
Abstract
Inhibition of phosphodiesterase-4 (PDE4), an enzyme that catalyzes the hydrolysis of cyclic AMP (cAMP), increases phosphorylation of the cAMP response element binding protein (pCREB) and hippocampal neurogenesis, and produces antidepressant-like effects on behavior; however, causal links among these actions have not been established. In this study, chronic administration of rolipram (0.31–1.25 mg/kg, 16–23 days) produced antidepressant- and anxiolytic-like effects on behavior in mice. It also increased cAMP and pCREB levels in the hippocampus and prefrontal cortex, but increased Sox2, a marker for mitotic progenitor cells, only in the hippocampus. Chronic rolipram treatment also increased hippocampal neurogenesis, as evidenced by increased bromodeoxyuridine (BrdU)-positive cells in the hippocampal dentate gyrus. Methylazoxymethanol (MAM), which is toxic to proliferating cells, reversed rolipram-induced increases in BrdU-positive cells and pCREB in the hippocampus and partially blocked its behavioral effects. Approximately 84% of BrdU-positive cells became newborn neurons, 93% of which co-expressed pCREB; these proportions were not altered by rolipram or MAM, either alone or in combination. Finally, 3 weeks after the end of the MAM treatment, when neurogenesis was no longer inhibited, rolipram again increased hippocampal pCREB and its antidepressant- and anxiolytic-like effects were restored. Overall, these results suggest that rolipram produces its effects on behavior in a manner that at least partially depends on its neurogenic action in the hippocampus, targeting mitotic progenitor cells rather than newborn or mature neurons; cAMP/CREB signaling in hippocampal newborn neurons is critical for neurogenesis and contributes to the behavioral effects of rolipram.
Similar content being viewed by others


INTRODUCTION
Hippocampal neurogenesis is the process of generating new neurons in the dentate gyrus (Duman et al, 2001; Emsley et al, 2005; Gage 2000; Gould et al, 1999; Van Praag et al, 2002). Specifically, progenitor cells residing in the subgranular zone (SGZ) of the dentate gyrus proliferate to form postmitotic daughter cells, which migrate into the granular cell layer and mature into neurons and other cell types. Neurogenesis is not only an important source of new neurons in the adult mammalian brain, but also an extremely dynamic process that can be up- or downregulated by a variety of pharmacological, environmental, and endocrinogical factors (Bruel-Jungerman et al, 2007; Dranovsky and Hen 2006; Duman et al, 2001; Eisch et al, 2000; Schmidt and Duman 2007). Hippocampal neurogenesis has been shown to be required for antidepressant-like behavior in some circumstances (Santarelli et al, 2003); however, some behavioral effects of certain antidepressants appear to be neurogenesis-independent (Holick et al, 2008).
Phosphodiesterase-4 (PDE4), an enzyme that catalyzes the hydrolysis of cyclic AMP (cAMP) and plays a critical role in the control of its intracellular levels, has been implicated in antidepressant activity and memory. Administration of rolipram, a prototypic, selective PDE4 inhibitor, increases cAMP signaling and produces a variety of behavioral actions, including antidepressant-like (O'Donnell and Frith 1999; Zhang, 2009; Zhang et al, 2002, 2006) and memory-enhancing effects in rodents (Barad et al, 1998; Zhang et al, 2000, 2004, 2005), which are associated with the increased phosphorylation of cAMP response element binding protein (pCREB) in the hippocampus (Blendy 2006; Monti et al, 2006; Sairanen et al, 2007). Chronic, but not acute, treatment with rolipram also increases hippocampal neurogenesis, as evidenced by an increased cell proliferation and survival of newborn neurons in the dentate gyrus (Nakagawa et al, 2002a, 2002b; Sasaki et al, 2007); this effect is accompanied by the activation of CREB (Fujioka et al, 2004; Nakagawa et al, 2002b; Nibuya et al, 1996; Zhu et al, 2004). However, it has been reported that mice deficient in CREB or expressing dominant negative CREB display antidepressant-like effects on behavior and increased hippocampal neurogenesis (Gur et al, 2007; Newton et al, 2002). Further studies need to be carried out to clarify these unexpected effects.
Compared with the role of PDE4 in antidepressant activity and memory, little is known about the regulation of PDE4 in anxiety. It has been reported in a preliminary study that acute treatment with rolipram produces an anxiolytic-like effect on behavior in rats (Silvestre et al, 1999a). However, anxiogenic-like behavior induced by PDE4 inhibitors including rolipram has been reported (Heaslip and Evans 1995; Imaizumi et al, 1994). Given that mice deficient in CREB display an anxiety-like effect on behavior (Gur et al, 2007), rolipram may produce an anxiolytic-like effect.
Using tests sensitive to antidepressant and anxiolytic drugs, we determined the behavioral effects of chronic treatment with rolipram (0.31–1.25 mg/kg, 16–23 days). We also examined hippocampal neurogenesis and pCREB expression. In addition, we assessed whether increased neurogenesis was required for the behavioral effects of rolipram by co-administrating methylazoxymethanol (MAM), an antimitotic DNA-methylating agent that has been shown to reduce hippocampal neurogenesis (Johnston and Coyle 1979; Shors et al, 2001).
MATERIALS AND METHODS
Animals
Male ICR mice (Harlan, Indianapolis, IN), initially weighing 21–23 g, were housed in a temperature-controlled room with a 12-h light–dark cycle (lights on from 0600–1800 hours). Animals had access to food and water ad libitum. All experiments were carried out according to the ‘NIH Guide for the Care and Use of Laboratory Animals’ (NIH Publications No. 80–23, revised 1996). The procedures were approved by the Animal Care and Use Committee of West Virginia University Health Sciences Center.
Drugs
Rolipram, fluoxetine, diazepam, BrdU (bromodeoxyuridine) (Sigma-Aldrich, St Louis, MO), and MAM (Midwest Research Institute, Kansas City, MO) were dissolved in saline (for BrdU and MAM) or saline containing 5% dimethyl sulfoxide (DMSO). Injections were i.p. except for MAM (s.c.) at a volume of 10 ml/kg body weight.
For testing the effects of chronic drug treatment on behavior, rolipram (0.31, 0.62, 1.25 mg/kg), fluoxetine (10 mg/kg), diazepam (1 mg/kg), or vehicle was given once per day for 16–23 days. All the behavioral tests were performed 1 h after drug injections following the treatment schedules (Figure 1).

Schedules of drug treatments and tests. (a) Treatment schedule and test order for blockade of neurogenesis by MAM. BrdU was injected (i.p.) on days 10, 12, and 14. Behavioral tests were carried out on days 16–22 (1 h after the injection of rolipram or vehicle). (b) Treatment schedule and test order for recovery from MAM-induced inhibition of neurogenesis. BrdU was injected on days 24, 26, and 28 (ie, 10–14 days after MAM treatment termination). Behavioral tests were carried out on days 33–35. MAM or saline (s.c.) and rolipram or its vehicle (saline containing 5% DMSO; i.p.) were injected once per day for 14 days (ie, days 1–14), after which only rolipram and vehicle were continued until the end of the tests (day 23 or 37) when the animals were killed. OF, open-field; EPM, elevated-plus maze; FST, forced-swim test; NSF, novelty suppressed feeding; LTD, light–dark transition; HB, holeboard test; TST, tail-suspension test; SAC, sacrifice.
Behavioral Experiments
Three batches of mice were used for testing behavioral and neurochemical effects of rolipram, MAM with or without rolipram, and termination of MAM (eight to nine mice per group) following this sequence: open-field, elevated-plus maze, forced-swim, novelty suppressed feeding, light–dark transition, holeboard, and tail-suspension tests.
Open-field test
This test was performed as described previously with minor modifications (Zhang et al, 2008). On days 16 and 19 (for two separate groups; see Results below), mice were placed individually in a white Plexiglas box (60 × 60 × 15 cm) with the floor divided into four identical squares. Line crossings (with all four paws placed into a new square) and rears (with both front paws raised from the floor) were recorded in a 5-min period.
Elevated plus-maze test
The mouse was placed in the center of the maze (40 cm above the floor) facing an enclosed arm. The number of entries into (with all four paws) and time spent in both open and enclosed arms (30 × 5 cm and 30 × 5 × 15 cm high, respectively) were recorded for 5 min as described previously (Masood et al, 2008). The percentages of entries and time spent in open arms were calculated as open-arm entries and time divided by total arm entries and total time, respectively.
Light–dark transition test
Each mouse was placed in the dark compartment (15 × 23 cm) of the light–dark chamber. The latency to cross through an opening (8 × 6 cm) into the light compartment (30 × 23 cm; illuminated with a 60-W bulb positioned 50 cm above), the time spent on the light side, and transitions (ie, the number of crossings from the dark to light compartments) were recorded for 5 min as described previously (Zhang et al, 2008).
Holeboard test
Mice were placed individually in the center of an open Plexiglas box (40 × 40 × 30 cm), which had four holes (3 cm in diameter, 2 cm in depth) in the floor. The number of head-dips and the time spent in head-dipping were recorded for 5 min as described previously (Zhang et al, 2008).
