
Abstract
The demonstration that dopamine loss is the key pathological feature of Parkinson's disease (PD), and the subsequent introduction of levodopa have revolutionalized the field of PD therapeutics. This review will discuss the significant progress that has been made in the development of new pharmacological and surgical tools to treat PD motor symptoms since this major breakthrough in the 1960s. However, we will also highlight some of the challenges the field of PD therapeutics has been struggling with during the past decades. The lack of neuroprotective therapies and the limited treatment strategies for the nonmotor symptoms of the disease (ie, cognitive impairments, autonomic dysfunctions, psychiatric disorders, etc.) are among the most pressing issues to be addressed in the years to come. It appears that the combination of early PD nonmotor symptoms with imaging of the nigrostriatal dopaminergic system offers a promising path toward the identification of PD biomarkers, which, once characterized, will set the stage for efficient use of neuroprotective agents that could slow down and alter the course of the disease.
Similar content being viewed by others
INTRODUCTION
Since James Parkinson described the disease that is named after him in 1817 (Parkinson, 1817), there have been tremendous advances in our understanding of the etiology, pathophysiology, and genetics of this disorder, which have led to major breakthroughs in the development of novel and highly effective therapies for Parkinson's disease (PD) (Table 1). The goal of this review is to highlight the impact some of these developments have had during the past 50 years, and discuss the challenges emerging antiparkinsonian therapies may face in the years to come.
The pioneering works of Carlsson and colleagues identified striatal dopamine depletion as the main cause of parkinsonian motor symptoms (Carlsson et al, 1957; Carlsson and Waldeck, 1958; see also Bertler and Rosengren, 1959). Since then, PD treatments have largely focused on ‘correcting’ the dopaminergic deficit, thereby alleviating the cardinal motor symptoms of the disease (ie, bradykinesia, rigidity, rest tremor, and gait disturbances—Box 1). Treatment with the dopamine precursor levodopa, introduced soon after the discovery of nigral dopamine cell loss in PD (Birkmayer and Hornykiewicz, 1961; Barbeau et al, 1962), truly revolutionized the treatment of PD. For the past five decades, levodopa has been the gold-standard therapy for the motor symptoms of the disease. However, the introduction of levodopa also led to new and significant challenges in the clinical management of patients with PD, specifically the development of long-term motor complications, such as involuntary movements (dyskinesias; Cotzias et al, 1969; Godwin-Austen, 1973; Simuni and Hurtig, 2008) (Table 1).
Appreciation of the complications of levodopa therapy led to the use of dopamine receptor agonists early in the course of the disease because they offer antiparkinsonian effects with a lower risk of developing troublesome dyskinesias (Corrodi et al, 1973; Calne et al, 1974a, 1974b). This was later confirmed in several large-scale randomized controlled trials of second-generation agents such as ropinirole and pramipexole (Parkinson Study Group, 2000; Rascol et al, 2000). However, it has become clear that the use of dopamine receptor agonists is not free of motor complications and, most importantly, they often result in more severe nonmotor side effects (psychiatric disorders including psychosis and impulse control disorders, nausea, vomiting, orthostatic hypotension, increased somnolencesleep attacks, fatigue, and ankle edema) than levodopa (for a review, see Nisipeanu and Korczyn, 2008).
Thus, the development of these motor and nonmotor side effects in response to all types of dopamine replacement therapy clearly highlights the fact that symptomatic therapies that rely entirely on ‘normalizing’ dopaminergic transmission may have limited benefits along the course of the disease. The search for additional sites, which could be targeted alone or in combination with dopaminergic drugs, such as adenosine A2A receptors and metabotropic glutamate receptors, will be discussed in this review.
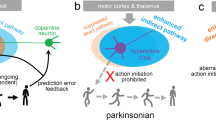
Another major breakthrough that has had a significant impact on the treatment of patients with advanced PD was the introduction of the functional model of the basal ganglia circuitry (Albin et al, 1989; Crossman, 1989; DeLong, 1990), which quickly led to renewed interest in surgical therapies for PD (Bergman et al, 1990; Laitinen et al, 1992; Limousin et al, 1995a, 1995b) (Figure 1). Although surgeons originally opted for use of ablative techniques, targeting the internal pallidal segment (internal globus pallidus (GPi)) or the subthalamic nucleus (STN), the standard surgical treatment most commonly used in some patients with advanced PD is now deep brain stimulation (DBS) of the same targets (Benabid et al, 2009a, 2009b). In spite of obvious therapeutic benefits toward PD motor symptoms, evidence for the significant nonmotor side effects of STN DBS published in recent years (including depression, psychosis, confusion, and impulse control disorders) has raised concerns about the inappropriate choice of patients and misplacement of DBS electrodes within nonmotor regions of the STN. In this review we will critically examine the rationale, significance, therapeutic benefits, and adverse effects of STN DBS over other potential targets.

Schematic diagram of the direct (Dir.) and indirect (Indir.) pathways of the basal ganglia motor circuits in normal and parkinsonian states. Red arrows indicate inhibitory projections, and blue arrows indicate excitatory projections. The changes in the thickness of the arrows in the parkinsonian state indicate the proposed increase (larger arrow) or decrease (thinner arrow) in firing-rate activity of specific connections. The dashed arrows used to label the dopaminergic projection from the SNc to the putamen in parkinsonism indicate partial lesion of that system in this condition. Note that many connections have been purposefully omitted from this diagram. CM, centromedian nucleus; CMA, cingulate motor area; GPe, globus pallidus, external segment; GPi, globus pallidus, internal segment; M1, primary motor cortex; PMC, pre-motor cortex; PPN, pedunculopontine nucleus; SMA, supplementary motor area; SNc, substantia nigra pars compacta; SNr, substantia nigra pars reticulata; STN, subthalamic nucleus; VA/VL, ventral anterior/ventral lateral nucleus (modified from Galvan and Wichmann, 2008).
Transplantation of dopaminergic tissue has long been considered as another potential therapy for PD (Brundin et al, 2010). However, the future of this approach is in doubt due to the lack of effectiveness and the appearance of unexpected side effects in large controlled clinical trials (Freed et al, 2001; Olanow et al, 2003). Similar to the general approach, there is significant interest in the use of stem cells as a treatment for PD. At the time of this writing, the use of these methods in humans remains limited by safety concerns and regulatory issues (Li et al, 2008a), which will be discussed below. Finally, this review will examine the current status of a variety of gene-therapy approaches, using viral vector-mediated enzyme replacement or growth factors delivery in specific brain regions of PD patients (Bjorklund and Kirik, 2009; Bjorklund et al, 2010a, 2010b; Bjorklund and Kordower, 2010; Rangasamy et al, 2010).
All of the therapeutic approaches outlined above aim at treating the dopaminergic motor symptoms of PD, which remain the current focus of therapy development. However, it is clear that PD pathology extends far beyond the dopaminergic nigrostriatal system, and that nonmotor symptoms such as cognitive impairment, dementia, depression, psychosis, and autonomic dysfunction significantly contribute to the complex battery of deficits PD patients face, even at early stages of the disease (Chaudhuri et al, 2006, 2011; Chaudhuri and Schapira, 2009; Kasten et al, 2010; Lim and Lang, 2010; Poewe, 2010; Wood et al, 2010; Bassetti, 2011) (Table 3). Because most of these symptoms do not respond to, but are often exacerbated by, the traditional dopamine replacement therapy and STN DBS, the development of pharmacotherapy that could alleviate these nonmotor symptoms, while being effective in reducing parkinsonian motor signs, represents one of the most important challenges both basic and clinical PD scientists may face in the years to come. In this review, we will present and discuss the results of the double-blind randomized placebo-controlled trials for treatments of some of these nonmotor symptoms (psychosis, depression, dementia), but significant work remains to be done in this field (Table 4).
Another important feature to consider is the fact that current PD therapies discussed in this review do not reduce the rate or extent of dopaminergic cell loss, and thereby do not affect the course of the disease progression. Thus far, attempts to generate a neuroprotective therapy for PD have failed in humans, in spite of the results of promising preclinical animal studies (Siderowf and Stern, 2008; Schapira, 2009a, 2009b). While this reflects in part the inadequacies of the available animal models, progress in this field is also significantly hindered by the lack of reliable biomarkers that would allow early (preclinical) diagnosis of the disease. Neuroprotective treatment trials are also complicated by the absence of endpoints that are biologically meaningful, and not confounded by the symptomatic effects of antiparkinsonian treatments (Schapira and Olanow, 2004; Siderowf and Stern, 2008). However, we will discuss recent evidence obtained in the Parkinson-Associated Risk Study (PARS) indicating that the combination of early nonmotor clinical markers of PD and dopaminergic imaging may provide a sensitive method to diagnose at-risk individuals before the development of motor symptoms and extensive brain degeneration.
This brief review will attempt at summarizing the recent clinical trials and preclinical studies that may impact the therapy of patients with PD in the future. Because of space limitations, we will not provide an extensive account of the previous literature in this field, but rather focus on a few promising research avenues that may have a significant impact in the improvement of PD therapeutics in the years to come. Reflecting the limited availability of specific treatments for the nonmotor features in PD (Chaudhuri et al, 2006; Chaudhuri and Schapira, 2009; Lim and Lang, 2010; Wood et al, 2010), this review will mainly focus on the treatment of the parkinsonian motor symptoms induced by degeneration of midbrain dopaminergic neurons. A summary of the main PD therapeutic approaches that will be discussed in this review is illustrated in Figure 2.

Summary of Parkinson's disease therapeutics discussed in this review. *Indicates agents that have not yet been tested through human double-blind trials.
PD PHARMACOTHERAPEUTICS
Dopamine Replacement Therapy
Pros and cons of levodopa vs dopamine receptor agonists
Dopaminergic drugs have clearly been disease-modifying, bringing about improved daily function, quality of life, and survival (Box 2). Approval of the dopamine precursor LD in 1970 initiated the dopaminergic era (Table 1), followed by the development of numerous dopamine receptor agonists, as well as inhibitors of dopamine-metabolizing enzymes, including monoamine oxidase (MAO) B and (peripheral) catechol-O-methyltransferase (COMT). COMT inhibitors reduce the metabolism of LD to 3-O-methyldopa in the periphery, which extends the duration of the central effects of LD. In the brain, MAO B inhibitors can also enhance the effects of both endogenous and exogenous dopamine. Overall, the currently available agents include three forms of carbidopa/LD (immediate release, controlled release, and orally disintegrating), two orally active dopamine receptor agonists, pramipexole and ropinirole, one injectable dopamine receptor agonist (apomorphine), two MAO-B inhibitors, ie, selegiline (oral, and orally disintegrating) and rasagiline, and two peripheral COMT inhibitors, ie, tolcapone and entacapone. It is beyond the scope of this paper to review these agents in detail, but their characteristics, therapeutic benefits, and limitations have been discussed in previous reviews (Gottwald and Aminoff, 2008; Simuni and Hurtig, 2008; Rajput et al, 2008; Waters, 2008). A list of double-blind randomized placebo-controlled trials for dopamine replacement therapies achieved since 2000 is shown in Table 2.
There has not been a great deal of change regarding dopaminergic agents in the last 5–10 years, but there are a few points worth making about our current view of dopamine replacement therapy. LD remains the most potent symptomatic therapy available. Although the discovery of dopamine and LD is accepted as the most important breakthrough in the field of PD research and therapeutics (Hornykiewicz, 2002a, 2002b), the use of this drug has been controversial for 25 years, because of concern that LD may be toxic to dopaminergic neurons, and that the chronic use of this drug may increase the rate of nigral degeneration (Fahn, 1996a, 1996b, Agid, 1998; Simuni and Hurtig, 2008).
This notion was based on a body of evidence, indicating that decreased glutathione, increased Fe2+, increased malondialdehyde, and decreased mitochondrial complex I activity occurred in the SN of parkinsonian patients, thereby suggesting an important role of free radicals in the pathogenesis and apoptotic death of midbrain dopaminergic neurons in PD (Fahn and Cohen, 1992; Blandini et al, 2003; Olanow et al, 2004). Dopamine, when metabolized by MAO or auto-oxidized, forms H2O2, a precursor to the toxic hydroxyl radical. In PD, after loss of a substantial number of nigral cells, surviving neurons increase their dopamine metabolism, thus, possibly increasing the risk of further free-radical formation and neurodegeneration, especially in an environment where protective mechanisms, such as glutathione activity, are diminished, and iron has accumulated (Ahlskog, 2005; Olanow, 1990). Thus, under such conditions, the rise in cytosolic LD was thought to lead to an increase in dopamine formation, and thereby to a further increase in dopamine metabolism with greater free-radical formation (Ahlskog, 2005; Olanow, 1990). These considerations, combined with the adverse side effects commonly associated with long-term LD treatment (see below), led many physicians to delay the use of this drug in PD patients, and relegate it to ‘second line’ after dopamine receptor agonists (for reviews, see Watts, 1997; Nisipeanu and Korczyn, 2008; Simuni and Hurtig, 2008). However, data from cell culture studies as well as in vivo animal and human studies led to conflicting results regarding the notion that LD is toxic (Agid, 1998; Agid et al, 1999; Simuni and Hurtig, 2008).
The most recent and prominent evidence for this lack of toxicity came from the ELLDOPA (Early vs Later Levodopa Therapy in Parkinson's Disease) trial (Fahn et al, 2004), a multicenter, placebo-controlled, randomized, double-blind clinical trial in which PD patients with a disease duration of <2 years and a Hoehn & Yahr stage <3 were randomized to receive either placebo, or 50, 100, or 200 mg of LD three times daily. Of the 361 enrolled patients, 311 completed the study, which involved 40 weeks of therapy, including a 3-day taper and a 2-week withdrawal period. The primary outcome was the change in severity of PD symptoms between baseline and the end of the study, as measured by a standard clinical rating scale, the United Parkinson's Disease Rating Scale (UPDRS). The clinical results demonstrated that LD improves PD in a dose-dependent manner, beginning at week 9, and that this improvement lasts through the entire treatment period. Interestingly, after the 2-week withdrawal period, subjects receiving the higher dose of LD were less severely impaired than the other three groups. None of the active treatment groups deteriorated to the level of the placebo group after washout. The main conclusion of this trial was that there is no clinical evidence that LD accelerates PD progression, although imaging data showed an accelerated striatal dopaminergic denervation in LD-treated patients (Fahn et al, 2004).
Since then, LD is once again recommended as a therapeutic intervention to be used early in the course of the disease (Obeso and Schapira, 2009; Schapira, 2009a, 2009c; Schapira et al, 2009; Sethi, 2010), a recommendation that is supported by the American Academy of Neurology Practice Guidelines (Miyasaki et al, 2002). Currently, LD, oral dopamine receptor agonists, and MAO B inhibitors are approved by the Federal Drug Administration (FDA) for use in early and advanced PD, while COMT inhibitors and the broad-spectrum injectable dopamine agonist, apomorphine, are approved only for fluctuating PD symptoms. Clinical trials have demonstrated the usefulness of these drugs under their approved conditions (Factor, 2008).
Side effects of dopamine replacement therapies
Despite their obvious benefits for PD motor symptoms, both LD and dopamine receptor agonists produce important side effects, which, in the case of LD, generally appear after several years of chronic use. The development of motor fluctuations, drug-induced dyskinesia, and psychosis represent some of the key challenges faced by physicians treating PD patients with LD following the long-term use of the drug. Because it can affect as many as 50% of patients chronically treated with LD, the etiology and management of LD-induced dyskinesias have been the topic of a tremendous amount of literature since the early 1970s. However, in spite of these efforts, the underlying substrates of LD-induced dyskinesia remain poorly understood (Nutt, 2000; Gunzler and Nutt, 2008).
Although the use of dopamine receptor agonists represents a reasonable approach to reduce the prevalence of dyskinesias following chronic dopamine replacement therapy in PD, these drugs are not completely free of dyskinesias (5–10%) (Parkinson Study Group, 2000; Rascol et al, 2000) and, most importantly, induce disruptive nonmotor side effects to a greater degree than does LD (Antonini et al, 2009). Two main categories of dopamine receptor agonist drugs have been available as PD therapeutics, ie, D3/D2 non-ergot dopamine receptor agonists, such as ropinirole and pramipexole, and D2 ergot derivatives, such as bromocriptine, lisuride, cabergoline, and pergolide, which, in addition to their interactions with D2 family receptors, also affect D1 receptors, as well as specific serotonin and adrenergic receptors. Because of their different chemical nature and pharmacological characteristics, D2 dopamine receptor agonists display distinctive adverse side-effect profiles.
The general pharmacological profiles of dopamine receptor agonists are sufficient to explain many of the differences in the clinical efficacy and side effects between these drugs and LD. In order to mediate their effects, dopamine receptor agonists bypass the degenerating dopaminergic neurons and directly stimulate the intact, although denervated, postsynaptic dopamine receptors in the striatum, and other cortical and subcortical brain regions. In addition, these drugs are not affected by the pharmacokinetic limitation of the short elimination half-life of LD; ie, 1.5 h for LD vs 8 and 6 h for pramipexole and ropinirole, respectively. These particular properties allow for a more prolonged stimulation of dopamine receptors when treated with agonists than with LD. Because dopamine receptor agonists target particular receptor subtypes, while LD impacts all dopamine receptors, agonists may mediate more specific therapeutic benefits, and eliminate certain side effects that could result from the broad-spectrum dopamine receptor activation induced by LD. Dopamine agonists may also provide a wider therapeutic window with a decrease in risk of dyskinesias, perhaps because of their longer half-life. Finally, these drugs may diminish the metabolism of dopamine and therefore decrease the formation of free radicals in the remaining dopaminergic neurons and striatum (Factor, 1999). Some of the most common problems that led to the dropout of dopamine receptor agonist therapy include marked peripheral effects such as nausea and orthostatic hypotension, most likely due to direct dopaminergic modulation of the chemoreceptor trigger zone in the vomiting center of the area postrema, and inhibition of the sympathetic nervous system combined with autonomic dysfunction frequently seen in PD. Other unwanted central side effects likely to be generated by dopamine receptor agonists include increased somnolence, sleep attacks, REM sleep disorder, and a variety of psychiatric symptoms (depression, euphoria, hypomania, hallucinations, delusions, paranoia, psychosis, pathologic gambling/shopping, hypersexuality) (for reviews, see Nisipeanu and Korczyn, 2008; Wood, 2010).
On the other hand, because retroperitoneal, pericardial, and pleuropulmonary fibrosis have been associated with the use of ergot derivatives (Rinne, 1987; Tintner et al, 2005), the use of this specific group of dopamine-related drugs as treatment for PD was ended, and pergolide in particular was removed from the market.
Sustained levels of circulating dopaminergic drugs: a key to reduce motor fluctuations and dyskinesias
Although the underlying substrate of motor fluctuations and dyskinesias remains unknown, there is good evidence that fluctuating levels (high peaks, low valleys) of circulating dopaminergic drugs favor the development of dyskinesias (Obeso et al, 2000; Olanow et al, 2006a, 2006b; Antonini and Odin, 2009). The finding that LD, with its short (1.5 h) half-life and sharp peaks, is associated with a substantially higher frequency of dyskinesia than dopamine agonists, which have longer (6–8 h) half-lives and flatter pharmacokinetic curves, supports this notion. Furthermore, the finding that COMT inhibitors, which increase the half-life of LD, decrease motor fluctuations also suggests that longer-acting agents can treat or prevent these complications. Thus, the development of new delivery technology that provides more sustained levels of the therapeutic dopaminergic agents may be a useful approach to minimize the unwanted side effects (Nutt et al, 2000; Olanow et al, 2006a, 2006b; Olanow, 2008; Odin et al, 2008). We will discuss two different approaches here.
One approach to accomplish this goal is through the continous infusion of a short-acting dopamine receptor agonist. Apomorphine hydrochloride has been used for this purpose. The drug is a non-ergot compound that has full dopamine receptor agonist properties, is one of the oldest of all dopamine receptor agonists, but also the most recently FDA-approved PD therapeutic agent in the United States. Its affinities to D1, D2, D3, and D4 dopamine receptors are similar to those of dopamine (Factor, 1999; Antonini and Tolosa, 2009). It is the only dopamine receptor agonist with antiparkinsonian efficacy similar to LD. Because the oral use of apomorphine is not effective due to extensive hepatic metabolism, other routes of administration must be used. The drug is rapidly absorbed after subcutaneous injection, and has a half-life of 30–60 min. Clinical improvements start within 20 min of administration when the drug appears in the cerebrospinal fluid. Double-blind clinical trials have shown the effectiveness of subcutaneously injected apomorphine in improving PD motor symptoms (Cotzias et al, 1970; Dewey et al, 2001). Despite these benefits, the long-term use of subcutaneous apomorphine infusions in the United Kingdom has been associated with significant technical difficulties and cutaneous adverse effects (Hughes et al, 1993). Because of these issues, infusions of apomorphine have not been very actively pursued in the United States. The development of other infusion methods (such as sublingual, intranasal, rectal, transdermal, or intravenous administration) may make apomorphine treatment a more attractive choice in the treatment of PD (Factor, 2004; LeWitt, 2004). The main autonomic side effects of apomorphine are the same as those described above for other dopamine receptor agonists (ie, nausea, hypotension, and drowsiness/sleep problems), although psychiatric problems appear to be less prevalent with intermittent apomorphine injections than with other agonists perhaps because of the short-term effect (Dewey et al, 2001, Factor, 2004).
Another approach to avoid the effects of pulsatile LD dosing is to deliver the drug in a continuous manner. A recently introduced method has been to deliver LD through direct duodenal infusion. This therapeutic approach requires surgical placement of a percutaneous enteroduodenal tube. Its use has been reserved for the most advanced patients, and particularly those who are not candidates for surgical therapy. Early attempts to use LD in this way were found to be cumbersome and complicated by solubility issues (Nyholm and Aquilonius, 2004). After its approval in Europe, a recently introduced formulation and delivery device that relies on duodenal infusions of LD in a viscous gel (Duodopa) is being studied in a phase 3 trial in the United States (Annic et al, 2009; Westin et al, 2011). The gel medium prevents LD from coming out of the solution and avoids clogging of administration tubes. This form of treatment is safe as monotherapy and appears to provide significant improvement in motor fluctuations over various oral dopamine-related drug combinations (Nilsson et al, 2001). However, the common incidence of adverse effects such as movement of the cannula from the duodenum, irritation, and erosion of the skin around the port warrants further investigations before this approach can be safely used as routine PD treatment. A summary of the key developments about dopamine replacement therapy is shown in Box 2.
Non-Dopaminergic Therapies
Anticholinergic drugs: current cholinergic therapies
Anticholinergic drugs are the oldest therapeutic agents utilized in PD, dating back to the late 1800s and Charcot (Adler, 2008; Box 3). Initially, naturally occurring belladonna alkaloids were used, including atropine and scopolamine, but in the 1950s, synthetic formulations of muscarinic receptor blockers arrived on the scene, including benztropine, trihexyphenidyl, and ethopromazine. In the early 1960s, it was theorized that the dopaminergic deficit would lead to an increase in striatal cholinergic activity, contributing to tremor and other symptoms (Barbeau, 1962; Barbeau et al, 1962; Adler, 2008). There are surprisingly few trials of anticholinergic drugs in PD, and most of them have been carried out over 30 years ago (for a review, see Adler, 2008). In general, the clinical effects were found to be modest and much less robust than LD. The effect is believed to be most notable for rigidity and tremor (Doshay et al, 1956, Koller, 1986). In practice, the use of these drugs is fairly limited. They are associated with a range of adverse effects, including memory loss, confusion, hallucinations, constipation, urinary symptoms, dry mouth, dry eyes, and blurred vision. Because the anticholinergic drugs are poorly tolerated by the elderly, they are most often used in young patients with tremor-predominant disease and dystonia. The latter use is based on the effectiveness of these drugs in idiopathic dystonia (Chuang et al, 2002). In PD patients, these drugs can cause or increase dyskinesia as well. A summary of the key developments about dopamine replacement therapy is shown in Box 2.
Anticholinergic drugs: specific muscarinic cholinergic agents
Acetylcholine mediates its central effects through activation of two main groups of receptors, the ionotropic fast-acting nicotinic receptors and the metabotropic G protein-coupled muscarinic cholinergic receptors (mAChRs) that comprise five subtypes, namely M1–M5. M1, M3, and M5 couple to Gq and activate phospholipase C, whereas M2 and M4 couple to Gi/o and related ion channels and adenylyl cyclase. There is strong evidence that the main autonomic adverse effects of cholinergic drugs are mediated by the activation of peripheral M2 and M3 receptors, while cognitive effects may be related to M1 receptor activation (Anagnostaras et al, 2003; Bymaster et al, 2003a, 2003b, 2003c; Hasselmo, 2006; Fisher, 2008; Langmead et al, 2008; Conn et al, 2009a). Given this evidence, drugs acting specifically at M4 or M5 receptors may have fewer side effects than the current non-specific drugs. Unfortunately, previous efforts to develop selective ligands for individual mAChR subtypes have failed. However, recent studies have led to the discovery and characterization of ligands for individual mAChR subtypes, including M1, M4, and M5 (Conn et al, 2009a; Weaver et al, 2009a; Digby et al, 2010), that display a high specificity profile, at least when tested in vitro. The development of these compounds was achieved by targeting allosteric sites on the mAChRs that are not as highly conserved as the orthosteric acetylcholine binding site. These agents provide new opportunities to determine the physiological roles of individual mAChRs in the basal ganglia circuitry and to assess the antiparkinsonian efficacy of highly selective mAChR antagonists (Conn et al, 2009a, 2009b), but to do so the full specificity profile of these compounds must be characterized in vivo. In this regard, recent in vivo studies indicate that the allosteric modulators display favorable pharmacokinetic properties and blood–brain barrier permeability, and have confirmed their potential therapeutic benefits in rodent models of Alzheimer's disease and schizophrenia (Caccamo et al, 2006; May et al, 2007; Brady et al, 2008; Chan et al, 2008; Shekhar et al, 2008; Conn et al, 2009a, 2009b; Bridges et al, 2010; Digby et al, 2010).
