
Abstract
By analyzing the exomes of 12,332 unrelated Swedish individuals, including 4,877 individuals affected with schizophrenia, in ways informed by exome sequences from 45,376 other individuals, we identified 244,246 coding-sequence and splice-site ultra-rare variants (URVs) that were unique to individual Swedes. We found that gene-disruptive and putatively protein-damaging URVs (but not synonymous URVs) were more abundant among individuals with schizophrenia than among controls (P = 1.3 × 10−10). This elevation of protein-compromising URVs was several times larger than an analogously elevated rate for de novo mutations, suggesting that most rare-variant effects on schizophrenia risk are inherited. Among individuals with schizophrenia, the elevated frequency of protein-compromising URVs was concentrated in brain-expressed genes, particularly in neuronally expressed genes; most of this elevation arose from large sets of genes whose RNAs have been found to interact with synaptically localized proteins. Our results suggest that synaptic dysfunction may mediate a large fraction of strong, individually rare genetic influences on schizophrenia risk.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others


References
McGrath, J., Saha, S., Chant, D. & Welham, J. Schizophrenia: a concise overview of incidence, prevalence, and mortality. Epidemiol. Rev. 30, 67–76 (2008).
Sullivan, P.F., Kendler, K.S. & Neale, M.C. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch. Gen. Psychiatry 60, 1187–1192 (2003).
Lichtenstein, P. et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet 373, 234–239 (2009).
Bundy, H., Stahl, D. & MacCabe, J.H. A systematic review and meta-analysis of the fertility of patients with schizophrenia and their unaffected relatives. Acta Psychiatr. Scand. 123, 98–106 (2011).
Power, R.A. et al. Fecundity of patients with schizophrenia, autism, bipolar disorder, depression, anorexia nervosa, or substance abuse vs their unaffected siblings. JAMA Psychiatry 70, 22–30 (2013).
Zuk, O. et al. Searching for missing heritability: designing rare variant association studies. Proc. Natl. Acad. Sci. USA 111, E455–E464 (2014).
Stefansson, H. et al. CNVs conferring risk of autism or schizophrenia affect cognition in controls. Nature 505, 361–366 (2014).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Purcell, S.M. et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190 (2014).
MacArthur, D.G. et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science 335, 823–828 (2012).
Maquat, L.E. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 5, 89–99 (2004).
Fromer, M. et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184 (2014).
Iossifov, I. et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221 (2014).
Ionita-Laza, I., Lee, S., Makarov, V., Buxbaum, J.D. & Lin, X. Sequence kernel association tests for the combined effect of rare and common variants. Am. J. Hum. Genet. 92, 841–853 (2013).
Purcell, S., Cherny, S.S. & Sham, P.C. Genetic power calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 19, 149–150 (2003).
Takata, A. et al. Loss-of-function variants in schizophrenia risk and SETD1A as a candidate susceptibility gene. Neuron 82, 773–780 (2014).
Singh, T. et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat. Neurosci. 19, 571–577 (2016).
Rujescu, D. et al. Disruption of the neurexin 1 gene is associated with schizophrenia. Hum. Mol. Genet. 18, 988–996 (2009).
Xu, B. et al. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat. Genet. 44, 1365–1369 (2012).
Millar, J.K. et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum. Mol. Genet. 9, 1415–1423 (2000).
Pinto, D. et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 94, 677–694 (2014).
Samocha, K.E. et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 46, 944–950 (2014).
Fagerberg, L. et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 13, 397–406 (2014).
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Finucane, H.K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015).
Cahoy, J.D. et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 28, 264–278 (2008).
Darnell, J.C. et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261 (2011).
Wagnon, J.L. et al. CELF4 regulates translation and local abundance of a vast set of mRNAs, including genes associated with regulation of synaptic function. PLoS Genet. 8, e1003067 (2012).
Lee, J.-A. et al. Cytoplasmic Rbfox1 regulates the expression of synaptic and autism-related genes. Neuron 89, 113–128 (2016).