Forced-swim test
The test was carried out as described previously (Zhang et al, 2002). Mice were placed individually in a plastic cylinder (45 cm high × 20 cm diameter) filled with water (23–24°C; 28 cm in depth), allowing for free swimming. The duration of immobility, which was defined as floating in an upright position without additional movement other than that necessary for the animal to keep its head above the water, was recorded during the last 4 min of the 6-min test period.
Tail-suspension test
Each mouse was suspended 40 cm above the floor using adhesive tape placed approximately 1 cm from the tip of the tail, as described previously (Zhang et al, 2002). The duration of immobility was recorded for 6 min. Mice were considered immobile only when they hung motionless.
Novelty Suppressed Feeding
The test was performed as described previously (Santarelli et al, 2003) with minor modification. After 24 h of food deprivation, mice were individually placed in a corner of the white, plastic, open chamber (35 × 28 × 16 cm); the floor was covered by a 1-cm thick layer of wooden bedding; four pellets of food (regular chow) were placed in the center of the floor. The latency to begin to chew the pellets was recorded for up to 5 min.
Neurogenesis Blockade by MAM
This was performed as described previously (Bruel-Jungerman et al, 2005; Shors et al, 2001) with some modifications. Mice were treated once daily with (1) vehicle (5% DMSO)+saline, (2) vehicle+MAM (5 mg/kg; chosen based on our preliminary study), (3) rolipram (1.25 mg/kg)+saline, or (4) rolipram+MAM. MAM treatment was terminated after 14 days, while rolipram continued to be administered throughout the behavioral testing and until animals were killed. For evaluation of neurogenesis, BrdU (100 mg/kg) was administered to four to five mice in each group on days 10, 12, and 14 (Figure 1a). Beginning from day 16, mice were tested for locomotor activity and anxiolytic- and antidepressant-like behaviors. On day 23, 1 h after the last rolipram injection, the mice were anesthetized with pentobarbital (100 mg/kg) and perfused transcardially with 4% paraformaldehyde. The brains were post-fixed with cold paraformaldehyde overnight and dehydrated with 30% sucrose before coronal sections (30 μm) using a freezing microtome. The remaining mice (4–5 mice/group) were decapitated and the hippocampi and prefrontal cortices dissected and stored at −80°C for immunoblotting analysis or cAMP assay.
Recovery of Neurogenesis Following MAM Inhibition
The treatment assignment and strategy were the same as described above. BrdU was given (also to one-half of the mice in each group) on days 24, 26, and 28 (ie, 10–14 days after the termination of MAM treatment; Figure 1b). Behavioral tests were performed on days 33–35. On day 37, BrdU-treated mice were killed for neurogenesis analysis, whereas the remaining mice were used for immunoblotting analysis.
Cyclic AMP Assay
The samples extracted by RIPA lysis buffer were diluted with 0.1 N HCl to a final protein concentration of 1 mg/ml. Cyclic AMP levels were determined by ELISA following the instructions provided by the company (Assay Designs, Ann Arbor, MI).
Immunoblotting Analyses
Brain tissues were sonicated in RIPA lysis buffer (Upstate, Temecula, CA) containing protease and phosphatase inhibitors (Pierce Biotechnology, Rockford, IL) and centrifuged at 16 000 × g for 30 min. Samples (75 μg protein each) were separated using SDS-PAGE before transferring to nitrocellulose membranes, which were then incubated with rabbit anti-pCREB (Ser133), anti-CREB, anti-Sox2, or anti-β-actin antibodies (1 : 1000; Chemicon, Temecula, CA) at 4 °C overnight except for the anti-β-actin antibody, which was incubated for 60 min. The membranes were then incubated with Alexa Fluor 700 conjugated goat anti-rabbit antibody (1 : 20 000; Invitrogen, Eugene, OR) for 30 min. The detection and quantification of specific bands were carried out using a fluorescence scanner (Odyssey Infrared Imaging System, LI-COR Biotechnology, Lincoln, NE). For band stripping, the membranes were incubated with stripping buffer (Chemicon) for 15 min.
Immunohistochemical Analyses
This was performed as described previously (Nakagawa et al, 2002a, 2002b; Fujioka et al, 2004) with minor modifications. For immunofluorescent staining of co-expression of pCREB and calbindin, a selective marker of mature neurons (Encinas et al, 2006), free-floating sections were incubated in 0.01 M PBS containing 0.2% Triton X-100 and 5% normal goat serum (PBS-plus) for 60 min followed by primary antibodies rabbit anti-pCREB IgG (1 : 200; Chemicon) and mouse anti-calbindin IgG (1 : 50; GeneTex, San Antonio, TX) at 4°C for 1 day. The sections were incubated with anti-mouse rhodamine Red-X (1 : 200) and anti-rabbit Cy5 (1 : 400; Jackson ImmunoResearch, West Grove, PA) in PBS for 2 h before mounting on slides using Vectashield (Vector Laboratories, Burlingame, CA).
For immunofluorescent staining of BrdU and its colocalization, free-floating brain sections were incubated in 2 × SSC/50% formamide at 65°C for 2 h, followed by 2 N HCl at 37°C for 30 min and 0.1 M boric acid (pH 8.5) for 10 min before blocking with PBS-plus for 60 min. The sections were then incubated at 4°C for 1 day with rat anti-BrdU IgG, mouse anti-polysialic acid-neural cell adhesion molecule (PSA-NCAM, an immature neuron marker; Nakagawa et al, 2002a, 2002b) IgM, rabbit anti-pCREB IgG (1 : 200), mouse anti-neuron-specific nuclear protein (NeuN) IgG (1 : 1000; Chemicon), or rabbit anti-S100β IgG (1 : 100; GeneTex, San Antonio, TX), followed by anti-rat FITC (1 : 200), anti-mouse rhodamine Red-X (1 : 200), and anti-rabbit Cy5 (1 : 400; Jackson ImmunoResearch) for 2 h before mounting with Vectashield (Vector Laboratories). Fluorescence analyses were performed using confocal laser microscopy (Zeiss LSM510, Thornwood, NY). To determine the percentage of newborn neurons labeled by PSA-NCAM in BrdU-labeled cells and examine pCREB in the newborn neurons (ie, cells labeled with both BrdU and PSA-NCAM), at least 50 BrdU-positive cells in the dentate gyrus were randomly identified from each animal. The number of cells in each category for each animal was determined.
The BrdU-positive cells were counted using a modified stereological protocol (Gould et al, 1999; Eisch et al, 2000; Nakagawa et al, 2002a, 2002b; West et al, 1991; Malberg et al, 2000). In brief, every ninth section throughout the entire hippocampus was processed for BrdU immunohistochemistry. All BrdU-labeled cells in the granular cell layer and hilus were counted through a × 60 objective lens in order to distinguish individual cells and multiplied by 10, recording as the total number of labeled cells in the dentate gyrus.
Statistical Analyses
Data shown represent means±SEM; they were analyzed using one-way analysis of variance (ANOVA) followed by Bonferroni's multiple comparison tests. The r2 values were calculated using linear regression.
RESULTS
Anxiolytic-Like Effects of Rolipram
To determine whether rolipram affected anxiety-associated behavior, we examined the performance of mice repeatedly treated with different doses of rolipram in tests sensitive to anxiolytic drugs. In the elevated plus-maze test, a well-established model to detect both anxiolytic- and axiogenic-like behavior, rolipram (0.31–1.25 mg/kg for 17 days) increased the percentages of both entries (F4,36=3.04; p<0.05) into and time (F4,35=3.69; p=0.01) spent in the open arms; it was significant at the dose of 1.25 mg/kg for the former and at all the doses for the latter, compared with the vehicle control (p<0.05; Figure 2a). A similar effect was produced by the proven anxiolytic diazepam at 1 mg/kg for 17 days (p<0.05 and p<0.01 for the percentages of entries and time, respectively). Neither rolipram nor diazepam, at the doses used, altered the total arm entries or the total time spent in arm exploration (Table 1). In the light–dark transition test, another paradigm widely used for anxiolytic testing, rolipram (0.31–1.25 mg/kg for 20 days) decreased the latency to enter the light compartment (F4,36=4.41; p<0.01) and increased the duration on the light side (F4,36=6.24; p<0.001) in a dose-dependent manner; it was significant at the dose of 1.25 mg/kg (p<0.05; Figure 2b). Again, diazepam produced similar effects (p<0.05). The anxiolytic-like effects of rolipram were further confirmed using the holeboard test. Similar to diazepam, the same doses of rolipram administered for 21 days increased the number of head-dips (F4,35=3.58; p=0.01) and the time spent in head-dipping (F4,35=3.72; p=0.01) in a dose-dependent manner (Figure 2c). The effects of rolipram were statistically significant at the dose of 1.25 mg/kg relative to the vehicle control (p<0.05 for head-dips and p<0.01 for the head-dipping time). The treatment did not alter locomotor activity in the open-field test (data not shown). These results show that repeated rolipram treatment produces anxiolytic-like effects on behavior.