Adenosine A2A receptor antagonists: general overview
A potentially new symptomatic and/or neuroprotective approach in the treatment of PD is the use of adenosine A2A receptor antagonists (Schwarzschild et al, 2006; Menon and Stacy, 2008; Morelli et al, 2010; Shah and Hodgson, 2010). Adenosine is a ubiquitous purine with signaling properties that mediate its effects through four subtypes of G-protein-coupled adenosine receptors: A1, A2A, A2B, and A3 (Schwarzschild et al, 2006; Menon and Stacy, 2008; Morelli et al, 2007, 2009, 2010; Shah and Hodgson, 2010).
A1 and A2A receptors are expressed in the brain, with A2A being primarily localized to the dorsal striatum, nucleus accumbens, olfactory tubercle, and external globus pallidus (GPe) (Schwarzschild et al, 2006). In the striatum, A2A receptors are co-localized with dopamine D2 receptors, the peptide enkephalin, and the metabotropic glutamate receptor 5 (mGluR5), with which they functionally interact in dendrites and spines of medium spiny striatopallidal neurons within the indirect pathway of the basal ganglia (Fuxe et al, 2003, 2007a, 2007b, 2010a, 2010b; Tanganelli et al, 2004; Kachroo et al, 2005; Simola et al, 2008; Agnati et al, 2010; Tozzi et al, 2011) (Figure 1). D2 and A2A receptors have opposite effects on the activity of striatal neurons (Ferré et al, 1997). Because of their preferential localization on the D2 receptor containing striatofugal neurons of the basal ganglia, combined with basic electrophysiological data showing that A2A receptor activation regulates GABAergic transmission at striatopallidal synapses and modulates both glutamatergic and GABAergic transmission in the striatum, these receptors have generated significant interest as potential non-dopaminergic targets for PD therapy (Schwarzschild et al, 2006; Morelli et al, 2007; Simola et al, 2008; Shah and Hodgson, 2010). This interest stems from the idea that blockade of A2A receptors at the striatopallidal GABAergic synapse or at corticostriatal glutamatergic synapses, both known to be overactive in PD, may help to restore the balance between the striatofugal pathways and may thereby alleviate the motor symptoms of PD (DeLong, 1990; Wichmann and DeLong, 2003; Hauser and Schwarzschild, 2005; Schwarzschild et al, 2006) (Figure 1).
While the symptomatic anti-parkinsonian and anti-dyskinetic benefits of the drugs (see below) have generated most interest, the targeting of adenosine transmission as PD therapy is reinforced by epidemiological data suggesting that caffeine (a non-selective adenosine receptor antagonist) may protect nigral dopaminergic neurons against degeneration in PD (Jiménez-Jiménez et al, 1992; Ross et al, 2000; Ascherio et al, 2001; Hernan et al, 2002; Powers et al, 2008; McCulloch et al, 2008).
Adenosine A2A receptor antagonists: clinical trials in PD
So far, three A2A receptor antagonists have been developed for use as antiparkinsonian or anti-dyskinetic agents in PD. The first generation of these compounds was the xanthine istradefylline (previously known as KW-6002). Preclinical data from rat and nonhuman primate models of PD were very promising, showing that this drug can alleviate parkinsonian symptoms and reduce the incidence of LD-induced dyskinesias (Kanda et al, 1998; Grondin et al, 1999; Bibbiani et al, 2003; Lundblad et al, 2003; Jenner et al, 2009). However, four phase III double-blind, randomized, placebo-controlled trials in humans have not met these expectations. In these studies, istradefylline was utilized as add-on therapy to 1500 PD patients with motor fluctuations and dyskinesia. These studies demonstrated a statistically significant, but modest decrease of 0.7–1.2 h ‘off’ time, associated with a similar increase in ‘on’ time (LeWitt et al, 2008; Stacy et al, 2008). The frequency of dyskinesia in the treated group was actually greater than that observed in the group treated with placebo. The improvement in ‘off’ time and ‘on’ time lasted up to 52 weeks (Factor et al, 2010). A negative outcome was recently reported when istradefylline was used as first-line therapy (Fernandez et al, 2010). Together, these outcomes resulted in a ‘nonapprovable’ letter from the FDA.
However, it is important to notice that none of these studies examined the potential of istradefylline to prevent dyskinesia as done in the preclinical primate studies (Grondin et al, 1999), most likely because this would have required a forced decrease of LD, risking the development of severe parkinsonism in advanced patients.
A second-generation drug under examination in humans is the non-xanthine preladenant (previously known as SCH420814). The potency, affinity, and selectivity of this A2A receptor antagonist are higher than those of the other available A2A receptor antagonists (Hodgson et al, 2009). Like istradefylline, this agent has antiparkinsonian and antidyskinetic effects in rat and monkey models of PD (Hodgson et al, 2009, 2010). So far, the results of one double-blind, placebo-controlled phase II trial in PD subjects have been reported with preladenant, but, as with istradefylline, the antiparkinsonian effects were modest (Hauser et al, 2011). However, in contrast to istradefylline, preledenant does not increase dyskinesia, as demonstrated in the preclinical MPTP-treated monkey study (Hodgson et al, 2010). Phase III trials are currently underway for early untreated, as well as advanced PD patients.
Finally, human investigations of the two additional A2A antagonists, SYN 115 and BII014, have just begun. In a small blinded, crossover trial, perfusion magnetic resonance imaging (MRI) demonstrated that SYN 115 reduces cerebral blood flow in the thalamus of PD subjects, consistent with a potential reduction of overinhibition from the indirect pathway (Black et al, 2010). Phase II trials are underway.
Glutamate receptor-related compounds: ionotropic glutamate receptor antagonists
In the CNS, glutamate mediates its effects through two broad categories of receptors, namely the fast-acting ionotropic receptors, or slow modulatory G-protein-coupled metabotropic glutamate receptors (mGluRs). The family of ionotropic receptors comprises three main subtypes: the alpha-amino-3-hydroxy-5-methyl-4-isoxzazole-propionic acid (AMPA), the N-methyl-D-aspartate (NMDA), and the kainate (KA) receptors. These receptors, made up of combinations of different subunits, are coupled with sodium or calcium ion channels, and participate in fast excitatory neurotransmission, while the mGluRs, pooled into three main groups (called groups I, II, and III) based on their sequence homology and pharmacological properties, are coupled with different G proteins and mediate slow modulatory effects on postsynaptic neurons via either pre- or post-synaptic mechanisms (Pin and Duvoisin, 1995; Conn and Pin, 1997; Nakanishi et al, 1998; Anwyl, 1999).
PD is thought to be associated with increased glutamatergic transmission at corticostriatal as well as subthalamofugal synapses in the basal ganglia circuitry, resulting in substantial interest in the use of glutamate receptor blockers as treatment in patients with PD (DeLong, 1990; Blandini and Greenamyre, 1998; Chase et al, 1998; Oh and Chase, 2002; Greene, 2008) (Figure 1). Although various antagonists of ionotropic glutamate receptors have proven to be good antiparkinsonian agents to reduce motor symptoms in preclinical studies, most of these compounds were found to induce debilitating nonmotor side effects in humans that were not assessed in animal studies, most likely due to the fact that AMPA and NMDA receptors are widely distributed and essential for normal brain functioning (Starr, 1995; Blandini and Greenamyre, 1998). Apart from amantadine, a serendipitously discovered NMDA receptor antagonist with good anti-dyskinetic properties (Factor and Molho, 1999), no other glutamate receptor antagonists are clinically used to treat PD. In contrast to amantadine, the non-selective glutamate receptor antagonists memantine, riluzole, and remacemide failed to reduce dyskinesias (Merello et al, 1999; Braz et al, 2004). Infenprodil and related compounds, which belong to a family of polyamine channel blockers with high affinity for specific NMDA receptors that comprise the NR2B subunit, have anti-parkinsonian and anti-dyskinetic effects in rodent and monkey models of PD (Papa and Chase, 1996; Blanchet et al, 1999; Nash et al, 1999, 2000). However, preliminary human studies of these compounds led to negative outcomes (Montastruc et al, 1992). To date, there are no clinical trials of ionotropic glutamate receptor-active agents.
Metabotropic glutamate receptor ligands: MGluR5 antagonists
In general, the group I mGluRs (mGluR1 and mGluR5) are expressed postsynaptically and mediate excitatory effects on their postsynaptic targets, whereas group II (mGluR2/3) and group III (mGluR4,6,7,8) are mainly expressed presynaptically, where they act as inhibitory regulators of glutamatergic or GABAergic transmission (Schoepp and Conn, 1993; Pin and Duvoisin, 1995; Conn and Pin, 1997; Cartmell and Schoepp, 2000; DeBlasi et al, 2001; Galvan et al, 2006; Ferraguti et al, 2008; Niswender and Conn, 2010). Apart from mGluR6, which is confined to the retina, mGluRs are expressed to varying degrees in different brain regions, including the basal ganglia (Conn and Pin, 1997; Conn et al, 2005; Galvan et al, 2006). Most mGluRs are predominantly found in neurons, but mGluR2 and a significant contingent of mGluR5 are also expressed in glial cells. The mGluRs regulate neuronal activity via modulation of ion channels, release of intracellular calcium, and functional interactions with ionotropic glutamate receptors and other G-protein-coupled receptors (including D2 dopamine receptors and A2A adenosine receptors). Because of their modulatory nature, different pharmacological properties, enrichment in specific brain regions, and drug specificity, the mGluRs are considered as highly relevant potential drug targets for the treatment of various brain diseases, including PD (Ossowska et al, 2002; Conn et al, 2005; Swanson et al, 2005; Marino and Conn, 2006; Johnson et al, 2009; Lindsley et al, 2009; Dolen et al, 2010; Duty, 2010; Niswender and Conn, 2010; Spooren et al, 2010; Nicoletti et al, 2011).
All three groups of mGluRs are significantly enriched in the basal ganglia. Based on their distribution, physiological effects on synaptic transmission, and availability of specific drugs, mGluR5 and mGluR4 are currently being investigated as potentially relevant targets for PD therapy. Although group II mGluRs are also widely distributed throughout the basal ganglia circuitry, the modulation of these receptors does not offer significant benefit in animal models of PD (Smith et al, 2000, 2001; Conn et al, 2005; Niswender and Conn, 2010). The mGluR5 is heavily distributed postsynaptically in key basal ganglia nuclei, including the striatum, the globus pallidus, and the STN (Hanson and Smith, 1999; Smith et al, 2000, 2001; Hubert et al, 2001; Paquet and Smith, 2003; Kuwajima et al, 2004; Conn et al, 2005; Kuwajima et al, 2007; Poisik et al, 2007; Niswender and Conn, 2010). In each of these structures, the localization and physiological properties of this receptor have been carefully studied in rodents and nonhuman primates (Smith et al, 2000, 2001; Awad et al, 2000; Marino et al, 2001; Poisik et al, 2003; Conn et al, 2005; Galvan et al, 2006).
mGluR5 activation mediates slow excitatory effects upon basal ganglia nuclei. There is also evidence that the expression and function of mGluR5 vary in acute and chronic models of dopamine denervation (Samadi et al, 2008; Ouattara et al, 2010, 2011). The mGluR5 antagonists, MPEP (2-methyl-6-(phenlethenyl)-pyridine) and MTEP (3-(2-methyl-1,3-thiazol-4-yl(ethynyl)pyridine)), are among the potential antiparkinsonian therapeutic agents that have been tested in rodent and nonhuman primate models of PD. Although the antiparkinsonian effects of acute administration of these drugs are modest, behavioral data suggest that chronic exposure to MPEP may be more effective than acute administration in alleviating parkinsonian motor signs in partially dopamine-depleted rats (Breysse et al, 2002, 2003; Coccurello et al, 2004; Ossowska et al, 2005). In addition, antidyskinetic effects of the mGluR5 antagonist have been seen in both rat and monkey models of PD (Mela et al, 2007; Rylander et al, 2009; DeKundy et al, 2010; Johnston et al, 2010; Gregoire et al, 2011; Marin et al, 2011). Interestingly, mGluR5 and A2A receptor antagonists exert synergistic antiparkinsonian effects in rat models of PD (Coccurello et al, 2004). It is noteworthy that MPEP and MTEP are also neuroprotective toward degeneration of midbrain dopaminergic neurons in mice and monkey models of PD (Flor et al, 2002; Battaglia et al, 2004; Masilamoni et al, 2011). In MPTP-treated nonhuman primates, MTEP also protects locus coeruleus noradrenergic neurons from degeneration (Masilamoni et al, 2011). The translation of these preclinical studies in animal models to patients with PD must be interpreted with caution.
Although MTEP and MPEP are mGluR5 antagonists with high specificity and affinity, their pharmacological profile is not suitable for chronic therapeutic use in humans because of their short half-life and rapid clearance from the brain (Gasparini et al, 2008; Rodriguez et al, 2010). However, the recent development of new negative allosteric modulators of mGluR5 that offer better pharmacokinetic properties for this purpose may provide the necessary tools to further assess the clinical relevance of these drugs in humans (Gasparini et al, 2008; Zhou et al, 2009; Rodriguez et al, 2010; Nicoletti et al, 2011).
Metabotropic glutamate receptor ligands: MGluR4 agonists
Another mGluR that has generated significant interest as a potential antiparkinsonian target is mGluR4. Its strong expression at striatal GABAergic synapses in the GP and cortical glutamatergic synapses in the striatum, combined with electrophysiological in vitro data showing that its activation significantly reduces synaptic transmission at these key synapses of the basal ganglia circuitry (Valenti et al, 2003; Conn et al, 2005; Marino and Conn, 2006; Beurrier et al, 2009), has provided the rationale to examine the antiparkinsonian effects of mGluR4 agonists in rodent models of PD. Because of the lack of group III mGluR-related compounds that could cross the blood–brain barrier, the original evidence that mGluR4 activation would alleviate PD symptoms came from studies using intracerebroventricular or intrapallidal administration of either the broad-spectrum group III mGluR agonist L-AP4 (L-2-amino-4-phosphono-butanoate) or the mGluR4 allosteric potentiator PHCCC (N-phenyl-7-(hydroxylimino)cyclopropa[b]chromen-1a-carboxamide) (Marino et al, 2003; Valenti et al, 2003; Marino and Conn, 2006; Lopez et al, 2007, 2008; Johnson et al, 2009; Nicoletti et al, 2011).
The recent development of a series of allosteric mGluR4 potentiators that display good pharmacokinetic properties, cross the blood–brain barrier, and have significant antiparkinsonian effects in various rodent models of PD provides a promising avenue toward the development of mGluR4-related therapeutic agents in PD (Niswender et al, 2008; Hopkins et al, 2009; Johnson et al, 2009; Williams et al, 2009; Niswender and Conn, 2010; Engers et al, 2011).
Other potential non-dopaminergic PD pharmacotherapeutics
Other agents that are being considered as non-dopaminergic PD therapeutics include modulators of the noradrenergic system. In small open-label studies, the epinephrine precursor L-threo-DOPS was found to have beneficial effects on freezing and gait disturbances in PD patients (Tohgi et al, 1993; Giladi, 2008; Devos et al, 2010). Furthermore, methylphenidate, a mixed dopamine and noradrenaline reuptake inhibitor, was found to show benefit on gait and freezing scores in a randomized controlled trial of 17 advanced PD patients (Devos et al, 2007), but results of a more recent trial in 27 PD patients revealed that methylphenidate did not improve gait and tended to worsen measures of motor functions, sleepiness, and quality of life (Espay et al, 2011). Other trials using lower doses of this compound in PD patients with gait impairment are currently underway. Another category of noradrenaline-related agents being considered as a potential anti-dyskinetic therapy in PD are alpha 2c adrenoreceptor antagonists, such as idazoxan and fipamezole. A recent phase IIa study in 21 PD patients showed a significant beneficial effect of fipamezole on dyskinesia (Bara-Jimenez et al, 2004). A more extensive trial of this compound is underway.
Serotonin and serotonin receptors (5HT1a, 5HT 1b, d, 5HT 2a, c) are widely distributed throughout the basal ganglia, where they modulate GABAergic, glutamatergic, and dopaminergic transmission (Di Matteo et al, 2008). Changes in serotonergic transmission are usually not considered in models of the pathophysiology of PD, but the 5HT1a agonist, sarizotan, was recently found to reduce dyskinesias and extend the duration of action of LD in a recent phase II study in 18 patients with advanced PD (Bara-Jimenez et al, 2005). However, further studies failed to confirm these beneficial effects, most likely due to the additional dopamine D2 receptor antagonist properties of sarizotan (Goetz et al, 2007, 2008). Another, more selective, 5HT1A agonist, piclozotan, has also been developed, and is currently being tested in a multi-center, phase II trial in dyskinetic PD patients. There is also preliminary evidence that non-selective 5HT-2a, c receptor antagonists may have anti-dyskinetic effects (Oh et al, 2002; Durif et al, 2004), although additional randomized trials are needed to confirm these observations (Katzenschlager et al, 2004). A summary of the key recent findings related to non-dopaminergic pharmacotherapies for Parkinson's disease is discussed in Box 3.
THERAPEUTICS FOR NONMOTOR SYMPTOMS OF PD
Nonmotor symptoms are very common in PD and often result in significant disability and decrease in quality of life of these patients, often more so than the motor features (Box 4). The key nonmotor symptoms of PD are listed in Table 3. Unfortunately, most of these symptoms, which in some cases can occur at early stages of the disease, years prior to the appearance of motor deficits, are often poorly recognized by the clinicians and remain inadequately managed. Nonmotor symptoms are classified under four main categories: neuropsychiatric, sleep, autonomic, and sensory. Many of these symptoms, which are unresponsive, exacerbated, or even induced by conventional PD therapy, are very prevalent, affecting the vast majority of patients, most particularly those who survive for many years with the disease (Hely et al, 2005). For these reasons, nonmotor symptoms have become one of the greatest challenges to the current clinical management of PD. There have been a small number of placebo-controlled trials of treatment for specific PD nonmotor symptoms (Table 4), but this field remains largely unexplored and will deserve significant attention in the years to come. Treatments of hallucinations psychosis, depression, and dementia have received the greatest attention in recent years because they occur in such a large proportion of PD patients.
The efficacy and safety of clozapine for psychosis in PD has been confirmed in two randomized, placebo-controlled, double-blind trials, but owing to the small (<1%) but serious risk of developing agranulocytosis and the requirement for white blood cell monitoring, clinicians are hesitant in using this drug as a first-line antipsychotic agent. Quetiapine, which is not associated with this side effect and does not require long-term blood monitoring, is generally utilized first line because of its ease of use. Unfortunately, two recent small randomized controlled trials of quetiapine were negative, but owing to the small number of patients enrolled, additional larger-scale studies would be required to clearly assess the antipsychotic efficacy of quetiapine in PD (Table 4). Despite these trials, quetiapine continues to be used because it does not worsen motor features (this is also true of clozapine) as seen with other atypical antipsychotics such as olanzapine, rispiridone, aripirazole, and others. If quetiapine fails, then clozapine is generally prescribed.
Although variable degrees of depression afflict as many as 40–70% of PD patients, only a small proportion suffers from major depressive disorder (Wood et al, 2010). There is no clear correlation between the severity of motor symptoms and depression, nor is there any obvious relationship with the PD age of onset and family history of depression in these patients (Wood et al, 2010). Large randomized control trials in depressed PD patients are rare. The small and short trials completed thus far, which compared the relative efficacies of tricyclic antidepressants (TCAs) vs selective serotonin reuptake inhibitors (SSRIs), led to conflicting results that are hard to interpret because of the limited power of these studies. The largest published trial to date, which involved 52 patients with PD and depression, showed that that the TCA nortriptyline, known as a non-specific norepinephrine reuptake blocker (SNRI), was more efficacious than the SSRI paroxetine CR in reducing depression in PD (Menza et al, 2009) (Table 4). However, another small trial (N=55 subjects) assessing the antidepressant efficacy of a more selective SNRI, atomoxetine, led to negative results, but showed that this drug was associated with improvement in global cognitive performance and daytime sleepiness (Weintraub et al, 2010). Finally, the most recent and largest trial (N=115 subjects) that compared the antidepressant effects of SSRI (paroxetine) and SNRI (venlafaxine XR) concluded that both drugs significantly improve depression in PD patients (personal communication, Stewart A Factor). Additional larger multi-center trials, using depression-rating scales validated in PD, are needed to fully address this issue, and come up with clear recommendations about the best treatment strategies to treat depression in PD.
Cognitive impairment and dementia are often associated with PD. In contrast to dementia, which usually occurs at late stages of the disease, and causes significant impairment in social and occupational functioning, cognitive decline is often noted much earlier, and usually does not significantly hamper social and occupational activities. This early form is referred to as mild cognitive impairment, a terminology utilized in the Alzheimer literature as well. The reported prevalence of cognitive decline in PD is highly variable, ranging from 10 to 90%, while dementia affects about 30–40% of PD patients, although some studies indicate that dementia is as frequent as nearly 80% (Aarsland et al, 2002; Wood et al, 2010). The risk of dementia significantly increases with age. Cholinergic deficits and cortical Lewy bodies have been associated with the occurrence of PD dementia (PDD), while cognitive impairments are most likely due to the early and progressive degeneration of monoaminergic systems to associative cortical and subcortical regions (Rinne et al, 2000; Emre, 2003). Acetylcholinesterase inhibitors are first-line therapy in patients with PDD. Rivastigmine is the only FDA-approved acetylcholinesterase inhibitor for PDD, but donepezil and galantamine are two other commonly used agents. Although each of these drugs has significant benefits on global functioning (Table 4), they suffer from adverse effects (gastrointestinal distress, worsening of motor symptoms, nausea, vomiting). They have also been shown to improve psychotic symptoms associated with dementia. The outcomes of double-blind randomized trials that assess the efficacy of various acetylcholinesterase inhibitors in PDD are presented in Table 4.
To our knowledge, there are no randomized double-blind controlled trials that have assessed the efficacy of drug treatments for other nonmotor symptoms of PD, except for the recent assessment of the efficacy of amantadine toward pathological gambling (Thomas et al, 2010). There is little literature on the possible role of dopamine pathophysiology in anxiety and apathy, but no significant trials have yet been achieved to assess treatment for these disorders (Chaudhuri and Schapira, 2009). The management of autonomic and sleep disorders is merely symptomatic (Wood et al, 2010). Because of the morbidity and significant impact of nonmotor manifestations on PD patients’ quality of life, it is imperative that better diagnostic and therapeutic tools are developed to recognize and alter the course of these symptoms. A summary of the recent findings related to the therapeutic of nonmotor symptoms of PD is presented in Box 4.
SURGICAL THERAPIES FOR PD
Current Surgical Targets
In the 1950s and 60s, stereotactic ablative approaches targeting the GPi and the ventrolateral thalamus (VL) were commonly used to treat PD patients (Box 5). These procedures were abandoned in the late 1960s with the introduction of LD as an effective pharmacological treatment of PD. Over the past two decades, however, based on a better understanding of the pathophysiological basis of PD (Albin et al, 1989; Crossman, 1989; Bergman et al, 1990; DeLong, 1990; Galvan and Wichmann, 2008) (Figure 1), studies of the effects of ablation in animal models of PD, and the introduction of DBS, there has been a virtual renaissance of neurosurgical treatments of intractable and advanced PD.
In these procedures, nodes of the basal ganglia–thalamocortical motor circuit are targeted, specifically the STN and GPi (Figure 1). Based on a long historical record, ablative procedures at these locations became very popular in the 1990s, but have now been largely abandoned in developed countries in favor of DBS. DBS involves implantation of electrodes into STN or GPi, guided by imaging and electrophysiological techniques. The patients are also implanted with an externally programmable stimulator that is connected to the electrodes. The system can then be used to deliver continuous high-frequency electrical stimulation (most commonly in the 100–150-Hz range) to the implanted brain areas. Implantation of DBS electrodes is associated with a small surgical risk, which includes complications such as intracerebral hemorrhages, infection, or stroke. A list of the main DBS trials that have been performed since 2000 appears in Table 5.
