderson LP (2012). Anabolic androgenic steroid abuse: multiple mechanisms of regulation of GABAergic synapses in neuroendocrine control regions of the rodent forebrain. J Neuroendocrinol 24 (1): 202–214.
O'Connor LH, Cicero TJ (1993). Anabolic steroids: misuse or abuse? In: Schulkin, J (ed). Hormonally Induced Changes in Mind and Brain. Academic Press: San Diego, CA, pp 129–156.
Olive MF, Koenig HN, Nannini MA, Hodge CW (2002). Elevated extracellular CRF levels in the bed nucleus of the stria terminalis during ethanol withdrawal and reduction by subsequent ethanol intake. Pharmacol Biochem Behav 72: 213–220.
Pagonis TA, Angelopoulos NV, Koukoulis GN, Hadjichristodoulou CS (2006). Psychiatric side effects induced by supraphysiological does of combinations of anabolic steroids correlate to the severity of abuse. Eur Psychiatry 21: 551–562.
Paulmyer-Lacroix O, Héry M, Pugeat M, Grino M (1996). The modulatory role of estrogens on corticotropin-releasing factor gene expression in the hypothalamic paraventricular nucleus of ovariectomized rats: the role of the adrenal gland. J Neuroendocrinol 8 (7): 515–519.
Pelleymounter MA, Joppa M, Carmouche M, Cullen MJ, Brown B, Murphy B et al. (2000). Role of corticotropin-releasing factor (CRF) receptors in the anorexic syndrome induced by CRF. J Pharmacol Exp Ther 293: 799–806.
Penatti CA, Porter DM, Henderson LP (2009a). Chronic exposure to anabolic androgenic steroids alters neuronal function in the mammalian forebrain via androgen receptor- and estrogen receptor-mediated mechanisms. J Neurosci 29: 12484–12496.
Penatti CAA, Costine BA, Porter DM, Henderson LP (2009b). Effects of chronic exposure to an anabolic androgenic steroid mixture on α5-receptor-mediated GABAergic transmission and neural signaling in the forebrain of female mice. Neuroscience 161: 526–537.
Penatti CAA, Davis MA, Porter DM, Henderson LP (2010). Altered GABAA receptor-mediated synaptic transmission disrupts the firing of gonadotropin-releasing hormone neurons in male mice under conditions of steroid abuse. J Neurosci 30 (19): 6497–6506.
Penatti CAA, Henderson LP (2009). Androgen Actions on Receptors and Channels: Regulation of Electrical Excitability and Synaptic Transmission. In Pfaff DW, Arnold A, Etgen A, Fahrbach S, Rubin R (eds). Hormones, Brain and Behavior, 2nd edn. Elsevier/Academic Press: Amsterdam, pp 1245–1274.
Penatti CAA, Oberlander JG, Davis MC, Porter DM, Henderson LP (2011). Chronic exposure to anabolic androgenic steroids alters activity and synaptic function in neuroendocrine control regions of the female mouse. Neuropharmacology 61 (4): 653–664.
Penatti CAA, Porter DM, Jones BL, Henderson LP (2005). Sex-specific effects of chronic anabolic androgenic steroid treatment on GABAA receptor expression and function in adolescent mice. Neuroscience 135 (2): 533–543.
Peng IF, Berke BA, Zhu Y, Lee WH, Chen M, Wu CF (2007). Temperature-dependent developmental plasticity of Drosophila neurons: cell-autonomous roles of membrane excitability, Ca2+ influx, and cAMP signaling. J Neurosci 27 (46): 12611–12622.
Perry PJ, Lund BC, Deninger MJ, Kutscher EC, Schneider J (2005). Anabolic steroid use in weightlifters and bodybuilders. Clin J Sport Med 15 (5): 326–330.
Petrovich GD, Swanson LW (1997). Projections from the lateral part of the central amygdalar nucleus to the postulated fear conditioning circuit. Brain Res 763 (2): 247–254.
Pinna G, Agis-Balboa RC, Pibiri F, Nelson M, Guidotti A, Costa E (2008). Neurosteroid biosynthesis regulates sexually dimorphic fear and aggressive behavior in mice. Neurochem Res 33: 1990–2007.
Pitkanen A (2000). Connectivity of the rat amygdaloid complex. In Aggleton JP (ed). The Amygdala: A Functional Analysis. 2nd edn. Oxford University Press: Oxford, UK, pp 31–116.
Polston EK, Gu G, Simerly RB (2004). Neurons in the principal nucleus of the bed nuclei of the stria terminalis provide a sexually dimorphic GABAergic input to the anteroventral periventricular nucleus of the hypothalamus. Neuroscience 123 (3): 793–803.