Effects of rolipram on anxiolytic- and antidepressant-like behavior in mice. Treatment with rolipram (Rol) or diazepam (Diaz) increased percentages of entries into and the time spent in the open arms in the elevated plus-maze test (a), decreased latency to enter the light compartment and increased duration on the light side in the light–dark transition test (b), and increased head-dips and the time spent in head-dipping in the holeboard test (c). These suggest anxiolytic-like effects of the drug treatment on behavior. (d) Treatment with rolipram or fluoxetine (Flu) decreased immobility in the forced-swim and tail-suspension tests (FST and TST, respectively), which is indicative of antidepressant-like behavior. Rolipram (0.31–1.25 mg/kg), diazepam (1 mg/kg), fluoxetine (10 mg/kg), or vehicle (Veh) was administered (i.p.) once a day for 17 (a), 20 (b), 21 (c), or 18 days for the FST or 22 days for the TST (d). Tests were performed 1 h after the injection of drugs or vehicle on each testing day. Values shown are means±SEM of eight to nine mice per group. *p<0.05, **p<0.01 vs vehicle.
Antidepressant-Like Effect of Rolipram
Previous studies have shown that rolipram (0.5 mg/kg for 8 days) produces antidepressant-like behavior in the forced-swim test (FST) and tail-suspension test (TST) in a mixed background (C57 BL/6 × 129 ola) of mice (Zhang et al, 2002, 2008). Given that antidepressant-like behavior has strain differences (Lucki et al, 2001), it was necessary to determine whether rolipram produced an antidepressant-like effect in ICR mice using the current treatment strategy. Mice treated with rolipram or the proven antidepressant fluoxetine, a selective inhibitor of serotonin reuptake, were tested for the duration of immobility in the FST and TST. Chronic treatment with fluoxetine (10 mg/kg) decreased immobility of mice in both tests (p<0.05; Figure 2d). Similar effects were produced by rolipram at doses of 0.31–1.25 mg/kg for 18 days (FST; F4,35=5.36; p<0.01) or 22 days (TST; F4,36=2.74; p<0.05). Specifically, rolipram decreased immobility at all the doses in the FST (p<0.01) and at the doses of 0.62 and 1.25 mg/kg in the TST (p<0.05), indicating an antidepressant-like effect of rolipram on behavior under the present conditions.
Effects of Rolipram on cAMP, pCREB, and Sox2
The cAMP/CREB pathway has an important role in the mediation of antidepressant activity (D'Sa and Duman 2002). Fluoxetine (10 mg/kg for 23 days) increased cAMP levels (Figure 3a) and pCREB expression (Figure 3c, d) in both the hippocampus (p<0.05) and the prefrontal cortex (p<0.01 for cAMP and p<0.05 for pCREB), without altering CREB levels. Similarly, chronic treatment with rolipram (0.62–1.25 mg/kg for 23 days; doses chosen based on the behavioral results) also increased cAMP and pCREB in the hippocampus (F3,15=3.37; p<0.05 and F3,12=5.55; p=0.01, respectively) and prefrontal cortex (F3,13=6.27; p<0.01 and F3,12=5.21; p<0.05, respectively); it was significant at a dose of 1.25 mg/kg. By contrast, rolipram did not alter CREB levels in either brain region (F3,12=0.32; p=0.81 for hippocampus and F3,12=0.43; p=0.73 for prefrontal cortex; Figure 3c and d). The effects of rolipram on cAMP and pCREB paralleled its anxiolytic- and antidepressant-like effects, suggesting that the cAMP/CREB cascade in the brain is important for the mediation of the behavioral effects of rolipram.

Effects of rolipram (Rol) on the levels of cAMP, pCREB, CREB, and Sox2 in the mouse brain. (a) Rolipram and fluoxetine (Flu) increased cAMP levels in the hippocampus and prefrontal cortex. (b) Rolipram and fluoxetine increased Sox2 levels in the hippocampus, but not the prefrontal cortex. (c, d) Rolipram and fluoxetine increased the levels of pCREB, but not CREB, in the hippocampus (c) and prefrontal cortex (d). Lower panels (b–d) are representative immunoblots of the measures detected by Western blotting; upper panels are quantification of Sox2, pCREB, or CREB. Rolipram (0.62 and 1.25 mg/kg), fluoxetine (10 mg/kg), or vehicle (Veh) was administered (i.p.) once a day for 23 days; 1 h after the last drug injection, mice were decapitated and hippocampi and prefrontal cortices were dissected for cAMP assay or immunoblotting analyses. Values shown are means±SEM of four to five mice per group; the immunoblotting data (b–d) are expressed as percentages of corresponding optical density (normalized to β-actin) in the vehicle control samples. *p<0.05, **p<0.01 vs vehicle.
To determine the effects of drug treatment on neural progenitors, we examined the expression of Sox2, a transcription factor and an in vivo marker for mitotic neural progenitor cells (Encinas et al, 2006; Episkopou 2005; Graham et al, 2003; Masui et al, 2007; Bani-Yaghoub et al, 2006), in the brain. Fluoxetine increased Sox2 levels in the hippocampus (p<0.05), but not the prefrontal cortex (Figure 3b). Similarly, chronic treatment with rolipram (0.62–1.25 mg/kg for 23 days) increased Sox2 levels only in the hippocampus (F3,12=4.45; p<0.05 in contrast to F3,12=0.40; p>0.05 in the prefrontal cortex). These results suggest that rolipram increases progenitor cell proliferation in the hippocampus; the effect appears to be region-specific.
Effects of MAM Alone or in Combination with Rolipram on Body Weights and Locomotor Activity
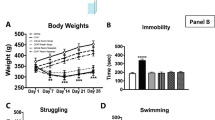
The DNA-methylating agent MAM is used for inhibiting neurogenesis (Shors et al, 2001; Bruel-Jungerman et al, 2005). To evaluate the potential, toxic effect of MAM, body weights of mice were monitored throughout the behavioral tests. Fourteen days after continuous administration of MAM (5 mg/kg) or rolipram (1.25 mg/kg), the body weights tended to be decreased, but were not statistically different from those in the vehicle controls. Combined treatment with both drugs led to a slower gain of body weight compared with the vehicle controls; this was significant during days 14–26 (Figure 4a). Beginning from day 28 (ie, 14 days after termination of MAM), body weights recovered to the levels that were no longer significantly different from the controls. At higher doses (7.5 and 15 mg/kg), MAM alone significantly reduced the gain of body weights and led to some animal deaths (data not shown).

Effects of MAM and/or rolipram (Rol) on body weights and behaviors. (a) Changes in body weights of mice treated with MAM and rolipram alone or in combination. Although the drugs used alone did not significantly alter the body weights, combination of both drugs led to a slower gain of body weights during days 14–26, compared with the corresponding controls. (b) Effects of MAM and/or rolipram on locomotor activity in the open-field test. None of the treatments altered locomotor activity, as assessed by line crossings and rears in the open-field test 2 days after termination of MAM treatment (day 16). (c, d) Attenuation by MAM of the anxiolytic-like effects of rolipram on behavior. Rolipram-induced increases in the percentages of entries into and time spent in open arms in the elevated plus-maze test (c) and the number of head-dips in the holeboard test (d) were attenuated by co-administration of MAM. (e, f) Attenuation by MAM of the antidepressant-like effects of rolipram on behavior. Rolipram-induced decreases in immobility in the forced-swim (e) and tail-suspension (f) tests were attenuated by co-administration of MAM. (g) Effects of rolipram and/or MAM on latency to feed in the novelty suppressed feeding (NSF) test. Rolipram (1.25 mg/kg) or vehicle (Veh) was injected (i.p.) once a day for 30 (a), 16 (b), 17 (c), 21 (d), 18 (e), 22 (f), or 19 (g) days. MAM (5 mg/kg) or saline (Sal) was co-administered (s.c.) with rolipram or vehicle for the first 14 days, after which MAM treatment was terminated. The tests were performed 1 h after the daily administration of rolipram or vehicle. Values shown are means±SEM of eight to nine (a, b, and g) or 16–18 (c–f) mice per group. *p<0.05, **p<0.01, ***p<0.001 vs control (saline+vehicle); #p<0.05, ##p<0.01, ###p<0.001 vs MAM+vehicle; $p<0.05 vs rolipram+saline.
Locomotor activity was examined in mice treated with MAM and rolipram alone or in combination in the open-field test. The test was carried out twice by splitting the mice (16–18 per group) into two groups, which were tested on days 16 and 19, ie, 2 and 5 days, respectively, after the last injection of MAM. During this period, the combination of MAM and rolipram led to the slowest gain of body weights (Figure 4a). However, none of the treatments, regardless of whether drugs were given alone or in combination, altered locomotor activity, as evidenced by unchanged crossings and rears in the open-field test after drug administration (day 16: F3,27=0.7; p>0.05 for crossings and F3,27=0.16; p<0.05 for rears; Figure 4b; day19: F3,28=0.37; p>0.05 for crossings and F3,28=0.08; p>0.05 for rears; data not shown). These results suggest that chronic treatment with MAM and/or rolipram at the doses used does not affect the general health nor decrease overall motor activity in mice.