The most common indications for surgery in PD are the presence of intractable tremor and drug-induced motor fluctuations or dyskinesias. Candidates for DBS treatment in PD should have documented LD responsiveness and should be free of significant dementia, psychiatric co-morbidities (Chang and Chou, 2006; Bronstein et al, 2011), and signs of atypical parkinsonism. DBS of the STN is most often performed bilaterally, although unilateral DBS of STN or GPi can be highly effective in some cases of asymmetric parkinsonism. In most patients, DBS alleviates parkinsonian motor signs, shortens ‘off’ periods, and reduces drug-induced dyskinesias, dystonia, and motor fluctuations (Rodriguez-Oroz et al, 2004; Anderson et al, 2005; Weaver et al, 2005; Portman et al, 2006; Bronstein et al, 2011). In general, both GPi- and STN-DBS are more effective than medical management alone to alleviate motor deficits in patients with advanced PD (Just and Ostergaard, 2002; Martinez-Martin et al, 2002; Troster et al, 2003; Lezcano et al, 2004; Diamond and Jankovic, 2005; Erola et al, 2005; Halbig et al, 2005; Lyons and Pahwa, 2005; Deuschl et al, 2006; Rodrigues et al, 2007a, 2007b; Montel and Bungener, 2009; Weaver et al, 2009b; Zahodne et al, 2009). In contrast to patients with GPi-DBS, those with STN-DBS are often able to substantially reduce the medication doses (Breit et al, 2004; Rodriguez-Oroz et al, 2004; Anderson et al, 2005; Erola et al, 2005; Follett et al, 2010; Moro et al, 2010).
Non-Motor Side Effects of STN and GPi-DBS
Both STN- and GPi-DBS can have non-motor side effects, especially affecting verbal fluency, cognition and mood (Kumar et al, 1999a, 1999b; Dujardin et al, 2001; Schupbach et al, 2005; Smeding et al, 2005, 2006, 2011; Castelli et al, 2006; De Gaspari et al, 2006, Merello et al, 2008). Verbal fluency and cognition problems are more often seen in old patients (Hariz et al, 2000; Saint-Cyr et al, 2000; Funkiewiez et al, 2004; Smeding et al, 2011), or in those with poor cognition or depression at baseline (De Gaspari et al, 2006). Specific cognition deficits include impairments of working memory (Saint-Cyr et al, 2000; Higginson et al, 2009; Okun et al, 2009), cognitive processing, visuo-spatial skills and set-shifting (Saint-Cyr et al, 2000; Alegret et al, 2001), response inhibition (Witt et al, 2004), or the decoding of facial expressions (Dujardin et al, 2004; Schroeder et al, 2004; Biseul et al, 2005; Drapier et al, 2008). Even when present, the impact of changes in verbal fluency on the quality of life appears to be relatively small (Alegret et al, 2004; Morrison et al, 2004; Montel and Bungener, 2009; Zahodne et al, 2009). Although almost half patients with DBS experience variable degrees of cognitive changes (Higginson et al, 2009), these deficits become ‘relevant’ in less than 10% treated patients (Castelli et al, 2006; Tir et al, 2007). However, because DBS is now widely used, even for patients with early PD in some centers, these adverse effects remain a major concern.
There is evidence that DBS may also worsen depression and mania, increase apathy, affect emotional lability, and increase the risk of suicide (Berney et al, 2002; Doshi et al, 2002; Okun et al, 2003, 2009; Funkiewiez et al, 2004; Smeding et al, 2005, 2006; Drapier et al, 2008; Voon et al, 2008). Mood problems are more common in patients treated with STN-DBS than those treated with GPi-DBS (Rodriguez-Oroz et al, 2005; Follett et al, 2010; Moro et al, 2010; Bronstein et al, 2011). Although the underlying substrate of these side effects remains to be characterized, it has been suggested that they may be induced by stimulation in non-motor areas of STN or GPi, inadvertent involvement of limbic structures outside of the target regions (Bejjani et al, 1999; Krack and Vercueil, 2001; Kulisevsky et al, 2002; Romito et al, 2002; Herzog et al, 2003a; Okun et al, 2003; Stefurak et al, 2003), and pre-existing psychiatric conditions (Lilleeng and Dietrichs, 2008). Although significant unpleasant mood side effects following STN or GPi DBS are relatively rare (Funkiewiez et al, 2004; Castelli et al, 2006; Tir et al, 2007), their occurrence significantly disrupts patients’ quality of life.
STN vs GPi as Targets for DBS in PD?
Most neurosurgeons currently prefer the STN over the GPi as a target for DBS in PD, because of the perceived greater antiparkinsonian benefit of STN-DBS (Anderson et al, 2005; Moro et al, 2010). However, there is no clear evidence that the STN is, in fact, a better target than GPi. Small clinical studies and a recent large randomized controlled trial have compared GPi- and STN-DBS, and found no significant differences in terms of the motor outcome of the procedures (Table 5). In fact, these studies have documented that patients with GPi-DBS decline less in terms of visuomotor processing speed, and have better depression scores than patients with STN-DBS (Follett et al, 2010). Another recent report concluded that, although patients experience improvement in health-related quality of life measures following STN-DBS, scale items concerning general life issues (eg, occupational function, interpersonal relationships or leisure activities) do not improve (Ferrara et al, 2010). Given these findings, the pendulum may swing towards the pallidal target in a greater proportion of patients in the future, based on the slightly lesser incidence of side effects rather than differences in efficacy.
New Targets for DBS in PD
The effects of DBS in brain areas other than STN or GPi are much less studied in PD patients. For the treatment of patients with severe tremor-predominant PD, thalamic DBS of the ventral intermediate nucleus (Vim) can be considered (Obeso et al, 1997; Ondo et al, 1998), but is not routinely used because it does not have much effect on the other cardinal symptoms of PD, such as bradykinesia and rigidity, which may subsequently develop. Recently, DBS of the centre median/parafascicular (CM/Pf) complex has been used in a small number of PD patients (Benabid, 2009). Although preliminary, some reports have suggested that CM/Pf stimulation may have therapeutic benefits in some PD patients towards dyskinesia (Caparros-Lefebvre et al, 1999), freezing of gait (Stefani et al, 2009), and rest tremor (Krauss et al, 2002; Peppe et al, 2008; Stefani et al, 2009). However, the mechanism of action, specific target sites and stimulation parameters of this surgical approach must be further characterized. Recent studies indicate widespread basal ganglia cellular responses associated with efficient anti-parkinsonian effects of Pf DBS in rat models of PD (Goff et al, 2009; Jouve et al, 2010).
Another DBS target that has recently generated some interest is the pedunculopontine nucleus (PPN), a conglomerate of cholinergic, glutamatergic and GABAergic neurons in the upper brainstem, tightly linked with the basal ganglia, thalamus and lower tegmental regions (Mena-Segovia et al, 2004). Inactivation of the PPN induces akinesia, while electrical low-frequency stimulation has antiparkinsonian properties in animals (Kojima et al, 1997; Nandi et al, 2002a, 2002b). In light of these data, small experimental trials of low frequency PPN stimulation in humans were carried out. These studies have lead to variable results (Plaha and Gill, 2005; Stefani et al, 2007, 2010; Mazzone et al, 2007, 2008; Pereira et al, 2008; Pierantozzi et al, 2008; Alessandro et al, 2010; Peppe et al, 2010; Rauch et al, 2010). In some patients, PPN DBS reduces gait and freezing problems unresponsive to drug and conventional stimulation approaches directed at subthalamic and pallidal targets, reduces falls and occasionally improves patients’ state of vigilance and quality of sleep (Pereira et al, 2008; Ferraye et al, 2010). The magnitude of the effects, the exact location of the stimulation electrode and best technique of implantation of DBS leads into this nucleus are still being debated (Aravamuthan et al, 2007, 2008, 2009; Weinberger et al, 2008; Zrinzo and Zrinzo, 2008; Zrinzo et al, 2008; Fu et al, 2009; Shimamoto et al, 2010).
There have also been several small experimental trials of extradural motor cortex stimulation in human patients (Benvenuti et al, 2006; Strafella et al, 2007; Cilia et al, 2007, 2008; Arle et al, 2008; Lefaucheur, 2009; Pagni et al, 2008). This procedure results in small improvements of motor performance, and reductions in dyskinesias and psychiatric symptoms, perhaps mostly because of reductions in medication requirements. In contrast to DBS, extradural cortical stimulation is easy to perform and could be an alternative surgical therapy for PD. However, the development of new subdural leads and further efficacy tests of this approach in larger cohorts of PD patients are needed before one could consider cortical stimulation as a potential PD therapy (Lefaucheur, 2009). The notion that cortical stimulation may be an effective strategy to treat PD has recently been further strengthened by animal experiments that suggested that some of the antiparkinsonian effects of STN-DBS may, in fact, be mediated by antidromic stimulation of the motor cortex (Li et al, 2007; Dejean et al, 2009; Gradinaru et al, 2009).
Stimulation of the caudal ZI may provide beneficial effects on parkinsonian tremor, and lesser benefits for rigidity and akinesia (Kitagawa et al, 2005; Plaha et al, 2006), most likely through its effects on ZI itself, but also through stimulation of ascending cerebellar projections and pallidofugal fibers to the thalamus that pass through the stimulated areas.
Opportunities for Further Development of Neurosurgical Therapies for PD
The impact of the development of DBS for PD has been likened to that of the introduction of LD in the 1960s. However, while it is true that DBS can be highly effective for the cardinal motor features of PD, tremor, rigidity, and bradykinesia, it does not affect the non-motor features of the disorder or the non-dopaminergic motor features, such as speech, swallowing, gait, and balance difficulties. The search for new surgical targets that may allow treatment of other symptoms is therefore a high priority. Other areas of development involve the use of smaller, rechargeable stimulators, feedback-controlled on-demand stimulation, and the development of stimulation electrodes that allow ‘sculpting’ of the electrical field, which may then help to reduce stimulation side effects further. Long-over-due changes in stimulator design should also allow the use of more flexible stimulation regimes. An example for this would be the use of short episodes of ‘desynchronizing’ stimulation, which may result in therapeutic effects that outlast the stimulation for long period of time, thus saving battery energy, and potentially also reducing the incidence of side effects of DBS (Hauptmann and Tass, 2010). Another technical development in DBS would be a better integration of imaging techniques, such as intraoperative MRI, which offers the potential of solely image-guided placement of DBS electrodes and lesioning without the use of intraoperative microelectrode recordings (Martin et al, 2005; Shahlaie et al, 2011).
DBS vs Ablative Surgeries for PD
A specific challenge for neurosurgical treatments is that, as for any treatment for lifelong progressive diseases, they must be affordable in order to be practical for large numbers of patients. In the United States and other developed countries, DBS procedures have been embraced by both physicians and the public despite their high up-front costs, the substantial costs arising from the need to test and adjust stimulation parameters in the postoperative period, replacement of batteries, and the need for a high level of medical expertise throughout the pre-, peri-, and postoperative phases of the treatment. Similar conditions do not exist in developing countries, amounting to a continued need to optimize and develop less-costly alternatives, including ablative procedures. An important advancement in this field has been the demonstration that STN lesions are effective in the treatment of PD (Alvarez et al, 2001, 2005, 2009; Parkin et al, 2001; Su et al, 2002; Patel et al, 2003). A significant complication is, however, the development of persistent hemi-chorea in a small percentage of patients, requiring subsequent pallidotomy. The need for future long-term studies that explore the clinical effectiveness of refined and more selective methods of subthalamotomy, and the development of surgical techniques to reduce the occurrence of dyskinesias, is warranted. A summary of the main findings related surgical therapies for PD is presented in Box 5.
NEURAL TRANSPLANTATION IN PD
Ventral Midbrain Neural Transplants in the Striatum
Neural transplantation has been considered as a potential therapy for PD for the past 30 years (Box 6). However, despite highly promising results from preclinical studies and open-label trials of the effects of grafted embryonic human dopaminergic neurons into the striatum in the 1990s (Brundin et al, 2010), the more recent results from double-blind placebo-controlled trials of fetal ventral midbrain intrastriatal transplantation have failed to meet the primary endpoints and raised significant concerns about the use and safety of this approach (Freed et al, 2001; Olanow et al, 2003). The first of these trials involved 40 patients with advanced PD who were randomly assigned to receive either a transplant of fetal ventral midbrain tissue or sham surgery (Freed et al, 2001). Thirty-three patients received the transplant. The primary outcome, ie, global rating of clinical improvement 1 year post-transplant, did not reveal any significant difference between the two groups. In addition to this disappointing result, these trials described for the first time the development of graft-induced, medication-independent dyskinesias. A second trial involving 34 patients with advanced PD showed similar results (Olanow et al, 2003).
Another important set of data that further complicated the use of ventral midbrain transplant in PD was the recent demonstration that some of the transplanted cells in PD patients who died 10–16 years after having received their transplants contained alpha-synuclein aggregates, suggesting that grafted neurons may be affected by the neurodegenerative process in the host's brain (Kordower et al, 2008a, 2008b; Li et al, 2008b, 2010). However, controversy remains about the extent and functional relevance of Lewy body-like structures in surviving grafted dopaminergic neurons (Isacson and Kordower, 2008; Mendez et al, 2008; Cooper et al, 2009; Isacson and Mendez, 2010).
In recent years, Bjorklund and colleagues showed in rodent studies that graft-induced dyskinesias may result from the inclusion of serotonergic neurons into the transplants that lead to dysregulated release of dopamine as a ‘false transmitter’ from serotonergic terminals (Lane et al, 2006, 2010). These observations are further supported by the fact that systemic administration of buspirone, an agonist of the inhibitory 5HT1A autoreceptors that dampens serotonin neuron activity, alleviates graft-induced dyskinesias in PD patients and rat models of PD (Lane et al, 2006, 2010; Politis et al, 2010). The efficacy of such an approach to prevent the development of graft-induced dyskinesias in PD remains to be determined.
Stem Cell-derived Neurons for PD Therapy?
In the past decade, PD has often been mentioned as one of the neurodegenerative disorders that could benefit from stem cell-derived transplant therapy. Various stem cell types are currently being considered as sources of dopamine neurons, including lineage-specific stem cells, pluripotent stem cells, and re-programmed somatic cells (Astradsson et al, 2008; Isacson and Kordower, 2008; Sanchez-Pernaute et al, 2008; Pruszak and Isacson, 2009; Hargus et al, 2010). However, the transplantation of proliferative populations of neurons into the brain suffers from major safety concerns, most importantly the possibility that stem cell transplants may give rise to unchecked proliferation of tissue, eventually resulting in the formation of brain tumors (Li et al, 2008a; Amariglio et al, 2009; Brundin et al, 2010). Despite the severe safety and regulatory issues that will have to be overcome before PD patients can be treated with stem cell therapy, the increased knowledge gained in human embryonic stem cell biology is likely to result in the development of new stem cell-based therapies for PD (Isacson, 2009; Allan et al, 2010; Arenas, 2010; Fricker-Gates and Gates, 2010; Xu et al, 2010; Kim, 2011). A summary of the key findings about neural transplantation in PD presented in Box 6.
NEURORESTORATIVE GROWTH FACTOR THERAPIES FOR PARKINSON’S DISEASE
Trophic factors are members of a class of proteins that promote development, growth, survival, and restoration of neurons in the CNS (Box 7). Because of these properties, they are often seen as key therapeutic tools for neurorestorative therapies in the CNS. The first of these trophic factors was nerve growth factor (NGF), with many others following in recent decades (Levi-Montalcini and Hamburger, 1951; Rangasamy et al, 2010; Aron and Klein, 2011). It is now well known that these factors display a wide range of structural, biochemical, pharmacological and biological properties in the CNS.
Growth factors are grouped into two main families: the neurotrophin family, which includes NGF, brain-derived neurotrophic factor (BDNF), neurotrophin-3, and neurotrophin-4/5, and the glial cell-derived neurotrophic factor (GDNF) family, which comprises four main trophic factors, GDNF, neurturin (NTN), artemin, and persephin (Rangasamy et al, 2010; Aron and Klein, 2011).
Neurotrophic factor therapy is an approach that has generated significant interest in the field of PD therapeutics for almost 20 years. GDNF has been most particularly scrutinized because of its powerful effects toward growth, survival, and protection of midbrain dopaminergic neurons against toxic insults (Lin et al, 1993; Tomac et al, 1995a, 1995b; Bjorklund et al, 2000; Kordower et al, 2000). In light of promising preclinical data, the first human double-blinded trial with GDNF protein administration into the lateral ventricles was launched in 1996 (Nutt et al, 2003). Fifty patients suffering from advanced idiopathic PD were enrolled. The results were disappointing. In addition to a lack of significant improvement of parkinsonian motor signs, many patients suffered from side effects, including nausea, anorexia, vomiting, and in some cases, depression, for several days after GDNF administration. The mechanisms underlying these negative effects remained unclear, but it is possible that the poor penetration of GDNF from the lateral ventricles into the striatum may have contributed. To examine this possibility, another double-blind placebo-controlled trial using intraputamenal convection-enhanced infusion of GDNF or placebo was undertaken in 34 subjects, half of whom received GDNF (Lang et al, 2006). Unfortunately, the outcome of this trial corroborated the disappointing findings of the first trial (Lang et al, 2006; Penn et al, 2006).
Most recently, interest has shifted towards NTN, which displays pharmacological features closely similar to GDNF, and has powerful effects on midbrain dopaminergic cell growth (Kotzbauer et al, 1996). Following the development of a recombinant adeno-associated viral vector (AAV)2-based vector encoding the human NTN (known as CERE-120), and its successful application in rodent and nonhuman primate models of PD (Kordower et al, 2006; Gasmi et al, 2007a, 2007b; Herzog et al, 2007, 2008) combined with the promising results of human open trial studies (Marks et al, 2008), a phase II double-blinded trial was initiated in 59 parkinsonian patients, two-thirds of whom received CERE-120 injections into their putamen (Marks et al, 2010). As in the GDNF trials, the primary endpoint was not met. Positron emission tomography (PET) 18F-DOPA scans, examining the dopamine metabolism in the striatum, also showed no difference. However, an 18-month analysis in 30 of the subjects demonstrated a significant difference between groups in the UPDRS scores. Postmortem pathology of two patients who died during or after the study, unrelated to the treatment, indicated that NTN expression was present in the striatum (15% of total putamen), but was minimal in the SNc, as opposed to that seen in preclinical non-human primate studies (Kordower et al, 2006; Herzog et al, 2007; Marks et al, 2010).
It is unclear why such disparity exists between the results of these studies, but some authors have suggested that it may be related to the lack of efficient retrograde transport of the gene and protein from the striatum to the cell bodies in the SNc (Bartus et al, 2011). This could relate to species-specific differences in protein transport, but most likely to the nature and severity of dopaminergic cell loss between the MPTP-treated non-human primate models used in preclinical studies and the advanced PD patients who were enrolled in the trial (Kordower et al, 2006; Marks et al, 2010; Bartus et al, 2011). In light of these disappointing data, it was suggested that direct nigral injections may more effectively deliver NTN to its primary location of action in PD (Bartus et al, 2011). A second phase II trial is currently underway, in which a larger dose of NTN is directly injected into the SNc, and in which patients are observed for longer periods of time (up to 15 months). A six-patient open-label safety trial was completed, which did not identify significant adverse effects. The double-blind, sham-controlled study has now been initiated with a target of 52 subjects.
BDNF and mesencephalic astrocyte-derived neurotrophic factor (MANF) are two other targets of interest for the possible development of growth factor-based therapies for PD patients (Rangasamy et al, 2010; Aron and Klein, 2011). A summary of the key findings related to neurorestorative therapies in PD is shown in Box 7.
GENE THERAPY FOR PD
Gene therapy using viral vector-mediated enzyme replacement is another area of interest for future promising developments in the field of PD therapeutics (Baekelandt et al, 2000; Bjorklund et al, 2000; Feng and Maguire-Zeiss, 2010; Box 8). The basic principle behind this approach is to render neurons capable of producing another neurotransmitter and displaying a different chemical phenotype. Studies have aimed at two main anatomical targets, the STN, in which an AAV vector has been used to deliver the enzyme glutamic acid decarboxylase (GAD) with the hope of transforming some glutamatergic STN neurons into GABA-releasing cells (Kaplitt et al, 2007; Lewitt et al, 2011), and the striatum, in which AAV or lentiviral vectors are used to generate striatal neurons that produce dopamine either by themselves or from dietary tyrosine or LD administered as a drug (Eberling et al, 2008; Christine et al, 2009; Jarraya et al, 2009). Ongoing trials are in progress to assess the efficacy of these approaches in advanced PD patients.
Viral Vector-Mediated GAD in STN Neurons
The development of the AAV2-GAD vector and its implantation into the STN cells was meant to be a non-dopaminergic alternative to STN DBS. The rationale behind this approach is the possibility of converting the chemical phenotype of STN neurons into GABAergic cells that would release GABA, instead of, or in addition to, glutamate, into the GPi, thereby reducing the presumed pathological over-excitation of basal ganglia output neurons (During et al, 2001; Luo et al, 2002). Based on pre-clinical rodent data (During et al, 2001), and the results of a positive open phase I trial in 12 patients with moderately advanced PD who were unilaterally infused (Kaplitt et al, 2007), a phase II double-blind, randomized, sham-controlled trial was recently completed at seven US centers (Lewitt et al, 2011), enrolling 45 patients diagnosed with PD for at least 5 years. The subjects were randomized 1:1 for bilateral STN AAV2-GAD infusion or sham surgery (a partial thickness burr hole). Assessments were carried out at baseline, and at 1, 3, and 6 months post surgery. The primary endpoint was a change in UPDRS motor scores from baseline to 6 months. Of the 45 patients, 8 were excluded on the basis of infusion catheter placement outside of the predetermined target zone or infusion pump failure (5 in the active group and 3 in the sham group). This study resulted in a significant, though modest, change in the primary endpoint, consisting of a 23.1% change in UPDRS motor scores in the AAV2-GAD group compared with 12.7% in the sham group, at all postoperative time points (ie, 1, 3, 6 months). The main adverse events, more commonly found in the active group, were nausea, headache, and depression. While the benefits demonstrated by this study are relatively small compared with those of conventional drug or surgical therapies, this is, nevertheless, the first phase II controlled, blinded gene therapy trial to be positive. A larger phase III study is currently being planned.
Viral Vector-Mediated Striatal Dopamine Replacement
Approaches using transfection of dopamine-synthesizing enzymes and related factors into striatal neurons in order to convert them into dopamine-producing cells have also generated significant interest. In light of findings from various in vitro preparations and rat models of PD (Azzouz et al, 2002; Bankiewicz et al, 2006a, 2006b; Forsayeth et al, 2006; Jarraya et al, 2009), two gene therapy approaches to render striatal neurons as sources of dopamine or DOPA are currently being explored in clinical trials. In the first trial, the utility of a triple enzyme transfer is examined. Striatal neurons are transfected with a lentiviral vector of non-human origin, the equine infectious anemia virus, that delivers three genes essential in dopamine biosynthesis, namely tyrosine hydroxylase (TH), aromatic acid decarboxylase (AADC), and guanosine 5′-triphosphate (GTP) cyclohydrolase 1 (GCH1). After encouraging results were obtained in rat and monkey models of PD with intrastriatal administration of this vector (Azzouz et al, 2002; Jarraya et al, 2009), a phase I/II clinical trial was launched in 2007.
A second trial currently in progress uses striatal AAV-based delivery of AADC, thereby making striatal neurons capable of producing dopamine from peripherally administered LD. This approach is based on the fact that there is a significant reduction in striatal AADC in patients with advanced PD (Lloyd and Hornykiewicz, 1970; Nagatsu et al, 1979), and that the intrastriatal conversion of LD into dopamine should, therefore, be enhanced if additional AADC is expressed, which may then eventually result in reduced need for LD. In this case, active neurotransmitter production is expected only after the patients take LD, so that the magnitude of the functional effects could be adjusted by regulating the oral LD dose (Bjorklund et al, 2010b). Testing in MPTP-treated monkeys (Bankiewicz et al, 2006a, 2006b; Forsayeth et al, 2006) showed stable long-term expression of the vector for up to 6 years, which was accompanied by improvement in clinical rating scores and a reduction of LD-associated side effects (Bankiewicz et al, 2006a, 2006b; Forsayeth et al, 2006). A phase I clinical trial, started in 2005 in five patients who received bilateral intrastriatal vector administration, showed only modest efficacy in ‘off’ state 6 months post transfection. In a second cohort of patients, who received a higher concentration of vector, there was also no significant improvement in the UPDRS rating scale despite increased 18F-fluoro-l-m-tyrosine binding in PET studies (Christine et al, 2009).
