
Abstract
The agents causing viral hemorrhagic fever (VHF) are a taxonomically diverse group of viruses that may share commonalities in the process whereby they produce systemic and frequently fatal disease. Significant progress has been made in understanding the biology of the Ebola virus, one of the best known examples. This knowledge has guided our thinking about other VHF agents, including Marburg, Lassa, the South American arenaviruses, yellow fever, Crimean-Congo and Rift Valley fever viruses. Comparisons among VHFs show that a common pathogenic feature is their ability to disable the host immune response by attacking and manipulating the cells that initiate the antiviral response. Of equal importance, these comparisons highlight critical gaps in our knowledge of these pathogens.
Similar content being viewed by others
Main
Our understanding of Ebola hemorrhagic fever pathogenesis has seen substantial developments over the past few years, dwarfing our comprehension of other viral hemorrhagic fever (VHF) infections. These infections occur in underdeveloped areas of the world, and much of our knowledge of naturally occurring disease is derived from sporadic outbreaks that occurred decades ago. Recently conducted experimental animal studies have provided insight into VHF pathogenesis and currently guide clinical investigations of disease. A better understanding of the relevant host and viral factors that influence clinical outcome will be critical to our ability to combat these aggressive pathogens. Here, we review data on the pathogenesis of these diseases, with special emphasis on their associated hematological and immunological abnormalities, and highlight some of the remaining questions that need to be addressed by future research.
Historical clinical studies of VHFs have provided important descriptive information on the pathogenesis of these agents, but the data are usually incomplete and occasionally contradictory or paradoxical. Here, we summarize data obtained from these clinical studies and draw parallels from experimental infections of nonhuman primates. Rodent models are also available for many of the VHFs. Although rodents are useful as a first screen for evaluating antiviral drugs and vaccine strategies, and genetically engineered mice can help in understanding specific host-pathogen interactions, the disease course in rodents is far less faithful to the human condition than in primate models1,2.
The agents
Exotic emerging viruses include Hendra and Nipah viruses and the West Nile virus, all of which lead to acute encephalitic syndromes. Here, we will focus on the most diverse and deadly group of exotic emerging viruses, the VHF agents. As a group, these viruses cause an illness characterized by fever, a bleeding diathesis and circulatory shock. The viruses are classified into four different families of RNA viruses—Arenaviridae, Bunyaviridae, Filoviridae, and Flaviviridae (Table 1). All of the viruses causing VHF have a single-stranded RNA genome and a lipid envelope, but vary considerably in morphology from typical small isometric or moderately sized spherical virions to highly unusual pleomorphic or filamentous particles (Fig. 1).

Junin virus (top left). Arenavirus particles range in morphology from highly pleomorphic, as shown in this field, to mainly spherical. Virion sizes range from 50 to 300 nm with a mean of 100–130 nm. Rift valley fever virus (top center). Bunyavirus particles are roughly spherical and range in diameter from 90 to 120 nm. Yellow fever virus (top right). Flavivirus particles are essentially isometric and consistent in size ranging from 40 to 50 nm in diameter. Ebola virus (bottom). Filovirus particles are mostly filamentous and vary in length up to 14,000 nm with a uniform diameter of 80 nm. Mean unit length is about 1,000 nm. Other forms of filovirus particles include U-shaped, '6'-shaped or circular configurations; branching of filamentous particles can also occur.
Transmission
Under natural conditions, members of the Arenaviridae, Bunyaviridae, Filoviridae and Flaviviridae that cause VHF have specific geographic distribution and diverse modes of transmission. Despite exhaustive efforts by several groups, the natural reservoir for Filoviridae remains unknown. As a group, the other VHF agents are linked to the ecology of their vectors or reservoirs, whether rodents or arthropods. Human-to-human transmission is possible for all VHF viruses. The majority of the person-to-person transmission for the arenaviruses and filoviruses has been attributed to direct contact with infected blood and body fluids. The potential for airborne transmission of the VHF agents appears to be an infrequent event, but cannot be categorically excluded as a mode of transmission.
Clinical picture of VHF infections
The overall incubation period for VHF ranges from 2 to 21 days. There is a wide constellation of clinical manifestations with varying degrees of severity in patients infected with these viruses, and not all patients develop classic VHF syndrome. The exact nature of the disease depends on viral virulence, routes of exposure, dose and host factors. Patients with VHF generally have nonspecific symptoms such as fever, myalgia and prostration; clinical examination may show only conjunctival injection, mild hypotension, flushing and petechial hemorrhages. Illness caused by filoviruses, flaviviruses and Rift Valley fever (RVF) viruses tends to have an abrupt onset, whereas the pathology triggered by arenaviruses has a more insidious onset. For Lassa fever patients, hemorrhagic manifestations are not pronounced; and neurological complications are infrequent, develop late and manifest only in the most severely ill group. Deafness is a frequent long-term consequence of severe Lassa fever. For the South American arenaviruses, neurological and hemorrhagic manifestations are much more prominent. Hepatic involvement is common for all VHFs, but only a small percentage of patients with RVF, Crimean-Congo HF (CCHF), Marburg hemorrhagic fever, Ebola hemorrhagic fever and yellow fever manifest a clinical picture dominated by jaundice and other evidence of hepatic failure. VHF mortality may be substantial, ranging from 5% to 20% or higher in recognized cases. Ebola outbreaks in Africa have had particularly high case fatality rates, from 50% up to 90%3,4,5.
Laboratory findings for VHFs may include thrombocytopenia (or abnormal platelet function) and leukopenia (except for Lassa fever, in which there is leukocytosis). Neutrophilia is prominent in Lassa-infected rhesus monkeys6, RVF-infected rhesus monkeys7 and Ebola-infected nonhuman primates8,9,10,11; however, neutropenia is more prominent in Argentine hemorrhagic fever (AHF)12,13 and yellow fever14. Some VHF patients have anemia, others have hemoconcentration, but most have elevated liver enzymes. Bilirubin is elevated in RVF and yellow fever. Prothrombin time, activated partial thromboplastin time (APTT) and bleeding time are often prolonged. Patients in disseminated intravascular coagulation (DIC) have elevated fibrin degradation products (FDPs) and decreased fibrinogen. Urine tests may show proteinuria and hematuria; patients with renal failure may have oliguria or azotemia. Blood may be present in stools.
Pathogenesis
The specific mechanisms underlying the pathogenesis of the VHF viruses have not been clearly explained, although recent progress has been made on Ebola virus. A central theme common to all VHFs, with the possible exception of yellow fever, is that lesions are not severe enough to account for terminal shock and death of the host. Yet a fulminant shock-like syndrome characterizes VHFs in fatal cases, suggesting that inflammatory mediators may have an important role in disease pathogenesis. Fatal VHF infections are generally characterized by high viremia and immunosuppression.
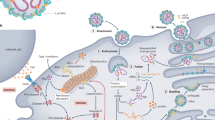
A model representing our current understanding of VHF pathogenesis in primates is shown in Figure 2. VHF in humans and nonhuman primates is characterized by deleterious changes in lymphoid tissues and defects in the coagulation system (Table 1). Another common thread among these viruses is the observation that all of the hemorrhagic fever viruses seem to target and impair the cells that initiate the antiviral immune response, probably leading to the overwhelming viral burdens that are commonly seen. We next review the interactions between VHF viruses and the cells they target, as well as factors contributing to the immunological and hematological imbalances that are associated with VHF infection.

(a) Virus spreads from the initial infection site to regional lymph nodes, liver and spleen. At these sites, the virus infects tissue macrophages (including Kupffer cells) and dendritic cells. Soluble factors released from virus-infected monocytes and macrophages act locally and systemically. Release of chemokines from these virus-infected cells recruits additional macrophages to sites of infection, making more target cells available for viral exploitation and further amplifying the dysregulated host response. Although none of these viruses infects lymphocytes, their rapid loss by apoptosis is a prominent feature of disease. The direct interaction of lymphocytes with viral proteins cannot be discounted as having a role in their destruction, but the marked loss of lymphocytes is likely to result from a combination of factors including virus infection of dendritic cells and release of soluble factors from virus-infected monocytes and macrophages. For example, virus infection of dendritic cells impairs their function by interfering with the upregulation of costimulatory molecules, which are important in providing rescue signals to T lymphocytes. Additionally, release of soluble factors from infected monocytes and macrophages results in deletion of lymphocytes, both directly by release of mediators such as nitric oxide and indirectly by contributing to upregulation of proapoptotic proteins such as Fas and TRAIL. The coagulation abnormalities vary in nature and magnitude among the VHFs. For example, Ebola virus induces the overexpression of tissue factor, which results in activation of the clotting pathway and the formation of fibrin in the vasculature. In contrast, coagulation disorders are less marked in Lassa fever, and impairment of endothelial function contributes to edema, which seems to be a more prominent finding in Lassa fever than in other VHFs. (b) The hemodynamic and coagulation disorders common among all of the VHFs are exacerbated by infection of hepatocytes and adrenal cortical cells. Infection of hepatocytes impairs synthesis of important clotting factors. At the same time, reduced synthesis of albumin by hepatocytes results in a reduced plasma osmotic pressure and contributes to edema. Impaired secretion of steroid-synthesizing enzymes by hemorrhagic fever virus–infected adrenal cortical cells leads to hypotension and sodium loss with hypovolemia. Macular rashes are often seen in VHFs.
Target cells and tissues
In general, VHF agents infect a wide range of cell types. Immunohistochemistry and in situ hybridization analyses of tissues from fatal human cases or experimentally infected nonhuman primates show that monocytes, macrophages, dendritic cells, endothelial cells, hepatocytes and adrenal cortical cells generally support replication of these viruses10,11,14,15,16,17,18,19,20,21,22,23,24. The sequence of infection, however, is largely unknown. Recent temporal studies in nonhuman primates experimentally infected with Ebola virus suggest that monocytes, macrophages and dendritic cells are early and preferred targets of these viruses, whereas endothelial cells are infected much later during the course of disease, proximal to death11,24.