Hamada, N. et al. Biochemical and morphological characterization of A2BP1 in neuronal tissue. J. Neurosci. Res. 91, 1303–1311 (2013).
Weyn-Vanhentenryck, S.M. et al. HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep. 6, 1139–1152 (2014).
Pirooznia, M. et al. SynaptomeDB: an ontology-based knowledgebase for synaptic genes. Bioinformatics 28, 897–899 (2012).
Mo, A. et al. Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron 86, 1369–1384 (2015).
Kirov, G. et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 17, 142–153 (2012).
Bayés, A. et al. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat. Neurosci. 14, 19–21 (2011).
Betel, D., Koppal, A., Agius, P., Sander, C. & Leslie, C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 11, R90 (2010).
Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 43, 969–976 (2011).
Robinson, E.B. et al. Autism spectrum disorder severity reflects the average contribution of de novo and familial influences. Proc. Natl. Acad. Sci. USA 111, 15161–15165 (2014).
Robinson, E.B., Neale, B.M. & Hyman, S.E. Genetic research in autism spectrum disorders. Curr. Opin. Pediatr. 27, 685–691 (2015).
Robinson, E.B. et al. Genetic risk for autism spectrum disorders and neuropsychiatric variation in the general population. Nat. Genet. 48, 552–555 (2016).
Moeschler, J.B. Genetic evaluation of intellectual disabilities. Semin. Pediatr. Neurol. 15, 2–9 (2008).
Gécz, J., Shoubridge, C. & Corbett, M. The genetic landscape of intellectual disability arising from chromosome X. Trends Genet. 25, 308–316 (2009).
McRae, J.F. et al. Prevalence, phenotype and architecture of developmental disorders caused by de novo mutation. bioRxiv http://dx.doi.org/10.1101/049056 (2016).
Jensen, L.R. et al. Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am. J. Hum. Genet. 76, 227–236 (2005).
Xiang, Y. et al. JARID1B is a histone H3 lysine 4 demethylase up-regulated in prostate cancer. Proc. Natl. Acad. Sci. USA 104, 19226–19231 (2007).
Matsumoto, M. et al. Ataxia and epileptic seizures in mice lacking type 1 inositol 1,4,5-trisphosphate receptor. Nature 379, 168–171 (1996).
Richards, A.L. et al. Exome arrays capture polygenic rare variant contributions to schizophrenia. Hum. Mol. Genet. 25, 1001–1007 (2016).
Iossifov, I. et al. De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299 (2012).
De Rubeis, S. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014).
Ripke, S. et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 45, 1150–1159 (2013).
Purcell, S.M. et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–752 (2009).
Bergen, S.E. et al. Genome-wide association study in a Swedish population yields support for greater CNV and MHC involvement in schizophrenia compared with bipolar disorder. Mol. Psychiatry 17, 880–886 (2012).
Szatkiewicz, J.P. et al. Copy number variation in schizophrenia in Sweden. Mol. Psychiatry 19, 762–773 (2014).
Genovese, G. et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 2477–2487 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The Genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
DePristo, M.A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Van der Auwera, G.A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 1–33 (2013).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Chang, C.C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Sekar, A. et al. Schizophrenia risk from complex variation of complement component 4. Nature 530, 177–183 (2016).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92 (2012).
Cingolani, P. et al. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Front. Genet. 3, 35 (2012).
Liu, X., Jian, X. & Boerwinkle, E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 32, 894–899 (2011).
Liu, X., Jian, X. & Boerwinkle, E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum. Mutat. 34, E2393–E2402 (2013).
Hsu, F. et al. The UCSC known genes. Bioinformatics 22, 1036–1046 (2006).
Kumar, P., Henikoff, S. & Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 (2009).
Adzhubei, I.A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Chun, S. & Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 19, 1553–1561 (2009).
Schwarz, J.M., Rödelsperger, C., Schuelke, M. & Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576 (2010).