Both these viral vector approaches rely on the assumption that striatal neurons can be converted into dopamine-releasing cells, despite the fact that these neurons do not express mechanisms of vesicular storage and release of dopamine. The lack of these basic features may potentially prove harmful to striatal neurons because of an overproduction of cytosolic dopamine, which may lead to oxidative stress in transfected cells (Chen et al, 2008), and the possibility that non-regulated release of dopamine by striatal neurons aggravates dyskinesias, as seen in MPTP-treated monkeys (Bankiewicz et al, 2006a, 2006b) and some patients enrolled in the safety trial (Christine et al, 2009). Thus, a third approach being considered that could overcome these problems is a continuous LD delivery strategy, achieved by delivery of TH and GTP cyclohydrolase 1 (a key co-factor needed for TH function), resulting in LD production without interference with endogenous AADC, and without the production of the potentially toxic dopamine. If dyskinesias partly develop because of fluctuations of synaptic dopamine levels due to intermittent administration of LD (see above), it is possible that striatal serotonergic terminals partly contribute to the swings in the release of LD-derived dopamine because they express AADC, which make them capable of converting exogenous LD to dopamine and release it in an uncontrolled fashion as a false transmitter (Lane et al, 2010; see above). Such problem could be overcome if LD was continuously produced as expected with the AAV-mediated TH/GCH1 delivery. In fact, preclinical testing of this method in rat models of LD-induced dyskinesias has resulted in promising results (Bjorklund et al, 2009).
There are still many unanswered critical questions regarding the use of gene therapy in PD. For instance, it is not clear how the proper regulation of the viral vector to induce the production of a physiologically relevant amount of transmitter can be ensured. The combination of genes that will produce the best results and fewest side effects remains to be determined. Finally, the possibility of reversing the approach in order to adjust the amount of gene product delivery to the patient's needs is another key criterion to consider in the successful development of this therapeutic approach for PD and other brain diseases. A summary of the key findings related to gene therapy for PD is shown in Box 8.
THE SEARCH FOR BIOMARKERS AND NEUROPROTECTIVE THERAPIES: FUTURE CHALLENGES IN PD
The therapeutic approaches discussed so far highlight the progress that has been made toward the development of symptomatic PD therapies. However, one of the most important challenges PD researchers and physicians have faced has not been successfully overcome, ie, the discovery of biomarkers that could predict disease onset, and, thus, guide the use of neuroprotective treatments. Knowing that PD motor symptoms develop only when the dopaminergic denervation of the striatum has reached 70–80%, and that as much as 50% of SNc dopaminergic neurons are lost, the identification of such biomarkers is absolutely essential for the effective use of neuroprotective therapies that could alter the progression and course of the disease. The current lack of such markers most likely explains the numerous disappointing failures of neuroprotective clinical trials that have been completed during the past 10 years (Schapira, 2004; Lang, 2009; Olanow, 2009; Rascol, 2009).
There are many potential biomarkers and early nonmotor clinical signs of PD that are currently being considered in combination to identify at-risk PD patients (Box 9). In the final section of this review, we will discuss the current status of knowledge of each of these approaches, and critically examine their limitations in their application toward early PD diagnosis.
Early Anosmia
The first evidence for an association between olfactory dysfunction and PD was published more than 30 years ago (Ansari and Johnson, 1975). Since then, there have been numerous reports confirming this association, and most importantly, suggesting that the loss of smell may precede the onset of motor disorders (Haehner et al, 2007, 2009; Morley and Duda, 2010). Although the reported prevalence of olfactory loss in PD varies across studies, it appears that as much as 50–90% of PD patients experience varying degrees of olfactory dysfunction, and that there is no correlation between the degree of olfactory dysfunction and the duration or clinical severity of the disease (Doty et al, 1988; Stern et al, 1994; Hawkes et al, 1997; Hawkes, 2003). The pathological substrate for this olfactory loss is not clear, some studies reporting conflicting information about damage to the olfactory epithelium or changes in olfactory bulb volumes as potential sources of these deficits (Muller et al, 2005; Witt et al, 2009; Wang et al, 2011). However, various studies have now reported an increase in the total number of periglomerular dopaminergic cells in the olfactory bulb of parkinsonian patients (Huisman et al, 2004; Haehner et al, 2009; Morley and Duda, 2010).
A number of studies have provided evidence that PD patients commonly report a loss of their sense of smell prior to the development of motor PD symptoms (Doty et al, 1988; Tissingh et al, 2001; Muller et al, 2002a, 2002b; Ponsen et al, 2004; Sommer et al, 2004). The results of a recent longitudinal study in 2267 elderly men in the Honolulu Heart Study demonstrate that loss of olfaction can precede PD motor symptoms by at least 4 years, thereby suggesting that it could serve as a screening tool to predict the future development of PD (Ross et al, 2008). There is also evidence that olfactory disturbances could be used to differentiate PD from other movement disorders such as progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD), but not from multiple system atrophy (MSA) (Wenning et al, 1995; Hummel et al, 1997; Muller et al, 2002b). In light of these observations, the American Academy of Neurology recommends the use of olfactory testing to differentiate PD from PSP and CBD, but not from MSA (McKinnon et al, 2007). It is noteworthy that olfactory impairment is not restricted to PD and MSA, but also occurs in Lewy body disease and Alzheimer's disease, old age, and as a side effect of the use of numerous medications (Liberini et al, 2000; Hawkes, 2003; Williams et al, 2009; Goldstein and Sewell, 2009). The possible role of Lewy body pathology in PD olfactory loss has been suggested (Braak et al, 2003; Morley and Duda, 2010).
Thus, although olfactory dysfunction alone cannot be used as a reliable early diagnostic tool to predict the development of PD, it can be added to a battery of other premotor deficits and imaging techniques, which, together, can serve as biomarkers for early PD diagnosis, thereby allowing neuroprotective therapies to be initiated prior to the development of motor disorders and extensive dopaminergic cell loss (Haehner et al, 2009; Marek and Jennings, 2009; Morley and Duda, 2010).
Neuroimaging
Three main imaging methods are being considered as tools for early (preclinical) diagnosis of PD. These include PET, single-photon emission computed tomography (SPECT) and MRI. PET and SPECT, which can be used to map changes in the abundance and function of dopamine terminals in the striatum, are considered as the two most sensitive approaches to identify patients with PD (Piccini and Whone, 2004; Lang and Mikulis, 2009). However, the high cost of PET scans limits considerably the use of this imaging method to identify PD biomarkers. SPECT dopamine transporter (DAT) imaging has received most attention because it is less costly and sensitive enough to detect early striatal dopaminergic deficit (Kagi et al, 2010). Various DAT ligands (123IFB-CIT, 123I-β-CIT, 99mTc-TRODAT-1, 18F-FECNT) have been developed and successfully applied to human PD studies. The use of these methods can facilitate accurate PD diagnosis in certain patients with non-conventional PD symptomatology. It can also help rule out PD diagnosis in drug-induced, psychogenic, and other forms of parkinsonism that do not rely on nigrostriatal dopaminergic dysfunction. 123Ioflupane, an analog of 123I-β-CIT, has recently been approved by the FDA to be used to image DAT distribution in the diagnosis of PD.
Another critical benefit of DAT imaging is its potential use as a biomarker that could help identify future PD subjects at an early stage of dopaminergic degeneration before the appearance of any motor symptoms. However, although attractive, this approach must be combined with other early signs of PD, such as olfactory loss, autonomic dysfunctions (constipation, hypotension), cognitive decline, cardiac sympathetic dysfunction, or sleep disturbances (REM behavior disorder; RBD) to identify at-risk subjects who have not yet been diagnosed (Langston, 2006; Tolosa, 2007; Tolosa et al, 2007). The combination of smell loss or RBD with DAT imaging has already been successfully used to identify groups of individuals with increased risk of developing PD (Ponsen et al, 2004; Stiasny-Kolster et al, 2005). These observations have led to the development of an extensive clinical effort called the PARS to generate a strategy that could help detect parkinsonism in a cohort of 30 000 first-degree relatives of PD patients using combined changes in olfaction and DAT imaging as biomarkers (Stern, 2004; Siderowf and Stern, 2006; Blekher et al, 2009; Marek and Jennings, 2009). Longitudinal clinical and imaging evaluations will help assess the progression of deficits and the state of DAT imaging in these individuals, and determine if these changes predict the eventual development of PD signs in a subset of patients. Should this be the case, neuroprotective studies could then be initiated in a presymptomatic PD cohort, in order to assess the efficacy of therapies that could slow down neuronal degeneration and delay the onset of symptomatic PD (Marek and Jennings, 2009). It is noteworthy that chronic LD treatment may modify DAT expression, leading some imaging centers to use tetrabenazine (vesicular monoamine transporter ligand) as a ligand for striatal dopamine innervation.
The recent identification of multiple genes associated with PD provides another ‘biomarker’ approach to identify at-risk PD individuals (Farrer, 2006; Hardy et al, 2006; Klein and Schlossmacher, 2007; Pankratz and Foroud, 2007; Cookson, 2010; Dachsel and Farrer, 2010). Although most monogenic mutations account for only a very small proportion of the PD population, the mutation of specific genes like LRRK2 may be associated with as much as 30% of PD patients in some populations like Ashkenazi Jews (Ozelius et al, 2006; Saunders-Pullman et al, 2006; Cookson, 2010; Dachsel and Farrer, 2010). Some imaging studies have demonstrated abnormal dopaminergic function in the striatum in small groups of patients with LRRK2 or other Park gene mutations (Khan et al, 2002; Adams et al, 2005). Various large-scale consortium efforts are currently underway to characterize the status of striatal dopamine imaging in larger cohorts of LRRK2 family relatives (Healy et al, 2008; Cookson, 2010; Dachsel and Farrer, 2010).
The use of brain imaging as a PD biomarker can extend beyond the dopaminergic system to include other markers indicative of PD pathology, such as inflammation, mitochondrial dysfunction, alpha-synuclein deposition, protein misfolding, and others. Inflammatory changes have received significant attention as an early marker of PD risk (Chen et al, 2005; 2008; Hirsch and Hunot, 2009). Imaging tracers that target activated microglia are significantly increased in the brain of PD patients (Ouchi et al, 2005; Gerhard et al, 2006; Hirsch and Hunot, 2009). The usefulness of these additional imaging targets in combination with early signs of PD as useful biomarkers relies on the development of specific ligands that offer high sensitivity and suitable longitudinal time-course assessment.
Proteomics and Related ‘Omics’ Approaches
The recent emergence of highly sensitive ‘omics’ methods to identify possible pathways associated with the progressive development of PD is another promising avenue for the identification of PD biomarkers (Antoniades and Barker, 2008; Caudle et al, 2010). The identification of changes in gene expression (transcriptomics), protein levels (proteomics), or metabolites (metabolomics) in various biological specimens (brain tissue, cerebrospinal fluid, blood) may allow us to characterize dysfunctional pathways specifically involved in the progressive degenerative process in PD, such as mitochondrial dysfunction, oxidative stress, axon guidance, and synaptogenesis. Together, the findings gathered through these different methods will provide a comprehensive analysis of the molecular mechanisms and functional pathways affected in PD. A summary of the main findings related to biomarkers for PD is discussed in Box 9.
CONCLUSION
In this review we have discussed some of the major breakthroughs that have characterized the field of PD therapeutics since the discovery of LD in the 1960s. Although much remains to be known about the etiology of PD, the refinement of symptomatic therapies and the recent start of large-scale studies of PD biomarkers are highly exciting initiatives that may lead to the introduction of novel neuroprotective therapies that will eventually alter the course of the disease, and provide PD patients with a better quality of life. The main challenge PD researchers and physicians have to overcome in the coming decades is to further characterize the underpinnings of the complex nonmotor symptoms in PD in order to develop appropriate treatment strategies for the complex array of autonomic, psychiatric, and cognitive disturbances that affect these patients.
References
Aarsland D, Laake K, Larsen JP, Janvin C (2002). Donepezil for cognitive impairment in Parkinson's disease: a randomised controlled study. J Neurol Neurosurg Psychiatry 72: 708–712.
Aarsland D, Ballard C, Walker Z, Bostrom F, Alves G, Kossakowski K et al (2009). Memantine in patients with Parkinson's disease dementia or dementia with Lewy bodies: a double-blind, placebo-controlled, multicentre trial. Lancet Neurol 8: 613–618.
Adams JR, van Netten H, Schulzer M, Mak E, McKenzie J, Strongosky A et al (2005). PET in LRRK2 mutations: comparison to sporadic Parkinson's disease and evidence for presymptomatic compensation. Brain 128: 2777–2785.
Adler CH (2008). Amantadine and anticholinergics. In: Factor SA, Weiner WJ (eds). Parkinson's Disease-Diagnostic and Clinical Management. Demos: New York. pp 491–497.
Agid Y (1998). Levodopa: is toxicity a myth? Neurology 50: 858–863.
Agid Y, Ahlskog E, Albanese A, Calne D, Chase T, De Yebenes J et al (1999). Levodopa in the treatment of Parkinson's disease: a consensus meeting. Mov Disord 14: 911–913.
Agnati LF, Guidolin D, Leo G, Carone C, Genedani S, Fuxe K (2010). Receptor-receptor interactions: a novel concept in brain integration. Prog Neurobiol 90: 157–175.
Ahlskog JE (2005). Challenging conventional wisdom: the etiologic role of dopamine oxidative stress in Parkinson's disease. Mov Disord 20: 271–282.
Albin RL, Young AB, Penney JB (1989). The functional anatomy of basal ganglia disorders. Trends Neurosci 12: 366–375. One of the first descriptions of the direct and indirect pathway models of the basal ganglia circuitry in normal and diseased states.
Alegret M, Junque C, Valldeoriola F, Vendrell P, Pilleri M, Rumia J et al (2001). Effects of bilateral subthalamic stimulation on cognitive function in Parkinson disease. Arch Neurol 58: 1223–1227.
Alegret M, Valldeoriola F, Marti M, Pilleri M, Junque C, Rumia J et al (2004). Comparative cognitive effects of bilateral subthalamic stimulation and subcutaneous continuous infusion of apomorphine in Parkinson's disease. Mov Disord 19: 1463–1469.
Alessandro S, Ceravolo R, Brusa L, Pierantozzi M, Costa A, Galati S et al (2010). Non-motor functions in parkinsonian patients implanted in the pedunculopontine nucleus: focus on sleep and cognitive domains. J Neurol Sci 289: 44–48.
Allan LE, Petit GH, Brundin P (2010). Cell transplantation in Parkinson's disease: problems and perspectives. Curr Opin Neurol 23: 426–432. Concise overview of the limitations and future developments in the use of cell transplantation for Parkinson's disease.
Alvarez L, Macias R, Guridi J, Lopez G, Alvarez E, Maragoto C et al (2001). Dorsal subthalamotomy for Parkinson's disease. Mov Disord 16: 72–78.
Alvarez L, Macias R, Lopez G, Alvarez E, Pavon N, Rodriguez-Oroz MC et al (2005). Bilateral subthalamotomy in Parkinson's disease: initial and long-term response. Brain 128: 570–583.
Alvarez L, Macias R, Pavon N, Lopez G, Rodriguez-Oroz MC, Rodriguez R et al (2009). Therapeutic efficacy of unilateral subthalamotomy in Parkinson's disease: results in 89 patients followed for up to 36 months. J Neurol Neurosurg Psychiatry 80: 979–985.
Amariglio N, Hirshberg A, Scheithauer BW, Cohen Y, Loewenthal R, Trakhtenbrot L et al (2009). Donor-derived brain tumor following neural stem cell transplantation in an ataxia telangiectasia patient. PLoS Med 6: e1000029.
Anagnostaras SG, Murphy GG, Hamilton SE, Mitchell SL, Rahnama NP, Nathanson NM et al (2003). Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nat Neurosci 6: 51–58.
Anderson VC, Burchiel KJ, Hogarth P, Favre J, Hammerstad JP (2005). Pallidal vs subthalamic nucleus deep brain stimulation in Parkinson disease. Arch Neurol 62: 554–601.
Annic A, Devos D, Seguy D, Dujardin K, Destee A, Defebvre L (2009). Continuous dopaminergic stimulation by Duodopa in advanced Parkinson's disease: Efficacy and safety. Rev Neurol (Paris) 165: 718–727.
Ansari KA, Johnson A (1975). Olfactory function in patients with Parkinson's disease. J Chronic Dis 28: 493–497. First direct association between olfactory dysfunctions and Parkinson's disease.
Antoniades CA, Barker RA (2008). The search for biomarkers in Parkinson's disease: a critical review. Expert Rev Neurother 8: 1841–1852.
Antonini A, Odin P (2009). Pros and cons of apomorphine and L-dopa continuous infusion in advanced Parkinson's disease. Parkinsonism Relat Disord 15 (Suppl 4): S97–S100.
Antonini A, Tolosa E (2009). Apomorphine and levodopa infusion therapies for advanced Parkinson's disease: selection criteria and patient management. Expert Rev Neurother 9: 859–867.
Antonini A, Tolosa E, Mizuno Y, Yamamoto M, Poewe WH (2009). A reassessment of risks and benefits of dopamine agonists in Parkinson's disease. Lancet Neurol 8: 929–937. A comprehensive review of the therapeutic benefits and adverse effects of dopamine receptor agonists as antiparkinsonian therapy.
Anwyl R (1999). Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Rev 29: 83–120.
Aravamuthan BR, McNab JA, Miller KL, Rushworth M, Jenkinson N, Stein JF et al (2009). Cortical and subcortical connections within the pedunculopontine nucleus of the primate Macaca mulatta determined using probabilistic diffusion tractography. J Clin Neurosci 16: 413–420.
Aravamuthan BR, Muthusamy KA, Stein JF, Aziz TZ, Johansen-Berg H (2007). Topography of cortical and subcortical connections of the human pedunculopontine and subthalamic nuclei. Neuroimage 37: 694–705.
Aravamuthan BR, Stein JF, Aziz TZ (2008). The anatomy and localization of the pedunculopontine nucleus determined using probabilistic diffusion tractography [corrected]. Br J Neurosurg 22 (Suppl 1): S25–S32.
Arenas E (2010). Towards stem cell replacement therapies for Parkinson's disease. Biochem Biophys Res Commun 396: 152–156.
Arle JE, Apetauerova D, Zani J, Deletis DV, Penney DL, Hoit D et al (2008). Motor cortex stimulation in patients with Parkinson disease: 12-month follow-up in 4 patients. J Neurosurg 109: 133–139.
Aron L, Klein R (2011). Repairing the parkinsonian brain with neurotrophic factors. Trends Neurosci 34: 88–100.
Ascherio A, Zhang SM, Hernan MA, Kawachi I, Colditz GA, Speizer FE et al (2001). Prospective study of caffeine consumption and risk of Parkinson's disease in men and women. Ann Neurol 50: 56–63.
Astradsson A, Cooper O, Vinuela A, Isacson O (2008). Recent advances in cell-based therapy for Parkinson's disease. Neurosurg Focus 24: E6.
Awad H, Hubert GW, Smith Y, Levey AI, Conn PJ (2000). Activation of metabotropic glutamate receptor 5 has direct excitatory effects and potentiates NMDA receptor currents in neurons of the subthalamic nucleus. J Neurosci 20: 7871–7879.
Azzouz M, Martin-Rendon E, Barber RD, Mitrophanous KA, Carter EE, Rohll JB et al (2002). Multicistronic lentiviral vector-mediated striatal gene transfer of aromatic L-amino acid decarboxylase, tyrosine hydroxylase, and GTP cyclohydrolase I induces sustained transgene expression, dopamine production, and functional improvement in a rat model of Parkinson's disease. J Neurosci 22: 10302–10312.
Baekelandt V, De Strooper B, Nuttin B, Debyser Z (2000). Gene therapeutic strategies for neurodegenerative diseases. Curr Opin Mol Ther 2: 540–554.
Bankiewicz KS, Daadi M, Pivirotto P, Bringas J, Sanftner L, Cunningham J et al (2006a). Focal striatal dopamine may potentiate dyskinesias in parkinsonian monkeys. Exp Neurol 197: 363–372.
Bankiewicz KS, Forsayeth J, Eberling JL, Sanchez-Pernaute R, Pivirotto P, Bringas J et al (2006b). Long-term clinical improvement in MPTP-lesioned primates after gene therapy with AAV-hAADC. Mol Ther 14: 564–570. Pre-clinical evidence for the successful use of gene therapy to restore striatal dopamine and alleviate parkinsonian motor symptoms in MPTP-treated monkeys.
Bara-Jimenez W, Bibbiani F, Morris MJ, Dimitrova T, Sherzai A, Mouradian MM et al (2005). Effects of serotonin 5-HT1A agonist in advanced Parkinson's disease. Mov Disord 20: 932–936.
Bara-Jimenez W, Dimitrova T, Sherzai A, Favit A, Mouradian MM, Chase TN (2004). Effect of monoamine reuptake inhibitor NS 2330 in advanced Parkinson's disease. Mov Disord 19: 1183–1186.
Barbeau A (1962). The pathogenesis of Parkinson's disease: a new hypothesis. Can Med Assoc J 87: 802–807.
Barbeau A, Sourkes TL, Murphy CF (1962). Les catecholamines de la maladie de Parkinson. In: de Ajuriaguerra J (ed). Monoamines et systeme Nerveux Central. Goerg & Cie SA: Geneva. pp 247–262. First evidence that oral L-DOPA is an effective treatment for Parkinson's disease.
Bartus RT, Herzog CD, Chu Y, Wilson A, Brown L, Siffert J et al (2011). Bioactivity of AAV2-neurturin gene therapy (CERE-120): differences between Parkinson's disease and nonhuman primate brains. Mov Disord 26: 27–36.
Bassetti CL (2011). Nonmotor disturbances in Parkinson's disease. Neurodegener Dis 8: 95–108.
Battaglia G, Busceti CL, Molinaro G, Biagioni F, Storto M, Fornai F et al (2004). Endogenous activation of mGlu5 metabotropic glutamate receptors contributes to the development of nigro-striatal damage induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. J Neurosci 24: 828–835.
Bejjani BP, Damier P, Arnulf I, Thivard L, Bonnet AM, Dormont D et al (1999). Transient acute depression induced by high-frequency deep-brain stimulation. N Engl J Med 340: 1476–1480.
Benabid AL (2009). Targeting the caudal intralaminar nuclei for functional neurosurgery of movement disorders. Brain Res Bull 78: 109–112.
Benabid AL, Chabardes S, Mitrofanis J, Pollak P (2009a). Deep brain stimulation of the subthalamic nucleus for the treatment of Parkinson's disease. Lancet Neurol 8: 67–81. A comprehensive review of the befenitial and adverse effects of deep brain stimulation of the STN in patients with Parkinson's disease.
Benabid AL, Chabardes S, Torres N, Piallat B, Krack P, Fraix V et al (2009b). Functional neurosurgery for movement disorders: a historical perspective. Prog Brain Res 175: 379–391.
Benabid AL, Pollak P, Hommel M, Gaio JM, de Rougemont J, Perret J (1989). Treatment of Parkinson tremor by chronic stimulation of the ventral intermediate nucleus of the thalamus. Rev Neurol (Paris) 145: 320–323.
Benvenuti E, Cecchi F, Colombini A, Gori G (2006). Extradural motor cortex stimulation as a method to treat advanced Parkinson's disease: new perspectives in geriatric medicine. Aging Clin Exp Res 18: 347–348.
Bergman H, Wichmann T, DeLong MR (1990). Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science 249: 1436–1438. The first direct evidence that lesion of the subthalamic nucleus in the MPTP-treated monkey model of Parkinson's disease alleviates parkinsonian motor symptoms.
Berney A, Vingerhoets F, Perrin A, Guex P, Villemure JG, Burkhard PR et al (2002). Effect on mood of subthalamic DBS for Parkinson's disease: a consecutive series of 24 patients. Neurology 59: 1427–1429.
Bertler A, Rosengren E (1959). Occurrence and distribution of catecholamines in brain. Acta Physiol Scand 47: 350–361.
Beurrier C, Lopez S, Revy D, Selvam C, Goudet C, Lherondel M et al (2009). Electrophysiological and behavioral evidence that modulation of metabotropic glutamate receptor 4 with a new agonist reverses experimental parkinsonism. FASEB J 23: 3619–3628.
Bibbiani F, Oh JD, Petzer JP, Castagnoli Jr N, Chen JF, Schwarzschild MA et al (2003). A2A antagonist prevents dopamine agonist-induced motor complications in animal models of Parkinson's disease. Exp Neurol 184: 285–294.
Birkmayer W, Hornykiewicz O (1961). (The L-3,4-dioxyphenylalanine (DOPA)-effect in Parkinson-akinesia). Wien Klin Wochenschr 73: 787–788. First evidence that L-DOPA may be a therapeutic agent for Parkinson's disease.
Biseul I, Sauleau P, Haegelen C, Trebon P, Drapier D, Raoul S et al (2005). Fear recognition is impaired by subthalamic nucleus stimulation in Parkinson's disease. Neuropsychologia 43: 1054–1059.
Bjorklund A, Kirik D, Rosenblad C, Georgievska B, Lundberg C, Mandel RJ (2000). Towards a neuroprotective gene therapy for Parkinson's disease: use of adenovirus, AAV and lentivirus vectors for gene transfer of GDNF to the nigrostriatal system in the rat Parkinson model. Brain Res 886: 82–98.