The entry mechanisms of the hemorrhagic fever viruses into host cells have not been well characterized, but entry is not thought to occur by direct fusion with the plasma membrane. Instead, it is thought that these viruses exploit the host cell's endocytic machinery to access the cytoplasm. Different types of cell-surface receptors have been proposed to participate in the entry of the viruses that cause VHF. For example, the asialoglycoprotein receptor of hepatocytes was postulated to serve as a receptor for Marburg virus25, whereas the folate receptor α and the dendritic cell–specific intercellular adhesion molecule-3-grabbing nonintegrin (DC-SIGN) and DC-SIGN-related factors have also been associated with the entry of Marburg and Ebola viruses26,27,28,29. Moreover, the β1 integrin receptors and a human macrophage galactose- and N-acetylgalactosamine-specific C-type lectin are associated with the entry of Ebola virus30,31. Alpha-dystroglycan was identified as an important receptor for Lassa virus32, but does not appear to be a receptor for the South American arenaviruses33. As these viruses have such a broad cell tropism, it is likely that they exploit several molecules for entry into host cells. In support of this view, it was recently proposed that Ebola virus uses a variety of different C-type lectins for efficient entry into host cells31.
The similarity of cell and tissue tropism among VHF agents suggests commonalities in entry mechanisms. Notably, findings in a number of laboratories have shown that the transmembrane proteins of a number of RNA viruses including Ebola and Lassa have common structural and functional elements essential for virus entry34. These include a hydrophobic region termed 'fusion peptide,' usually at or near the amino terminus, generated by cleavage of a precursor protein, together with a fibrous structure defined by two antiparallel alpha helices. These general principles may apply to other VHF viruses.
The role of the endothelium in the pathogenesis of the VHFs has been particularly controversial. Vascular damage can be induced by immunological mechanisms and/or by direct infection of the vascular tissue. Impairment of endothelial cell function can cause a wide range of vascular effects that lead to changes in vascular permeability or hemorrhage. Recent studies suggest that the Ebola virus glycoprotein is the main determinant of vascular cell injury and consequently that direct structural damage of endothelial cells induced by Ebola virus replication triggers the hemorrhagic diathesis35. But other studies suggest that Ebola infection of endothelial cells does not trigger cell death and that endothelial cell infection occurs only late24. Similarly, in vitro studies have shown that the Lassa virus can replicate in human endothelial cells without causing damage36.
Researchers investigated the etiology of the hemorrhagic diatheses in fatal cases caused by Ebola virus and Marburg virus in tissues from the initial outbreaks in 1967 and 1976, but did not identify any vascular lesions37,38. In a recent study, and consistent with the original histological observations in fatal human cases, it was shown that Ebola virus infection of endothelial cells does not extensively disrupt the architecture of the vascular endothelium in Ebola-infected cynomolgus monkeys24. Although Ebola virus replicated in endothelial cells of these animals, endothelial cell infection was only seen focally at late stages of disease, after the onset of the hemorrhagic abnormalities that characterize Ebola hemorrhagic fever. Although ultrastructural evidence of endothelial cell activation and disruption was observed, it was postulated that the vasoactive effects on endothelial cells were indirect, as these changes were not associated with the presence of intracytoplasmic Ebola viral antigens.
For other VHFs, the endothelium is only minimally involved. No specific vascular lesions were found in 12 fatal cases of AHF12, nor were specific vascular lesions observed in Rhesus monkeys infected with Machupo virus39. The endothelium was only minimally infected in Rhesus monkeys infected with Lassa virus and overt endothelial necrosis was not observed histologically2.
Circulating mediators regulate endothelial cell functions, and it has been shown that the hemorrhagic fever viruses induce mediator release during infection of target cells. Few studies have dissected the mediator–endothelial cell or immune cell–endothelial cell interactions, particularly with regard to the VHFs. But a classic study clearly showed that mediator release from virus-infected target cells can have deleterious effects on the integrity of the endothelium and may contribute to vascular instability40. In this study, fluids from human monocyte/macrophage cultures infected with Marburg virus increased the permeability of cultured human endothelial cell monolayers. The increase in endothelial permeability correlated temporally with tumor necrosis factor (TNF)-α release and was inhibited by a TNF-α specific monoclonal antibody. The effect of TNF-α could be primed in the presence of agents such as H2O2. Although changes in endothelial permeability would ostensibly contribute to the hemostatic impairments noted among the VHFs, they can also lead to interstitial edema, which is a noted feature in severe cases of Lassa fever41,42.
In addition to the macrophage-rich lymphoid tissues, the liver and the adrenal gland are important target organs for all of the VHF agents, and this tropism likely has an equally important role in disease pathogenesis (Fig. 2b). Various degrees of hepatocellular necrosis were reported in VHF infections of humans and nonhuman primates10,11,38,39,43,44,45,46,47. As previously noted, the hepatocellular lesions are generally not significant enough to explain the cause of death. The exception is yellow fever where the extent of direct liver injury in some cases is severe enough to account for the disease. In fact, markers of hepatocellular injury and fulminant hepatic dysfunction, such as circulating liver enzymes (such as aspartate aminotransferase and alanine aminotransferase) directly correlate with severity of yellow fever infection and prognosis48. Elevations in liver enzymes are prominent findings in most VHF infections7,11,14,16,49,50,51,52,53,54,55,56,57,58. Notably, hemorrhagic tendencies could be related to decreased synthesis of coagulation and other plasma proteins as a result of severe hepatocellular necrosis. In addition, reduced synthesis of albumin may lead to a reduction in plasma osmotic pressure and contribute to edema, which seems to be a recurrent feature of severe cases of Lassa fever41,42.
Various degrees of adrenocortical infection and necrosis were reported in VHF infections of humans and nonhuman primates11,16,21,46,59. The adrenal cortex has an important role in controlling blood pressure homoeostasis. Impaired secretion of steroid-synthesizing enzymes leads to hypotension and sodium loss with hypovolemia, which are important elements that have been reported in nearly all cases of VHF14,43,53,56,60. This suggests that impairment of adrenocortical function by viral infection may have an important role in the evolution of shock that typifies late stages of VHF.
Immunosuppression
For nearly all of the VHFs, various degrees of lymphoid depletion and necrosis are seen in spleen and lymph nodes of fatal cases and in experimentally infected nonhuman primates6,11,14,15,17,22,37,39,44,45,47,52,53,61,62,63,64,65,66,67. Although lymphoid tissues are primary targets for virus infection, there is usually little inflammatory cellular response in these tissues or other infected tissues. Lymphopenia is the most consistent finding among VHF infections of humans and nonhuman primates6,7,8,9,10,11,13,53,66,68,69,70,71,72,73,74. Despite the substantial loss of lymphocytes during VHF infection, none of the VHF agents replicate in lymphocytes. For Ebola and Marburg viruses, large numbers of lymphocytes undergo apoptosis in human and nonhuman primates11,75,76,77, explaining in part the progressive lymphopenia and lymphoid depletion at death. The prominence of tingible body macrophages in lymphoid tissues of rhesus monkeys experimentally infected with Junin virus47 suggests that apoptosis is also a primary factor in the loss of lymphocytes noted for other VHFs.
The mechanism for the underlying apoptosis and loss of bystander lymphocytes during the course of VHF is unclear, but is likely to depend on multiple pathways. Analysis of peripheral blood mononuclear cell gene expression in Ebola-infected cynomolgus monkeys showed temporal increases in TNF-related apoptosis-inducing ligand (TRAIL) and Fas transcripts, revealing possible proapoptotic mechanisms11,78. In addition, bystander apoptosis might be associated with dysfunction of dendritic cells induced by VHF. Also, abnormal production of soluble mediators such as nitric oxide (NO) has been associated with apoptosis of peripheral blood lymphocytes79; increased blood levels of NO were reported in humans and nonhuman primates infected with Ebola virus11,74,78.
We speculate that the predilection of the hemorrhagic fever viruses for dendritic cells has an important role in disease pathogenesis and in the immunosuppression that characterizes these infections. Indeed, it has been shown that Ebola virus infects human monocyte-derived dendritic cells and impairs their function80. Specifically, monocyte-derived dendritic cells exposed to Ebola virus do not secrete proinflammatory cytokines, do not upregulate costimulatory molecules including CD80 and CD86 and stimulate T cells poorly. Apoptosis of bystander lymphocytes during Ebola infections might result from the lack of costimulatory signals or from the engagement of death receptors or ligands such as Fas or TRAIL. As an example, dendritic cells prevent Fas-mediated T-cell apoptosis through costimulatory rescue signals81. Therefore, it is possible that Ebola-induced dysfunction of dendritic cells impairs costimulatory signals that are important for both rescue of activated T cells and/or for the proper development of T-cell responses. In addition to Ebola, Lassa virus also impairs dendritic cell function. Like Ebola, Lassa virus fails to induce the expression of CD86, and other costimulatory molecules on dendritic cells, and Lassa-infected dendritic cells stimulate T cells poorly80. In support of these findings, it was recently shown that dendritic cells do not mature in response to Lassa virus infection nor do they secrete proinflammatory cytokines82. So, it appears that downregulation of dendritic cell function contributes to the immune suppression observed in Ebola and Lassa virus infections, and likely has a role in the immunosuppression seen in other VHFs.
Several viruses have evolved mechanisms to impair the function of dendritic cells, thereby enhancing their chance to escape immune surveillance. It has been proposed that, in addition to inducing immunosuppression by downregulating costimulatory molecules, viral infection of dendritic cells induces immunosuppression through a multilayered defense strategy that also includes upregulating apoptosis-inducing ligands CD95L and TRAIL to delete activated T lymphocytes, complemented by a nondeletional mechanism involving undefined viral proteins that suppresses surviving T lymphocytes83. Of note, increased TRAIL expression was associated with Ebola infection of immature dendritic cells in vitro78. Furthermore, the discovery of an immunosuppressive motif in the C-terminal region of filoviral glycoproteins84,85 lends some additional credence to speculation that undefined viral proteins may suppress surviving T cells.
In summary, the rapid loss of lymphocytes during VHF likely results in a failure to control viral replication, resulting in enormous systemic viral burdens.
Inflammatory response
Cytokines, chemokines and other inflammatory mediators function in a pleiotropic manner, acting on many different cell types to modulate the host's immune response. Whereas cytokines and chemokines typically apply their antimicrobial actions locally (for example, in areas of infection), cytokines and chemokines might also act systemically, and they commonly induce many of the symptoms of infection (such as fever and myalgia). When present in high concentrations, cytokines and chemokines can have toxic or even lethal effects. Indeed, studies of septic shock have associated abnormal production of proinflammatory cytokines with disease severity and fatal outcome86,87,88.