Reva, B., Antipin, Y. & Sander, C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 39, e118 (2011).
Choi, Y., Sims, G.E., Murphy, S., Miller, J.R. & Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS One 7, e46688 (2012).
Berman, H.M. et al. The protein data bank. Nucleic Acids Res. 28, 235–242 (2000).
Shihab, H.A. et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 34, 57–65 (2013).
Abecasis, G.R. et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012).
Yang, J., Lee, S.H., Goddard, M.E. & Visscher, P.M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
Szatmari, P. et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 39, 319–328 (2007).
Sebat, J. et al. Strong association of de novo copy number mutations with autism. Science 316, 445–449 (2007).
Marshall, C.R. et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 82, 477–488 (2008).
Pinto, D. et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372 (2010).
Itsara, A. et al. De novo rates and selection of large copy number variation. Genome Res. 20, 1469–1481 (2010).
Sanders, S.J. et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70, 863–885 (2011).
Levy, D. et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 70, 886–897 (2011).
Gilman, S.R. et al. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70, 898–907 (2011).
Xu, B. et al. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet. 40, 880–885 (2008).
Malhotra, D. et al. High frequencies of de novo CNVs in bipolar disorder and schizophrenia. Neuron 72, 951–963 (2011).
Noor, A. et al. Copy number variant study of bipolar disorder in Canadian and UK populations implicates synaptic genes. Am. J. Med. Genet. 165B, 303–313 (2014).
Georgieva, L. et al. De novo CNVs in bipolar affective disorder and schizophrenia. Hum. Mol. Genet. 23, 6677–6683 (2014).
Neale, B.M. et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245 (2012).
Jiang, Y.H. et al. Detection of clinically relevant genetic variants in autism spectrum disorder by whole-genome sequencing. Am. J. Hum. Genet. 93, 249–263 (2013).
Allen, A.S. et al. De novo mutations in epileptic encephalopathies. Nature 501, 217–221 (2013).
EuroEPINOMICS-RES Consortium; Epilepsy Phenome/Genome Project; Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am. J. Hum. Genet. 95, 360–370 (2014).
Zaidi, S. et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature 498, 220–223 (2013).
de Ligt, J. et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 367, 1921–1929 (2012).
Rauch, A. et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 380, 1674–1682 (2012).
Gilissen, C. et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 511, 344–347 (2014).
Hamdan, F.F. et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet. 10, e1004772 (2014).
Girard, S.L. et al. Increased exonic de novo mutation rate in individuals with schizophrenia. Nat. Genet. 43, 860–863 (2011).
Gulsuner, S. et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 154, 518–529 (2013).
McCarthy, S.E. et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol. Psychiatry 19, 652–658 (2014).
Hamosh, A., Scott, A.F., Amberger, J.S., Bocchini, C.A. & McKusick, V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 33, D514–D517 (2005).
Stevenson, R.E., Schwartz, C.E., Rogers, R.C. & Rogers, R.C. Atlas of X-linked Intellectual Disability Syndromes (Oxford University Press, 2012).
Moeschler, J.B., Shevell, M. & American Academy of Pediatrics Committee on Genetics. Clinical genetic evaluation of the child with mental retardation or developmental delays. Pediatrics 117, 2304–2316 (2006).
Rauch, A. et al. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am. J. Med. Genet. A. 140, 2063–2074 (2006).
Cotton, A.M. et al. Analysis of expressed SNPs identifies variable extents of expression from the human inactive X chromosome. Genome Biol. 14, R122 (2013).
Crow, T.J. The XY gene hypothesis of psychosis: origins and current status. Am. J. Med. Genet. 162B, 800–824 (2013).
Ji, B., Higa, K.K., Kelsoe, J.R. & Zhou, X. Over-expression of XIST, the master gene for X chromosome inactivation, in females with major affective disorders. EBioMedicine 2, 909–918 (2015).
Crow, T.J. Is psychosis a disorder of XY epigenetics? EBioMedicine 2, 794–795 (2015).