Bjorklund T, Carlsson T, Cederfjall EA, Carta M, Kirik D (2010a). Optimized adeno-associated viral vector-mediated striatal DOPA delivery restores sensorimotor function and prevents dyskinesias in a model of advanced Parkinson's disease. Brain 133: 496–511.
Bjorklund T, Cederfjall EA, Kirik D (2010b). Gene therapy for dopamine replacement. Prog Brain Res 184: 221–235. A detailed review of the use of gene therapy for striatal dopamine replacement in Parkinson's disease.
Bjorklund T, Hall H, Breysse N, Soneson C, Carlsson T, Mandel RJ et al (2009). Optimization of continuous in vivo DOPA production and studies on ectopic DA synthesis using rAAV5 vectors in Parkinsonian rats. J Neurochem 111: 355–367.
Bjorklund T, Kirik D (2009). Scientific rationale for the development of gene therapy strategies for Parkinson's disease. Biochim Biophys Acta 1792: 703–713.
Bjorklund T, Kordower JH (2010). Gene therapy for Parkinson's disease. Mov Disord 25 (Suppl 1): S161–S173.
Black KJ, Koller JM, Campbell MC, Gusnard DA, Bandak SI (2010). Quantification of indirect pathway inhibition by the adenosine A2a antagonist SYN115 in Parkinson disease. J Neurosci 30: 16284–16292.
Blanchet PJ, Konitsiotis S, Whittemore ER, Zhou ZL, Woodward RM, Chase TN (1999). Differing effects of N-methyl-D-aspartate receptor subtype selective antagonists on dyskinesias in levodopa-treated 1-methyl-4-phenyl-tetrahydropyridine monkeys. J Pharmacol Exp Ther 290: 1034–1040.
Blandini F, Greenamyre JT (1998). Prospects of glutamate antagonists in the therapy of Parkinson's disease. Fundam Clin Pharmacol 12: 4–12.
Blandini F, Mangiagalli A, Consentino M, Marino F, Samuele A, Rasini E et al (2003). Peripheral markers of apoptosis in Parkinson's disease: the effects of dopaminergic drugs. Ann NY Acad Sci 1010: 675–678.
Blekher T, Weaver M, Rupp J, Nichols WC, Hui SL, Gray J et al (2009). Multiple step pattern as a biomarker in Parkinson disease. Parkinsonism Relat Disord 15: 506–510.
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003). Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24: 197–211. Introduction of the Braak and Braak stages of Parkinson's disease from the brainstem to the cerebral cortex based on the presence of Lewy body pathology. This paper had a significant impact on the recognition of nonmotor symptoms in PD.
Brady AE, Jones CK, Bridges TM, Kennedy JP, Thompson AD, Heiman JU et al (2008). Centrally active allosteric potentiators of the M4 muscarinic acetylcholine receptor reverse amphetamine-induced hyperlocomotor activity in rats. J Pharmacol Exp Ther 327: 941–953.
Braz CA, Borges V, Ferraz HB (2004). Effect of riluzole on dyskinesia and duration of the on state in Parkinson disease patients: a double-blind, placebo-controlled pilot study. Clin Neuropharmacol 27: 25–29.
Breit S, Schulz JB, Benabid AL (2004). Deep brain stimulation. Cell Tissue Res 318: 275–288.
Breysse N, Amalric M, Salin P (2003). Metabotropic glutamate 5 receptor blockade alleviates akinesia by normalizing activity of selective basal-ganglia structures in parkinsonian rats. J Neurosci 23: 8302–8309.
Breysse N, Baunez C, Spooren W, Gasparini F, Amalric M (2002). Chronic but not acute treatment with a metabotropic glutamate 5 receptor antagonist reverses the akinetic deficits in a rat model of parkinsonism. J Neurosci 22: 5669–5678.
Bridges TM, LeBois EP, Hopkins CR, Wood MR, Jones CK, Conn PJ et al (2010). The antipsychotic potential of muscarinic allosteric modulation. Drug News Perspect 23: 229–240.
Bronstein JM, Tagliati M, Alterman RL, Lozano AM, Volkmann J, Stefani A et al (2011). Deep brain stimulation for Parkinson disease: an expert consensus and review of key issues. Arch Neurol 68: 165.
Brundin P, Barker RA, Parmar M (2010). Neural grafting in Parkinson's disease problems and possibilities. Prog Brain Res 184: 265–294. A comprehensive assessment of the current knowledge of the benefits and adverse effects of neural transplantation in Parkinson's disease.
Burn D, Emre M, McKeith I, De Deyn PP, Aarsland D, Hsu C et al (2006). Effects of rivastigmine in patients with and without visual hallucinations in dementia associated with Parkinson's disease. Mov Disord 21: 1899–1907.
Bymaster FP, Carter PA, Yamada M, Gomeza J, Wess J, Hamilton SE et al (2003a). Role of specific muscarinic receptor subtypes in cholinergic parasympathomimetic responses, in vivo phosphoinositide hydrolysis, and pilocarpine-induced seizure activity. Eur J Neurosci 17: 1403–1410.
Bymaster FP, Felder CC, Tzavara E, Nomikos GG, Calligaro DO, McKinzie DL (2003b). Muscarinic mechanisms of antipsychotic atypicality. Prog Neuropsychopharmacol Biol Psychiatry 27: 1125–1143.
Bymaster FP, McKinzie DL, Felder CC, Wess J (2003c). Use of M1-M5 muscarinic receptor knockout mice as novel tools to delineate the physiological roles of the muscarinic cholinergic system. Neurochem Res 28: 437–442.
Caccamo A, Oddo S, Billings LM, Green KN, Martinez-Coria H, Fisher A et al (2006). M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron 49: 671–682.
Calne DB, Teychenne PF, Claveria LE, Eastman R, Greenacre JK, Petrie A (1974a). Bromocriptine in Parkinsonism. Br Med J 4: 442–444.
Calne DB, Teychenne PF, Leigh PN, Bamji AN, Greenacre JK (1974b). Treatment of parkinsonism with bromocriptine. Lancet 2: 1355–1356.
Caparros-Lefebvre D, Blond S, Feltin MP, Pollak P, Benabid AL (1999). Improvement of levodopa induced dyskinesias by thalamic deep brain stimulation is related to slight variation in electrode placement: possible involvement of the centre median and parafascicularis complex. J Neurol Neurosurg Psychiatry 67: 308–314.
Carlsson A, Lindqvist M, Magnusson T (1957). 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature 180: 1200. First evidence that Parkinson s disease may be due to loss of striatal dopamine.
Carlsson A, Lindqvist M, Magnusson T, Waldeck B (1958). On the presence of 3-hydroxytyramine in brain. Science 127: 471–472.
Cartmell J, Schoepp DD (2000). Regulation of neurotransmitter release by metabotropic glutamate receptors. J Neurochem 75: 889–907.
Castelli L, Perozzo P, Zibetti M, Crivelli B, Morabito U, Lanotte M et al (2006). Chronic deep brain stimulation of the subthalamic nucleus for Parkinson's disease: effects on cognition, mood, anxiety and personality traits. Eur Neurol 55: 136–144.
Caudle WM, Bammler TK, Lin Y, Pan S, Zhang J (2010). Using ‘omics’ to define pathogenesis and biomarkers of Parkinson's disease. Expert Rev Neurother 10: 925–942.
Chan WY, McKinzie DL, Bose S, Mitchell SN, Witkin JM, Thompson RC et al (2008). Allosteric modulation of the muscarinic M4 receptor as an approach to treating schizophrenia. Proc Natl Acad Sci USA 105: 10978–10983.
Chang VC, Chou KL (2006). Deep brain stimulation for Parkinson's disease: patient selection and motor outcomes. Med Health R I 89: 142–144.
Chase TN, Oh JD, Blanchet PJ (1998). Neostriatal mechanisms in Parkinson's disease. Neurology 51: S30–S35.
Chaudhuri KR, Healy DG, Schapira AHV (2006). Non-motor symptoms of Parkinson's disease: diagnosis and management. Lancet Neurol 5: 235–245.
Chaudhuri KR, Odin P, Antonini A, Martinez-Martin P (2011). Parkinson's disease: the non-motor issues. Parkinsonism Relat Disord; e-pub ahead of print 7 July 2011 (in press).
Chaudhuri KR, Schapira AHV (2009). Non-motor symptoms of Parkinson's disease: dopaminergic pathophysiology and treatment. Lancet Neurol 8: 464–474.
Charcot J-M (1879). Lectures on Diseases of the Nervous System. HC Lea: Philadelphia.
Charcot J-M, Vulpian A (1861). La paralysie agitante. Gaz Hebdom Med Chir 8: 765–767, 816–820; 9: 54–59.
Chen L, Ding Y, Cagniard B, Van Laar AD, Mortimer A, Chi W et al (2008). Unregulated cytosolic dopamine causes neurodegeneration associated with oxidative stress in mice. J Neurosci 28: 425–433.
Chen LW, Yung KL, Chan YS (2005). Reactive astrocytes as potential manipulation targets in novel cell replacement therapy of Parkinson's disease. Curr Drug Targets 6: 821–833.
Christine CW, Starr PA, Larson PS, Eberling JL, Jagust WJ, Hawkins RA et al (2009). Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 73: 1662–1669.
Chuang C, Fahn S, Frucht SJ (2002). The natural history and treatment of acquired hemidystonia: report of 33 cases and review of the literature. J Neurol Neurosurg Psychiatry 72: 59–67.
Cilia R, Landi A, Vergani F, Sganzerla E, Pezzoli G, Antonini A (2007). Extradural motor cortex stimulation in Parkinson's disease. Mov Disord 22: 111–114.
Cilia R, Marotta G, Landi A, Isaias IU, Vergani F, Benti R et al (2008). Cerebral activity modulation by extradural motor cortex stimulation in Parkinson's disease: a perfusion SPECT study. Eur J Neurol 15: 22–28.
Coccurello R, Breysse N, Amalric M (2004). Simultaneous blockade of adenosine A2A and metabotropic glutamate mGlu5 receptors increase their efficacy in reversing Parkinsonian deficits in rats. Neuropsychopharmacology 29: 1451–1461.
Conn PJ, Battaglia G, Marino MJ, Nicoletti F (2005). Metabotropic glutamate receptors in the basal ganglia motor circuit. Nat Rev Neurosci 6: 787–798.
Conn PJ, Christopoulos A, Lindsley CW (2009b). Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov 8: 41–54.
Conn PJ, Jones CK, Lindsley CW (2009a). Subtype-selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends Pharmacol Sci 30: 148–155. A concise review of the advantages of developing allosteric modulators of GPCRs for the treatment of brain disorders.
Conn PJ, Pin JP (1997). Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 37: 205–237.
Cookson MR (2010). Unravelling the role of defective genes. Prog Brain Res 183: 43–57.
Cooper O, Astradsson A, Hallett P, Robertson H, Mendez H, Isacson O (2009). Lack of functional relevance of isolated cell damage in transplants of Parkinson's disease patients. J Neurol 256 (Suppl 3): 310–316.
Corrodi H, Fuxe K, Hokfelt T, Lidbrink P, Ungerstedt U (1973). Effect of ergot drugs on central catecholamine neurons: evidence for a stimulation of central dopamine neurons. J Pharm Pharmacol 25: 409–412.
Cotzias GC, Papavasiliou PS, Gellene R (1969). Modification of parkinsonism: chronic treatment with L-Dopa. N Engl J Med 280: 337–345.
Cotzias GC, Papavasiliou PS, Fehling C, Kaufman B, Mena I (1970). Similarities between neurologic effects of L-dopa and of apomorphine. N Engl J Med 282: 31–33.
Cotzias GC, Van Woert MH, Schiffer LM (1967). Aromatic amino acids and modification of parkinsonism. N Engl J Med 276: 374–379.
Crossman AR (1989). Neural mechanisms in disorders of movement. Comp Biochem Physiol A Comp Physiol 93: 141–149.
Dachsel JC, Farrer MJ (2010). LRRK2 and Parkinson disease. Arch Neurol 67: 542–547.
De Blasi A, Conn PJ, Pin J, Nicoletti F (2001). Molecular determinants of metabotropic glutamate receptor signaling. Trends Pharmacol Sci 22: 114–120.
De Gaspari D, Siri C, Landi A, Cilia R, Bonetti A, Natuzzi F et al (2006). Clinical and neuropsychological follow up at 12 months in patients with complicated Parkinson's disease treated with subcutaneous apomorphine infusion or deep brain stimulation of the subthalamic nucleus. J Neurol Neurosurg Psychiatry 77: 450–453.
Dejean C, Hyland B, Arbuthnott G (2009). Cortical effects of subthalamic stimulation correlate with behavioral recovery from dopamine antagonist induced akinesia. Cereb Cortex 19: 1055–1063.
Dekundy A, Gravius A, Hechenberger M, Pietraszek M, Nagel J, Tober C et al (2010). Pharmacological characterization of MRZ-8676, a novel negative allosteric modulator of subtype 5 metabotropic glutamate receptor (mGluR5): focus on L-DOPA induced dyskinesia. J Neural Transm (e-pub ahead of print 16 December 2010).
DeLong MR (1990). Primate models of movement disorders of basal ganglia origin. Trends Neurosci 13: 281–285.
Deuschl G, Schade-Brittinger C, Krack P, Volkmann J, Schafer H, Botzel K et al (2006). A randomized trial of deep-brain stimulation for Parkinson's disease. N Engl J Med 355: 896–908.
Devos D, Defebvre L, Bordet R (2010). Dopaminergic and non-dopaminergic pharmacological hypotheses for gait disorders in Parkinson's disease. Fundam Clin Pharmacol 24: 407–421.
Devos D, Dujardin K, Poirot I, Moreau C, Cottencin O, Thomas P et al (2008). Comparison of desipramine and citalopram treatments for depression in Parkinson's disease: a double-blind, randomized, placebo-controlled study. Mov Disord 23: 850–857.
Devos D, Krystkowiak P, Clement F, Dujardin K, Cottencin O, Waucquier N et al (2007). Improvement of gait by chronic, high doses of methylphenidate in patients with advanced Parkinson's disease. J Neurol Neurosurg Psychiatry 78: 470–475.
Dewey Jr RB, Hutton JT, LeWitt PA, Factor SA (2001). A randomized, double-blind, placebo-controlled trial of subcutaneously injected apomorphine for parkinsonian off-state events. Arch Neurol 58: 1385–1392.
Di Matteo V, Pierucci M, Esposito E, Crescimanno G, Benigno A, Di Giovanni G (2008). Serotonin modulation of the basal ganglia circuitry: therapeutic implication for Parkinson's disease and other motor disorders. Prog Brain Res 172: 423–463.
Diamond A, Jankovic J (2005). The effect of deep brain stimulation on quality of life in movement disorders. J Neurol Neurosurg Psychiatry 76: 1188–1193.
Digby GJ, Shirey JK, Conn PJ (2010). Allosteric activators of muscarinic receptors as novel approaches for treatment of CNS disorders. Mol Biosyst 6: 1345–1354.
Dobkin RD, Menza M, Bienfait KL, Gara M, Marin H, Mark MH et al (2011). Depression in Parkinson's disease: symptom improvement and residual symptoms after acute pharmacologic management. Am J Geriatr Psychiatry 19: 222–229.
Doshi PK, Chhaya N, Bhatt MH (2002). Depression leading to attempted suicide after bilateral subthalamic nucleus stimulation for Parkinson's disease. Mov Disord 17: 1084–1085.
Dolen G, Carpenter RL, Ocain TD, Bear MF (2010). Mechanism-based approaches to treating fragile X. Pharmacol Ther 127: 78–93.
Doshay LJ, Constable K, Agate Jr FJ (1956). Ethopropazine (parsidol) hydrochloride in treatment of paralysis agitans; posology, method of administration, and effects. J Am Med Assoc 160: 348–351.
Doty RL, Deems DA, Stellar S (1988). Olfactory dysfunction in parkinsonism: a general deficit unrelated to neurologic signs, disease stage, or disease duration. Neurology 38: 1237–1244.
Drapier D, Peron J, Leray E, Sauleau P, Biseul I, Drapier S et al (2008). Emotion recognition impairment and apathy after subthalamic nucleus stimulation in Parkinson's disease have separate neural substrates. Neuropsychologia 46: 2796–2801.
Dujardin K, Blairy S, Defebvre L, Krystkowiak P, Hess U, Blond S et al (2004). Subthalamic nucleus stimulation induces deficits in decoding emotional facial expressions in Parkinson's disease. J Neurol Neurosurg Psychiatry 75: 202–208.
Dujardin K, Defebvre L, Krystkowiak P, Blond S, Destee A (2001). Influence of chronic bilateral stimulation of the subthalamic nucleus on cognitive function in Parkinson's disease. J Neurol 248: 603–611.
Durif F, Debilly B, Galitzky M, Morand D, Viallet F, Borg M et al (2004). Clozapine improves dyskinesias in Parkinson disease: a double-blind, placebo-controlled study. Neurology 62: 381–388.
Durif F, Lemaire JJ, Debilly B, Dordain G (2002). Long-term follow-up of globus pallidus chronic stimulation in advanced Parkinson's disease. Mov Disord 17: 803–807.
During MJ, Kaplitt MG, Stern MB, Eidelberg D (2001). Subthalamic GAD gene transfer in Parkinson disease patients who are candidates for deep brain stimulation. Hum Gene Ther 12: 1589–1591.
Duty S (2010). Therapeutic potential of targeting group III metabotropic glutamate receptors in the treatment of Parkinson's disease. Br J Pharmacol 161: 271–287.
Eberling JL, Jagust WJ, Christine CW, Starr P, Larson P, Bankiewicz KS et al (2008). Results from a phase I safety trial of hAADC gene therapy for Parkinson disease. Neurology 70: 1980–1983.
Ehringer H, Hornykiewicz O (1960). Vertilung von Noradrenalin und Dopamin (3-hydroxytyramin) im Gerhirn des Menschens und ihr Verhalten bei Erkrankugen des extrapyramidalen Systems. Klin Wochenschr 38: 1236–1239.
Emre M (2003). What causes mental dysfunction in Parkinson's disease? Mov Disord 18 (Suppl 6): S63–S71.
Emre M, Aarsland D, Albanese A, Byrne EJ, Deuschl G, De Deyn PP et al (2004). Rivastigmine for dementia associated with Parkinson's disease. N Engl J Med 351: 2509–2518.
Emre M, Tsolaki M, Bonuccelli U, Destee A, Tolosa E, Kutzelnigg A et al (2010). Memantine for patients with Parkinson's disease dementia or dementia with Lewy bodies: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 9: 969–977.
Engers DW, Field JR, Le U, Zhou Y, Bolinger JD, Zamorano R et al (2011). Discovery, synthesis, and structure-activity relationship development of a series of N-(4-acetamido)phenylpicolinamides as positive allosteric modulators of metabotropic glutamate receptor 4 (mGlu(4)) with CNS exposure in rats. J Med Chem 54: 1106–1110.
Entacapone-Tolcapone Switch Study Investigators (2007). Entacapone to tolcapone switch: multicenter double-blind, randomized, active-controlled trial in advanced Parkinson's disease. Mov Disord 22: 14–19.
Erola T, Karinen P, Heikkinen E, Tuominen J, Haapaniemi T, Koivukangas J et al (2005). Bilateral subthalamic nucleus stimulation improves health-related quality of life in Parkinsonian patients. Parkinsonism Relat Disord 11: 89–94.
Espay AJ, Dwivedi AK, Payne M, Gaines L, Vaughan JE, Maddux BN et al (2011). Methylphenidate for gait impairment in Parkinson disease: a randomized clinical trial. Neurology 76: 1256–1262.
Factor SA (1999). Dopamine agonists. Med Clin North Am 83: 415–443, vi–vii.
Factor SA (2004). Literature review: intermittent subcutaneous apomorphine therapy in Parkinson's disease. Neurology 62: S12–S17.
Factor SA (2008). Current status of symptomatic medical therapy in Parkinson's disease. Neurotherapeutics 5: 164–180.
Factor S, Mark MH, Watts R, Struck L, Mori A, Ballerini R, et al, Istradefylline 6002-US-007 Study Group (2010). A long-term study of istradefylline in subjects with Parkinson's disease. Parkinsonism Relat Disorders 16: 423–426.
Factor SA, Molho ES (1999). Transient benefit of amantadine in Parkinson's disease: the facts about the myth. Mov Disord 14: 515–517.
Fahn S (1989). The history of parkinsonism. Mov Disord 4 (Suppl 1): S2–S10.
Fahn S (1996a). Controversies in the therapy of Parkinson's disease. Adv Neurol 69: 477–486.
Fahn S (1996b). Is levodopa toxic? Neurology 47: S184–S195.
Fahn S, Cohen G (1992). The oxidant stress hypothesis in Parkinson's disease: evidence supporting it. Ann Neurol 32: 804–812.
Fahn S, Oakes D, Shoulson I, Kieburtz K, Rudolph A, Lang A et al (2004). Levodopa and the progression of Parkinson's disease. N Engl J Med 351: 2498–2508. This paper presents the results of the ELLDOPA trial that assessed the efficacy of L-DOPA towards PD motor symptoms. This paper also demonstrates that L-DOPA therapy does not accelerate PD progression.
Fahn S, Parkinson Study Group (2005). Does levodopa slow or hasten the rate of progression of Parkinson's disease? J Neurol 252 (Suppl 4): IV37–IV42.
Farrer MJ (2006). Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet 7: 306–318.
Feng LR, Maguire-Zeiss KA (2010). Gene therapy in Parkinson's disease: rationale and current status. CNS Drugs 24: 177–192.
Fernandez HH, Greeley DR, Zweig RM, Wojcieszek J, Mori A, Sussman NM (2010). Istradefylline as monotherapy for Parkinson disease: results of the 6002-US-051 trial. Parkinsonism Relat Disord 16: 16–20.
Ferraguti F, Crepaldi L, Nicoletti F (2008). Metabotropic glutamate receptor 1: current cocepts and perspectives. Pharmacol Rev 60: 536–581.
Ferrara J, Diamond A, Hunter C, Davidson A, Almaguer M, Jankovic J (2010). Impact of STN-DBS on life and health satisfaction in patients with Parkinson's disease. J Neurol Neurosurg Psychiatry 81: 315–319.
Ferraye MU, Debu B, Fraix V, Goetz L, Ardouin C, Yelnik J et al (2010). Effects of pedunculopontine nucleus area stimulation on gait disorders in Parkinson's disease. Brain 133: 205–214.
Ferré S, Fredholm BB, Morelli M, Popoli P, Fuxe K (1997). Adenosine-dopamine receptor-receptor interactions as an integrative mechanism in the basal ganglia. Trends Neurosci 20: 482–487.
Fisher A (2008). M1 muscarinic agonists target major hallmarks of Alzheimer's disease: The pivotal role of brain M1 receptors. Neurodegener Dis 5: 237–240.
Flor PJ, Battaglia G, Nicoletti F, Gasparini F, Bruno V (2002). Neuroprotective activity of metabotropic glutamate receptor ligands. Adv Exp Med Biol 513: 197–223.
Follett KA, Weaver FM, Stern M, Hur K, Harris CL, Luo P et al (2010). Pallidal vs subthalamic deep-brain stimulation for Parkinson's disease. N Engl J Med 362: 2077–2091. A comprehensive comparative analysis of the benefits and adverse effects of GPi vs STN DBS in Parkinson's disease.
Forsayeth JR, Eberling JL, Sanftner LM, Zhen Z, Pivirotto P, Bringas J et al (2006). A dose-ranging study of AAV-hAADC therapy in Parkinsonian monkeys. Mol Ther 14: 571–577.
Freed CR, Greene PE, Breeze RE, Tsai WY, DuMouchel W, Kao R et al (2001). Transplantation of embryonic dopamine neurons for severe Parkinson's disease. N Engl J Med 344: 710–719.
French Clozapine Parkinson Study Group (1999). Clozapine in drug-induced psychosis in Parkinson's disease. Lancet 353: 2041–2042.
Fricker-Gates RA, Gates MA (2010). Stem cell-derived dopamine neurons for brain repair in Parkinson's disease. Regen Med 5: 267–278.
Fu Y, Gao W, Zhu M, Chen X, Lin Z, Wang S (2009). Computer-assisted automatic localization of the human pedunculopontine nucleus in T1-weighted MR images: a preliminary study. Int J Med Robot 5: 309–318.
Funk C (1911). Synthese des d, 1-3-4, Dioxyphenylalanins. Chem Zentralbl.
Funkiewiez A, Ardouin C, Caputo E, Krack P, Fraix V, Klinger H et al (2004). Long term effects of bilateral subthalamic nucleus stimulation on cognitive function, mood, and behaviour in Parkinson's disease. J Neurol Neurosurg Psychiatry 75: 834–839.
Fuxe K, Agnati LF, Jacobsen K, Hillion J, Canals M, Torvinen M et al (2003). Receptor heteromerization in adenosine A2A receptor signaling: relevance for striatal function and Parkinson's disease. Neurology 61: S19–S23.