VHF infection of humans and nonhuman primates triggers the expression of a number of inflammatory mediators including the interferons, interleukin (IL)-6, IL-8, IL-10, IL-12, interferon-inducible protein (IP)-10, monocyte chemoattractant protein-1 (MCP-1), regulated upon activation, normal T-cell expressed and secreted (RANTES), TNF-α and nitric oxide11,57,75,78,89,90,91,92,93,94,95,96. Infection of various primary human cells in vitro also shows that VHF infection can trigger the production of many of these same inflammatory mediators24,40,78,97,98. Overall, it seems that virus-induced expression of these mediators results in an immunological imbalance that contributes to the progression of disease. But information regarding the inflammatory response after infection with the VHF agents is incomplete, and the data available are often contradictory. For example, high levels of circulating interferon (IFN)-α were noted in acute-phase sera of patients infected with Ebola virus93, but were not detected in a subsequent study75. The differences in profiles of circulating cytokines and chemokines among the VHFs may result from factors other than the differences among the viruses, such as genetic differences among patients and differences on the disease phase when the samples were obtained.
It has been postulated that, for patients with asymptomatic, nonfatal Ebola virus infection, the infection is controlled by an initial increase in cytokines including IL-1β, IL-6 and TNF-α that is followed by a return to baseline levels99. Protection against fatal outcome for other VHFs may likewise depend on an early and robust cytokine response, but this remains to be established. Conversely, disease severity may also be increased by an inappropriate proinflammatory response early in the disease course. So, the balance between protective and deleterious cytokine and chemokine responses remains to be defined for all the VHF agents.
In general, the type I IFN response seems to have an important role in VHF pathogenesis. The IFN response is particularly important in RVF. A delayed IFN response was correlated with increased mortality in a rhesus monkey model of RVF7. In AHF patients, circulating levels of IFN-α are unusually high, and are substantially higher in fatal cases than in survivors during the acute phase90. As noted above, there are inconsistencies in circulating levels of IFN-α in Ebola-infected patients; however, high plasma levels of IFN-α were observed in experimentally infected monkeys11,78. The role of IFNs in Lassa fever is uncertain. Several studies have evaluated resistance of Lassa virus to IFNs in vitro. In one study, Lassa viruses were resistant to IFN-α100, whereas more recently IFN-α and IFN-γ were shown to inhibit viral replication101. A concern when interpreting and comparing results from VHF studies is the observation that there are 12 different subtypes of IFN-α, and previous studies have shown that the antiviral activities of the subtypes vary greatly102. All of the work on VHF agents has only evaluated total levels of IFN-α, and there have been no efforts to dissect out which subtypes are represented in the plasma of infected patients and which subtypes may have antiviral properties.
The ability of these viruses to directly modulate the host inflammatory response has yet to be fully delineated. The Ebola viral protein VP35 reportedly functions as a type I IFN antagonist103,104,105. Recent studies show that VP35 prevents interferon regulatory factor (IRF-3) activation by inhibiting phosphorylation, and it is likely that VP35 prevents Ebola-induced transcription of IFN-β104. Other studies suggest that Ebola viral protein VP24 might also interfere with type I IFN signaling105. The RVF virus also has a viral protein—NSs, or nonstructural proteins—that acts as a type 1 IFN antagonist106. Little is known about whether other VHF viruses have analogous proteins that interfere with type I IFNs.
Although not extensively reported, one consistency in the proinflammatory response among the VHFs noted so far is the potential importance of NO in disease pathogenesis. Increased blood levels of NO were reported in nonhuman primates experimentally infected with Ebola virus11,78 and more recently in Ebola-infected patients74. Notably, increased blood levels of NO have been associated with mortality74. Considerable blood levels of NO have also been shown in AHF patient sera98. Abnormal NO production has been associated with several pathological conditions including apoptosis of bystander lymphocytes, tissue damage and loss of vascular integrity, which may contribute to virus-induced shock. NO has both protective and negative effects, and this autotoxic overproduction may represent the host's endogenous counter-regulatory mechanism of protection against noxious agents. In general, microbes induce monocytes and macrophages to produce NO in attempt to control infection. But in the case of VHFs, monocytes and macrophages are preferred target cells for viral replication. Enhanced replication in these cells may in turn exacerbate disease by producing large amounts of NO, resulting in deleterious effects such as suppressive effects on lymphocyte proliferation and damage to other cells. NO is an important mediator of hypotension107 and, as noted previously, hypotension is a prominent finding among most of the VHFs14,43,53,56,60. Together, the data suggest that an ineffective immune response leads to high viral loads and high levels of proinflammatory mediators in the late stages of disease.
Coagulation abnormalities
Abnormalities in blood coagulation and fibrinolysis during VHF infection are manifested as petechiae, ecchymoses, mucosal hemorrhages and uncontrolled bleeding at venipuncture sites. But massive loss of blood is atypical and, when present, is largely restricted to the gastrointestinal tract. In fact, even in these cases, blood volume loss is insufficient to account for death. DIC is a syndrome characterized by systemic intravascular activation of coagulation leading to widespread deposition of fibrin in the vasculature, which contributes to the development of multiple organ failure108,109. DIC is associated with both bleeding and thrombotic abnormalities, and, widespread thrombosis and bleeding commonly occur simultaneously. The occurrence of DIC in VHF infection is the subject of much debate, and information that supports or refutes the presence of DIC is inconclusive. In general, DIC seems to be more prominent in Ebola and CC hemorrhagic fevers than in other VHFs, while it has little or no role in Lassa fever. In fact, it has even been suggested that Lassa fever is pathologically not a hemorrhagic fever at all46. The presence of DIC in any of the VHFs strongly correlates with poor outcome. Nonetheless, regardless of whether DIC is an important and consistent feature among all VHFs, impairment of the coagulation system ostensibly contributes to the disease pathogenesis. It appears that both coagulation and fibrinolysis are activated by VHF infection, and the degree of impairment of the coagulation system seems to be associated with the balance between these counteracting processes by the host.
Cutaneous flushing or macular rashes characterize most VHF infections in humans and in nonhuman primate models; however, the characteristics of these rashes vary among VHFs. For example, nonpruritic petechial skin rashes on the axillae and groins, forehead and chest are seen in up to 50% of patients infected with Ebola or Marburg viruses and is more evident in patients with light-colored skin3,4,5,49,110,111. This type of rash evolves in more than 50% of nonhuman primates experimentally infected with Ebola or Marburg viruses9,10,11,54,68,112,113. Petechial skin rashes are also associated with cases of yellow fever14. In general, arenavirus infections in human and nonhuman primates are typically characterized by flushed, erythematous rashes on the face and thorax39,47,114,115, but oral and axillary petechiae are frequently observed in human cases of AHF116. For many VHFs, petechia are sometimes observed on visceral organs2,11,14. In addition, congestion of various organs is a frequent finding at autopsy10,11,41,43,45,63.
Thrombocytopenia is a consistent finding among VHF infections of human and nonhuman primates7,8,12,13,14,37,43,49,54,58,66,68,74,113,117,118,119,120,121,122 with the notable exception of Lassa fever2,6,16,123,124. Moderate thrombocytopenia was reported in patients with severe Lassa fever, but the most notable changes were observed in platelet function, which was markedly depressed53. Marked changes in platelet function have also been observed in Lassa6 and yellow fever14 infected rhesus monkeys. It has been postulated that the thrombocytopenia seen in the South American VHFs is in part to the result of maturation arrest of megakaryocytes attributable to the high levels of IFN present in these patients90. Similar inferences have been made for yellow fever14.
Histological and biochemical evidence of impairment of the coagulation system has been shown for a number of the VHFs, but the data are incomplete and paradoxical. More is known about Ebola and Marburg viruses than about other VHFs. Fibrin deposition has been documented for Marburg hemorrhagic fever38,125, and clinical laboratory data suggest that DIC is an important feature of human Ebola and Marburg fevers4,126. Numerous studies have shown histological and biochemical evidence of DIC during Ebola infection in a variety of nonhuman primate species including significant changes in markers of blood coagulation and fibrinolysis including various clotting factors, FDPs, D-dimers, protein C, tissue plasminogen activator and urokinase plasminogen activator9,10,11,55,113,122. Fibrin and fibrinocellular thrombi in vessels in numerous tissues and fibrin deposits in red pulp and marginal zone of spleen are frequent findings in Ebola virus–infected cynomolgus and rhesus macaques1,10,19,113,122.
AHF infections are characterized by substantial changes in coagulation and fibrinolysis markers, including thrombin-antithrombin complexes, prothrombin fragments, protein C, D-dimers, tissue plasminogen activator, and plasminogen activator inhibitor-1 (ref. 121). Increased fibrinogen levels were detected in a study of 32 AHF patients, but FDPs were not detected, and DIC was not a relevant factor in these cases116. Three of twelve AHF cases showed intravascular fibrin thrombi and clinical features consistent with DIC12. Neotropical primates experimentally infected with Junin virus showed an increase in prothrombin time and increases in circulating levels of fibrinogen and FDPs119. Prolongation of the APTT was noted in rhesus monkeys experimentally infected with Machupo virus, but evidence for DIC was inconclusive118. Microscopic evidence of DIC was noted in only one of ten rhesus monkeys experimentally infected with Machupo virus39.
Evidence of DIC has been reported in fatal cases of CCHF. Values for prothrombin ratio, prothrombin time, APTT and FDPs were elevated in 15 fatal cases, whereas fibrinogen and hemoglobin levels were depressed54. Many of these clinical pathologic changes were evident at an early stage of disease and had a highly predictive value for fatal outcome of the 35 patients monitored. Experimental infection of rhesus monkeys with RVF virus does not produce a uniformly lethal disease. However, as in human cases, the degree of hemorrhagic manifestations is associated with fatal outcome. Not surprisingly, changes in circulating levels of clotting factors, APTT, prothrombin time, FDPs, and evidence of DIC was more prominent in three of 17 RVF virus–infected monkeys that succumbed to challenge than in animals that survived7,52. Multiple fibrin thrombi were present within the glomeruli and small intertubular vessels of these experimentally infected rhesus monkeys. In addition, fibrillar material that stained positive for fibrinogen was abundant in the spleen. DIC also seems to have a role in yellow fever; changes in clotting and prothrombin times, clotting factors, fibrinogen levels and FDPs have been reported14,127.