Vilhjálmsson, B.J. et al. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am. J. Hum. Genet. 97, 576–592 (2015).
Acknowledgements
We thank C. Usher for comments on the manuscript and work on the figures. This study was supported by grants from the National Human Genome Research Institute (U54 HG003067, R01 HG006855 to S.A.M.), the National Institute of Mental Health (R01 MH077139 to P.F.S., R01 MH095034 to P.S., and RC2 MH089905 to S.M.P. and P.S.), the Stanley Center for Psychiatric Research, the Alexander and Margaret Stewart Trust, and the Sylvan C. Herman Foundation.
Author information
Authors and Affiliations
Contributions
G.G. and S.A.M. designed the analyses and wrote early drafts of the manuscript. G.G. performed the analyses. M.F. contributed to analyses of de novo mutated genes, D.M.R. and E.A.S. contributed with the specific design of the analyses. K.C. contributed with sample processing and data management. M.L., J.L.M., S.M.P., P.S., P.F.S. and C.M.H. contributed with sample and phenotype collection. All of the authors contributed to interpretation of the findings and revisions of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Missense damaging predictions as a function of allele frequency
Percentage of missense variants classified as damaging by eight different classifiers and a classifier consisting of the intersection of classifiers SIFT, PolyPhen-2 HDIV, PolyPhen-2 HVAR, LRT, Mutation Taster, Mutation Assessor, and PROVEAN as a function of minor allele count across 12,332 unrelated individuals from Sweden. Gray and black colors indicate, respectively, variants never observed and variants already observed in the ExAC cohort of 45,376 individuals.
Supplementary Figure 2 Correlations across ultra-rare variant types counts
Relationships between four separate types of coding-sequence and splice-site URV counts across 12,332 unrelated individuals from Sweden: (a) disruptive vs. damaging; (b) disruprive vs. missense non-damaging; (c) damaging vs. missense non-damaging; (d) disruptive vs. synonymous; (e) damaging vs. synonymous; (f) missense non-damaging vs synonymous. Black dots indicate individuals with less than or equal to 100 URVs detected, and red crosses indicate individuals with more than 100 URVs detected. Pearson correlation coefficients are indicated on the top left of each panel.
Supplementary Figure 3 Enrichment in schizophrenia cases across ultra-rare variant types
Observed enrichment in 4,877 schizophrenia cases compared to 6,203 controls for (a) URVs across all annotations, with non-coding including both intronic and untranslated region variants, (b) missense URVs classified as damaging by classifiers PolyPhen-2 HDIV, PolyPhen-2 HVAR, SIFT, LRT, PROVEAN, FATHMM, Mutation Taster, and Mutation Assessor, as well as missense damaging, in-frame indel, protein-protein-contact, splice-acceptor, splice-donor, stop-gained, and frameshift URVs. Enrichment and P values were computed using a linear regression model (left panels) and a logistic regression model (right panels) and correcting for covariates (a) with and (b) without the exclusion of URV individual count. Horizontal bars indicate 95% confidence intervals.
Supplementary Figure 4 Single gene burden tests for disruptive and damaging variants at different allele frequency thresholds
Quantile-quantile plots for burden tests for association with schizophrenia of all Ensembl genes for disruptive and damaging variants. Burden test for association was performed using SKAT software.
Supplementary Figure 5 Enrichment of dURVs in schizophrenia cases across selected gene sets stratified per variant types and cohorts
Burden of dURVs across selected gene sets analyzed in this study stratified across disruptive and damaging URVs (left panel) and across previously analyzed exomes and newly available exomes (right panel). Enrichment and P values were computed using a logistic regression model using exome-wide dURV count as a covariate to correct for average exome-wide burden (dot-dashed line). Horizontal bars indicate 95% confidence intervals.