Fuxe K, Ferre S, Genedani S, Franco R, Agnati LF (2007a). Adenosine receptor-dopamine receptor interactions in the basal ganglia and their relevance for brain function. Physiol Behav 92: 210–217.
Fuxe K, Marcellino D, Borroto-Escuela DO, Guescini M, Fernandez-Duenas V, Tanganelli S et al (2010a). Adenosine-dopamine interactions in the pathophysiology and treatment of CNS disorders. CNS Neurosci Ther 16: e18–e42.
Fuxe K, Marcellino D, Genedani S, Agnati L (2007b). Adenosine A(2A) receptors, dopamine D(2) receptors and their interactions in Parkinson's disease. Mov Disord 22: 1990–2017.
Fuxe K, Marcellino D, Leo G, Agnati LF (2010b). Molecular integration via allosteric interactions in receptor heteromers. A working hypothesis. Curr Opin Pharmacol 10: 14–22.
Gallagher DA, Lees AJ, Schrag A (2010). What are the most important nonmotor symptoms in patients with Parkinson's disease and are we missing them? Mov Disord 25: 2493–2500.
Galvan A, Kuwajima M, Smith Y (2006). Glutamate and GABA receptors and transporters in the basal ganglia: what does their subsynaptic localization reveal about their function? Neuroscience 143: 351–375.
Galvan A, Wichmann T (2008). Pathophysiology of parkinsonism. Clin Neurophysiol 119: 1459–1474.
Gasmi M, Brandon EP, Herzog CD, Wilson A, Bishop KM, Hofer EK et al (2007a). AAV2-mediated delivery of human neurturin to the rat nigrostriatal system: long-term efficacy and tolerability of CERE-120 for Parkinson's disease. Neurobiol Dis 27: 67–76.
Gasmi M, Herzog CD, Brandon EP, Cunningham JJ, Ramirez GA, Ketchum ET et al (2007b). Striatal delivery of neurturin by CERE-120, an AAV2 vector for the treatment of dopaminergic neuron degeneration in Parkinson's disease. Mol Ther 15: 62–68.
Gasparini F, Bilbe G, Gomez-Mancilla B, Spooren W (2008). mGluR5 antagonists: discovery, characterization and drug development. Curr Opin Drug Discov Devel 11: 655–665.
Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A et al (2006). In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson's disease. Neurobiol Dis 21: 404–412.
Germano IM, Gracies JM, Weisz DJ, Tse W, Koller WC, Olanow CW (2004). Unilateral stimulation of the subthalamic nucleus in Parkinson disease: a double-blind 12-month evaluation study. J Neurosurg 101: 36–42.
Giladi N (2008). Medical treatment of freezing of gait. Mov Disord 23 (Suppl 2): S482–S488.
Godwin-Austen RB (1973). The long term therapeutic effect of levodopain the treatment of parkinsonism. Adv Neurol 3: 23–27.
Goetz CG, Damier P, Hicking C, Laska E, Muller T, Olanow CW et al (2007). Sarizotan as a treatment for dyskinesias in Parkinson's disease: a double-blind placebo-controlled trial. Mov Disord 22: 179–186.
Goetz CG, Laska E, Hicking C, Damier P, Müller T, Nutt J et al (2008). Placebo influences on dyskinesias in Parkinson's disease. Mov Disord 23: 700–707.
Goff LK, Jouve L, Melon C, Salin P (2009). Rationale for targeting the thalamic centre-median parafascicular complex in the surgical treatment of Parkinson's disease. Parkinsonism Relat Disord 15 (Suppl 3): S167–S170.
Goldstein DS, Sewell L (2009). Olfactory dysfunction in pure autonomic failure: implications for the pathogenesis of Lewy body diseases. Parkinsonism Relat Disord 15: 516–520.
Gottwald MD, Aminoff MJ (2008). New frontiers in the pharmacological management of Parkinson's disease. Drugs Today (Barc) 44: 531–545.
Gradinaru V, Mogri M, Thompson KR, Henderson JM, Deisseroth K (2009). Optical deconstruction of parkinsonian neural circuitry. Science 324: 354–359.
Greene JG (2008). Excitatory amino acid antagonists. In: Factor SA, Weiner WJ (eds). Parkinson's Disease-Diagnostic and Clinical Management. Demos: New York. pp 545–555.
Gregoire L, Morin N, Ouattara B, Gasparini F, Bilbe G, Johns D et al (2011). The acute antiparkinsonian and antidyskinetic effect of AFQ056, a novel metabotropic glutamate receptor type 5 antagonist, in l-Dopa-treated parkinsonian monkeys. Parkinsonism Relat Disord 17: 270–276.
Grondin R, Bedard PJ, Hadj Tahar A, Gregoire L, Mori A, Kase H (1999). Antiparkinsonian effect of a new selective adenosine A2A receptor antagonist in MPTP-treated monkeys. Neurology 52: 1673–1677.
Gunzler SA, Nutt JG (2008). Motor fluctuations with dyskinesia. In: Factor SA, Weiner WJ (eds). Parkinson's Disease-Diagnostic and Clinical Management. Demos: New York. pp 575–584.
Haehner A, Boesveldt S, Berendse HW, Mackay-Sim A, Fleischmann J, Silburn PA et al (2009). Prevalence of smell loss in Parkinson's disease—a multicenter study. Parkinsonism Relat Disord 15: 490–494.
Haehner A, Hummel T, Hummel C, Sommer U, Junghanns S, Reichmann H (2007). Olfactory loss may be a first sign of idiopathic Parkinson's disease. Mov Disord 22: 839–842.
Haehner A, Hummel T, Reichmann H (2009). Olfactory dysfunction as a diagnostic marker for Parkinson's disease. Expert Rev Neurother 9: 1773–1779.
Halbig TD, Gruber D, Kopp UA, Schneider GH, Trottenberg T, Kupsch A (2005). Pallidal stimulation in dystonia: effects on cognition, mood, and quality of life. J Neurol Neurosurg Psychiatry 76: 1713–1716.
Hanson JE, Smith Y (1999). Group I metabotropic glutamate receptors at GABAergic synapses in monkeys. J Neurosci 19: 6488–6496.
Hardy J, Cai H, Cookson MR, Gwinn-Hardy K, Singleton A (2006). Genetics of Parkinson's disease and parkinsonism. Ann Neurol 60: 389–398.
Hargus G, Cooper O, Deleidi M, Levy A, Lee K, Marlow E et al (2010). Differentiated Parkinson patient-derived induced pluripotent stem cell grow in the adult rodent brain and reduce motor asymmetry in parkinsonian rats. Proc Natl Acd Sci USA 107: 15921–15927.
Hariz MI, Johansson F, Shamsgovara P, Johansson E, Hariz GM, Fagerlund M (2000). Bilateral subthalamic nucleus stimulation in a parkinsonian patient with preoperative deficits in speech and cognition: persistent improvement in mobility but increased dependency: a case study. Mov Disord 15: 136–139.
Hasselmo ME (2006). The role of acetylcholine in learning and memory. Curr Opin Neurobiol 16: 710–715.
Hauptmann C, Tass PA (2010). Restoration of segregated, physiological neuronal connectivity by desynchronizing stimulation. J Neural Eng 7: 056008.
Hauser RA, Cantillon M, Pourcher E, Micheli F, Mok V, Onofrj M et al (2011). Preladenant in patients with Parkinson's disease and motor fluctuations: a phase 2, double-blind, randomised trial. Lancet Neurol 10: 221–229.
Hauser RA, Schwarzschild MA (2005). Adenosine A2A receptor antagonists for Parkinson's disease: rationale, therapeutic potential and clinical experience. Drugs Aging 22: 471–482.
Hawkes C (2003). Olfaction in neurodegenerative disorder. Mov Disord 18: 364–372.
Hawkes CH, Shephard BC, Daniel SE (1997). Olfactory dysfunction in Parkinson's disease. J Neurol Neurosurg Psychiatry 62: 436–446.
Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S et al (2008). Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet Neurol 7: 583–590.
Hely MA, Morris JG, Reid WG, Trafficante R (2005). Sydney Multicenter Study of Parkinson's disease: non-L-dopa-responsive problems dominate at 15 years. Mov Disord 20: 190–199. Extensive survey showing the high prevalence of nonmotor symptoms that are unresponsive to dopamine therapy in PD.
Hernan MA, Takkouche B, Caamano-Isorna F, Gestal-Otero JJ (2002). A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson's disease. Ann Neurol 52: 276–284.
Herzog CD, Dass B, Gasmi M, Bakay R, Stansell JE, Tuszynski M et al (2008). Transgene expression, bioactivity, and safety of CERE-120 (AAV2-neurturin) following delivery to the monkey striatum. Mol Ther 16: 1737–1744.
Herzog CD, Dass B, Holden JE, Stansell III J, Gasmi M, Tuszynski MH et al (2007). Striatal delivery of CERE-120, an AAV2 vector encoding human neurturin, enhances activity of the dopaminergic nigrostriatal system in aged monkeys. Mov Disord 22: 1124–1132.
Herzog J, Reiff J, Krack P, Witt K, Schrader B, Muller D et al (2003a). Manic episode with psychotic symptoms induced by subthalamic nucleus stimulation in a patient with Parkinson's disease. Mov Disord 18: 1382–1384.
Herzog J, Volkmann J, Krack P, Kopper F, Pötter M, Lorenz D et al (2003b). Two-year follow-up of subthalamic deep brain stimulation in Parkinson's disease. Mov Disord 18: 1332–1337.
Higginson CI, Wheelock VL, Levine D, King DS, Pappas CT, Sigvardt KA (2009). The clinical significance of neuropsychological changes following bilateral subthalamic nucleus deep brain stimulation for Parkinson's disease. J Clin Exp Neuropsychol 31: 65–72.
Hirsch EC, Hunot S (2009). Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol 8: 382–397. A comprehensive review of evidence that neuroinflammation contributes to PD pathogenesis.
Hodgson RA, Bedard PJ, Varty GB, Kazdoba TM, Di Paolo T, Grzelak ME et al (2010). Preladenant, a selective A(2A) receptor antagonist, is active in primate models of movement disorders. Exp Neurol 225: 384–390.
Hodgson RA, Bertorelli R, Varty GB, Lachowicz JE, Forlani A, Fredduzzi S et al (2009). Characterization of the potent and highly selective A2A receptor antagonists preladenant and SCH 412348 [7-[2-[4-2,4-difluorophenyl]-1-piperazinyl]ethyl]-2-(2-furanyl)-7H-pyrazol o[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine] in rodent models of movement disorders and depression. J Pharmacol Exp Ther 330: 294–303.
Holloway RG, Shoulson I, Fahn S, Kieburtz K, Lang A, Marek K et al, Parkinson Study Group (2004). Pramipexole vs levodopa as initial treatment for Parkinson disease: a 4-year randomized controlled trial. Arch Neurol 61: 1044–1053.
Hopkins CR, Lindsley CW, Niswender CM (2009). mGluR4-positive allosteric modulation as potential treatment for Parkinson's disease. Future Med Chem 1: 501–513.
Hornykiewicz O (2002a). Dopamine miracle: from brain homogenate to dopamine replacement. Mov Disord 17: 501–508.
Hornykiewicz O (2002b). L-DOPA: from a biologically inactive amino acid to a successful therapeutic agent. Amino Acids 23: 65–70.
Hubert GW, Paquet M, Smith Y (2001). Differential subcellular localization of mGluR1a and mGluR5 in the rat and monkey Substantia nigra. J Neurosci 21: 1838–1847.
Hughes AJ, Bishop S, Kleedorfer B, Turjanski N, Fernandez W, Lees AJ et al (1993). Subcutaneous apomorphine in Parkinson's disease: response to chronic administration for up to five years. Mov Disord 8: 165–170.
Huisman E, Uylings HB, Hoogland PV (2004). A 100% increase of dopaminergic cells in the olfactory bulb may explain hyposmia in Parkinson's disease. Mov Disord 19: 687–692.
Hummel T, Sekinger B, Wolf SR, Pauli E, Kobal G (1997). ‘Sniffin sticks’: olfactory performance assessed by the combined testing of odor identification, odor discrimination and olfactory threshold.. Chem Senses 22: 39–52.
Isacson O (2009). Cell therapy ahead for Parkinson's disease. Science 326: 1060.
Isacson O, Kordower JF (2008). Future of cell and gene therapies for Parkinson's disease. Ann Neurol 64 (Suppl 2): S122–S138.
Isacson O, Mendez I (2010). Being too inclusive about synuclein inclusions. Nat Med 16: 960–961.
Jarraya B, Boulet S, Ralph GS, Jan C, Bonvento G, Azzouz M et al (2009). Dopamine gene therapy for Parkinson's disease in a nonhuman primate without associated dyskinesia. Sci Transl Med 1: 2ra4.
Jenner P, Mori A, Hauser R, Morelli M, Fredholm BB, Chen JF (2009). Adenosine, adenosine A 2A antagonists, and Parkinson's disease. Parkinsonism Relat Disord 15: 406–413.
Jiménez-Jiménez FJ, Mateo D, Giménez-Roldan S (1992). Premorbid smoking, alcohol consumption, and coffee drinking habits in Parkinson's disease: a case-control study. Mov Disord 7: 339–344.
Johnson KA, Conn PJ, Niswender CM (2009). Glutamate receptors as therapeutic targets for Parkinson's disease. CNS Neurol Disord Drug Targets 8: 475–491.
Johnston TH, Fox SH, McIldowie MJ, Piggott MJ, Brotchie JM (2010). Reduction of L-DOPA-induced dyskinesia by the selective metabotropic glutamate receptor 5 antagonist 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned macaque model of Parkinson's disease. J Pharmacol Exp Ther 333: 865–873.
Jouve L, Salin P, Melon C, Kerkerian-Le Goff L (2010). Deep brain stimulation of the center median-parafascicular complex of the thalamus has efficient anti-parkinsonian action associated with widespread cellular responses in the basal ganglia network in a rat model of Parkinson's disease. J Neurosci 30: 9919–9928.
Just H, Ostergaard K (2002). Health-related quality of life in patients with advanced Parkinson's disease treated with deep brain stimulation of the subthalamic nuclei. Mov Disord 17: 539–545.
Kachroo A, Orlando LR, Grandy DK, Chen JF, Young AB, Schwarzschild MA (2005). Interactions between metabotropic glutamate 5 and adenosine A2A receptors in normal and parkinsonian mice. J Neurosci 25: 10414–10419.
Kagi G, Bhatia KP, Tolosa E (2010). The role of DAT-SPECT in movement disorders. J Neurol Neurosurg Psychiatry 81: 5–12.
Kanda T, Jackson MJ, Smith LA, Pearce RK, Nakamura J, Kase H et al (1998). Adenosine A2A antagonist: a novel antiparkinsonian agent that does not provoke dyskinesia in parkinsonian monkeys. Ann Neurol 43: 507–513.
Kaplitt MG, Feigin A, Tang C, Fitzsimons HL, Mattis P, Lawlor PA et al (2007). Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson's disease: an open label, phase I trial. Lancet 369: 2097–2105.
Kapp W (1992). The history of drugs for the treatment of Parkinson's disease. J Neural Transm 38: 1–6.
Kasten M, Kertelge L, Bruggemann N, van der Vegt J, Schmidt A, Tadic V et al (2010). Nonmotor symptoms in genetic Parkinson's disease. Arch Neurol 67: 670–676.
Katzenschlager R, Manson AJ, Evans A, Watt H, Lees AJ (2004). Low dose quetiapine for drug induced dyskinesias in Parkinson's disease: a double blind cross over study. J Neurol Neurosurg Psychiatry 75: 295–297.
Khan NL, Brooks DJ, Pavese N, Sweeney MG, Wood NW, Lees AJ et al (2002). Progression of nigrostriatal dysfunction in a parkin kindred: an [18F]dopa PET and clinical study. Brain 125: 2248–2256.
Kim HJ (2011). Stem cell potential in Parkinson's disease and molecular factors for the generation of dopamine neurons. Biochim Biophys Acta 1812: 1–11.
Kitagawa M, Murata J, Uesugi H, Kikuchi S, Saito H, Tashiro K et al (2005). Two-year follow-up of chronic stimulation of the posterior subthalamic white matter for tremor-dominant Parkinson's disease. Neurosurgery 56: 281–289; discussion 281–289.
Klein C, Schlossmacher MG (2007). Parkinson disease, 10 years after its genetic revolution: multiple clues to a complex disorder. Neurology 69: 2093–2104.
Kojima J, Yamaji Y, Matsumura M, Nambu A, Inase M, Tokuno H et al (1997). Excitotoxic lesions of the pedunculopontine tegmental nucleus produce contralateral hemiparkinsonism in the monkey. Neurosci Lett 226: 111–114.
Koller WC (1986). Pharmacologic treatment of parkinsonian tremor. Arch Neurol 43: 126–127.
Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW (2008a). Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med 14: 504–506. Case reports of evidence for Lewy body pathology in transplanted dopaminergic neurons in the striatum of parkinsonian patients.
Kordower JH, Chu Y, Hauser RA, Olanow CW, Freeman TB (2008b). Transplanted dopaminergic neurons develop PD pathologic changes: a second case report. Mov Disord 23: 2303–2306.
Kordower JH, Emborg ME, Bloch J, Ma SY, Chu Y, Leventhal L et al (2000). Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson's disease. Science 290: 767–773.
Kordower JH, Herzog CD, Dass B, Bakay RA, Stansell III J, Gasmi M et al (2006). Delivery of neurturin by AAV2 (CERE-120)-mediated gene transfer provides structural and functional neuroprotection and neurorestoration in MPTP-treated monkeys. Ann Neurol 60: 706–715.
Kotzbauer PT, Lampe PA, Heuckeroth RO, Golden JP, Creedon DJ, Johnson Jr EM et al (1996). Neurturin, a relative of glial-cell-line-derived neurotrophic factor. Nature 384: 467–470.
Krack P, Vercueil L (2001). Review of the functional surgical treatment of dystonia. Eur J Neurol 8: 389–399.
Krauss JK, Pohle T, Weigel R, Burgunder JM (2002). Deep brain stimulation of the centre median-parafascicular complex in patients with movement disorders. J Neurol Neurosurg Psychiatry 72: 546–548.
Kulisevsky J, Berthier ML, Gironell A, Pascual-Sedano B, Molet J, Pares P (2002). Mania following deep brain stimulation for Parkinson's disease. Neurology 59: 1421–1424.
Kumar R, Krack P, Pollak P (1999a). Transient acute depression induced by high-frequency deep-brain stimulation. N Engl J Med 341: 1003–1004; discussion 1004.
Kumar R, Lozano AM, Sime E, Halket E, Lang AE (1999b). Comparative effects of unilateral and bilateral subthalamic nucleus deep brain stimulation. Neurology 53: 561–566.
Kuwajima M, Dehoff MH, Furuichi T, Worley PF, Hall RA, Smith Y (2007). Localization and expression of group I metabotropic glutamate receptors in the mouse striatum, globus pallidus, and subthalamic nucleus: regulatory effects of MPTP treatment and constitutive Homer deletion. J Neurosci 27: 6249–6260.
Kuwajima M, Hall RA, Aiba A, Smith Y (2004). Subcellular and subsynaptic localization of group I metabotropic glutamate receptors in the monkey subthalamic nucleus. J Comp Neurol 474: 589–602.
Laitinen LV, Bergenheim AT, Hariz MI (1992). Ventroposterolateral pallidotomy can abolish all parkinsonian symptoms. Stereotact Funct Neurosurg 58: 14–21. Clinical evidence that launched the rejuvenation of the use of pallidotomy as treatment for Parkinson's disease.
Lane EL, Bjorklund A, Dunnett SB, Winkler C (2010). Neural grafting in Parkinson's disease unraveling the mechanisms underlying graft-induced dyskinesia. Prog Brain Res 184: 295–309.
Lane EL, Winkler C, Brundin P, Cenci MA (2006). The impact of graft size on the development of dyskinesia following intrastriatal grafting of embryonic dopamine neurons in the rat. Neurobiol Dis 22: 334–345.
Lang AE (2009). When and how should treatment be started in Parkinson disease? Neurology 72: S39–S43.
Lang AE, Gill S, Patel NK, Lozano A, Nutt JG, Penn R et al (2006). Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol 59: 459–466.
Lang AE, Langston JW, Stoessl AJ, Brodsky M, Brooks DJ, Dhawan V et al (2006). GDNF in treatment of Parkinson's disease: response to editorial. Lancet Neurol 5: 200–202.
Lang AE, Mikulis D (2009). A new sensitive imaging biomarker for Parkinson disease? Neurology 72: 1374–1375.
Langmead CJ, Watson J, Reavill C (2008). Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol Ther 117: 232–243.
Langston JW (2006). The Parkinson's complex: parkinsonism is just the tip of the iceberg. Ann Neurol 59: 591–596.
Langston JW, Ballard Jr PA (1983). Parkinson's disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N Engl J Med 309: 310–315.
Lefaucheur JP (2009). Treatment of Parkinson's disease by cortical stimulation. Expert Rev Neurother 9: 1755–1771.
Leroi I, Brandt J, Reich SG, Lyketsos CG, Grill S, Thompson R et al (2004). Randomized placebo-controlled trial of donepezil in cognitive impairment in Parkinson's disease. Int J Geriatr Psychiatry 19: 1–8.
Leroi I, Overshott R, Byrne EJ, Daniel E, Burns A (2009). Randomized controlled trial of memantine in dementia associated with Parkinson's disease. Mov Disord 24: 1217–1221.
Lim SY, Lang AE (2010). The nonmotor symptoms of Parkinson's disease—an overview. Mov Disord 25 (Suppl 1): S123–S130.
Levi-Montalcini R, Hamburger V (1951). Selective growth stimulating effects of mouse sarcoma on the sensory and sympathetic nervous system of the chick embryo. J Exp Zool 116: 321–361.
LeWitt PA (2004). Subcutaneously administered apomorphine: pharmacokinetics and metabolism. Neurology 62: S8–S11.
LeWitt PA, Guttman M, Tetrud JW, Tuite PJ, Mori A, Chaikin P et al (2008). Adenosine A2A receptor antagonist istradefylline (KW-6002) reduces “off” time in Parkinson's disease: a double-blind, randomized, multicenter clinical trial (6002-US-005). Ann Neurol 63: 295–302.
LeWitt PA, Rezai AR, Leehey MA, Ojemann SG, Flaherty AW, Eskandar EN et al (2011). AAV2-GAD gene therapy for advanced Parkinson's disease: a double-blind, sham-surgery controlled, randomised trial. Lancet Neurol 10: 309–319. First report of the results of a double-blind placebo controlled trial for gene therapy in PD.
Lewy FH (1912). Paralysis agitans. Pathologishe anatomie. In: Lewandowsky M (ed). Handbook der Neurologie. Springer: Berlin. pp 920–933.
Lezcano E, Gomez-Esteban JC, Zarranz JJ, Lambarri I, Madoz P, Bilbao G et al (2004). Improvement in quality of life in patients with advanced Parkinson's disease following bilateral deep-brain stimulation in subthalamic nucleus. Eur J Neurol 11: 451–454.
Li JY, Christophersen NS, Hall V, Soulet D, Brundin P (2008a). Critical issues of clinical human embryonic stem cell therapy for brain repair. Trends Neurosci 31: 146–153. Concise assessment of the potential promises and problems stem cell therapy faces as treatment for Parkinson's disease.
Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ et al (2008b). Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med 14: 501–503.
Li JY, Englund E, Widner H, Rehncrona S, Bjorklund A, Lindvall O et al (2010). Characterization of Lewy body pathology in 12- and 16-year-old intrastriatal mesencephalic grafts surviving in a patient with Parkinson's disease. Mov Disord 25: 1091–1096.
Li S, Arbuthnott GW, Jutras MJ, Goldberg JA, Jaeger D (2007). Resonant antidromic cortical circuit activation as a consequence of high-frequency subthalamic deep-brain stimulation. J Neurophysiol 98: 3525–3537.
Liberini P, Parola S, Spano PF, Antonini L (2000). Olfaction in Parkinson's disease: methods of assessment and clinical relevance. J Neurol 247: 88–96.
Lilleeng B, Dietrichs E (2008). Unmasking psychiatric symptoms after STN deep brain stimulation in Parkinson's disease. Acta Neurol Scand Suppl 188: 41–45.
Limousin P, Pollak P, Benazzouz A, Hoffmann D, Broussolle E, Perret JE et al (1995a). Bilateral subthalamic nucleus stimulation for severe Parkinson's disease. Mov Disord 10: 672–674. Early evidence that deep brain stimulation of the STN alleviates motor symptoms in PD.
Limousin P, Pollak P, Benazzouz A, Hoffmann D, Le Bas JF, Broussolle E et al (1995b). Effect of parkinsonian signs and symptoms of bilateral subthalamic nucleus stimulation. Lancet 345: 91–95.
Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F (1993). GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 260: 1130–1132.
Lindsley CW, Niswender CM, Engers DW, Hopkins CR (2009). Recent progress in the development of mGluR4 positive allosteric modulators for the treatment of Parkinson's disease. Curr Top Med Chem 9: 949–963.