Pope HG, Katz DL (1988). Affective and psychotic symptoms associated with anabolic steroid use. Am J Psychiatry 145: 487–490.
Pope HG, Katz DL (1994). Psychiatric and medical effects of anabolic-androgenic steroid use. A controlled study of 160 athletes. Arch Gen Psychiatry 51 (5): 375–382.
Potter E, Behan DP, Linton EA, Lowry PJ, Sawchenko PE, Vale WW (1992). The central distribution of a corticotropin-releasing factor (CRF)- binding protein predicts multiple sites and modes of interaction with CRF. Proc Natl Acad Sci USA 89: 4192–4196.
Rashid W (2000). Testosterone abuse and affective disorders. J Subst Abuse Treat 18 (2): 179–184.
Rassnick S, Koob GF, Geyer MA (1992). Responding to acoustic startle during chronic ethanol intoxication and withdrawal. Psychopharmacology (Berl) 106 (3): 351–358.
Rebaudo R, Melani R, Balestrino M, Izvarina N (2001). Electrophysiological effects of sustained delivery of CRF and its receptor agonists in hippocampal slices. Brain Res 922 (1): 112–117.
Reddy DS (2010). Neurosteroids: endogenous role in the human brain and therapeutic potentials. Prog Brain Res 186: 113–137.
Resstel LBM, Alves FHF, Reis DG, Crestani CC, Corrêa FMA, Guimarães FS (2008). Anxiolytic-like effects induced by reversible inactivation of the bed nucleus of stria terminalis. Neuroscience 154: 869–876.
Reul JM, Holsboer F (2002). Corticotropin-releasing factor receptors 1 and 2 in anxiety and depression. Curr Opin Pharmacol 2 (1): 23–33.
Ricci LA, Rasakham K, Grimes JM, Melloni Jr RH (2006). Serotonin-1A receptor activity and expression modulate adolescent anabolic/androgenic steroid-induced aggression in hamsters. Pharmacol Biochem Behav 85 (1): 1–11.
Risbrough VB, Hauger RL, Pelleymounter MA, Geyer MA (2003). Role of corticotropin releasing factor (CRF) receptors 1 and 2 in CRF-potentiated acoustic startle in mice. Psychopharmacology 170: 178–187.
Risbrough VB, Hauger RL, Roberts AL, Vale WW, Geyer MA (2004). Corticotropin-releasing factor receptors CRF1 and CRF2 exert both additive and opposing influences on defensive startle behavior. J Neurosci 24: 6545–6552.
Risbrough VB, Geyer MA (2005). Anxiogenic treatments do not increase fear-potentiated startle in mice. Biol Psychiatry 57: 33–43.
Roberto M, Cruz MT, Gilpin NW, Sabino V, Schweitzer P, Bajo M et al. (2010). Corticotropin releasing factor-induced amygdala gamma-aminobutyric Acid release plays a key role in alcohol dependence. Biol Psychiatry 67 (9): 831–839.
Roberto M, Madamba SG, Stouffer DG, Parsons LH, Siggins GR (2004). Increased GABA release in the central amygdala of ethanol-dependent rats. J Neurosci 24 (45): 10159–10166.
Rocha VM, Calil CM, Ferreira R, Moura MJCS, Marcondes FK (2007). Influence of anabolic steroid on anxiety levels in sedentary male rats. Stress 10: 326–331.
Rojas-Ortiz YA, Rundle-González V, Rivera-Ramos I, Jorge JC (2005). Modulation of elevated plus maze behavior after chronic exposure to the anabolic steroid 17alpha-methyltestosterone in adult mice. Horm Behav 49 (1): 123–128.
Sara Y, Virmani T, Deák F, Liu X, Kavalali ET (2005). An isolated pool of vesicles recycles at rest and drives spontaneous neurotransmission. Neuron 45 (4): 563–573.
Sato SM, Schulz KM, Sisk CL, Wood RI (2008). Adolescents and androgens, receptors and rewards. Horm Behav 53: 647–658.
Schmued LC (1994). Diagonal ventral forebrain continuum has overlapping telencephalic inputs and brainstem outputs which may represent loci for limbic/autonomic integration. Brain Res 667: 175–191.
Schulz KM, Zehr JL, Salas-Ramirez KY, Sisk CL (2009). Testosterone programs adult social behavior before and during, but not after, adolescence. Endocrinology 150 (8): 3690–3698.