Supplementary Figure 6 Enrichment of dURVs in schizophrenia cases across constrained and unconstrained genes
Burden of dURVs across loss-of-function intolerant (LoF-intolerant) genes, missense constrained genes and their complementary sets, stratified across disruptive and damaging URVs. Enrichment and P values were computed using a linear regression model (left panels) and a logistic regression model (right panels) and, contrary to most other analyses in this study, without using exome-wide dURV count to correct for average exome-wide burden. Horizontal bars indicate 95% confidence intervals. As LoF-intolerant and missense constrained genes were defined outside this study, different enrichments observed within and outside these gene sets cannot be attributed to differential false positive rates between cases and controls.
Supplementary Figure 7 Enrichment of dURVs in schizophrenia cases across gene sets from different tissues
Burden of dURVs across 27 tissue expression specific gene sets. Each gene set was generated selecting genes for which expression in a given tissue was at least 5 times the median expression across 27 different human organs and tissues ascertained from 95 individual. Enrichment and P values were computed using a logistic regression model using exome-wide dURV count as a covariate to correct for average exome-wide burden (dot-dashed line). Horizontal bars indicate 95% confidence intervals. Brain specific genes were significantly more enriched for dURVs in schizophrenia cases than the average gene.
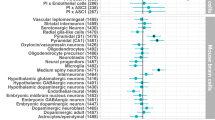
Supplementary Figure 8 Enrichment of dURVs in schizophrenia cases across gene sets from different brain and neuronal cell types
Burden analysis for dURVs across genes defined from (a) 11 brain cell types and (b) 3 neuron cell types: excitatory pyramidal neurons (Exc), parvalbumin (PV)-expressing fast-spiking interneurons, vasoactive intestinal peptide (VIP)-expressing interneurons, both for expressed genes and cell-type specific genes. Brain cell type gene sets were generated selecting genes for which log-expression in a given cell type was 0.5 greater than the median log-expression across 11 central nervous system cell types ascertained from developing and mature mouse forebrain. Neuron cell type expressed gene sets were defined as those with more than 50 observed transcripts per million (TPM). Neuron cell type specific gene sets were defined as those observed more than 5 times the minimum expression across the 3 different cell types. Expression profiles for neurons were ascertained from nuclei isolated from adult (8–11 weeks) mouse neocortex. Enrichment and P values were computed using a logistic regression model using exome-wide dURV count as a covariate to correct for average exome-wide burden (dot-dashed line). Horizontal bars indicate 95% confidence intervals.
Supplementary Figure 9 Enrichment of dURVs in schizophrenia cases across different X linked intellectual disability gene sets
Burden analysis for dURVs across X linked intellectual disability (XLID) genes and developmental disorder genes. X linked ID genes correspond to the union of X linked ID sets of genes as defined from OMIM, GCC, and Chicago. Enrichment was computed separately for males, females, and both groups together. Enrichment and P values were computed using a logistic regression model using exome-wide dURV count as a covariate to correct for average exome-wide burden (dot-dashed line). Horizontal bars indicate 95% confidence intervals. Larger confidence intervals for males reflect the smaller number of variants observed in males due to carrying half as much X chromosome DNA.
Supplementary Figure 10 Average ultra-rare variant types count across sequencing waves
Average number of URVs detected in controls and in schizophrenia cases used in this analysis across several classes of variants and sequencing waves. Numbers of controls and cases within each wave is indicated in parentheses. Wave 1 is denoted in red as for this batch, accounting for approximately 1% of the whole cohort, an earlier version of the hybrid-capture procedure was used which captured approximately 10% less of the exome. Vertical bars indicate 95% confidence intervals.
Supplementary Figure 11 Duplicate and first degree relationship estimates across cohort
Estimates of percentage of genome shared IBD (PI_HAT) and percentage of genome shared IBD1 (Z1) among all pairs from 12,384 samples for which PI_HAT>.35. Estimates were generated with plink command “--genome full”.