Lindvall O, Rehncrona S, Brundin P, Gustavii B, Astedt B, Widner H et al (1989). Human fetal dopamine neurons grafted into the striatum in two patients with severe Parkinson's disease. A detailed account of methodology and a 6-month follow-up. Arch Neurol 46: 615–631.
Lloyd K, Hornykiewicz O (1970). Parkinson's disease: activity of L-dopa decarboxylase in discrete brain regions. Science 170: 1212–1213.
Lopez S, Turle-Lorenzo N, Acher F, De Leonibus E, Mele A, Amalric M (2007). Targeting group III metabotropic glutamate receptors produces complex behavioral effects in rodent models of Parkinson's disease. J Neurosci 27: 6701–6711.
Lopez S, Turle-Lorenzo N, Johnston TH, Brotchie JM, Schann S, Neuville P et al (2008). Functional interaction between adenosine A2A and group III metabotropic glutamate receptors to reduce parkinsonian symptoms in rats. Neuropharmacology 55: 483–490.
Lundblad M, Vaudano E, Cenci MA (2003). Cellular and behavioural effects of the adenosine A2a receptor antagonist KW-6002 in a rat model of l-DOPA-induced dyskinesia. J Neurochem 84: 1398–1410.
Luo J, Kaplitt MG, Fitzsimons HL, Zuzga DS, Liu Y, Oshinsky ML et al (2002). Subthalamic GAD gene therapy in Parkinson's disease rat model. Science 298: 425–429.
Lyons KE, Pahwa R (2005). Long-term benefits in quality of life provided by bilateral subthalamic stimulation in patients with Parkinson disease. J Neurosurg 103: 252–255.
Lyons KE, Wilkinson SB, Tröster AI, Pahwa R (2002). Long-term efficacy of globus pallidus stimulation for the treatment of Parkinson's disease. Stereotact Funct Neurosurg 79: 214–220.
Madrazo I, León V, Torres C, Aguilera MC, Varela G, Alvarez F et al (1988). Transplantation of fetal substantia nigra and adrenal medulla to the caudate nucleus in two patients with Parkinson's disease. N Engl J Med 318: 51–60.
Marek K, Jennings D (2009). Can we image premotor Parkinson disease? Neurology 72: S21–S26.
Marin C, Bonastre M, Aguilar E, Jimenez A (2011). The metabotropic glutamate receptor antagonist 2-methyl-6-(phenylethynyl) pyridine decreases striatal VGlut2 expression in association with an attenuation of L-dopa-induced dyskinesias. Synapse 65: 1080–1086.
Marino MJ, Conn PJ (2006). Glutamate-based therapeutic approaches: allosteric modulators of metabotropic glutamate receptors. Curr Opin Pharmacol 6: 98–102.
Marino MJ, Williams Jr DL, O’Brien JA, Valenti O, McDonald TP, Clements MK et al (2003). Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach of Parkinson's disease treatment. Proc Natl Acad Sci 100: 13668–13673.
Marino MJ, Wittmann M, Bradley SR, Hubert GW, Smith Y, Conn PJ (2001). Activation of group I metabotropic glutamate receptors produces a direct excitation and disinhibition of GABAergic projection neurons in the substantia nigra pars reticulata. J Neurosci 21: 7001–7012.
Marks Jr WJ, Bartus RT, Siffert J, Davis CS, Lozano A, Boulis N et al (2010). Gene delivery of AAV2-neurturin for Parkinson's disease: a double-blind, randomised, controlled trial. Lancet Neurol 9: 1164–1172.
Marks Jr WJ, Ostrem JL, Verhagen L, Starr PA, Larson PS, Bakay RA et al (2008). Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson's disease: an open-label, phase I trial. Lancet Neurol 7: 400–408.
Martin AJ, Larson PS, Ostrem JL, Keith Sootsman W, Talke P, Weber OM et al (2005). Placement of deep brain stimulator electrodes using real-time high-field interventional magnetic resonance imaging. Magn Reson Med 54: 1107–1114.
Martinez-Martin P, Valldeoriola F, Tolosa E, Pilleri M, Molinuevo JL, Rumia J et al (2002). Bilateral subthalamic nucleus stimulation and quality of life in advanced Parkinson's disease. Mov Disord 17: 372–377.
Masilamoni G, Bogenpohl JW, Alagille D, Delevich K, Tamagnan G, Votaw JR et al (2011). Metabotropic glutamate receptor 5 antagonist protects dopaminergic and noradrenergic neurons from degeneration in MPTP-treated monkeys. Brain 134: 2057–2073.
May LT, Avlani VA, Langmead CJ, Herdon HJ, Wood MD, Sexton PM et al (2007). Structure-function studies of allosteric agonism at M2 muscarinic acetylcholine receptors. Mol Pharmacol 72: 463–476.
Mazzone P, Insola A, Lozano A, Galati S, Scarnati E, Peppe A et al (2007). Peripeduncular and pedunculopontine nuclei: a dispute on a clinically relevant target. Neuroreport 18: 1407–1408.
Mazzone P, Sposato S, Insola A, Dilazzaro V, Scarnati E (2008). Stereotactic surgery of nucleus tegmenti pedunculopontine [corrected]. Br J Neurosurg 22 (Suppl 1): S33–S40.
McCulloch CC, Kay DM, Factor SA, Samii A, Nutt JG, Higgins DS et al (2008). Exploring gene-environment interactions in Parkinson's disease. Hum Genet 123: 257–265.
McKinnon JH, Demaerschalk BM, Caviness JN, Wellik KE, Adler CH, Wingerchuk DM (2007). Sniffing out Parkinson disease: can olfactory testing differentiate parkinsonian disorders? Neurologist 13: 382–385.
Mela F, Marti M, Dekundy A, Danysz W, Morari M, Cenci MA (2007). Antagonism of metabotropic glutamate receptor type 5 attenuates l-DOPA-induced dyskinesia and its molecular and neurochemical correlates in a rat model of Parkinson's disease. J Neurochem 101: 483–497.
Mena-Segovia J, Bolam JP, Magill PJ (2004). Pedunculopontine nucleus and basal ganglia: distant relatives or part of the same family? Trends Neurosci 27: 585–588.
Mendez I, Viñuela A, Astradsson A, Mukhida K, Hallett P, Robertson H et al (2008). Dopamine neurons implanted into people survive without pathology for 14 years. Nat Med 14: 507–509.
Menon R, Stacy MA (2008). Adenosine A2A receptor antagonists. In: Factor SA, Weiner WJ (eds). Parkinson's Disease-Diagnostic and Clinical Management. Demos: New York. pp 557–562.
Menza M, Dobkin RD, Marin H, Mark MH, Gara M, Buyske S et al (2009). A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 72: 886–892.
Merello M, Nouzeilles MI, Cammarota A, Leiguarda R (1999). Effect of memantine (NMDA antagonist) on Parkinson's disease: a double-blind crossover randomized study. Clin Neuropharmacol 22: 273–276.
Merello M, Tenca E, Pérez Lloret S, Martín ME, Bruno V, Cavanagh S et al (2008). Prospective randomized 1-year follow-up comparison of bilateral subthalamotomy verus bilateral subthalamic stimulation and the combination of both in Parkinson's disease patients: a pilot study. Br J Neurosurg 22: 415–422.
Meyers R (1940). Surgical procedure for postencephalitic tremor, with notes on the physiology of premotor fibres. Arch Neurol Psychiatry 44: 455–459.
Miyasaki JM, Martin W, Suchowersky O, Weiner WJ, Lang AE (2002). Practice parameter: initiation of treatment for Parkinson's disease: an evidence-based review: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 58: 11–17.
Mizuno Y, Hasegawa K, Kondo T, Kuno S, Yamamoto M (2010). Clinical efficacy of istradefylline (KW-6002) in Parkinson's disease: a randomized, controlled study. Mov Disord 25: 1437–1443.
Montastruc JL, Rascol O, Senard JM, Rascol A (1992). A pilot study of N-methyl-D-aspartate (NMDA) antagonist in Parkinson's disease. J Neurol Neurosurg Psychiatry 55: 630–631.
Montel SR, Bungener C (2009). Coping and quality of life of patients with Parkinson disease who have undergone deep brain stimulation of the subthalamic nucleus. Surg Neurol 72: 105–110; discussion 110–111.
Morelli M, Carta AR, Jenner P (2009). Adenosine A2A receptors and Parkinson's disease. Handb Exp Pharmacol 589–615. A review of the anatomical, neurochemical and functional evidence supporting the use of adenosine A2A receptor antagonists as antiparkinsonian therapy.
Morelli M, Carta AR, Kachroo A, Schwarzschild MA (2010). Pathophysiological roles for purines: adenosine, caffeine and urate. Prog Brain Res 183: 183–208.
Morelli M, Di Paolo T, Wardas J, Calon F, Xiao D, Schwarzschild MA (2007). Role of adenosine A2A receptors in parkinsonian motor impairment and l-DOPA-induced motor complications. Prog Neurobiol 83: 293–309.
Morley JF, Duda JE (2010). Olfaction as a biomarker in Parkinson's disease. Biomark Med 4: 661–670.
Moro E, Lozano AM, Pollak P, Agid Y, Rehncrona S, Volkmann J et al (2010). Long-term results of a multicenter study on subthalamic and pallidal stimulation in Parkinson's disease. Mov Disord 25: 578–586.
Morrison CE, Borod JC, Perrine K, Beric A, Brin MF, Rezai A et al (2004). Neuropsychological functioning following bilateral subthalamic nucleus stimulation in Parkinson's disease. Archives of Clinical Neuropsychology 19: 165–181.
Muller A, Mungersdorf M, Reichmann H, Strehle G, Hummel T (2002a). Olfactory function in Parkinsonian syndromes. J Clin Neurosci 9: 521–524.
Muller A, Reichmann H, Livermore A, Hummel T (2002b). Olfactory function in idiopathic Parkinson's disease (IPD): results from cross-sectional studies in IPD patients and long-term follow-up of de-novo IPD patients. J Neural Transm 109: 805–811.
Muller CM, de Vos RA, Maurage CA, Thal DR, Tolnay M, Braak H (2005). Staging of sporadic Parkinson disease-related alpha-synuclein pathology: inter- and intra-rater reliability. J Neuropathol Exp Neurol 64: 623–628.
Nagatsu I, Kondo Y, Inagaki S, Kojima H, Nagatsu T (1979). Immunofluorescent and biochemical studies on tyrosine hydroxylase and dopamine-beta-hydroxylase of the bullfrog sciatic nerves. Histochemistry 61: 103–109.
Nakanishi S, Nakajima Y, Masu M, Ueda Y, Nakahara K, Watanabe D et al (1998). Glutamate receptors: brain function and signal transduction. Brain Res Rev 26: 230–235.
Nandi D, Aziz TZ, Giladi N, Winter J, Stein JF (2002a). Reversal of akinesia in experimental parkinsonism by GABA antagonist microinjections in the pedunculopontine nucleus. Brain 125: 2418–2430.
Nandi D, Liu X, Winter JL, Aziz TZ, Stein JF (2002b). Deep brain stimulation of the pedunculopontine region in the normal non-human primate. J Clin Neurosci 9: 170–174.
Nash JE, Hill MP, Brotchie JM (1999). Antiparkinsonian actions of blockade of NR2B-containing NMDA receptors in the reserpine-treated rat. Exp Neurol 155: 42–48.
Nash JE, Fox SH, Henry B, Hill MP, Peggs D, McGuire S et al (2000). Antiparkinsonian actions of infenprodil in the MPTP-lesioned marmoset model of Parkinson's disease. Exp Neurol 165: 136–142.
Nicoletti F, Bockaert J, Collingridge GL, Conn PJ, Ferraguti F, Schoepp DD et al (2011). Metabotropic glutamate receptors: From the workbench to the bedside. Neuropharmacology 60: 1017–1041. A review of the preclinical studies that led to the development of mGluR-related drug therapies for brain diseases.
Nilsson D, Nyholm D, Aquilonius SM (2001). Duodenal levodopa infusion in Parkinson's disease—long-term experience. Acta Neurol Scand 104: 343–348.
Nisipeanu PF, Korczyn AD (2008). Dopamine agonists. In: Factor SA, Weiner WJ (eds). Parkinson's Disease-Diagnostic and Clinical Management. Demos: New York. pp 515–532.
Niswender CM, Conn PJ (2010). Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50: 295–322. A comprehensive review of the physiology, pharmacology and therapeutic relevance of metabotropic glutamate receptors as drug targets for brain disorders.
Niswender CM, Johnson KA, Weaver CD, Jones CK, Xiang Z, Luo Q et al (2008). Discovery, characterization, and antiparkinsonian effect of novel positive allosteric modulators of metabotropic glutamate receptor 4. Mol Pharmacol 74: 1345–1358.
Nutt JG (2000). Clinical pharmacology of levodopa-induced dyskinesia. Ann Neurol 47: S160–S164; discussion S164–S166.
Nutt JG, Burchiel KJ, Comella CL, Jankovic J, Lang AE, Laws Jr ER et al (2003). Randomized, double-blind trial of glial cell line-derived neurotrophic factor (GDNF) in PD. Neurology 60: 69–73.
Nutt JG, Obeso JA, Stocchi F (2000). Continuous dopamine-receptor stimulation in advanced Parkinson's disease. Trends Neurosci 23: S109–S115.
Nyholm D, Aquilonius SM (2004). Levodopa infusion therapy in Parkinson disease: state of the art in 2004. Clin Neuropharmacol 27: 245–256.
Obeso JA, Guridi J, DeLong M (1997). Surgery for Parkinson's disease. J Neurol Neurosurg Psychiatry 62: 2–8.
Obeso JA, Olanow CW, Nutt JG (2000). Levodopa motor complications in Parkinson's disease. Trends Neurosci 23: S2–S7. A detailed assessment of the main motor side effects of chronic L-DOPA therapy in PD.
Obeso JA, Schapira AH (2009). Compensatory mechanisms in Parkinson's disease. Mov Disord 24: 153–154.
Odin P, Wolters E, Antonini A (2008). Continuous dopaminergic stimulation achieved by duodenal levodopa infusion. Neurol Sci 29 (Suppl 5): S387–S388.
Oh JD, Bibbiani F, Chase TN (2002). Quetiapine attenuates levodopa-induced motor complications in rodent and primate parkinsonian models. Exp Neurol 177: 557–564.
Oh JD, Chase TN (2002). Glutamate-mediated striatal dysregulation and the pathogenesis of motor response complications in Parkinson's disease. Amino Acids 23: 133–139.
Okun MS, Fernandez HH, Wu SS, Kirsch-Darrow L, Bowers D, Bova F et al (2009). Cognition and mood in Parkinson's disease in subthalamic nucleus vs globus pallidus interna deep brain stimulation: the COMPARE trial. Ann Neurol 65: 586–595.
Okun MS, Green J, Saben R, Gross R, Foote KD, Vitek JL (2003). Mood changes with deep brain stimulation of STN and GPi: results of a pilot study. J Neurol Neurosurg Psychiatry 74: 1584–1586.
Olanow CW (1990). Oxidation reactions in Parkinson's disease. Neurology 40 (Suppl 3): S32–S37.
Olanow CW (2008). Levodopa/dopamine replacement strategies in Parkinson's disease—future directions. Mov Disord 23 (Suppl 3): S613–S622.
Olanow CW (2009). Can we achieve neuroprotection with currently available anti-parkinsonian interventions? Neurology 72: S59–S64.
Olanow CW, Goetz CG, Kordower JH, Stoessl AJ, Sossi V, Brin MF et al (2003). A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson's disease. Ann Neurol 54: 403–414. Results of a randomized double blind placebo-controlled trial showing the lack of efficacy of dopaminergic neurons striatal transplantation in PD.
Olanow CW, Agid Y, Mizuno Y, Albanese A, Bonuccelli U, Damier P et al (2004). Levodopa in the treatment of Parkinson's disease: current controversies. Mov Disord 19: 997–1005.
Olanow CW, Obeso JA, Stocchi F (2006a). Continuous dopamine-receptor treatment of Parkinson's disease: scientific rationale and clinical implications. Lancet Neurol 5: 677–687.
Olanow CW, Obeso JA, Stocchi F (2006b). Drug insight: Continuous dopaminergic stimulation in the treatment of Parkinson's disease. Nat Clin Pract Neurol 2: 382–392.
Ondo W, Jankovic J, Schwartz K, Almaguer M, Simpson RK (1998). Unilateral thalamic deep brain stimulation for refractory essential tremor and Parkinson's disease tremor. Neurology 51: 1063–1069.
Ondo WG, Sethi KD, Kricorian G (2007). Selegiline orally disintegrating tablets in patients with Parkinson disease and “wearing off” symptoms. Clin Neuropharmacol 30: 295–300.
Ondo WG, Shinawi L, Davidson A, Lai D (2011). Memantine for non-motor features of Parkinson's disease: a double-blind placebo controlled exploratory pilot trial. Parkinsonism Relat Disord 17: 156–159.
Ondo WG, Tintner R, Voung KD, Lai D, Ringholz G (2005). Double-blind, placebo-controlled, unforced titration parallel trial of quetiapine for dopaminergic-induced hallucinations in Parkinson's disease. Mov Disord 20: 958–963.
Ossowska K, Konieczny J, Wardas J, Golembiowska K, Wolfarth S, Pilc A (2002). The role of striatal metabotropic glutamate receptors in Parkinson's disease. Amino Acids 23: 193–198.
Ossowska K, Konieczny J, Wolfarth S, Pilc A (2005). MTEP, a new selective antagonist of the metabotropic glutamate receptor subtype 5 (mGluR5), produces antiparkinsonian-like effects in rats. Neuropharmacology 49: 447–455.
Ouattara B, Gasparini F, Morissette M, Gregoire L, Samadi P, Gomez-Mancilla B et al (2010). Effect of L-Dopa on metabotropic glutamate receptor 5 in the brain of parkinsonian monkeys. J Neurochem 113: 715–724.
Ouattara B, Gregoire L, Morissette M, Gasparini F, Vranesic I, Bilbe G et al (2011). Metabotropic glutamate receptor type 5 in levodopa-induced motor complications. Neurobiol Aging 32: 1286–1295.
Ouchi Y, Yoshikawa E, Sekine Y, Futatsubashi M, Kanno T, Ogusu T et al (2005). Microglial activation and dopamine terminal loss in early Parkinson's disease. Ann Neurol 57: 168–175.
Ozelius LJ, Senthil G, Saunders-Pullman R, Ohmann E, Deligtisch A, Tagliati M et al (2006). LRRK2 G2019S as a cause of Parkinson's disease in Ashkenazi Jews. N Engl J Med 354: 424–425.
Pagni CA, Albanese A, Bentivoglio A, Broggi G, Canavero S, Cioni B et al (2008). Results by motor cortex stimulation in treatment of focal dystonia, Parkinson's disease and post-ictal spasticity. The experience of the Italian Study Group of the Italian Neurosurgical Society. Acta Neurochir Suppl 101: 13–21.
Pankratz N, Foroud T (2007). Genetics of Parkinson disease. Genet Med 9: 801–811.
Papa SM, Chase TN (1996). Levodopa-induced dyskinesias improved by a glutamate antagonist in Parkinsonian monkeys. Ann Neurol 39: 574–578.
Paquet M, Smith Y (2003). Group I metabotropic glutamate receptors in the monkey striatum: subsynaptic association with glutamatergic and dopaminergic afferents. J Neurosci 23: 7659–7669.
Parkin S, Nandi D, Giladi N, Joint C, Gregory R, Bain P et al (2001). Lesioning the subthalamic nucleus in the treatment of Parkinson's disease. Stereotact Funct Neurosurg 77: 68–72.
Parkinson J (1817). An essay on the Shaking Palsy. Whiitngham and Rowland: London.
Parkinson Study Group (1999). Low-dose clozapine for the treatment of drug-induced psychosis in Parkinson's disease. N Engl J Med 340: 757–763.
Parkinson Study Group (2000). Pramipexole vs levodopa as initial treatment of Parkinson's disease: A randomized controlled trial. JAMA 284: 1931–1938.
Parkinson Study Group (2002). A controlled trial of rasagiline in early Parkinson disease: the TEMPO study. Arch Neurol 59: 1937–1943.
Parkinson Study Group (2005). A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: the PRESTO study. Arch Neurol 62: 241–248.
Patel NK, Heywood P, O’Sullivan K, McCarter R, Love S, Gill SS (2003). Unilateral subthalamotomy in the treatment of Parkinson's disease. Brain 126: 1136–1145.
Penn RD, Dalvi A, Slevin J, Young B, Gash D, Gerhardt G et al (2006). GDNF in treatment of Parkinson's disease: response to editorial. Lancet Neurol 5: 202–203.
Peppe A, Gasbarra A, Stefani A, Chiavalon C, Pierantozzi M, Fermi E et al (2008). Deep brain stimulation of CM/Pf of thalamus could be the new elective target for tremor in advanced Parkinson's disease. Parkinsonism Relat Disord 14: 501–504.
Peppe A, Pierantozzi M, Chiavalon C, Marchetti F, Caltagirone C, Musicco M et al (2010). Deep brain stimulation of the pedunculopontine tegmentum and subthalamic nucleus: effects on gait in Parkinson's disease. Gait Posture 32: 512–518.
Pereira EA, Muthusamy KA, De Pennington N, Joint CA, Aziz TZ (2008). Deep brain stimulation of the pedunculopontine nucleus in Parkinson's disease. Preliminary experience at Oxford. Br J Neurosurg 22 (Suppl 1): S41–S44.
Piccini P, Whone A (2004). Functional brain imaging in the differential diagnosis of Parkinson's disease. Lancet Neurol 3: 284–290.
Pierantozzi M, Palmieri MG, Galati S, Stanzione P, Peppe A, Tropepi D et al (2008). Pedunculopontine nucleus deep brain stimulation changes spinal cord excitability in Parkinson's disease patients. J Neural Transm 115: 731–735.
Pin JP, Duvoisin R (1995). The metabotropic glutamate receptors: Structure and functions. Neuropharmacology 34: 1–26.
Plaha P, Ben-Shlomo Y, Patel NK, Gill SS (2006). Stimulation of the caudal zona incerta is superior to stimulation of the subthalamic nucleus in improving contralateral parkinsonism. Brain 129: 1732–1747.
Plaha P, Gill SS (2005). Bilateral deep brain stimulation of the pedunculopontine nucleus for Parkinson's disease. Neuroreport 16: 1883–1887.
Poewe W (2010). Parkinson disease: treatment of the nonmotor symptoms of Parkinson's disease. Nat Rev Neurol 6: 417–418. Comprehensive overview of the current status of treatment for nonmotor symptoms of PD.
Poewe W, Wolters E, Emre M, Onofrj M, Hsu C, Tekin S et al (2006). Long-term benefits of rivastigmine in dementia associated with Parkinson's disease: an active treatment extension study. Mov Disord 21: 456–461.
Poisik OV, Mannaioni G, Traynelis S, Smith Y, Conn PJ (2003). Distinct functional roles of the metabotropic glutamate receptors 1 and 5 in the rat globus pallidus. J Neurosci 23: 122–130.
Poisik OV, Smith Y, Conn PJ (2007). D1- and D2-like dopamine receptors regulate signaling properties of group I metabotropic glutamate receptors in the globus pallidus. Eur J Neurosci 26: 852–862.
Politis M, Wu K, Loane C, Quinn NP, Brooks DJ, Rehncrona S et al (2010). Serotonergic neurons mediate dyskinesia side effects in Parkinson's patients with neural transplants. Sci Transl Med 2: 38ra46.
Pollak P, Tison F, Rascol O, Destee A, Pere JJ, Senard JM et al (2004). Clozapine in drug induced psychosis in Parkinson's disease: a randomised, placebo controlled study with open follow up. J Neurol Neurosurg Psychiatry 75: 689–695.
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A et al (1997). Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276: 2045–2047.
Ponsen MM, Stoffers D, Booij J, van Eck-Smit BL, Wolters E, Berendse HW (2004). Idiopathic hyposmia as a preclinical sign of Parkinson's disease. Ann Neurol 56: 173–181.
Portman AT, van Laar T, Staal MJ, Rutgers AW, Journee HL, Leenders KL (2006). Chronic stimulation of the subthalamic nucleus increases daily on-time without dyskinesia in advanced Parkinson's disease. Parkinsonism Relat Disord 12: 143–148.
Powers KM, Kay DM, Factor SA, Zabetian CP, Higgins DS, Samii A et al (2008). Combined effects of smoking, coffee, and NSAIDs on Parkinson's disease risk. Mov Disord 23: 88–95.
Pruszak J, Isacson O (2009). Molecular and cellular determinants for generating ES-cell derived dopamine neurons for cell therapy. Adv Exp Med Biol 651: 112–123.
Raab W, Gigee W (1951). Concentration and distribution of “encephalin” in the brain of humans and animals. Proc Soc Exp Biol Med 180: 1200.
Rabey JM, Prokhorov T, Miniovitz A, Dobronevsky E, Klein C (2007). Effect of quetiapine in psychotic Parkinson's disease patients: a double-blind labeled study of 3 months’ duration. Mov Disord 22: 313–318.