Effects of MAM on Rolipram-Induced Anxiolytic- and Antidepressant-Like Behavior
Repeated treatment with rolipram (1.25 mg/kg plus saline for 17 days) produced anxiolytic-like effects in the elevated-plus maze test, as evidenced by increased percentages of entries into and time spent in the open arms compared with the corresponding control (vehicle+saline; p<0.001; Figure 4c). The effects of rolipram were attenuated by MAM (5 mg/kg for 14 days; time%: F3,61=13.24; p<0.001; entries%: F3,61=13.11; p<0.001); post hoc comparison revealed significant decreases compared to rolipram alone (p<0.05). MAM alone had no effect. None of the treatments altered the total arm entries (F3,61=1.24; p>0.05) or the total time (F3,61=0.19; p>0.05) in arm exploration (Table 2). Similarly, the anxiolytic-like effect of rolipram in the holeboard test also was attenuated by MAM (F3,61=6.69; p<0.001; Figure 4d). In the FST and TST, MAM alone did not decrease immobility significantly. However, it partially, but significantly, blocked rolipram-induced antidepressant-like effects, ie, decreased immobility in the FST (F3,61=13.33; p<0.001; Figure 4e) and TST (F3,61=8.00; p<0.001; Figure 4f).
The mouse novelty suppressed feeding (NSF) test has been demonstrated to be sensitive to chronic antidepressant treatment (Santarelli et al, 2003). MAM increased the latency to feed (F3,30=10.12; p<0.0001; post hoc Newman–Keuls test, p<0.001 vs vehicle+saline), whereas rolipram did not alter the latency (Figure 4g). Nevertheless, the effect of MAM on latency was decreased to the control level in the presence of rolipram.
Effect of MAM on Hippocampal Neurogenesis in Mice Treated with Rolipram
To determine the effects of MAM and/or rolipram on hippocampal neurogenesis, mice were killed 13 days after the beginning of BrdU labeling. Cells in the hippocampal dentate gyrus labeled with BrdU were counted. BrdU-positive cells were predominantly localized in the SGZ and, to a much less extent, in the hilus (Figure 5a). Rolipram (1.25 mg/kg for 23 days) increased, whereas MAM (5 mg/kg for 14 days) decreased the number of BrdU-positive cells in the dentate gyrus (F3,13=11.02; p<0.001; Figure 5a and b). The rolipram-induced increase in BrdU-positive cells was reversed by co-administration of MAM (p<0.05).

Effects of MAM and/or rolipram (Rol) on BrdU-, PSA-NCAM-, and pCREB-labeled cells in the hippocampal dentate gyrus in mice. (a) Confocal micrographs of BrdU-labeled cells (green) in the dentate gyrus from mice repeatedly treated with vehicle (Veh), MAM, rolipram, or MAM+rolipram. The majority of the BrdU-labeled cells were located in the subgranular zone (SGZ, indicated by arrow). (b) Quantification of BrdU-positive cells following drug treatments. Rolipram increased, while MAM decreased, BrdU-positive cells in the SGZ; the effect of rolipram was reversed by MAM. (c and d) Phenotype of BrdU- positive cells in the dentate gyrus. Confocal micrographs of cells double-labeled for BrdU (green; left panels) and NeuN (red; middle-upper), or S100β (blue; middle-lower). The proportions of neuronal and glial cells (71.4 and 18.5%, respectively) were not altered by any of the treatments. (e) Colocalization of PSA-NCAM and pCREB in developing BrdU-positive cells. Representative confocal micrographs of cells triple-labeled for BrdU, PSA-NCAM (red), and pCREB (blue) in the dentate gyrus. (f) Effects of MAM and/or rolipram on PSA-NCAM-labeled cells, which constituted 84.2% of BrdU-positive cells. (g) Effects of MAM and/or rolipram on pCREB-labeled cells, which constituted 93.4% of PSA-NCAM- and BrdU-positive cells. These percentages were not altered by MAM or rolipram alone or in combination. BrdU (100 mg/kg) was injected (i.p.) once a day on days 10, 12, and 14 of rolipram treatment. Mice were perfused and brain sections were processed 9 days after the last of the three BrdU injections. Values shown are means±SEM of four to five mice per group. *p<0.05 vs vehicle; #p<0.05, ##p<0.01 vs MAM+vehicle; $p<0.05 vs rolipram+saline.
To examine the phenotypes of BrdU-positive cells in the dentate gyrus, BrdU staining was carried out 13 days after the first BrdU injection, during which time newborn cells develop differentiated phenotypes (Kempermann et al, 2003); double-labeling for BrdU and NeuN (a neuronal marker; Mullen et al, 1992), or S100β (a glial marker; Boyes et al, 1986) was performed. Confocal microscopy showed that BrdU-positive cells colocalized with NeuN- or S100β-labeled cells (Figure 5c and d), which consisted of 71 and 19% of BrdU-positive cells, respectively. Neither MAM nor rolipram altered the proportions of cells maturing into neurons or glia.
Newborn neurons start to be involved in the learning and memory processes when they are approximately 1–2 weeks of age (Shors et al, 2001; Bruel-Jungerman et al, 2005); they express PSA-NCAM approximately 2 weeks before maturing into granule neuronal cells (Encinas et al, 2006). To determine the proportion of newborn neurons among BrdU-positive cells and the proportion of neurons expressing pCREB among newborn neurons, triple staining for BrdU, PSA-NCAM, and pCREB was performed 13 days after the first injection of BrdU. Approximately 84% of BrdU-labeled cells were newborn neurons, as evidenced by colocalization with PSA-NCAM (Figure 5e and f); 93% of the BrdU-labeled newborn neurons co-expressed pCREB (Figure 5e and g). These proportions were not affected by any of the treatments (F3,12=1.67; p>0.05; Figure 5f and F3,12=0.25; p>0.05; Figure 5g), indicating that pCREB plays an important role in survival of newborn neurons, which appear not to be critical for the initial action of rolipram.
Effects of MAM on pCREB Levels in the Hippocampus and Prefrontal Cortex of Mice Treated with Rolipram
To determine the role of cAMP/CREB signaling in the effects of MAM and/or rolipram, we examined the expression of pCREB and CREB in the hippocampus and prefrontal cortex in mice repeatedly treated with each drug alone or in combination. One-way ANOVA revealed significant changes in the levels of pCREB among the treatment groups in the hippocampus (F3,12=14.71; p<0.001; Figure 6a) and prefrontal cortex (F3,12=15.76; p<0.001; Figure 6b). Compared with the control (vehicle+saline), rolipram (1.25 mg/kg for 23 days) increased pCREB levels in both the hippocampus (p<0.01) and the prefrontal cortex (p<0.001), whereas MAM (5 mg/kg for 14 days) decreased pCREB only in the hippocampus (p<0.05). In addition, MAM reversed the rolipram-induced increase in pCREB in the hippocampus (p<0.01; Figure 6a), but not prefrontal cortex (Figure 6b). Expression of CREB was not altered by any of the treatments (data not shown).

Effects of MAM and/or rolipram (Rol) on pCREB expression and pCREB- and calbindin-labeled cells in the mouse hippocampus and prefrontal cortex. (a) Drug-induced changes in pCREB in the hippocampus; (b) drug-induced changes in pCREB in the prefrontal cortex. Lower panels are representative immunoblots of pCREB detected by western blotting; upper panels are quantification of pCREB. MAM (5 mg/kg, s.c., 14 days) decreased pCREB in the hippocampus, but not the prefrontal cortex. By contrast, rolipram (1.25 mg/kg, i.p., 23 days) increased pCREB in both brain regions; the effect of rolipram was reversed by MAM only in the hippocampus. (c and d) Confocal micrographs of cells double-labeled for pCREB (blue; left panels) and calbindin (red; middle panels). Mature neurons (labeled by calbindin) in the dentate gyrus (c) did not show pCREB immunostaining; by contrast, calbindin-labeled neurons in the prefrontal cortex (d) all displayed pCREB immunostaining (right panels, indicated by arrows). Values shown are means ± SEM of four mice per group. *p<0.05, **p<0.01, ***p<0.001 vs vehicle; #p<0.05, ##p<0.01, ###p<0.001 vs MAM+vehicle; $$p<0.01 vs rolipam+saline.
To explore the potential relationship between the changes in hippocampal neurogenesis and pCREB, the r2 values were calculated using the same treatment groups (ie, MAM, MAM+Rol, and Rol). It was found that changes in pCREB levels in the hippocampus were highly correlated with those in BrdU-positive cells (r2=0.95). Interestingly, while mature neurons labeled by calbindin did not express pCREB in the hippocampal dentate gyrus (Figure 6c), almost all the mature neurons in the prefrontal cortex co-expressed pCREB (Figure 6d). These results suggest that the activation of CREB in the newborn neurons in the hippocampus is important in the mediation of hippocampal neurogenesis.