Scicli AP, Petrovich GD, Swanson LW, Thompson RF (2004). Contextual fear conditioning is associated with lateralized expression of the immediate early gene c-fos in the central and basolateral amygdalar nuclei. Behav Neurosci 118 (1): 5–14.
Schwartzer JJ, Ricci LA, Melloni Jr RH (2009). Adolescent anabolic-androgenic steroid exposure alters lateral anterior hypothalamic serotonin-2A receptors in aggressive male hamsters. Behav Brain Res 199 (2): 257–262.
Sharp FR, Sagar SM, Swanson RA (1993). Metabolic mapping with cellular resolution: c-fos vs. 2-deoxyglucose. Crit Rev Neurobiol 7: 205–228.
Sheffield ME, Best TK, Mensh BD, Kath WL, Spruston N (2011). Slow integration leads to persistent action potential firing in distal axons of coupled interneurons. Nat Neurosci 14 (2): 200–207.
Shekhar A, Truitt W, Rainnie D, Sajdyk T (2005). Role of stress, corticotrophin releasing factor (CRF) and amygdala plasticity in chronic anxiety. Stress 8 (4): 209–219.
Shen H, Gong HQ, Aoki C, Yuan M, Ruderman Y, Dattilo M et al. (2007). Reversal of neurosteroid effects at α4β2δ GABAA receptors triggers anxiety at puberty. Nat Neurosci 10 (4): 469–477.
Skórzewska A, Bidziński A, Hamed A, Lehner M, Turzyńska D, Sobolewska A et al. (2008). The influence of CRF and alpha-helical CRF(9-41) on rat fear responses, c-Fos and CRF expression, and concentration of amino acids in brain structures. Horm Behav 54 (5): 602–612.
Steensland P, Blakely G, Nyberg F, Fahlke C, Pohorecky LA (2005). Anabolic androgenic steroid affects social aggression and fear-related behaviors in male pair-housed rats. Horm Behav 48: 216–224.
Smith SS, Aoki C, Shen H (2009). Puberty, steroids, and GABAA receptor plasticity. Psychoneuroendocrinology 345: S91–S103.
Sommer WH, Rimondini R, Hansson AC, Hipskind PA, Gehlert DR, Barr CS et al. (2008). Upregulation of voluntary alcohol intake, behavioral sensitivity to stress, and amygdala Crhr1 expression following a history of dependence. Biol Psychiatry 63: 139–145.
Su TP, Pagliaro M, Schmidt PJ, Pickar D, Wolkowitz O, Rubinow DR (1993). Neuropsychiatric effects of anabolic steroids in male normal volunteers. JAMA 269: 2760–2764.
Sun N, Cassell MD (1993). Intrinsic GABAergic neurons in the rat central extended amygdala. J Comp Neurol 330: 381–404.
Sun N, Roberts L, Cassell MD (1991). Rat central Amygdaloid Nucleus Projections to the Bed Nucleus of the Stria Terminalis. Brain Res Bull 27: 651–662.
Swanson LW, Sawchenko PE, Rivier J, Vale WW (1983). Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology 36 (3): 165–186.
Swerdlow NR, Britton KT, Koob GF (1989). Potentiation of acoustic startle by corticotropin-releasing factor (CRF) and by fear are both reversed by α-helical CRF (9-41). Neuropsychopharmacology 2: 285–292.
Swerdlow NR, Geyer MA, Vale WW, Koob GF (1986). Corticotropin-releasing factor potentiates acoustic startle in rats: blockade by chlordiazepoxide. Psychopharmacology 88: 147–152.
Thiblin I, Petersson A (2005). Pharmacoepidemiology of anabolic androgenic steroids: a review. Fundam Clin Pharmacol 19 (1): 27–44.
Trenton AJ, Currier GW (2005). Behavioural manifestations of anabolic steroid use. CNS Drugs 19: 571–595.
van Pett K, Viau V, Bittencourt JC, Chan RKW, Li HY, Arias C et al. (2000). Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol 428: 191–212.
Veening JG, Böcker KB, Verdouw PM, Olivier B, De Jongh R, Groenink L (2009). Activation of the septohippocampal system differentiates anxiety from fear in startle paradigms. Neuroscience 163 (4): 1046–1060.
Veinante P, Freund-Mercier M (1998). Intrinsic and extrinsic connections of the rat central extended amygdala: an in vivo electrophysiological study of the central amygdaloid nucleus. Brain Res 794 (2): 188–198.
Veinante P, Stoeckel ME, Freund-Mercier MJ (1997). GABA- and peptide immunoreactivities co-localize in the rat central extended amygdala. Neuroreport 8: 2985–2989.