Rajput A, Zesiewicz TA, Hauser RA (2008). Monoamine oxidase inhibitors. In: Factor SA, Weiner WJ (eds). Parkinson's Disease-Diagnostic and Clinical Management. Demos: New York. pp. 499–513.
Rangasamy SB, Soderstrom K, Bakay RA, Kordower JH (2010). Neurotrophic factor therapy for Parkinson's disease. Prog Brain Res 184: 237–264.
Rascol O (2009). ‘Disease-modification’ trials in Parkinson disease: target populations, endpoints and study design. Neurology 72: S51–S58.
Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE (2000). A five-year study of the incidenceof dyskinesia in patients with early Parkinson's disease who were treated with ropinirole or levodopa. N Engl J Med 342: 1484–1491.
Rascol O, Brooks DJ, Melamed E, Oertel W, Poewe W, Stocchi F et al (2005). Rasagiline as an adjunct to levodopa in patients with Parkinson's disease and motor fluctuations (LARGO, Lasting effect in Adjunct therapy with Rasagiline Given Once daily, study): a randomised, double-blind, parallel-group trial. Lancet 365: 947–954.
Rauch F, Schwabe K, Krauss JK (2010). Effect of deep brain stimulation in the pedunculopontine nucleus on motor function in the rat 6-hydroxydopamine Parkinson model. Behav Brain Res 210: 46–53.
Ravina B, Putt M, Siderowf A, Farrar JT, Gillespie M, Crawley A et al (2005). Donepezil for dementia in Parkinson's disease: a randomised, double blind, placebo controlled, crossover study. J Neurol Neurosurg Psychiatry 76: 934–939.
Richard IH, McDermott M, McDonald WM (2010). A multicenter randomized, double-blind, placebo controlled trial of Paroxetine and Venlafaxine extended release for depression in Parkinson's disease. 2nd World Parkinson Congress Proceedings, P19.18.
Rinne JO (1987). Early combination of bromocriptine and levodopa in the treatment of Parkinson's disease: a 5-year follow-up. Neurology 37: 826–828.
Rinne JO, Portin R, Ruottinen H, Nurmi E, Bergman J, Haaparanta M et al (2000). Cognitive impairment and the brain dopaminergic system in Parkinson disease: [18F]fluorodopa positron emission tomographic study. Arch Neurol 57: 470–475.
Rodrigues JP, Walters SE, Watson P, Stell R, Mastaglia FL (2007a). Globus pallidus stimulation improves both motor and nonmotor aspects of quality of life in advanced Parkinson's disease. Mov Disord 22: 1866–1870.
Rodrigues JP, Walters SE, Watson P, Stell R, Mastaglia FL (2007b). Globus pallidus stimulation in advanced Parkinson's disease. J Clin Neurosci 14: 208–215.
Rodriguez AL, Grier MD, Jones CK, Herman EJ, Kane AS, Smith RL et al (2010). Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Mol Pharmacol 78: 1105–1123.
Rodriguez-Oroz MC, Obeso JA, Lang AE, Houeto JL, Pollak P, Rehncrona S et al (2005). Bilateral deep brain stimulation in Parkinson's disease. A multicenter study with 4 years follow-up. Brain 128: 2240–2249.
Rodriguez-Oroz MC, Zamarbide I, Guridi J, Palmero MR, Obeso JA (2004). Efficacy of deep brain stimulation of the subthalamic nucleus in Parkinson's disease 4 years after surgery: double blind and open label evaluation. J Neurol Neurosurg Psychiatry 75: 1382–1385.
Romito LM, Raja M, Daniele A, Contarino MF, Bentivoglio AR, Barbier A et al (2002). Transient mania with hypersexuality after surgery for high frequency stimulation of the subthalamic nucleus in Parkinson's disease. Mov Disord 17: 1371–1374.
Ross GW, Abbott RD, Petrovitch H, White LR, Tanner CM (2000). Relationship between caffeine intake and parkinson disease. JAMA 284: 1378–1379.
Ross GW, Petrovitch H, Abbott RD, Tanner CM, Popper J, Masaki K et al (2008). Association of olfactory dysfunction with risk for future Parkinson's disease. Ann Neurol 63: 167–173.
Rylander D, Recchia A, Mela F, Dekundy A, Danysz W, Cenci MA (2009). Pharmacological modulation of glutamate transmission in a rat model of L-DOPA-induced dyskinesia: effects on motor behavior and striatal nuclear signaling. J Pharmacol Exp Ther 330: 227–235.
Saint-Cyr JA, Trepanier LL, Kumar R, Lozano AM, Lang AE (2000). Neuropsychological consequences of chronic bilateral stimulation of the subthalamic nucleus in Parkinson's disease. Brain 123: 2091–2108.
Samadi P, Gregoire L, Morissette M, Calon F, Hadj Tahar A, Dridi M et al (2008). mGluR5 metabotropic glutamate receptors and dyskinesias in MPTP monkeys. Neurobiol Aging 29: 1040–1051.
Sanders WR (1865). Case of an unusual form of nervous disease, dystaxia or pseudo-paralysis agitans, with remarks. Edinburgh Med J 10: 987–997.
Sanchez-Pernaute R, Lee H, Patterson M, Reske-Nielsen C, Yoshizaki T, Sonntag KC et al (2008). Pathogenetic dopamine neurons from primate embryonic stem cells restore function in experimental Parkinson's disease. Brain 131: 2127–2139.
Saunders-Pullman R, Lipton RB, Senthil G, Katz M, Costan-Toth C, Derby C et al (2006). Increased frequency of the LRRK2 G2019S mutation in an elderly Ashkenazi Jewish population is not associated with dementia. Neurosci Lett 402: 92–96.
Schapira AH (2004). Disease modification in Parkinson's disease. Lancet Neurol 3: 362–368. Discussion of the current status of neuroprotective therapies in PD.
Schapira AH (2009a). Etiology and pathogenesis of Parkinson disease. Neurol Clin 27: 583–603, v.
Schapira AH (2009b). Molecular and clinical pathways to neuroprotection of dopaminergic drugs in Parkinson disease. Neurology 72: S44–S50.
Schapira AH (2009c). Neurobiology and treatment of Parkinson's disease. Trends Pharmacol Sci 30: 41–47.
Schapira AH, Agid Y, Barone P, Jenner P, Lemke MR, Poewe W et al (2009). Perspectives on recent advances in the understanding and treatment of Parkinson's disease. Eur J Neurol 16: 1090–1099.
Schapira AH, Olanow CW (2004). Neuroprotection in Parkinson disease: mysteries, myths, and misconceptions. JAMA 291: 358–364.
Schoepp DD, Conn PJ (1993). Metabotropic glutamate receptors in brain function and pathology. Trends Pharmacol Sci 14: 13–20.
Schroeder U, Kuehler A, Hennenlotter A, Haslinger B, Tronnier VM, Krause M et al (2004). Facial expression recognition and subthalamic nucleus stimulation. J Neurol Neurosurg Psychiatry 75: 648–650.
Schupbach WM, Chastan N, Welter ML, Houeto JL, Mesnage V, Bonnet AM et al (2005). Stimulation of the subthalamic nucleus in Parkinson's disease: a 5 year follow up. J Neurol Neurosurg Psychiatry 76: 1640–1644.
Schwab RS, England AC, Poskanzer DC, Young RR (1969). Amantadine in the treatment of Parkinson's disease. JAMA 208: 1168–1170.
Schwarzschild MA, Agnati L, Fuxe K, Chen JF, Morelli M (2006). Targeting adenosine A2A receptors in Parkinson's disease. Trends Neurosci 29: 647–654.
Sethi KD (2010). The impact of levodopa on quality of life in patients with Parkinson disease. Neurologist 16: 76–83.
Shah U, Hodgson R (2010). Recent progress in the discovery of adenosine A(2A) receptor antagonists for the treatment of Parkinson's disease. Curr Opin Drug Discov Devel 13: 466–480.
Shahlaie K, Larson PS, Starr PA (2011). Intraoperative computed tomography for deep brain stimulation surgery: technique and accuracy assessment. Neurosurgery 68: 114–124; discussion 124.
Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dube S, Mallinckrodt C et al (2008). Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry 165: 1033–1039.
Shimamoto SA, Larson PS, Ostrem JL, Glass GA, Turner RS, Starr PA (2010). Physiological identification of the human pedunculopontine nucleus. J Neurol Neurosurg Psychiatry 81: 80–86.
Siderowf A, Stern MB (2006). Preclinical diagnosis of Parkinson's disease: are we there yet? Curr Neurol Neurosci Rep 6: 295–301.
Siderowf A, Stern MB (2008). Premotor Parkinson's disease: clinical features, detection, and prospects for treatment. Ann Neurol 64 (Suppl 2): S139–S147.
Simola N, Morelli M, Pinna A (2008). Adenosine A2A receptor antagonists and Parkinson's disease: state of the art and future directions. Curr Pharm Des 14: 1475–1489.
Simuni T, Jaggi JL, Mulholland H, Hurtig HI, Colcher A, Siderowf AD et al (2002). Bilateral stimulation of the subthalamic nucleus in patients with Parkinson disease: a study of efficacy and safety. J Neurosurg 96: 666–672.
Simuni T, Hurtig H (2008). Levodopa: a pharmacologic miracle four decades later. In: Factor SA, Weiner WJ (eds). Parkinson's Disease-Diagnosis and Clinical Management. Demos: New York. pp 471–490.
Smeding HM, Esselink RA, Schmand B, Koning-Haanstra M, Nijhuis I, Wijnalda EM et al (2005). Unilateral pallidotomy vs bilateral subthalamic nucleus stimulation in PD—a comparison of neuropsychological effects. J Neurol 252: 176–182.
Smeding HM, Speelman JD, Huizenga HM, Schuurman PR, Schmand B (2011). Predictors of cognitive and psychosocial outcome after STN DBS in Parkinson Disease. J Neurol Neurosurg Psychiatry 82: 754–760.
Smeding HM, Speelman JD, Koning-Haanstra M, Schuurman PR, Nijssen P, van Laar T et al (2006). Neuropsychological effects of bilateral STN stimulation in Parkinson disease: a controlled study. Neurology 66: 1830–1836.
Smith Y, Charara A, Hanson JE, Paquet M, Levey AI (2000). GABA(B) and group I metabotropic glutamate receptors in the striatopallidal complex in primates. J Anat 196 (Part 4): 555–576.
Smith Y, Charara A, Paquet M, Kieval JZ, Pare JF, Hanson JE et al (2001). Ionotropic and metabotropic GABA and glutamate receptors in primate basal ganglia. J Chem Neuroanat 22: 13–42.
Soh SE, Morris ME, McGinley JL (2011). Determinants of health-related quality of life in Parkinson's disease: a systematic review. Parkinsonism Relat Disord 17: 1–9.
Sommer U, Hummel T, Cormann K, Mueller A, Frasnelli J, Kropp J et al (2004). Detection of presymptomatic Parkinson's disease: combining smell tests, transcranial sonography, and SPECT. Mov Disord 19: 1196–1202.
Spooren W, Lesage A, Lavreysen H, Gasparini F, Steckler T (2010). Metabotropic glutamate receptors: their therapeutic potential in anxiety. Curr Top Behav Neurosci 2: 391–413.
Stacy M, Silver D, Mendis T, Sutton J, Mori A, Chaikin P et al (2008). A 12-week, placebo-controlled study (6002-US-006) of istradefylline in Parkinson disease. Neurology 70: 2233–2240.
Starr MS (1995). Glutamate/dopamine D1/D2 balance in the basal ganglia and its relevance to Parkinson's disease. Synapse 19: 264–293.
Stefani A, Lozano AM, Peppe A, Stanzione P, Galati S, Tropepi D et al (2007). Bilateral deep brain stimulation of the pedunculopontine and subthalamic nuclei in severe Parkinson's disease. Brain 130: 1596–1607.
Stefani A, Peppe A, Pierantozzi M, Galati S, Moschella V, Stanzione P et al (2009). Multi-target strategy for Parkinsonian patients: the role of deep brain stimulation in the centromedian-parafascicularis complex. Brain Res Bull 78: 113–118.
Stefani A, Pierantozzi M, Ceravolo R, Brusa L, Galati S, Stanzione P (2010). Deep brain stimulation of pedunculopontine tegmental nucleus (PPTg) promotes cognitive and metabolic changes: a target-specific effect or response to a low-frequency pattern of stimulation? Clin EEG Neurosci 41: 82–86.
Stefurak T, Mikulis D, Mayberg H, Lang AE, Hevenor S, Pahapill P et al (2003). Deep brain stimulation for Parkinson's disease dissociates mood and motor circuits: a functional MRI case study. Mov Disord 18: 1508–1516.
Stern MB (2004). The preclinical detection of Parkinson's disease: ready for prime time? Ann Neurol 56: 169–171.
Stern MB, Doty RL, Dotti M, Corcoran P, Crawford D, McKeown DA et al (1994). Olfactory function in Parkinson's disease subtypes. Neurology 44: 266–268.
Stiasny-Kolster K, Doerr Y, Möller JC, Höffken H, Behr TM, Oertel WH et al (2005). Combination of idiopathic REM sleep behavior disorder and olfactory dysfunction as possible indicator for alpha-synucleopathy demonstrated by dopamine transporter FP-CIT-SPECT. Brain 128: 126–137.
Stocchi F, Rascol O, Kieburtz K, Poewe W, Jankovic J, Tolosa E et al (2010). Initiating levodopa/carbidopa therapy with and without entacapone in early Parkinson disease: the STRIDE-PD study. Ann Neurol 68: 18–27.
Strafella AP, Lozano AM, Lang AE, Ko JH, Poon YY, Moro E (2007). Subdural motor cortex stimulation in Parkinson's disease does not modify movement-related rCBF pattern. Mov Disord 22: 2113–2116.
Su PC, Tseng HM, Liu HM, Yen RF, Liou HH (2002). Subthalamotomy for advanced Parkinson disease. J Neurosurg 97: 598–606.
Swanson CJ, Bures M, Johnson MP, Linden AM, Monn JA, Schoepp DD (2005). Metabotropic glutamate receptors as novel targets for anxiety and stress disorders. Nat Rev Drug Discov 4: 131–144.
Taba HA, Wu SS, Foote KD, Hass CJ, Fernandez HH, Malaty IA et al (2010). A closer look at unilateral versus bilateral deep brain stimulation: results of the National Institutes of Health COMPARE cohort. J Neurosurg 113: 1224–1229.
Tanganelli S, Sandager Nielsen K, Ferraro L, Antonelli T, Kehr J, Franco R et al (2004). Striatal plasticity at the network level. Focus on adenosine A2A and D2 interactions in models of Parkinson's Disease. Parkinsonism Relat Disord 10: 273–280.
Temel Y, Kessels A, Tan S, Topdag A, Boon P, Visser-Vandewalle V (2006). Behavioural changes after bilateral subthalamic stimulation in advanced Parkinson disease: a systematic review. Parkinsonism Relat Disord 12: 265–272.
Temel Y, Visser-Vandewalle V (2006). Targets for deep brain stimulation in Parkinson's disease. Expert Opin Ther Targets 10: 355–362.
Thomas A, Bonanni L, Gambi F, Di Iorio A, Onofrj M (2010). Pathological gambling in Parkinson disease is reduced by amantadine. Ann Neurol 68: 400–404.
Tintner R, Manian P, Gauthier P, Jankovic J (2005). Pleuropulmonary fibrosis after long-term treatment with the dopamine agonist pergolide for Parkinson Disease. Arch Neurol 62: 1290–1295.
Tir M, Devos D, Blond S, Touzet G, Reyns N, Duhamel A et al (2007). Exhaustive, one-year follow-up of subthalamic nucleus deep brain stimulation in a large, single-center cohort of Parkinsonian patients. Neurosurgery 61: 297–304.
Tissingh G, Berendse HW, Bergmans P, DeWaard R, Drukarch B, Stoof JC et al (2001). Loss of olfaction in de novo and treated Parkinson's disease: possible implications for early diagnosis. Mov Disord 16: 41–46.
Tohgi H, Abe T, Takahashi S (1993). The effects of L-threo-3,4-dihydroxyphenylserine on the total norepinephrine and dopamine concentrations in the cerebrospinal fluid and freezing gait in parkinsonian patients. J Neural Transm Park Dis Dement Sect 5: 27–34.
Tolosa E (2007). Movement disorders: advances on many fronts. Lancet Neurol 6: 7–8.
Tolosa E, Compta Y, Gaig C (2007). The premotor phase of Parkinson's disease. Parkinsonism Relat Disord 13 (Suppl): S2–S7.
Tomac A, Lindqvist E, Lin LF, Ogren SO, Young D, Hoffer BJ et al (1995a). Protection and repair of the nigrostriatal dopaminergic system by GDNF in vivo. Nature 373: 335–339.
Tomac A, Widenfalk J, Lin LF, Kohno T, Ebendal T, Hoffer BJ et al (1995b). Retrograde axonal transport of glial cell line-derived neurotrophic factor in the adult nigrostriatal system suggests a trophic role in the adult. Proc Natl Acad Sci USA 92: 8274–8278.
Tozzi A, de Iure A, Di Filippo M, Tantucci M, Costa C, Borsini F et al (2011). The distinct role of medium spiny neurons and cholinergic interneurons in the DA receptor interaction in the striatum: implications for Parkinson's disease. J Neurosci 31: 1850–1862.
Troster AI, Fields JA, Wilkinson S, Pahwa R, Koller WC, Lyons KE (2003). Effect of motor improvement on quality of life following subthalamic stimulation is mediated by changes in depressive symptomatology. Stereotact Funct Neurosurg 80: 43–47.
Valenti O, Marino MJ, Wittmann M, Lis E, DiLella AG, Kinney GG et al (2003). Group III metabotropic glutamate receptor-mediated modulation of the striatopallidal synapse. J Neurosci 23: 7218–7226.
Volkmann J, Allert N, Voges J, Sturm V, Schnitzler A, Freund HJ (2004). Long-term results of bilateral pallidal stimulation in Parkinson's disease. Ann Neurol 55: 871–875.
Voon V, Krack P, Lang AE, Lozano AM, Dujardin K, Schupbach M et al (2008). A multicentre study on suicide outcomes following subthalamic stimulation for Parkinson's disease. Brain 131: 2720–2728.
Wang J, You H, Liu JF, Ni DF, Zhang ZX, Guan J (2011). Association of olfactory bulb volume and olfactory sulcus depth with olfactory function in patients with Parkinson's disease. AJNR Am J Neuroradiol 32: 677–681.
Waters C (2008). Catechol-O-Methyltransferase inhibitors. In: Factor SA, Weiner WJ (eds). Parkinson's disease-Diagnostic and Clinical Management. Demos: New York. pp 533–543.
Waters CH, Sethi KD, Hauser RA, Molho E, Bertoni JM, Zydis Selegiline Study Group (2004). Zydis selegiline reduces off time in Parkinson's disease patients with motor fluctuations: a 3-month, randomized, placebo-controlled study. Mov Disord 19: 426–432.
Watts RL (1997). The role of dopamine agonists in early Parkinson's disease. Neurology 49: S34–S48.
Weaver CD, Sheffler DJ, Lewis LM, Bridges TM, Williams R, Nalywajko NT et al (2009a). Discovery and development of a potent and highly selective small molecule muscarinic acetylcholine receptor subtype I (mAChR 1 or M1) antagonist in vitro and in vivo probe. Curr Top Med Chem 9: 1217–1226.
Weaver F, Follett K, Hur K, Ippolito D, Stern M (2005). Deep brain stimulation in Parkinson disease: a metaanalysis of patient outcomes. J Neurosurg 103: 956–967.
Weaver FM, Follett K, Stern M, Hur K, Harris C, Marks Jr WJ et al (2009b). Bilateral deep brain stimulation vs best medical therapy for patients with advanced Parkinson disease: a randomized controlled trial. JAMA 301: 63–73.
Weinberger M, Hamani C, Hutchison WD, Moro E, Lozano AM, Dostrovsky JO (2008). Pedunculopontine nucleus microelectrode recordings in movement disorder patients. Exp Brain Res 188: 165–174.
Weintraub D, Mavandadi S, Mamikonyan E, Siderowf AD, Duda JE, Hurtig HI et al (2010). Atomoxetine for depression and other neuropsychiatric symptoms in Parkinson disease. Neurology 75: 448–455.
Wenning GK, Shephard B, Hawkes C, Petruckevitch A, Lees A, Quinn N (1995). Olfactory function in atypical parkinsonian syndromes. Acta Neurol Scand 91: 247–250.
Westin J, Nyholm D, Palhagen S, Willows T, Groth T, Dougherty M et al (2011). A pharmacokinetic-pharmacodynamic model for duodenal levodopa infusion. Clin Neuropharmacol 34: 61–65.
Wichmann T, DeLong MR (2003). Functional neuroanatomy of the basal ganglia in Parkinson's disease. Adv Neurol 91: 9–18.
Williams SS, Williams J, Combrinck M, Christie S, Smith AD, McShane R (2009). Olfactory impairment is more marked in patients with mild dementia with Lewy bodies than those with mild Alzheimer disease. J Neurol Neurosurg Psychiatry 80: 667–670.
Witt K, Pulkowski U, Herzog J, Lorenz D, Hamel W, Deuschl G et al (2004). Deep brain stimulation of the subthalamic nucleus improves cognitive flexibility but impairs response inhibition in Parkinson disease. Arch Neurol 61: 697–700.
Witt M, Bormann K, Gudziol V, Pehlke K, Barth K, Minovi A et al (2009). Biopsies of olfactory epithelium in patients with Parkinson's disease. Mov Disord 24: 906–914.
Wood LD (2010). Clinical review and treatment of select adverse effects of dopamine receptor agonists in Parkinson's disease. Drugs Aging 27: 295–310.
Wood LD, Neumiller JJ, Setter SM, Dobbins EK (2010). Clinical review of treatment options for select nonmotor symptoms of Parkinson's disease. Am J Geriatr Pharmacother 8: 294–315.
Xu L, Tan YY, Ding JQ, Chen SD (2010). The iPS technique provides hope for Parkinson's disease treatment. Stem Cell Rev 6: 398–404.
Zahodne LB, Okun MS, Foote KD, Fernandez HH, Rodriguez RL, Kirsch-Darrow L et al (2009a). Cognitive declines one year after unilateral deep brain stimulation surgery in Parkinson's disease: a controlled study using reliable change. Clin Neuropsychol 23: 385–405.
Zahodne LB, Okun MS, Foote KD, Fernandez HH, Rodriguez RL, Wu SS et al (2009b). Greater improvement in quality of life following unilateral deep brain stimulation surgery in the globus pallidus as compared to the subthalamic nucleus. J Neurol 256: 1321–1329.
Zhou Y, Rodriguez AL, Williams R, Weaver CD, Conn PJ, Lindsley CW (2009). Synthesis and SAR of novel, non-MPEP chemotype mGluR5 NAMs identified by functional HTS. Bioorg Med Chem Lett 19: 6502–6506.
Zrinzo L, Zrinzo LV (2008). Surgical anatomy of the pedunculopontine and peripeduncular nuclei. Brit J Neurosurg 22 (Suppl 1): S19–S24.
Zrinzo L, Zrinzo LV, Tisch S, Limousin PD, Yousry TA, Afshar F et al (2008). Stereotactic localization of the human pedunculopontine nucleus: atlas-based coordinates and validation of a magnetic resonance imaging protocol for direct localization. Brain 131: 1588–1598.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Dr Smith declares that he has received a research grant from Oxford Biomedica in the past 3 years. Dr Factor declares that he has received research grant support from the following companies: Ceregene, TEVA Neurosciences, Ipsen and EMD Serono. He has also received educational grants from Allergan and Lundbeck, and personal compensation for consulting from Merz, Allergan. Dr DeLong declares over the past three years he has received compensation for consulting from Merck and for professional services from The Dystonia Medical Research Foundation. The other authors declare that this review was prepared in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Rights and permissions
About this article
Cite this article
Smith, Y., Wichmann, T., Factor, S. et al. Parkinson's Disease Therapeutics: New Developments and Challenges Since the Introduction of Levodopa. Neuropsychopharmacol 37, 213–246 (2012). https://doi.org/10.1038/npp.2011.212
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2011.212
Keywords
This article is cited by
-
Investigation of the mechanism of action of deep brain stimulation for the treatment of Parkinson’s disease
Cognitive Neurodynamics (2024)
-
O-GlcNAcylation in health and neurodegenerative diseases
Experimental & Molecular Medicine (2021)
-
Non-invasive delivery of levodopa-loaded nanoparticles to the brain via lymphatic vasculature to enhance treatment of Parkinson’s disease
Nano Research (2021)
-
Recent advances in dopaminergic strategies for the treatment of Parkinson’s disease
Acta Pharmacologica Sinica (2020)
-
Challenges and translational considerations of mesenchymal stem/stromal cell therapy for Parkinson’s disease
npj Regenerative Medicine (2020)