Recovery from MAM-Induced Changes in Hippocampal Neurogenesis, pCREB Expression, and Behavior
To verify the relationship among neurogenesis, pCREB, and behavioral alterations, the effects of MAM and rolipram alone or in combination on these measures were examined 19–23 days (BrdU was injected 10–14 days) after termination of MAM treatment (Figure 1b); this interval allowed a recovery of neurogenesis from MAM-induced inhibition (Figure 7a and b vs Figure 5a and b). Twenty-three days after the last MAM injection, one-way ANOVA revealed overall significant changes in BrdU-positive cells among treatments (F3,13=10.96; p<0.001). Repeated treatment with rolipram (1.25 mg/kg for 37 days) increased BrdU-positive cells in the dentate gyrus (p<0.01), whereas MAM-treated mice no longer displayed a decrease in BrdU-positive cells, compared with the control (vehicle+saline); reversal by MAM of the rolipram-induced increase in BrdU-positive cells (Figure 5a and b) was not observed after the termination of MAM treatment (Figure 7a and b), suggesting a recovery of hippocampal neurogenesis. In addition, expression of pCREB in the hippocampus also recovered from MAM inhibition in a similar pattern (F3,12=6.46; p<0.01; Figure 7c); ie, rolipram again increased pCREB (p<0.05). The rolipram-induced increase in pCREB in the prefrontal cortex was still not changed under this treatment condition (Figure 7d). Expression of CREB was not changed by any of the treatments (Figure 7c and d).

Effects of termination of MAM treatment on rolipram-induced changes in hippocampal neurogenesis and expression of pCREB and CREB in the mouse hippocampus and prefrontal cortex. (a) Confocal micrographs of BrdU-labeled cells (green) in the dentate gyrus from mice, which had been repeatedly, treated with vehicle (Veh), MAM, rolipram (Rol), MAM+rolipram, with a 3-week washout of MAM. (b) Quantification of BrdU-positive cells following the drug treatments. Rolipram-induced increases in BrdU-positive cells were no longer inhibited by MAM 3 weeks after termination of its treatment. (c and d) Rolipram-induced increases in pCREB in the hippocampus (c) and prefrontal cortex (d) were not changed after termination of MAM treatment; the treatments did not alter expression of CREB. Lower panels are representative immunoblots of pCREB or CREB detected by western blotting; upper panels are quantification of pCREB and CREB. Rolipram (1.25 mg/kg) was given (i.p.) for 37 days and MAM (5 mg/kg) was co-administered (s.c.) with rolipram or vehicle for the first 14 days. BrdU (100 mg/kg) was injected (i.p.) once per day on days 24, 26, and 28. Mice were perfused or killed 1 h after the final injection of rolipram or vehicle on day 37. Values shown are means ± S.E.M of four to five mice per group. *p<0.05, **p<0.01 vs vehicle+saline; #p<0.05, ##p<0.01 vs MAM+vehicle.
With the recovery from MAM-induced inhibition of neurogenesis and pCREB expression, anxiolytic- and antidepressant-like effects of rolipram on behavior were no longer attenuated by MAM, including the effects in the elevated plus-maze (entries%: F3,30=4.97; p<0.01 and time%: F3,30=8.33; p=0.001; Figure 8a), FST (F3,30=4.22; p=0.01), and TST (F3,30=4.29; p=0.01; Figure 8b). Total arm activity was not changed in the elevated plus-maze test (Table 3).

Effects of termination of MAM treatment on rolipram-induced anxiolytic- and antidepressant-like behavior in mice. (a) Rolipram-induced increases in the percentages of entries into and time spent in open arms in the elevated plus-maze were not altered after termination of MAM treatment. (b) Rolipram-induced decreases in immobility in the FST and TST were not changed after termination of MAM. Rolipram (1.25 mg/kg) was given (i.p.) for 33–35 days before the tests were carried out 1 h after the daily drug injection on days 33 (a), 34 (FST), or 35 (TST). MAM (5 mg/kg) was co-administered (s.c.) with rolipram or vehicle for the first 14 days. Values shown are means±SEM of eight to nine mice per group. *p<0.05, **p<0.01 vs vehicle+saline; #p<0.05, ##p<0.01 vs MAM+vehicle.
DISCUSSION
Chronic treatment with rolipram produced antidepressant- and anxiolytic-like effects on behavior in mice. It also increased neurogenesis and levels of cAMP, pCREB, and Sox2 in the hippocampus. The effects of rolipram on hippocampal neurogenesis and pCREB were completely blocked and those on behavior markedly reduced by co-administration of MAM, which methylates DNA and inhibits neurogenesis (Shors et al, 2001). The behavioral effects of rolipram were restored following the recovery from MAM-induced decreases in BrdU-positive cells and pCREB in the hippocampus. Overall, changes in hippocampal pCREB were highly correlated with neurogenesis and associated with antidepressant- and anxiolytic-like effects on behavior.
Hippocampal Neurogenesis and Anxiolytic- and Antidepressant-Like Behavior
It has been shown that cAMP/CREB signaling positively regulates anxiety-like behavior (Pandey et al, 2005; Wand 2005). As a critical controller of this signaling pathway, PDE4 was anticipated to play a role in the behavioral effects regulated by cAMP signaling. We found that chronic treatment with rolipram produced anxiolytic-like effects on behavior; this is supported by an earlier study showing that acute treatment with rolipram produces an anxiolytic-like effect (Silvestre et al, 1999a), although opposite results were reported in some other studies (Heaslip and Evans 1995; Imaizumi et al, 1994). The discrepancy may be at least partially because of the sedative effect of PDE4 inhibitors administered acutely (Griebel et al, 1991; Silvestre et al, 1999b), as sedation may be interpreted inappropriately as an anxiogenic-like effect in certain tests (Weiss et al, 1998). However, this was not the case in this study. Although acute administration of rolipram (1 mg/kg) produces a sedative effect (Silvestre et al, 1999b; Zhang and O'Donnell 2000), repeated treatment with rolipram at 1.25 mg/kg did not alter locomotor activity in the open-field test nor change the total arm exploration, an index of general motor activity (Zhang et al, 2008), in the elevated-plus maze test 1 h post-treatment, when anxiolytic behavior was assessed. The dissociation of anxiolytic-like and sedative effects indicates that, after repeated administration, animals may exhibit tolerance to the sedative effect of rolipram while sustaining sensitivity to its anxiolytic-like action. Although it is not known what caused the behavioral dissociation, a similar phenomenon has been observed after repeated benzodiazepine treatment, which produces a greater anxiolytic-like effect, but the sedative action is diminished, compared with that observed after acute administration (Bourin et al, 1992). Unfortunately, the emetic effect of rolipram appears not to be reduced by repeated treatment, leading to the withdrawal of rolipram from drug development (Scott et al, 1991). There is some reason for optimism given that some newly developed PDE4 inhibitors appear to produce therapeutic effects without emetic responses (Yamamoto et al, 2006).
The anxiolytic-like effects produced by chronic rolipram treatment were consistent among different tests sensitive to the proven anxiolytic diazepam. These results are in agreement with the downregulation of PDE4 induced by diazepam (Cherry et al, 2001) and nicotine (Polesskaya et al, 2007); the latter also exerts anxiolytic- and antidepressant-like effects (Biala and Budzynska 2006; Semba et al, 1998). Although these two drugs most likely produce their behavioral effects by mechanisms other than the regulation of PDE4 activity, the potential contribution of their inhibitory role in PDE4 cannot be completely excluded.
The forced-swim and tail-suspension tests are well-established measures for evaluation of antidepressant drugs; both are highly reliable and have the highest rates of predictive validity for antidepressants. In addition, both tests also have been used to study the correlation of antidepressant-like behavior and neurogenesis (Catts et al, 2008; Thakker-Varia et al, 2007). The forced-swim test has been demonstrated to be responsive to chronic, but not acute, treatment with rolipram (Zhang et al, 2002). Consistent with our previous studies (Zhang et al, 2002, 2006, 2008), chronic administration of rolipram also produced antidepressant-like effects on FST and TST behavior; it also increased hippocampal neurogenesis. A relationship between antidepressant/anxiolytic and neurogenic effects was indicated by results from co-administration of MAM with rolipram. Inhibition of neurogenesis by MAM attenuated rolipram-induced antidepressant- and anxiolytic-like effects on behavior. The effect of MAM was not due to general toxicity, as MAM at 5 mg/kg decreased neither body weight nor locomotor activity. These results are consistent with the latest findings that, at doses of 1–5 mg/kg that reduce hippocampal neurogenesis, MAM does not affect general health nor reduce body weights in mice (Ko et al, 2009). By contrast, MAM at higher doses (7.5 and 15 mg/kg) significantly decreased the gain of body weights and led to some animal deaths (data not shown); previous findings indicate that MAM at higher doses (7–14 mg/kg) decreases the gain of body weights and locomotor activity in rats (Dupret et al, 2005).