DIC does not seem to be involved in Lassa fever. Microscopic hemorrhagic diathesis is rare, and the absence of fibrin thrombi correlates with generally normal measurements of coagulation mechanisms2,123, although splenic necrosis that centered in the marginal zone was accompanied in one report by the deposition of fibrin2. In rhesus monkeys infected with Lassa virus, several groups reported no changes in circulating levels of clotting factors and no evidence of DIC6,16,124.
The mechanism behind the coagulation abnormalities seen in VHF has not been fully delineated. New studies on the Ebola virus have begun to shed light on this problem and suggest that development of coagulation abnormalities might occur much earlier than previously thought. Although it is likely that the coagulopathy seen in Ebola fever is caused by multiple factors, particularly during the later stages of disease, recent data strongly implicate tissue factor expression and release from Ebola-infected monocytes/macrophages as key for the induction of coagulation irregularities122. Interestingly, levels of tissue factor may also be affected by the production of proinflammatory cytokines. For example, IL-6 upregulates expression of tissue factor on monocytes128,129. Other factors speculated to contribute to the coagulopathy seen Ebola hemorrhagic fever include an impairment of the fibrinolytic system as evidenced by rapid declines in plasma levels of protein C during the course of infection in cynomolgus monkeys122. Interestingly, reduced plasma levels of protein C were also seen in AHF patients121.
Future directions in pathogenesis
Several recent developments will have major roles in elucidating the molecular mechanisms of VHFs. One key breakthrough has been the successful development of reverse genetics systems for the generation of infectious Ebola130,131, CCHF132 and yellow fever133 viruses. These systems will help identify regulatory elements and structure-function relationships in the viral life cycle.
Another technology that will facilitate our ability to dissect the pathogenesis of VHF infection is cDNA microarrays. A recent application of cDNA microarrays to experimental Ebola virus infections in primates showed prominent induction of NF-κB- and TNF-α-regulated genes for Ebola, in contrast with negligible expression during variola virus infection134.
Laser capture microdissection135,136, combined with gene profiling methods, will facilitate the identification of genes differentially expressed in each cell population, as well as numerous genes that might help explain the aggressive nature of VHF infection. Indeed, the use of reverse systems together with cDNA microarrays and laser capture microdissection can be used to understand the contribution of individual viral genes to pathogenicity. These efforts may provide a better understanding of the molecular mechanisms underlying VHF, as well as help to identify suitable targets for the development of novel treatment strategies. Comparative analysis among viral genomes may reveal differentially expressed genes unique to each agent, and have diagnostic utility by identifying markers of disease progression and predictors of outcome, as well as improving our understanding of disease pathogenesis.
Countermeasures
Vaccines. With the possible exception of yellow fever, outbreaks of VHF have been relatively infrequent and small in size, largely confined to remote geographic locales, and quarantine of sick individuals has been effective in controlling epidemics. In the past, this small market has generated little commercial interest for developing VHF vaccines. But heightened awareness of bioterrorism137 has profoundly changed perspectives regarding the need for vaccines against VHFs.
The only established virus-specific vaccine available against any of VHF is the yellow fever live attenuated 17D vaccine, which is mandatory for travelers to endemic areas of Africa and South America138. For prophylaxis against AHF, a live attenuated Junin vaccine strain (Candid #1) is available as an Investigational New Drug (IND). Candid #1 was proven to be effective in phase 3 clinical trials in Argentina139, and plans are proceeding to obtain a new drug license. Candid #1 elicits high levels of protective antibodies, lasting nine years in about 90% of the people vaccinated with a single dose. This vaccine also provides protection against Bolivian hemorrhagic fever in experimentally infected primates. Two IND vaccines were developed against RVF; a formalin-inactivated vaccine that requires three boosters, which has been in use for 20 years, and a live attenuated RVF virus strain (MP-12). The inactivated vaccine has been administered to laboratory workers and appears to be safe and efficacious, but the ability to produce this vaccine in the US no longer exists140.
For the remaining VHF agents, availability of effective vaccines is more distant. As with the VHFs noted above, early attempts to develop vaccines were based on classical approaches primarily directed at using inactivated whole virion preparations as vaccines141,142. Results from these studies were inconsistent and largely unsuccessful. The recent focus on VHF vaccine development has been concentrated on various recombinant vectors for expression of virus-encoded proteins in various combinations to induce protective immunity, and tested for protective efficacy in animal models of VHF. Vaccination using recombinant vaccinia viruses expressing Lassa viral proteins successfully protected cynomolgus and rhesus monkeys from lethal Lassa fever72,143. However, a similar strategy employing the recombinant vaccinia virus platform as a vaccine for Ebola virus failed to confer any protection to nonhuman primates against lethal Ebola hemorrhagic fever1. One especially promising strategy has been the use of adenovirus vectors expressing Ebola glycoprotein and nucleoprotein genes to protect monkeys against lethal Ebola virus challenge144,145. This platform should be readily adapted to other VHF agents, and a multivalent VHF vaccine is a plausible possibility.
An alternative presentation strategy utilizes an attenuated vesicular stomatitis virus vector expressing Ebola glycoprotein which has also successfully protected monkeys against lethal Ebola challenge146. Other immunization strategies are under investigation, including virus-like particles and alphavirus replicons. One technical obstacle to development of a multivalent VHF vaccine is the potential for prior immunity to the vector, either through natural exposure or prior immunization; such a prior encounter might inhibit immunogenicity.
Therapies. Currently, there are no antiviral drugs approved by US Food and Drug Administration (FDA) for treating the VHFs. There is a critical need for the development of effective therapies to respond to outbreaks of VHF in Africa and South America, and to counter potential acts of bioterrorism. In addition, the recent death of a Russian scientist after an accidental exposure to Ebola virus147 emphasizes the need for countermeasures for postexposure prophylaxis. Considering the aggressive nature of VHF infections, in particular the rapid and overwhelming viral burdens, early diagnosis will have a considerable role in determining the success of any intervention strategy.
Development of effective therapies has been slow for several reasons, including little commercial interest and the need for special containment facilities for safe research. In addition, development of antiviral drugs has been problematic because of the rapid and tremendous increase in viral loads during the acute phase of illness. For example, viremia during the acute phase of Ebola infection of humans or nonhuman primates typically exceeds 3.2 × 106 plaque-forming units (p.f.u.)/ml of sera4, and in nonhuman primates viremia can go from less than 1.0 × 102 p.f.u./ml to over 1.0 × 105 p.f.u./ml in 24 h11. So, a 50% inhibition of viral load may be insignificant in controlling infection, and it seems that compounds that substantially inhibit Ebola replication in vitro or in rodents148 may have little efficacy when employed in monkeys149.
Ribavirin is a nonimmunosuppressive nucleoside analog with broad antiviral properties150, and is of value against some VHF agents. Ribavirin reduces mortality from Lassa fever in high-risk subjects151,152, and presumably decreases morbidity in all patients with Lassa fever. In Argentina, ribavirin has been shown to reduce virological parameters of Junin virus infection and is now used routinely as an adjunct to immune plasma. Unfortunately, ribavirin does not cross the blood-brain barrier and is expected to protect only against the visceral and not the neurological phase of Junin infection153. Small studies investigating the use of ribavirin in treating Bolivian hemorrhagic fever and CCHF have been promising154,155,156, as have preclinical studies for RVF153. Conversely, ribavirin is ineffective against both filoviruses and flaviviruses. Ribavirin is approved for use in treating VHF caused by arenaviruses and bunyaviruses, but not filoviruses under the compassionate use provisions for investigational new drugs.
Different preparations of type I IFNs were used to determine their utility in treating VHFs, with little success157,158. At the moment, type I IFNs seem to have no role in therapy, with the possible exception of RVF, in which fatal fever has been associated with low IFN responses in experimental animals159. Exogenous IFN-γ was also shown to hold promise for treating RVF infections160; its role in treating other VHFs is unknown.
Passive immunization has been attempted for treating the diseases caused by VHFs. Studies and case reports describing successes and clinical utility52,161,162,163,164,165,166 are frequently tempered by more systematic studies, where efficacy is less obvious or of no benefit151,158,167,168. For all VHF agents, the benefit of passive immunization seems to correlate with the concentration of neutralizing antibodies, which are readily induced by some, but not all, of the viruses. AHF responds to antibody therapy with two or more units of convalescent plasma that contain adequate amounts of neutralizing antibody (or an equivalent amount of immune globulin), provided that treatment is initiated within eight days of onset164. Antibody therapy is also beneficial for treating Bolivian hemorrhagic fever. Efficacy of immune plasma for treating Lassa fever169 and CCHF170 is limited by low neutralizing antibody titers and the consequent need for careful donor selection. In the future, passive immunization strategies with recombinant human monoclonal antibodies may have utility against the VHF agents given the potential benefit of passive treatment described in a number of studies52,161,162,163,164,165,166.
New avenues of therapy should focus on some of the recently developing antiviral approaches. One technology that holds great promise and may have utility for treating VHF infections is RNA interference. Various strategies have been employed to test the efficacies of small interfering RNAs as potential therapeutic agents against RNA viruses. Whereas most studies showed inhibition of viral entry or production in vitro, recent studies documented that transgene expression can be suppressed in vivo. Specifically, the therapeutic potential of this technique was shown by effectively targeting conserved regions of influenza virus genes in mice171. Although there have been no published studies evaluating the efficacy of RNA interference as a strategy for suppressing replication of VHF agents in vitro or in vivo, development and optimization of this technology might lead to useful and effective treatments against VHF. The utility of this approach as an effective strategy to combat VHF will depend on overcoming a number of obstacles, in particular the ability to develop an efficient drug delivery system, and progress is being made in this area against other viruses171.