Walf AA, Frye CA (2007). The use of the elevated plus maze as an assay of anxiety-related behavior in rodents. Nat Protoc 2 (2): 322–328.
Walker D, Yang Y, Ratti E, Corsi M, Trist D, Davis M (2009b). Differential effects of the CRF-R1 antagonist GSK876008 on fear-potentiated, light- and CRF-enhanced startle suggest preferential involvement in sustained vs. phasic threat responses. Neuropsychopharmacology 34: 1533–1542.
Walker DL, Miles LA, Davis M (2009a). Selective participation of the bed nucleus of the stria terminalis and CRF in sustained anxiety-like versus phasic fear-like responses. Prog Neuropsychopharmacol Biol Psychiatry 33: 1291–1308.
Wasser CR, Ertunc M, Liu X, Kavalali ET (2007). Cholesterol-dependent balance between evoked and spontaneous synaptic vesicle recycling. J Physiol 579 (Pt 2): 413–429.
Wasser CR, Kavalali ET (2009). Leaky synapses: regulation of spontaneous neurotransmission in central synapses. Neuroscience 158 (1): 177–188.
Webster EL, Lewis DB, Torpy DJ, Zachman EK, Rice KC, Chrousos GP (1996). In vivo and in vitro characterization of antalarmin, a nonpeptide corticotropin-releasing hormone (CRH) receptor antagonist: suppression of pituitary ACTH release and peripheral inflammation. Endocrinology 137 (12): 5747–5750.
Westphal NJ, Seasholtz AF (2006). CRH-BP: the regulation and function of a phylogenetically conserved binding protein. Front Biosci 11: 1878–1891.
Wong ML, Webster EL, Spokes H, Phu P, Ehrhart-Bornstein M, Bornstein S et al. (1999). Chronic administration of the non-peptide CRH type 1 receptor antagonist antalarmin does not blunt hypothalamic-pituitary-adrenal axis responses to acute immobilization stress. Life Sci 65 (4): PL53–PL58.
Yang P, Jones BL, Henderson LP (2002). Mechanisms of anabolic androgenic steroid modulation of α1β3γ2L GABAA receptors. Neuropharmacology 43 (4): 619–633.
Yang P, Jones BL, Henderson LP (2005). Role of the α subunit in the modulation of GABAA receptors by anabolic androgenic steroids. Neuropharmacology 49 (3): 300–316.
Zender R, Olshansky E (2009). Women's mental health: depression and anxiety. Nurs Clin North Am 44: 355–364.
Zorrilla EP, Valdez GR, Nozulak J, Koob GF, Markou A (2002). Effects of antalarmin, a CRF type 1 receptor antagonist, on anxiety-like behavior and motor activation in the rat. Brain Res 952 (2): 188–199.
Acknowledgements
We thank Dr Robert N. Leaton for critical insights into the analysis for these data, and Ms. Marie Onakomaiya, Ms. Donna Porter, Dr Carlos Penatti, and Dr Allan Gulledge for comments on the manuscript. This work was supported by the NIH (DA022716 to LPH, DA014137 to LPH and training Grant DK07508 for JGO). We have also made use of the University of Virginia Ligand Assay Core Laboratory, which is supported by SCCPIR U54 HD28934 from the NIH, to conduct the hormone assays under a standard fee for service.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare that, except for income received from our primary employer, no financial support or compensation has been received from any individual or corporate entity over the past three years for research or professional services (with the exception of honoraria for invited talks at other universities and for service to the NSF and the NIH for LPH) and there are no personal financial holdings that could be perceived as constituting a potential conflict of interest.
Rights and permissions
About this article
Cite this article
Oberlander, J., Henderson, L. Corticotropin-Releasing Factor Modulation of Forebrain GABAergic Transmission has a Pivotal Role in the Expression of Anabolic Steroid-Induced Anxiety in the Female Mouse. Neuropsychopharmacol 37, 1483–1499 (2012). https://doi.org/10.1038/npp.2011.334
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2011.334
Keywords
This article is cited by
-
The Behavioral and Neurochemical Changes Induced by Boldenone and/or Tramadol in Adult Male Rats
Neurochemical Research (2023)
-
The Role of Anabolic Androgenic Steroids in Disruption of the Physiological Function in Discrete Areas of the Central Nervous System
Molecular Neurobiology (2018)
-
Mad men, women and steroid cocktails: a review of the impact of sex and other factors on anabolic androgenic steroids effects on affective behaviors
Psychopharmacology (2016)
-
The role of PKC signaling in CRF-induced modulation of startle
Psychopharmacology (2013)