The NSF test is a paradigm sensitive to anxiolytics and chronic antidepressants, which reduce feeding latency increased by a novel environment (Dulawa and Hen, 2005; Merali et al, 2003). It has been used to study the link between neurogenesis and antidepressant activity (Santarelli et al, 2003). Unexpectedly, rolipram administered chronically did not alter the latency in the NSF test. At least two possibilities may account for this. First, chronic rolipram treatment may inhibit food consumption to a certain extent, leading to unchanged feeding latency in the NSF. This might be true given that rolipram blocks clonidine-induced increase in food intake (Przegalinski and Jurkowska, 1987). Second, the NSF test may not be suitable for evaluating novel antidepressants such as rolipram. Further studies are needed to clarify this.
Another surprising result with NSF was the increase in feeding latency produced by chronic treatment with MAM, whereas MAM did not produce behavioral effects in other tests. The effects of MAM on NSF behavior and neurogenesis differ from those in a previous study, in which decreases in hippocampal neurogenesis by X-irradiation did not alter NSF behavior (Santarelli et al, 2003). Difference in the two approaches for reducing neurogenesis might play a role in this discrepancy. Nevertheless, the effect of MAM on NSF behavior was reversed by chronic treatment with rolipram. This appears to be consistent with the interactions between rolipram and MAM in the other behavioral tests. Therefore, the effect of MAM alone and the lack of an effect of rolipram alone on NSF suggest some dissociation between this test and the other behavioral tests.
When hippocampal neurogenesis recovered to control levels approximately 3 weeks after termination of MAM treatment, the behavioral effects of rolipram were restored. These results support the contribution of neurogenesis to the antidepressant- and anxiolytic-like effects of rolipram, which is consistent with the requirement of hippocampal neurogenesis for some behavioral effects of certain antidepressants (Santarelli et al, 2003). It appears that the reduced number of new neurons present following MAM treatment may alter the neural systems on which rolipram acts to produce its effects on behavior.
Mice treated with MAM and rolipram in combination displayed a significantly slower gain of body weight relative to vehicle-treated controls. Although the reason for this is not clear, it did not appear to be because of overt toxicity as the animals had normal behavior in terms of general motor activity in the open-field test and total arm exploration in the elevated-plus maze test.
It was noted that, although MAM significantly decreased hippocampal neurogenesis and completely blocked the neurogenic effect of rolipram, it did not produce effects opposite to the behavioral actions of rolipram nor completely block the anxiolytic- and antidepressant-like effects of rolipram. These results appear to be consistent with the finding that a decrease in neurogenesis is not necessary for the development of depression (Drew and Hen 2007; Reif et al, 2006; Vollmayr et al, 2003), although increased hippocampal neurogenesis may be required for some aspects of antidepressant activity (Santarelli et al, 2003). The results with the NSF test differed somewhat from the other behavioral tests in that MAM treatment did produce a depressive- or anxiety-like effect. Several reasons may account for the discrepant effects of MAM on hippocampal neurogenesis and behavior. First, other brain regions, such as the prefrontal cortex and amygdala that do not exhibit adult neurogenesis, also may contribute to the behavioral effects associated with PDE4-mediated cAMP/CREB signaling (Banasr and Duman, 2007). This is supported by the upregulation of PDE4 in the frontal cortex induced by learned helplessness, an animal model of depression (Itoh et al, 2003). Second, neurogenesis-independent mechanisms may be involved in the behavioral effects of rolipram; these mechanisms may include rolipram-induced upregulation of brain-derived neurotrophic factor (Nibuya et al, 1996) and increases in dendritic branching (Fujioka et al, 2004), which also are involved in the regulation of anxiety- and depression-associated behavior (Fujimaki et al, 2000; Vyas et al, 2002). Strain differences also may play a role. For instance, the antidepressant-like effect of chronic fluoxetine treatment is dependent on its neurogenic effect in 129/Sv mice (Santarelli et al, 2003), but not in BALB/cJ mice (Holick et al, 2008; Huang et al, 2008). For these reasons, changes in hippocampal neurogenesis may not fully parallel changes in behavior.
Rolipram also produces a potent anti-inflammatory effect (Zhu et al, 2001). However, this appears not to be involved in the interaction with MAM in terms of behavioral and neurogenic effects, given that MAM does not induce inflammation, which usually occurs with irradiation-induced decreases in neurogenesis (Monje et al, 2002).
Role of Hippocampal Progenitor Cells in Rolipram's Actions
Adult neurogenesis is characterized by DNA synthesis during the S phase of mitosis of dividing progenitor cells. In the mitotic phase of hippocampal progenitors, quiescent neural progenitors (QNPs) generate amplifying neural progenitors (ANPs) through asymmetric divisions (Encinas et al, 2006). Fluoxetine increases the rate of symmetric divisions of ANPs and subsequently increases newborn neurons in the dentate gyrus (Encinas et al, 2006). However, the type of cells that rolipram may target in the neuronal differentiation cascade has not been investigated. Similar to fluoxetine, rolipram, administered repeatedly at doses that increased pCREB and produced behavioral effects, increased expression of Sox2 in the hippocampus, a marker of neural progenitor cells (Episkopou, 2005; Graham et al, 2003). By contrast, neither drug altered Sox2 expression in the prefrontal cortex, suggesting that rolipram increases neural progenitor cells in a brain region-specific manner.
Rolipram-induced proliferation of progenitor cells appears to be dominated by ANPs. First, Sox2 is only expressed in QNPs and ANPs and BrdU mainly labels ANPs (Encinas et al, 2006). Second, chronic rolipram treatment increases cell proliferation, as evidenced by increased BrdU-labeled cells in the dentate gyrus 2 h after the BrdU injection (Nakagawa et al, 2002b); it increased Sox2 only in the hippocampus. Third, the unaltered proportion of newborn neurons in BrdU-positive cells or pCREB-expressing neurons in newborn neurons indicates that rolipram likely does not directly target the post-mitotic phase of progenitor cells. Thus, rolipram increases neurogenesis likely by originally targeting ANPs in the SGZ. This is supported by the unaltered phenotype of BrdU-positive cells after rolipram administration in the present and previous studies (Malberg et al, 2000; Nakagawa et al, 2002b).
Relationship Among cAMP/CREB, Neurogenesis, and Behavior
The effect of rolipram on pCREB was blocked by MAM in a brain region-specific manner, in that it was observed in the hippocampus but not the prefrontal cortex; the blockade disappeared 3 weeks after termination of MAM treatment. The changes in pCREB followed the same pattern by those of BrdU-positive cells in the dentate gyrus; these effects were highly correlated. The correlation between CREB phosphorylation and hippocampal neurogenesis is supported by previous studies showing that pCREB is restricted to the dentate gyrus in adult mice (Nakagawa et al, 2002a; Thome et al, 2000). By contrast, pCREB in newborn neurons in the dentate gyrus only partially contributed to the behavioral effects of rolipram.
It was interesting that almost all the BrdU-labeled, newborn neurons expressed pCREB, whereas mature neurons in the dentate gyrus labeled by calbindin, a selective marker of mature neurons (Encinas et al, 2006), did not express pCREB. This appears to be supported by previous findings that NeuN-labeled cells (primarily mature neurons) express very low pCREB in the dentate gyrus (Sasaki et al, 2007). By contrast, almost all mature neurons in the prefrontal cortex expressed pCREB, indicating that rolipram differentially affects CREB phosphorylation in the two brain regions. This and the different blocking effects of MAM on rolipram-induced increases in pCREB in the two regions support the brain region-specific profile of CREB function (Carlezon et al, 2005).
In conclusion, the antidepressant- and anxiolytic-like effects of rolipram on behavior are accompanied with increased pCREB and neurogenesis in the hippocampus. Inhibition of neurogenesis with MAM also reduced the effect of rolipram on both pCREB and behavior. Thus, cAMP/CREB signaling in the hippocampus appears to be critical for hippocampal neurogenesis, which appears to play a role in the mediation of behavioral effects of chronic administration of rolipram; this process likely is mediated by targeting on mitotic progenitor cells in the dentate gyrus. The combined antidepressant- and anxiolytic-like effects of rolipram could benefit in the treatment of comorbid disorders of anxiety and depression, which are thought to share some common genetics (Kendler et al, 1992; Roy et al, 1995).
References
Bani-Yaghoub M, Tremblay RG, Lei JX, Zhang D, Zurakowski B, Sandhu JK et al (2006). Role of Sox2 in the development of the mouse neocortex. Dev Biol 295: 52–66.
Banasr M, Duman RS (2007). Regulation of neurogenesis and gliogenesis by stress and antidepressant treatment. CNS Neurol Disord Drug Targets 6: 311–320.
Barad M, Bourtchouladze R, Winder DG, Golan H, Kandel E (1998). Rolipram, a type IV-specific phosphodiesterase inhibitor, facilitates the establishment of long-lasting long-term potentiation and improves memory. Proc Natl Acad Sci USA 95: 15020–15025.
Biala G, Budzynska B (2006). Effects of acute and chronic nicotine on elevated plus maze in mice: involvement of calcium channels. Life Sci 79: 81–88.
Blendy JA (2006). The role of CREB in depression and antidepressant treatment. Biol Psychiatry 59: 1144–1150.
Bourin M, Hascoet M, Mansouri B, Colombel MC, Bradwejn J (1992). Comparison of behavioral effects after single and repeated administrations of four benzodiazepines in three mice behavioral models. J Psychiatry Neurosci 17: 72–77.