In addition to antiviral therapies, strategies to modulate the host response or mitigate the effects of disease may have some utility and should be actively pursued. Two subjects infected with Marburg virus in 1975 were given vigorous supportive treatment and prophylactic heparin37. This apparent success inspired the use of heparin to treat one of the individuals infected with Ebola in the original 1976 outbreak in Zaire126; unfortunately, this was unsuccessful. An alternative strategy for Ebola is inhibition of the procoagulant tissue factor pathway. The basis for this speculation is that Ebola virus infection induces overexpression of tissue factor in primate monocytes/macrophages122. On the basis of these data, it was postulated that blocking factor VIIa–tissue factor might be beneficial after Ebola infection113. In a preliminary study, nine Ebola-infected monkeys were treated with a protein—recombinant nematode anticoagulant protein c2 (rNAPc2)—that prevents blood clotting. Three of the nine treated animals survived, whereas all control animals died. Importantly, Ebola infection is nearly 100% fatal in monkeys and kills up to 90% of humans infected with the virus, so a 33% survival rate for one of the most virulent diseases known is remarkable.
It is probable that the marked apoptosis of bystander lymphocytes shown for Ebola and Marburg hemorrhagic fevers contributes to the immunosuppression that characterizes VHF infection of primates. If this proves true for all of the VHFs, successful inhibition of bystander lymphocyte apoptosis offers an attractive target for mitigating the lethal effects of VHF. The exact mechanism for triggering apoptosis pathways activated during Ebola infection of primates remains unknown. Although it will be important to determine whether proteins from Ebola and other VHF agents exhibit proapoptotic activities, it is clear that increased expression of proapoptotic genes such as TRAIL and Fas during Ebola infections contribute to the observed lymphocyte apoptosis and are obvious targets for intervention strategies. Therapies directed to inhibit Fas and/or TRAIL function might have a beneficial effect. These therapeutic interventions are currently being tested in other diseases172. But the targeted blockade of apoptosis to lymphocytes, and not to cell populations that harbor hemorrhagic fever viruses, is a formidable hurdle that must be overcome before this therapeutic strategy can be fully realized.
Conclusions
The pathways to viral hemorrhagic fever involve some virus-specific attributes, but there are common threads of pathogenesis that are potential targets for generic, therapeutic interventions. All VHF viruses preferentially target monocytes, macrophages and dendritic cells of spleen and lymph nodes on their initial assault of the host organism. The ability of the host to control this first assault is critical to the eventual outcome. Some host responses might be deleterious. For example, infected monocytes and macrophages release an assortment of soluble factors that may exacerbate the course of infection by recruiting target cells to primary sites of infection.
Dendritic cells ignite adaptive immunity by priming naive T lymphocytes. At the same time, viral infection of dendritic cells impairs the host's ability to begin to generate an effective adaptive immune response. Upregulation of proapoptotic molecules on infected macrophages and the release of NO may have key roles in the apoptosis of bystander lymphocytes and the resulting lymphopenia and destruction of lymphoid tissues. So, early impairment of the innate response contributes to the pathogenesis by impeding the ability to suppress infection long enough for the host to mount an effective adaptive response. Early targeting of monocytes and macrophages may be an effective and important immune escape mechanism exploited by viruses causing VHF. Notably, there is no evidence that host cells infected with hemorrhagic fever viruses undergo apoptosis. On the contrary, the Ebola virus may upregulate protective transcripts in infected cells11. So, regulation of bystander and host cell apoptosis by hemorrhagic fever viruses may represent a very effective strategy for these viruses to evade host immune responses and enhance their survival.
VHF patients typically have very high levels of circulating IFN-α, yet these high levels of interferon in general do not seem to be effective in containing the infection. We speculate that the type I IFN response is triggered too late to control the infection, or is the wrong type of IFN response. Specifically, infection may induce IFN-α subtypes that have little or no antiviral activity while inhibiting the production of potentially beneficial subtypes. Moreover, overproduction of IFNs that have no antiviral activity may have detrimental side effects such as triggering thrombocytopenia by suppressing maturation of megakaryocytes173 and enhancing apoptosis of bystander lymphocytes by upregulating proapoptotic molecules174.
Coagulation disorders vary among VHFs. Overall, both coagulation and fibrinolysis are activated by hemorrhagic fever virus infection, and the degree of impairment of the coagulation system is associated with the ability of the host to effectively balance these counteracting effects. Macrophages have a key role in triggering or exacerbating these coagulation disorders. Tissue factor is upregulated by Ebola virus and seems to be important in the hemorrhagic diathesis. The potential role of tissue factor in other VHFs is worthy of further investigation. Future studies should also focus on defining other mechanisms that underlie the coagulation disorders that typify VHF infection.
If left unchecked, VHF infection extends to parenchymal cells such as hepatocytes and adrenal cortical cells. As has been pointed out for filoviruses, dissemination of virus to hepatocytes is facilitated by the anatomy of the liver175. Once infection is established in the these tissues, effects on their function contribute to the overall loss of homeostasis and at the terminal stages of infection. For example, dysfunction of hepatocytes as a result of the infection contributes to the coagulation disorders, as many important coagulation factors normally synthesized by hepatocytes are not produced. In addition, impaired secretion of steroid-synthesizing enzymes by virus-infected adrenal cortical cells may contribute to hypotension and sodium loss with hypovolemia. Little is know about adrenal function in VHF infection, and this is an area that warrants further investigation. In the end, the host cannot control the overwhelming viremia. The consequences of the high viral burden are primarily the diffuse coagulopathy and the marked hypotension; the multiple organ failure that ensues is similar to that seen in septic shock.
Although much has been learned in the last decade, particularly regarding Ebola hemorrhagic fever, major gaps still exist in our understanding of the way in which VHF agents induce lethal infections. Efforts should focus on understanding how these viruses impair the immune and coagulation systems. The fact that all of these viruses have early and devastating effects on lymphocytes but do not infect them is an area of research that particularly warrants further study.
With regard to vaccines, successful efforts such as the adenovirus-based platform for Ebola should be pursued for other VHFs. Therapies should focus on efforts to target the disease process in addition to replication of the virus itself. Combination approaches using inhibitors of viral replication in conjunction with drugs that modulate the disease process should be explored. Clearly, an increased understanding of the mechanisms of VHF pathogenesis will augment our ability to develop effective countermeasures against these aggressive pathogens.
References
Geisbert, T.W., Pushko, P., Anderson, K., Smith, J. & Davis, K.J. & Jahrling, P.B. Evaluation in nonhuman primates of vaccines against Ebola virus. Emerg. Infect. Dis. 8, 503–507 (2002).
Walker, D.H. & Murphy, F.A. Pathology and pathogenesis of arenavirus infections. Curr. Top. Microbiol. Immunol. 133, 89–113 (1987).
WHO. Ebola haemorrhagic fever in Sudan, 1976. Report of an international study team. Bull. World Health Organ. 56, 247–270 (1978).
WHO. Ebola haemorrhagic fever in Zaire, 1976. Report of an international commission. Bull. World Health Organ. 56, 271–293 (1978).
Khan, A.S. et al. The reemergence of Ebola hemorrhagic fever, Democratic Republic of the Congo, 1995. J. Infect. Dis. 179 Suppl 1, S76–S86 (1999).
Fisher-Hoch, S.P. et al. Physiological and immunologic disturbances associated with shock in a primate model of Lassa fever. J. Infect. Dis. 155, 465–474 (1987).
Morrill, J.C. et al. Pathogenesis of Rift Valley fever in rhesus monkeys: role of the interferon response. Arch. Virol. 110, 195–212 (1990).
Fisher-Hoch, S.P. et al. Haematological and biochemical monitoring of Ebola infection in rhesus monkeys: implications for patient management. Lancet 2, 1055–1058 (1983).
Fisher-Hoch, S.P. et al. Pathophysiology of shock and hemorrhage in a fulminating viral infection (Ebola). J. Infect. Dis. 152, 887–894 (1985).
Jaax, N.K. et al. Lethal experimental infection of rhesus monkeys with Ebola-Zaire (Mayinga) virus by the oral and conjunctival route of exposure. Arch. Pathol. Lab. Med. 120, 140–155 (1996).
Geisbert, T.W. et al. Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: evidence that dendritic cells are early and sustained targets of infection. Am. J. Pathol. 163, 2347–2370 (2003).
Elsner, B., Schwarz, E., Mando, O.G., Maiztegui, J. & Vilches, A. Pathology of 12 fatal cases of Argentine hemorrhagic fever. Am. J. Trop. Med. Hyg. 22, 229–236 (1973).
McKee, K.T., Jr, Mahlandt, B.G., Maiztegui, J.I., Green, D.E. & Peters, C.J. Virus-specific factors in Argentine hemorrhagic fever in rhesus macaques. J. Med. Virol. 22, 99–111 (1987).
Monath, T.P. & Barrett, A.D. Pathogenesis and pathophysiology of yellow fever. Adv. Virus Res. 60, 343–395 (2003).
Maiztegui, J.I. et al. Ultrastructural and immunohistochemical studies in five cases of Argentine hemorrhagic fever. J. Infect. Dis. 132, 35–53 (1975).
Jahrling, P.B. et al. Lassa virus infection of rhesus monkeys: pathogenesis and treatment with ribavirin. J. Infect. Dis. 141, 580–589 (1980).
Gonzalez, P.H., Cossio, P.M., Arana, R., Maiztegui, J.I. & Laugens, R.P. Lymphatic tissue in Argentine hemorrhagic fever. Pathologic features. Arch. Pathol. Lab. Med. 104, 250–254 (1980).
De Brito, T. et al. Human fatal yellow fever. Immunohistochemical detection of viral antigens in the liver, kidney and heart. Pathol. Res. Pract. 188, 177–181 (1992).
Geisbert, T.W., Jahrling, P.B., Hanes, M.A. & Zack, P.M. Association of Ebola-related Reston virus particles and antigen with tissue lesions of monkeys imported to the United States. J. Comp. Pathol. 106, 137–152 (1992).
Burt, F.J. et al. Immunohistochemical and in situ localization of Crimean-Congo hemorrhagic fever (CCHF) virus in human tissues and implications for CCHF pathogenesis. Arch. Pathol. Lab. Med. 121, 839–846 (1997).
Geisbert, T.W. & Jaax, N.K. Marburg hemorrhagic fever: report of a case studied by immunohistochemistry and electron microscopy. Ultrastruct. Pathol. 22, 3–17 (1998).