Supplementary Figure 12 Ultra-rare variants counts relationships with population stratification
Covariates within the Swedish exome cohort: (a) ultra-rare SNP and indel counts across cohort 12,334 individuals; (b) distribution of birth year across cohort individuals; (c) relationship of cohort individuals (in black) with 1000 Genomes project phase 1 individuals with respect to the first two principal components with removed outliers (in red); (d) relationship of cohort individuals with respect to the two main Swedish principal components; (e) relationship between principal components tracking Finnish ancestry and URV count; (f) relationship between principal components tracking Northern-Southern Swedish ancestry and URV count. Black crosses indicate individuals with less than or equal to 100 URVs detected, and red crosses indicate individuals with more than 100 URVs detected.
Supplementary Figure 13 Gender estimates from genotypes
X chromosome inbreeding coefficient (F) and Y-chromosome non-missing genotype calls (YCOUNT) for the entire cohort of 12,384 samples. Male individuals inferred with 47,XXY karyotype (Klinefelter syndrome) are indicated in green and individuals with mismatching reported and genotyped sex are indicated as yellow crosses. Estimates were generated with plink command ′--check-sex ycount′.
Supplementary Figure 14 Manhattan plot for association with schizophrenia of common exome variants
Manhattan plot for association of exonic variants with schizophrenia phenotype using a logistic regression model with sex and first five principal components as covariates. Variants with P value less than 10-6 include variant rs281766 on chromosome 2 in the UTR5 of genes TYW5 and C2orf47, and seven variants in the MHC region around the HLA genes.
Supplementary Figure 15 Missense predictors comparisons for association with schizophrenia
P values for enrichment of missense URVs classified as damaging by all 256 possible predictors defined as the combination of damaging definitions from any subset of Polyphen2_HDIV, Polyphen2_HVAR, SIFT, LRT, PROVEAN, FATHMM, Mutation Taster, and Mutation Assessor algorithms. P values were computed using a linear regression model and correcting for covariates including total URV count. Predictors including FATHMM performed significantly worse than predictors excluding FATHMM while 25 predictors performed better than the predictor chosen for the analyses in this manuscript (bright red). Both predictors including all algorithms but FATHMM (bright red) and including LRT, MutationTaster, PolyPhen2 HDIV, PolyPhen2 HVAR, and SIFT (green) performed better than each predictor based on a single algorithm.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–15 and Supplementary Tables 1, 2 and 8 (PDF 2266 kb)
Supplementary Methods Checklist
(PDF 129 kb)
Supplementary Table 3
List of dURVs identified across 4,877 schizophernia cases and 6,203 controls. (XLSX 2968 kb)
Supplementary Table 4
List of studies to define genes hit by de novo CNVs (Fig. 5a). (XLSX 6 kb)
Supplementary Table 5
List of de novo CNVs previously found in individuals with schizophrenia, bipolar disorder, and autism (Fig. 5a). (XLSX 34 kb)
Supplementary Table 6
List of studies to define genes hit by de novo non-synonymous variants (Fig. 5b). (XLSX 6 kb)
Supplementary Table 7
List of de novo variants found in individuals with schizophrenia, autism, epilepsy, intellectual disability, congenital heart disease, and controls (Fig. 5b). (XLSX 4529 kb)
Rights and permissions
About this article

Cite this article
Genovese, G., Fromer, M., Stahl, E. et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat Neurosci 19, 1433–1441 (2016). https://doi.org/10.1038/nn.4402
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nn.4402
This article is cited by
-
The molecular pathology of schizophrenia: an overview of existing knowledge and new directions for future research
Molecular Psychiatry (2023)
-
A cytoskeleton-membrane interaction conserved in fast-spiking neurons controls movement, emotion, and memory
Molecular Psychiatry (2023)
-
Clinical characteristics indexing genetic differences in schizophrenia: a systematic review
Molecular Psychiatry (2023)
-
The genetic architecture of schizophrenia: review of large-scale genetic studies
Journal of Human Genetics (2023)
-
Neurite outgrowth deficits caused by rare PLXNB1 mutation in pediatric bipolar disorder
Molecular Psychiatry (2023)