Boyes B, Kim SU, Lee V, Sung SC (1986). Immunohistochemical colocalization of S-100b and the glial fibrillary acidic protein in rat brain. Neuroscience 17: 857–865.
Bruel-Jungerman E, Laroche S, Rampon C (2005). New neurons in the dentate gyrus are involved in the expression of enhanced long-term memory following environmental enrichment. Eur J Neurosci 21: 513–521.
Bruel-Jungerman E, Rampon C, Laroche S (2007). Adult hippocampal neurogenesis, synaptic plasticity and memory: facts and hypotheses. Rev Neurosci 18: 93–114.
Carlezon WA, Duman RS, Nestler EJ (2005). The many faces of CREB. Trends Neurosci 28: 436–445.
Catts VS, Al-Menhali N, Burne TH, Colditz MJ, Coulson EJ (2008). The p75 neurotrophin receptor regulates hippocampal neurogenesis and related behaviours. Eur J Neurosci 28: 883–892.
Cherry JA, Thompson BE, Pho V (2001). Diazepam and rolipram differentially inhibit cyclic AMP-specific phosphodiesterases PDE4A1 and PDE4B3 in the mouse. Biochim Biophys Acta 1518: 27–35.
Dranovsky A, Hen R (2006). Hippocampal neurogenesis: regulation by stress and antidepressants. Biol Psychiatry 59: 1136–1143.
Drew MR, Hen R (2007). Adult hippocampal neurogenesis as target for the treatment of depression. CNS Neurol Disord Drug Targets 6: 205–218.
D'Sa C, Duman RS (2002). Antidepressants and neuroplasticity. Bipolar Disord 4: 183–194.
Dulawa SC, Hen R (2005). Recent advances in animal models of chronic antidepressant effects: the novelty-induced hypophagia test. Neurosci Biobehav Rev 29: 771–783.
Duman R, Malberg J, Nakagawa S (2001). Regulation of adult neurogenesis by psychotropic drugs and stress. J Pharmacol Exp Ther 299: 401–407.
Dupret D, Montaron MF, Drapeau E, Aurousseau C, Le Moal M, Piazza PV et al (2005). Methylazoxymethanol acetate does not fully block cell genesis in the young and aged dentate gyrus. Eur J Neurosci 22: 778–783.
Eisch A, Barrot M, Schad CA, Self DW, Nestler EJ (2000). Opiates inhibit neurogenesis in the adult rat hippocampus. Proc Natl Acad Sci USA 97: 7579–7584.
Emsley JG, Mitchell BD, Kempermann G, Macklis JD (2005). Adult neurogenesis and repair of the adult CNS with neural progenitors, precursors, and stem cells. Prog Neurobiol 75: 321–341.
Encinas JM, Vaahtokari A, Enikolopov G (2006). Fluoxetine targets early progenitor cells in the adult brain. Proc Natl Acad Sci USA 103: 8233–8238.
Episkopou V (2005). SOX2 functions in adult neural stem cells. Trends Neurosci 28: 219–221.
Fujioka T, Fujioka A, Duman RS (2004). Activation of cAMP signaling facilitates the morphological maturation of newborn neurons in adult hippocampus. J Neurosci 24: 319–328.
Fujimaki K, Morinobu S, Duman RS (2000). Administration of a cAMP phosphodiesterase 4 inhibitor enhances antidepressant-induction of BDNF mRNA in rat hippocampus. Neuropsychopharmacology 22: 42–51.
Gage F (2000). Mammalian neural stem cells. Science 287: 1433–1438.
Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ (1999). Learning enhances adult neurogenesis in the hippocampal formation. Nat Neurosci 2: 260–265.
Graham V, Khudyakov J, Ellis P, Pevny L (2003). SOX2 functions to maintain neural progenitor identity. Neuron 39: 749–765.
Griebel G, Misslin R, Vogel E, Bourguignon JJ (1991). Behavioral effects of rolipram and structurally related compounds in mice: behavioral sedation of cAMP phosphodiesterase inhibitors. Pharmacol Biochem Behav 39: 321–323.
Gur TL, Conti AC, Holden J, Bechtholt AJ, Hill TE, Lucki I et al (2007). cAMP response element-binding protein deficiency allows for increased neurogenesis and a rapid onset of antidepressant response. J Neurosci 27: 7860–7868.
Heaslip RJ, Evans DY (1995). Emetic, central nervous system, and pulmonary activities of rolipram in the dog. Eur J Pharmacol 286: 281–290.
Holick KA, Lee DC, Hen R, Dulawa SC (2008). Behavioral effects of chronic fluoxetine in BALB/cJ mice do not require adult hippocampal neurogenesis or the serotonin 1A receptor. Neuropsychopharmacology 33: 406–417.
Huang GJ, Bannerman D, Flint J (2008). Chronic fluoxetine treatment alters behavior, but not adult hippocampal neurogenesis, in BALB/cJ mice. Mol Psychiatry 13: 119–121.
Imaizumi M, Miyazaki S, Onodera K (1994). Effects of a non-xanthine adenosine antagonist, CGS 15943, and a phosphodiesterase inhibitor, Ro 20-1724, in a light/dark test in mice. Methods Find Exp Clin Pharmacol 16: 717–721.
Itoh T, Abe K, Tokumura M, Horiuchi M, Inoue O, Ibii N (2003). Different regulation of adenylyl cyclase and rolipram-sensitive phosphodiesterase activity on the frontal cortex and hippocampus in learned helplessness rats. Brain Res 991: 142–149.
Johnston MV, Coyle JT (1979). Histological and neurochemical effects of fetal treatment with methylazoxymethanol on rat neocortex in adulthood. Brain Res 170: 135–155.
Kempermann G, Gast D, Kronenberg G, Yamaguchi M, Gage FH (2003). Early determination and long-term persistence of adult-generated new neurons in the hippocampus of mice. Development 130: 391–399.
Kendler KS, Neale MC, Kessler RC, Heath AC, Eaves LJ (1992). Major depression and generalized anxiety disorder. Same genes, (partly) different environments? Arch Gen Psychiatry 49: 716–722.
Ko HG, Jang DJ, Son J, Kwak C, Choi JH, Ji YH et al (2009). Effect of ablated hippocampal neurogenesis on the formation and extinction of contextual fear memory. Mol Brain 2: 1–10.
Lucki I, Dalvi A, Mayorga AJ (2001). Sensitivity to the effects of pharmacologically selective antidepressants in different strains of mice. Psychopharmacology (Berl) 155: 315–322.
Malberg J, Eisch AJ, Nestler EJ, Duman RS (2000). Chronic antidepressant treatment increases neurogenesis in adult hippocampus. J Neurosci 20: 9104–9110.
Masood A, Nadeem A, Mustafa SJ, O'Donnell JM (2008). Reversal of oxidative stress-induced anxiety by inhibition of phosphodiesterase-2 in mice. J Pharmacol Exp Ther 326: 369–379.
Masui S, Nakatake Y, Toyooka Y, Shimosato D, Yagi R, Takahashi K et al (2007). Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat Cell Biol 9: 625–635.
Merali Z, Levac C, Anisman H (2003). Validation of a simple, ethologically relevant paradigm for assessing anxiety in mice. Biol Psychiatry 54: 552–565.
Monje ML, Mizumatsu S, Fike JR, Palmer TD (2002). Irradiation induces neural precursor-cell dysfunction. Nat Med 8: 955–962.
Monti B, Berteotti C, Contestabile A (2006). Subchronic rolipram delivery activates hippocampal CREB and arc, enhances retention and slows down extinction of conditioned fear. Neuropsychopharmacology 31: 278–286.
Mullen R, Buck CR, Smith AM (1992). NeuN, a neuronal specific nuclear protein in vertebrates. Development 116: 201–211.
Nakagawa S, Kim JE, Lee R, Chen J, Fujioka T, Malberg J et al (2002a). Localization of phosphorylated cAMP response element binding protein in immature neurons of adult hippocampus. J Neurosci 22: 9868–9876.
Nakagawa S, Kim JE, Lee R, Malberg JE, Chen J, Steffen C et al (2002b). Regulation of neurogenesis in adult mouse hippocampus by cAMP and the cAMP response element-binding protein. J Neurosci 22: 3673–3682.
Newton SS, Thome J, Wallace TL, Shirayama Y, Schlesinger L, Sakai N et al (2002). Inhibition of cAMP response element-binding protein or dynorphin in the nucleus accumbens produces an antidepressant-like effect. J Neurosci 22: 10883–10890.
Nibuya M, Nestler EJ, Duman RS (1996). Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci 16: 2365–2372.
O'Donnell JM, Frith S (1999). Behavioral effects of family-selective inhibitors of cyclic nucleotide phosphodiesterases. Pharmacol Biochem Behav 63: 185–192.
Pandey SC, Zhang H, Roy A, Xu T (2005). Deficits in amygdaloid cAMP-responsive element-binding protein signaling play a role in genetic predisposition to anxiety and alcoholism. J Clin Invest 115: 2762–2773.