Zaki, S.R. & Goldsmith, C.S. Pathologic features of filovirus infections in humans. Curr. Top. Microbiol. Immunol. 235, 97–116 (1999).
de Filippis, A.M. et al. Outbreak of jaundice and hemorrhagic fever in the Southeast of Brazil in 2001: detection and molecular characterization of yellow fever virus. J. Med. Virol. 68, 620–627 (2002).
Geisbert, T.W. et al. Pathogenesis of Ebola hemorrhagic fever in primate models: evidence that hemorrhage is not a direct effect of virus-induced cytolysis of endothelial cells. Am. J. Pathol. 163, 2371–2382 (2003).
Becker, S., Spiess, M. & Klenk, H.D. The asialoglycoprotein receptor is a potential liver-specific receptor for Marburg virus. J. Gen. Virol. 76, 393–399 (1995).
Chan, S.Y. et al. Folate receptor-alpha is a cofactor for cellular entry by Marburg and Ebola viruses. Cell 106, 117–126 (2001).
Alvarez, C.P. et al. C-type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J. Virol. 76, 6841–6844 (2002).
Simmons, G. et al. DC-SIGN and DC-SIGNR bind ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology 305, 115–123 (2003).
Marzi, A. et al. DC-SIGN and DC-SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J. Virol. 78, 12090–12095 (2004).
Takada, A. et al. Downregulation of beta1 integrins by Ebola virus glycoprotein: implication for virus entry. Virology 278, 20–26 (2000).
Takada, A. et al. Human macrophage C-type lectin specific for galactose and N-acetylgalactosamine promotes filovirus entry. J. Virol. 78, 2943–2947 (2004).
Cao, W. et al. Identification of α-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science 282, 2079–2081 (1998).
Spiropoulou, C.F., Kunz, S., Rollin, P.E., Campbell, K.P. & Oldstone, M.B. New World arenavirus clade C, but not clade A and B viruses, utilizes alpha-dystroglycan as its major receptor. J. Virol. 76, 5140–5146 (2002).
Gallaher, W.R., DiSimone, C. & Buchmeier, M.J. The viral transmembrane superfamily: possible divergence of Arenavirus and Filovirus glycoproteins from a common RNA virus ancestor. BMC Microbiol. 1, http://www.biomedcentral.com/1471-2180/1/1 (2001).
Yang, Z. et al. Identification of the Ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nat. Med. 6, 886–889 (2000).
Lukashevich, I.S. et al. Lassa and Mopeia virus replication in human monocytes/macrophages and in endothelial cells: different effects on IL-8 and TNF-alpha gene expression. J. Med. Virol. 59, 552–560 (1999).
Gear, J.S. et al. Outbreake of Marburg virus disease in Johannesburg. Br. Med. J. 4, 489–493 (1975).
Murphy, F.A. Pathology of Ebola virus infection, in Ebola Virus Haemorrhagic Fever (ed Pattyn, S.R.) (Elsevier/North-Holland Biomedical Press, New York, 1978).
Terrell, T.G., Stookey, J.L., Eddy, G.A. & Kastello, M.D. Pathology of Bolivian hemorrhagic fever in the rhesus monkey. Am. J. Pathol. 73, 477–494 (1973).
Feldmann, H. et al. Filovirus-induced endothelial leakage triggered by infected monocytes/macrophages. J. Virol. 70, 2208–2214 (1996).
Callis, R.T., Jahrling, P.B. & DePaoli, A. Pathology of Lassa virus infection in the rhesus monkey. Am. J. Trop. Med. Hyg. 31, 1038–1045 (1982).
Frame, J.D. Clinical features of Lassa fever in Liberia. Rev. Infect. Dis. 11 Suppl 4, S783–S789 (1989).
Child, P.L., MacKenzie, R.B., Valverde, L.R. & Johnson, K.M. Bolivian hemorrhagic fever: a pathological description. Arch. Pathol. 83, 434–445 (1967).
Gedigk, P., Bechtelsheimer, H. & Korb, G. Pathologic anatomy of the Marburg virus disease, in Marburg Virus Disease (eds Martini, G.A. & Siegert, R.) (Springer-Verlag, New York, 1971).
Winn, W.C., Jr & Walker, D.H. The pathology of human Lassa fever. Bull. World Health Organ. 52, 535–545 (1975).
Walker, D.H. et al. Pathologic and virologic study of fatal Lassa fever in man. Am. J. Pathol. 107, 349–356 (1982).
Green, D.E., Mahlandt, B.G. & McKee, K.T., Jr. Experimental Argentine hemorrhagic fever in rhesus macaques: virus-specific variations in pathology. J. Med. Virol. 22, 113–133 (1987).
Elton, N.W., Romero, A. & Trejos, A. Clinical pathology of yellow fever. Am. J. Clin. Pathol. 25, 135–146 (1955).
Martini, G.A. Marburg virus disease: clinical syndrome, in Marburg Virus Disease (eds Martini, G.A. & Siegert, R.) (Springer-Verlag, New York, 1971).
Joubert, J.R., King, J.B., Rossouw, D.J. & Cooper, R. A nosocomial outbreak of Crimean-Congo haemorrhagic fever at Tygerberg Hospital. Part III. Clinical pathology and pathogenesis. S. Afr. Med. J. 68, 722–728 (1985).
Peters, C.J., Jahrling, P.B., Liu, C.T., Kenyon, R.H. & McKee, K.T., Jr. & Barrera Oro, J.G. Experimental studies of arenaviral hemorrhagic fevers. Curr. Top. Microbiol. Immunol. 134, 5–68 (1987).
Peters, C.J. et al. Experimental Rift Valley fever in rhesus macaques. Arch. Virol. 99, 31–44 (1988).
Fisher-Hoch, S.P., McCormick, J.B., Sasso, D. & Craven, R.B. Hematologic dysfunction in Lassa fever. J. Med. Virol. 26, 127–135 (1988).
Swanepoel, R. et al. The clinical pathology of Crimean-Congo hemorrhagic fever. Rev. Infect. Dis. 11 Suppl 4, S794–S800 (1989).
Fisher-Hoch, S.P. et al. Pathogenic potential of filoviruses: role of geographic origin of primate host and virus strain. J. Infect. Dis. 166, 753–763 (1992).
Sanchez, A. et al. Filoviridae: Marburg and Ebola viruses, in Fields Virology (eds Knipe, D.S. & Howley, P.M.) (Lippincott Williams & Wilkins, Philadelphia, 2001).
Schmitz, H. et al. Monitoring of clinical and laboratory data in two cases of imported Lassa fever. Microbes Infect. 4, 43–50 (2002).
Ergonul, O. et al. Characteristics of patients with Crimean-Congo hemorrhagic fever in a recent outbreak in Turkey and impact of oral ribavirin therapy. Clin. Infect. Dis. 39, 284–287 (2004).
McLeod, C.G., Jr, Stookey, J.L., White, J.D., Eddy, G.A. & Fry, G.A. Pathology of Bolivian Hemorrhagic fever in the African green monkey. Am. J. Trop. Med. Hyg. 27, 822–826 (1978).
Peters, C.J. Arenaviruses, in Clincal Virology (Eds Richman, D.D., Whitley, R.J. & Hayden, F.G.) (Churchill Livingstone, New York, 1997).
Hudson, N. The pathology of experimental yellow fever in the Macaca rhesus. III. Comparison with the pathology of yellow fever in man. Am. J. Pathol. 4, 419–439 (1928).
Klotz, O. & Belt, T.H. Pathology in spleen in yellow fever. Am. J. Pathol. 6, 655–662 (1930).
Zlotnik, I. Marburg agent disease: Pathology. Trans. R. Soc. Trop. Med. Hyg. 63, 310–323 (1969).
Murphy, F.A., Simpson, D.I.H., Whitfield, S.G., Zlotnik, I. & Carter, G.B. Marburg virus infection in monkeys. Lab. Invest. 24, 279–291 (1971).
Edington, G.M. & White, H.A. The pathology of Lassa fever. Trans. R. Soc. Trop. Med. Hyg. 66, 381–389 (1972).
Monath, T.P., Brinker, K.R., Chandler, F.W., Kemp, G.E. & Cropp, C.B. Pathophysiologic correlations in a rhesus monkey model of yellow fever with special observations on the acute necrosis of B cell areas of lymphoid tissues. Am. J. Trop. Med. Hyg. 30, 431–443 (1981).
Baskerville, A., Satti, A., Murphy, F.A. & Simpson, D.I. Congo-Crimean haemorrhagic fever in Dubai: histopathological studies. J. Clin. Pathol. 34, 871–874 (1981).
Simpson, D.I.H. Marburg agent disease: in monkeys. Trans. R. Soc. Trop. Med. Hyg. 63, 303–309 (1969).
Havemann, K. & Schmidt, H.A. Haematological findings in Marburg virus disease: evidence for involvement of the immunological system, in Marburg Virus Disease (eds Martini, G.A. & Siegert, R.) (Springer-Verlag, New York, 1971).
Kastello, M.D., Eddy, G.A. & Kuehne, R.W. A rhesus monkey model for the study of Bolivian hemorrhagic fever. J. Infect. Dis. 133, 57–62 (1976).
Wagner, F.S., Eddy, G.A. & Brand, O.M. The African green monkey as an alternate primate host for studying Machupo virus infection. Am. J. Trop. Med. Hyg. 26, 159–162 (1977).
Fisher-Hoch, S.P. et al. Protection of rhesus monkeys from fatal Lassa fever by vaccination with a recombinant vaccinia virus containing the Lassa virus glycoprotein gene. Proc. Natl. Acad. Sci. USA 86, 317–321 (1989).
Vallejos, D.A., Ambrosio, A.M., Feuillade, M.R. & Maiztegui, J.I. Lymphocyte subsets alteration in patients with Argentine hemorrhagic fever. J. Med. Virol. 27, 160–163 (1989).
Sanchez, A. et al. Analysis of human peripheral blood samples from fatal and nonfatal cases of Ebola (Sudan) hemorrhagic fever: cellular responses, virus load, and nitric oxide levels. J. Virol. 78, 10370–10377 (2004).
Baize, S. et al. Defective humoral responses and extensive intravascular apoptosis are associated with fatal outcome in Ebola virus-infected patients. Nat. Med. 5, 423–426 (1999).
Geisbert, T.W. et al. Apoptosis induced in vitro and in vivo during infection by Ebola and Marburg viruses. Lab. Invest. 80, 171–186 (2000).
Reed, D.S., Hensley, L.E., Geisbert, J.B., Jahrling, P.B. & Geisbert, T.W. Depletion of peripheral blood T lymphocytes and NK cells during the course of Ebola hemorrhagic fever in cynomolgus macaques. Viral Immunol. 17, 390–400 (2004).
Hensley, L.E., Young, H.A., Jahrling, P.B. & Geisbert, T.W. Proinflammatory response during Ebola virus infection of primate models: possible involvement of the tumor necrosis factor receptor superfamily. Immunol. Lett. 80, 169–179 (2002).
Takabayashi, A. et al. Nitric oxide induces a decrease in the mitochondrial membrane potential of peripheral blood lymphocytes, especially in natural killer cells. Antioxid. Redox Signal. 2, 673–680 (2000).
Mahanty, S. et al. Cutting edge: impairment of dendritic cells and adaptive immunity by Ebola and Lassa viruses. J. Immunol. 170, 2797–2801 (2003).
Daniel, P.T., Scholz, C., Westermann, J., Dorken, B. & Pezzutto, A. Dendritic cells prevent CD95 mediated T lymphocyte death through costimulatory signals. Adv. Exp. Med. Biol. 451, 173–177 (1998).
Baize, S. et al. Lassa virus infection of human dendritic cells and macrophages is productive but fails to activate cells. J. Immunol. 172, 2861–2869 (2004).
Raftery, M.J. et al. Targeting the function of mature dendritic cells by human cytomegalovirus: a multilayered viral defense strategy. Immunity 15, 997–1009 (2001).
Volchkov, V.E., Blinov, V.M. & Netesov, S.V. The envelope glycoprotein of Ebola virus contains an immunosuppressive-like domain similar to oncogenic retroviruses. FEBS Lett. 305, 181–184 (1992).
Bukreyev, A., Volchkov, V.E., Blinov, V.M. & Netesov, S.V. The GP-protein of Marburg virus contains the region similar to the 'immunosuppressive domain' of oncogenic retrovirus P15E proteins. FEBS Lett. 323, 183–187 (1993).
Calandra, T., Gerain, J., Heumann, D. & Baumgartner, J.D. & Glauser MP. High circulating levels of interleukin-6 in patients with septic shock: evolution during sepsis, prognostic value, and interplay with other cytokines. The Swiss-Dutch J5 Immunoglobulin Study Group. Am. J. Med. 91, 23–29 (1991).
Damas, P. et al. Cytokine serum level during severe sepsis in human IL-6 as a marker of severity. Ann. Surg. 215, 356–362 (1992).
Marty, C. et al. Circulating interleukin-8 concentrations in patients with multiple organ failure of septic and nonseptic origin. Crit. Care Med. 22, 673–679 (1994).
Levis, S.C. et al. Endogenous interferon in Argentine hemorrhagic fever. J. Infect. Dis. 149, 428–433 (1984).
Levis, S.C. et al. Correlation between endogenous interferon and the clinical evolution of patients with Argentine hemorrhagic fever. J. Interferon Res. 5, 383–389 (1985).
Heller, M.V., Saavedra, M.C., Falcoff, R., Maiztegui, J.I. & Molinas, F.C. Increased tumor necrosis factor-alpha levels in Argentine hemorrhagic fever. J. Infect. Dis. 166, 1203–1204 (1992).
Marta, R.F. et al. Proinflammatory cytokines and elastase-α-1-antitrypsin in Argentine hemorrhagic fever. Am. J. Trop. Med. Hyg. 60, 85–89 (1999).
Villinger, F. et al. Markedly elevated levels of interferon (IFN)-gamma, IFN-alpha, interleukin (IL)-2, IL-10, and tumor necrosis factor-alpha associated with fatal Ebola virus infection. J. Infect. Dis. 179 Suppl 1, S188–S191 (1999).
Ignatiev, G.M., Dadaeva, A.A., Luchko, S.V. & Chepurnov, A.A. Immune and pathophysiological processes in baboons experimentally infected with Ebola virus adapted to guinea pigs. Immunol. Lett. 71, 131–140 (2000).
Mahanty, S. et al. Low levels of interleukin-8 and interferon-inducible protein-10 in serum are associated with fatal infections in acute Lassa fever. J. Infect. Dis. 183, 1713–1721 (2001).
Ter Meulen, J. et al. Activation of the cytokine network and unfavorable outcome in patients with yellow fever. J. Infect. Dis. 190, 1821–1827 (2004).
Stroher, U. et al. Infection and activation of monocytes by Marburg and Ebola viruses. J. Virol. 75, 11025–11033 (2001).
Gomez, R.M. et al. Endothelial cell function alteration after Junin virus infection. Thromb. Haemost. 90, 326–333 (2003).
Leroy, E.M. et al. Human asymptomatic Ebola infection and strong inflammatory response. Lancet 355, 2210–2215 (2000).
Peters, C.J., Liu, C.T., Anderson, G.W., Jr, Morrill, J.C. & Jahrling, P.B. Pathogenesis of viral hemorrhagic fevers: Rift Valley fever and Lassa fever contrasted. Rev. Infect. Dis. 11 Suppl 4, S743–S749 (1989).
Asper, M. et al. Inhibition of different Lassa virus strains by alpha and gamma interferons and comparison with a less pathogenic arenavirus. J. Virol. 78, 3162–3169 (2004).
Foster, G.R. & Finter, N.B. Are all type I human interferons equivalent? J. Viral Hepat. 5, 143–152 (1998).
Basler, C.F. et al. The Ebola virus VP35 protein functions as a type I IFN antagonist. Proc. Natl. Acad. Sci. USA 97, 12289–12294 (2000).
Basler, C.F. et al. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J. Virol. 77, 7945–7956 (2003).
Basler, C.F. & Palese, P. Modulation of innate immunity by filoviruses, in Ebola and Marburg viruses: molecular and cellular biology (eds Klenk, H.D. & Feldmann, H) (Horizon Bioscience, Norfolk, UK, 2004).
Bouloy, M. et al. Genetic evidence for an interferon-antagonist function of rift valley fever virus nonstructural protein NSs. J. Virol. 75, 1371–1377 (2001).
Li, H. & Forstermann, U. Nitric oxide in the pathogenesis of vascular disease. J. Pathol. 190, 244–254 (2000).
Mammen, E.F. Disseminated intravascular coagulation (DIC). Clin. Lab. Sci. 13, 239–245 (2000).
Levi, M. Current understanding of disseminated intravascular coagulation. Br. J. Haematol. 124, 567–576 (2004).
Martini, G.A., Knauff, H.G., Schmidt, H.A., Mayer, G. & Baltzer, G. A hitherto unknown infectious disease contracted from monkeys. “Marburg-virus” disease. Ger. Med. Mon. 13, 457–470 (1968).
Bwaka, M.A. et al. Ebola hemorrhagic fever in Kikwit, Democratic Republic of the Congo: clinical observations in 103 patients. J. Infect. Dis. 179 suppl. Suppl 1, S1–S7 (1999).
Bowen, E.T., Platt, G.S., Simpson, D.I., McArdell, L.B. & Raymond, R.T. Ebola haemorrhagic fever: experimental infection of monkeys. Trans. R. Soc. Trop. Med. Hyg. 72, 188–191 (1978).
Geisbert, T.W. et al. Treatment of Ebola virus infection with a recombinant inhibitor of factor VIIa/tissue factor: a study in rhesus monkeys. Lancet 362, 1953–1958 (2003).
Mackenzie, R.B., Beye, H.K. & Valverde, L. & Garr'on, H. Epidemic hemorrhagic fever in Bolivia. I. A preliminary report of the epidemiological and clinical findings in a new epidemic area in South America. Am. J. Trop. Med. Hyg. 13, 620–625 (1964).
Peters, C.J., Zaki, S.R. & Rollin, P.E. Viral hemorrhagic fevers, in Atlas of Infectious Diseases Vol. 3 (ed Feteky, R.) (Churchill Livingstone, Philadelphia, 1997).
Molinas, F.C., de Bracco, M.M. & Maiztegui, J.I. Coagulation studies in Argentine hemorrhagic fever. J. Infect. Dis. 143, 1–6 (1981).
Dennis, L.H., Reisberg, B.E., Crosbie, J., Crozier, D. & Conrad, M.E. The original haemorrhagic fever: yellow fever. Br. J. Haematol. 17, 455–462 (1969).
Scott, S.K. et al. Studies of the coagulation system and blood pressure during experimental Bolivian hemorrhagic fever in rhesus monkeys. Am. J. Trop. Med. Hyg. 27, 1232–1239 (1978).
Molinas, F.C. et al. Alteration of blood coagulation and complement system in neotropical primates infected with Junin virus. J. Med. Virol. 12, 281–292 (1983).
Salas, R. et al. Venezuelan haemorrhagic fever. Lancet 338, 1033–1036 (1991).
Heller, M.V. et al. Early markers of blood coagulation and fibrinolysis activation in Argentine hemorrhagic fever. Thromb. Haemost. 73, 368–373 (1995).
Geisbert, T.W. et al. Mechanisms underlying coagulation abnormalities in Ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key event. J. Infect. Dis. 188, 1618–1629 (2003).
Knobloch, J. et al. Clinical obersvations in 42 patients with Lassa fever. Tropenmed. Parasitol. 31, 389–398 (1980).
Lange, J.V. et al. Kinetic study of platelets and fibrinogen in Lassa virus-infected monkeys and early pathologic events in Mopeia virus-infected monkeys. Am. J. Trop. Med. Hyg. 34, 999–1007 (1985).
Rippey, J.J., Schepers, N.J. & Gear, J.H. The pathology of Marburg virus disease. S. Afr. Med. J. 66, 50–54 (1984).
Isaacson, M., Sureua, P., Courtielle, G. & Pattyn, S.R. Clinical aspects of Ebola virus disease at the Ngaliema hospital, Kinshasa, Zaire, 1976, in Ebola Virus Haemorrhagic Fever (ed. Pattyn, S.R.) (Elsevier/North-Holland Biomedical Press, New York, 1978).
Santos, F. et al. Coagulacao intravascular diseminada aguda na febre amarela: dosagem dos factores da coagulacao. Brasilia Med. 9, 9–16 (1973).