Polesskaya OO, Smith RF, Fryxell KJ (2007). Chronic nicotine doses down-regulate PDE4 isoforms that are targets of antidepressants in adolescent female rats. Biol Psychiatry 61: 56–64.
Przegalinski E, Jurkowska T (1987). Effect of repeated treatment with antidepressant drugs or electroconvulsive shock (ECS) on the increase in food intake induced by clonidine injected into the paraventricular nucleus. Arch Int Pharmacodyn Ther 290: 257–266.
Reif A, Fritzen S, Finger M, Strobel A, Lauer M, Schmitt A et al (2006). Neural stem cell proliferation is decreased in schizophrenia, but not in depression. Mol Psychiatry 11: 514–522.
Roy MA, Neale MC, Pedersen NL, Mathé AA, Kendler KS (1995). A twin study of generalized anxiety disorder and major depression. Psychol Med 25: 1037–1049.
Sairanen M, O'Leary OF, Knuuttila JE, Castrén E (2007). Chronic antidepressant treatment selectively increases expression of plasticity-related proteins in the hippocampus and medial prefrontal cortex of the rat. Neuroscience 144: 368–374.
Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S et al (2003). Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301: 805–809.
Sasaki T, Kitagawa K, Omura-Matsuoka E, Todo K, Terasaki Y, Sugiura S et al (2007). The phosphodiesterase inhibitor rolipram promotes survival of newborn hippocampal neurons after ischemia. Stroke 38: 1597–1605.
Schmidt HD, Duman RS (2007). The role of neurotrophic factors in adult hippocampal neurogenesis, antidepressant treatments and animal models of depressive-like behavior. Behav Pharmacol 18: 391–418.
Scott AI, Perini AF, Shering PA, Whalley LJ (1991). In-patient major depression: is rolipram as effective as amitriptyline. Eur J Clin Pharmacol 40: 127–129.
Semba J, Mataki C, Yamada S, Nankai M, Toru M (1998). Antidepressant-like effects of chronic nicotine on learned helplessness paradigm in rats. Biol Psychiatry 43: 389–391.
Shors TJ, Miesegaes G, Beylin A, Zhao MR, Rydel T, Gould E (2001). Neurogenesis in the adult is involved in the formation of trace memories. Nature 410: 372–376.
Silvestre JS, Fernandez AG, Palacios JM (1999a). Effects of rolipram on the elevated plus-maze test in rats: a preliminary study. J Psychopharmacol 13: 274–277.
Silvestre JS, Fernandez AG, Palacios JM (1999b). Preliminary evidence for an involvement of the cholinergic system in the sedative effects of rolipram in rats. Pharmacol Biochem Behav 64: 1–5.
Thakker-Varia S, Krol JJ, Nettleton J, Bilimoria PM, Bangasser DA, Shors TJ et al (2007). The neuropeptide VGF produces antidepressant-like behavioral effects and enhances proliferation in the hippocampus. J Neurosci 27: 12156–12167.
Thome J, Sakai N, Shin K, Steffen C, Zhang YJ, Impey S et al (2000). cAMP response element-mediated gene transcription is upregulated by chronic antidepressant treatment. J Neurosci 20: 4030–4036.
van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH (2002). Functional neurogenesis in the adult hippocampus. Nature 415: 1030–1034.
Vollmayr B, Simonis C, Weber S, Gass P, Henn F (2003). Reduced cell proliferation in the dentate gyrus is not correlated with the development of learned helplessness. Biol Psychiatry 54: 1035–1040.
Vyas A, Mitra R, Shankaranarayana Rao BS, Chattarji S (2002). Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J Neurosci 22: 6810–6818.
Wand G (2005). The anxious amygdala: CREB signaling and predisposition to anxiety and alcoholism. J Clin Invest 115: 2697–2699.
Weiss SM, Wadsworth G, Fletcher A, Dourish CT (1998). Utility of ethological analysis to overcome locomotor confounds in elevated maze models of anxiety. Neurosci Biobehav Rev 23: 265–271.
West M, Slomianka L, Gundersen H (1991). Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec 231: 482–497.
Yamamoto S, Sugahara S, Naito R, Ichikawa A, Ikeda K, Yamada T et al (2006). The effects of a novel phosphodiesterase 7A and -4 dual inhibitor, YM-393059, on T-cell-related cytokine production in vitro and in vivo. Eur J Pharmacol 541: 106–114.
Zhang HT (2009). Cyclic AMP-specific phosphodiesterase-4 as a target for the development of antidepressant drugs. Curr Pharm Design 15: 1688–1698.
Zhang HT, O'Donnell JM (2000). Effects of rolipram on scopolamine-induced impairment of working and reference memory in the radial-arm maze tests in rats. Psychopharmacology (Berl) 150: 311–316.
Zhang HT, Crissman AM, Dorairaj NR, Chandler LJ, O'Donnell JM (2000). Inhibition of cyclic AMP phosphodiesterase (PDE4) reverses memory deficits associated with NMDA receptor antagonism. Neuropsychopharmacology 23: 198–204.
Zhang HT, Huang Y, Jin SL, Frith SA, Suvarna N, Conti M et al (2002). Antidepressant-like profile and reduced sensitivity to rolipram in mice deficient in the PDE4D phosphodiesterase enzyme. Neuropsychopharmacology 27: 587–595.
Zhang HT, Zhao Y, Huang Y, Dorairaj NR, Chandler LJ, O'Donnell JM (2004). Inhibition of the phosphodiesterase 4 (PDE4) enzyme reverses memory deficits produced by infusion of the MEK inhibitor U0126 into the CA1 subregion of the rat hippocampus. Neuropsychopharmacology 29: 1432–1439.
Zhang HT, Huang Y, Suvarna NU, Deng C, Crissman AM, Hopper AT et al (2005). Effects of the novel PDE4 inhibitors MEM1018 and MEM1091 on memory in the radial-arm maze and inhibitory avoidance tests in rats. Psychopharmacology (Berl) 179: 613–619.
Zhang HT, Zhao Y, Huang Y, Deng C, Hopper AT, De Vivo M et al (2006). Antidepressant-like effects of PDE4 inhibitors mediated by the high-affinity rolipram binding state (HARBS) of the phosphodiesterase-4 enzyme (PDE4) in rats. Psychopharmacology (Berl) 186: 209–217.
Zhang HT, Huang Y, Masood A, Stolinski LR, Li Y, Zhang L et al (2008). Anxiogenic-like behavioral phenotype of mice deficient in phosphodiesterase 4B (PDE4B). Neuropsychopharmacology 33: 1611–1623.
Zhu J, Mix E, Winblad B (2001). The antidepressant and antiinflammatory effects of rolipram in the central nervous system. CNS Drug Rev 7: 387–398.
Zhu DY, Lau L, Liu SH, Wei JS, Lu YM (2004). Activation of cAMP-response-element-binding protein (CREB) after focal cerebral ischemia stimulates neurogenesis in the adult dentate gyrus. Proc Natl Acad Sci USA 101: 9453–9457.
Acknowledgements
This study was supported by research grants from NARSAD and NIA (AG031687 to HTZ) and the NIMH (MH051175, MH040697 to JMO). We thank Dr Albert S Berrebi for his advice and support and Mr Dennis Cole, Mr Jeffrey B Altemus, and Dr Karen H Martin for their technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
DISCLOSURE/CONFLICT OF INTEREST
Han-Ting Zhang and James M O'Donnell have received financial support for their research from Memory Pharmaceuticals, Lundbeck Pharmaceuticals, and Wyeth Pharmaceuticals. James M O'Donnell is on the Scientific Advisory Board of Fission Pharmaceuticals and Ivy Editing (both unpaid). The other authors do not have financial interests to disclose.
Rights and permissions
About this article
Cite this article
Li, YF., Huang, Y., Amsdell, S. et al. Antidepressant- and Anxiolytic-like Effects of the Phosphodiesterase-4 Inhibitor Rolipram on Behavior Depend on Cyclic AMP Response Element Binding Protein-Mediated Neurogenesis in the Hippocampus. Neuropsychopharmacol 34, 2404–2419 (2009). https://doi.org/10.1038/npp.2009.66
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2009.66
Keywords
This article is cited by
-
Effects of exercise-targeted hippocampal PDE-4 methylation on synaptic plasticity and spatial learning/memory impairments in D-galactose-induced aging rats
Experimental Brain Research (2024)
-
Pathophysiological basis and promise of experimental therapies for Gulf War Illness, a chronic neuropsychiatric syndrome in veterans
Psychopharmacology (2023)
-
Phosphodiesterase inhibitors in psychiatric disorders
Psychopharmacology (2023)
-
Inhibition of phosphodiesterase-4 mitigates stress-re-stress-paradigm induced mitochondrial perturbations in rats exhibiting PTSD-like symptoms
Neuroscience and Behavioral Physiology (2023)
-
Persimmon leaf extract alleviates chronic social defeat stress-induced depressive-like behaviors by preventing dendritic spine loss via inhibition of serotonin reuptake in mice
Chinese Medicine (2022)