Grignani, G. & Maiolo, A. Cytokines and hemostasis. Haematologica 85, 967–972 (2000).
Neumann, F-J. et al. Effect of human recombinant interleukin-6 and interleukin-8 on monocyte procoagulant activity. Arterioscler. Thromb. Vasc. Biol. 17, 3399–3405 (1997).
Volchkov, V.E. et al. Recovery of infectious Ebola virus from complementary DNA: RNA editing of the GP gene and viral cytotoxicity. Science 291, 1965–1969 (2001).
Neumann, G., Feldmann, H., Watanabe, S., Lukashevich, I. & Kawaoka, Y. Reverse genetics demonstrates that proteolytic processing of the Ebola virus glycoprotein is not essential for replication in cell culture. J. Virol. 76, 406–410 (2002).
Flick, R., Flick, K., Feldmann, H. & Elgh, F. Reverse genetics for Crimean-Congo hemorrhagic fever virus. J. Virol. 77, 5997–6006 (2003).
Bredenbeek, P.J. et al. A stable full-length yellow fever virus cDNA clone and the role of conserved RNA elements in flavivirus replication. J. Gen. Virol. 84, 1261–1268 (2003).
Rubins, K.H. et al. Systematic analysis of the gene expression program in peripheral blood cells in response to systemic smallpox infection. Proc. Natl. Acad. Sci. USA 101, 15190–15195 (2004).
Emmert-Buck, M.R. et al. Laser capture microdissection. Science 274, 998–1001 (1996).
Bonner, R.F. et al. Laser capture microdissection: molecular analysis of tissue. Science 278, 1481, 1483 (1997).
Alibek, K. & Handelman, S. Biohazard: The Chilling True Story of the Largest Covert Biological Weapons Program in the World. Told From the Inside by the Man Who Ran It (Random House, New York, 1999).
Monath, T.P. Yellow fever: an update. Lancet Infect. Dis. 1, 11–20 (2001).
Maiztegui, J.I. et al. (1998) Protective efficacy of a live attenuated vaccine against Argentine hemorrhagic fever. J. Infect. Dis. 177, 277–283 (1998).
Pittman, P.R. et al. Immunogenicity of an inactivated Rift Valley fever vaccine in humans. Vaccine 18, 181–189 (1999).
Geisbert, T.W. & Jahrling, P.B. Towards a vaccine against Ebola virus. Expert Rev. Vaccines 2, 777–789 (2003).
Fisher-Hoch, S.P. & McCormick, J.B. Lassa fever vaccine. Expert Rev. Vaccines 3, 189–197 (2004).
Fisher-Hoch, S.P., Hutwagner, L., Brown, B. & McCormick, J.B. Effective vaccine for Lassa fever. J. Virol. 74, 6777–6783 (2000).
Sullivan, N.J., Sanchez, A., Rollin, P.E., Yang, Z-Y. & Nabel, G.J. Development of a preventive vaccine for Ebola virus infection in primates. Nature 408, 605–609 (2000).
Sullivan, N.J. et al. Accelerated vaccination for Ebola virus haemorrhagic fever in non-human primates. Nature 424, 681–684.
Jones, S. et al. Replicating vectors for vaccine development. VRC Symposium on Viral Hemorrhagic Fevers, Bethesda, MD, October 14–17, 2003.
International Society for Infectious Diseases. Ebola, lab accident death - Russia (Siberia), May 22, 2004. Archive no. 20040522.1377. http://www.promedmail.org (2004).
Huggins, J., Zhang, Z.X. & Bray, M. Antiviral drug therapy of filovirus infections: S-adenosylhomocysteine hydrolase inhibitors inhibit Ebola virus in vitro and in a lethal mouse model. J. Infect. Dis. 179 Suppl 1, S240–S247 (1999).
Huggins, J.W., Zhang, Z.X., Davis, K. & Coulombe, R.A. Inhibition of Ebola virus by S-adenosylhomocysteine hydrolase inhibitors. Antiviral Res. 26, A301 (1995).
Canonico, P.G., Kende, M., Luscri, B.J. & Huggins, J.W. In-vivo activity of antivirals against exotic RNA viral infections. J. Antimicrob. Chemother. 14, 27–41 (1984).
McCormick, J.B. et al. Lassa fever. Effective therapy with ribavirin. N. Engl. J. Med. 314, 20–26 (1986).
Centers for Disease Control. Management of patients with suspected viral hemorrhagic fever. Morb. Mortal. Wkly. Rep. 37 Suppl 3, 1–16 (1988).
Huggins, J.W. Prospects for treatment of viral hemorrhagic fevers with ribavirin, a broad-spectrum antiviral drug. Rev. Infect. Dis. 11 Suppl 4, S750–S761 (1989).
Fisher-Hoch, S.P. et al. Crimean Congo-haemorrhagic fever treated with oral ribavirin. Lancet 346, 472–475 (1995).
Kilgore, P.E. et al. Treatment of Bolivian hemorrhagic fever with intravenous ribavirin. Clin. Infect. Dis. 24, 718–722 (1997).
Mardani, M., Jahromi, M.K., Naieni, K.H. & Zeinali, M. The efficacy of oral ribavirin in the treatment of crimean-congo hemorrhagic fever in Iran. Clin. Infect. Dis. 36, 1613–1618 (2003).
Ignat'ev, G.M., Strel'tsova, M.A., Agafonov, A.P., Kashentseva, E.A. & Prozorovskii, N.S. Experimental study of possible treatment of Marburg hemorrhagic fever with desferal, ribavirin, and homologous interferon. Vopr. Virusol. 41, 206–209 (1996).
Jahrling, P.B. et al. Evaluation of immune globulin and recombinant interferon-alpha2b for treatment of experimental Ebola virus infections. J. Infect. Dis. 179 Suppl 1, S224–S234 (1999).
Morrill, J.C., Jennings, G.B., Cosgriff, T.M., Gibbs, P.H. & Peters, C.J. (1989) Prevention of Rift Valley fever in rhesus monkeys with interferon-α. Rev. Infect. Dis. 11 Suppl 4, S815–S825 (1989).
Morrill, J.C., Czarniecki, C.W. & Peters, C.J. Recombinant human interferon-gamma modulates Rift Valley fever virus infection in the rhesus monkey. J. Interferon Res. 11, 297–304 (1991).
Leifer, E., Gocke, D.J. & Bourne, H. Lassa fever, a new virus disease in man from West Africa, II: report of a laboratory-acquired infection treated with plasma from a person recently recovered from the disease. Am. J. Trop. Med. Hyg. 19, 677–679 (1970).
Emond, R.T., Evans, B., Bowen, E.T. & Lloyd, G. A case of Ebola virus infection. Br. Med. J. 2, 541–544 (1977).
Maiztegui, J.I., Fernandez, N.J. & de Damilano, A.J. Efficacy of immune plasma in treatment of Argentine haemorrhagic fever and association between treatment and a late neurological syndrome. Lancet 2, 1216–1217 (1979).
Enria, D.A., Briggiler, A.M., Fernandez, N.J., Lewis, S.C. & Maiztegui, J.I. Importance of dose of neutralizing antibodies in treatment of Argentine hemorrhagic fever with immune plasma. Lancet 2, 255–256 (1984).
Frame, J.D., Verbrugge, G.P., Gill, R.G. & Pinneo, L. The use of Lassa fever convalescent plasma in Nigeria. Trans. R. Soc. Trop. Med. Hyg. 78, 319–324 (1984).
Mupapa, K. et al. Treatment of Ebola hemorrhagic fever with blood transfusions from convalescent patients. J. Infect. Dis. 179 Suppl 1, S18–S23 (1999).
White, H.A. Lassa fever: a study of 23 hospital cases. Trans. R. Soc. Trop. Med. Hyg. 66, 390–401 (1972).
Clayton, A.J. (1977) Lassa immune serum. Bull. World Health Organ. 55, 435–439 (1977).
Jahrling, P.B., Frame, J.D., Rhoderick, J.B. & Monson, M.H. Endemic Lassa fever in Liberia, IV: selection of optimally effective plasma for treatment by passive immunization. Trans. R. Soc. Trop. Med. Hyg. 79, 380–384 (1985).
Shepherd, A.J., Swanepoel, R. & Leman, P.A. Antibody response in Crimean-Congo hemorrhagic fever. Rev. Infect. Dis. 11 Suppl 4, S801–S806 (1989).
Ge, Q. et al. Inhibition of influenza virus production in virus-infected mice by RNA interference. Proc. Natl. Acad. Sci. USA 101, 8676–8681 (2004).
Miura, Y. et al. Critical contribution of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) to apoptosis of human CD4+ T cells in HIV-1-infected hu-PBL-NOD-SCID mice. J. Exp. Med. 193, 651–660 (2001).
Wang, Q., Miyakawa, Y., Fox, N. & Kaushansky, K. Interferon-alpha directly represses megakaryopoiesis by inhibiting thrombopoietin-induced signaling through induction of SOCS-1. Blood 96, 2093–2099 (2000).
Kayagaki, N., Yamaguchi, N., Nakayama, M., Eto, H. & Okumura, K. & Yagita, H. Type I interferons (IFNs) regulate tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression on human T cells: a novel mechanism for the antitumor effects of type I IFNs. J. Exp. Med. 189, 1451–1460 (1999).
Schnittler, H-J. & Feldmann, H. Marburg and Ebola hemorrhagic fevers: does the primary course of infection depend on the accessibility of organ-specific macrophages? Clin. Infect. Dis. 27, 404–406 (1998).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Geisbert, T., Jahrling, P. Exotic emerging viral diseases: progress and challenges. Nat Med 10 (Suppl 12), S110–S121 (2004). https://doi.org/10.1038/nm1142
Published:
Issue Date:
DOI: https://doi.org/10.1038/nm1142
This article is cited by
-
Rational design of universal immunotherapy for TfR1-tropic arenaviruses
Nature Communications (2020)
-
Structural basis for receptor recognition by Lujo virus
Nature Microbiology (2018)
-
T-bet, but not Gata3, overexpression is detrimental in a neurotropic viral infection
Scientific Reports (2017)
-
Post-exposure treatment of Ebola virus disease in guinea pigs using EBOTAb, an ovine antibody-based therapeutic
Scientific Reports (2016)
