
Abstract
Recognition of pathogens is mediated by a set of germline-encoded receptors that are referred to as pattern-recognition receptors (PRRs). These receptors recognize conserved molecular patterns (pathogen-associated molecular patterns), which are shared by large groups of microorganisms. Toll-like receptors (TLRs) function as the PRRs in mammals and play an essential role in the recognition of microbial components. The TLRs may also recognize endogenous ligands induced during the inflammatory response. Similar cytoplasmic domains allow TLRs to use the same signaling molecules used by the interleukin 1 receptors (IL-1Rs): these include MyD88, IL-1R–associated protein kinase and tumor necrosis factor receptor–activated factor 6. However, evidence is accumulating that the signaling pathways associated with each TLR are not identical and may, therefore, result in different biological responses.
Similar content being viewed by others

Main
Immunity can be broadly categorized into adaptive immunity and innate immunity. Adaptive immunity is mediated by clonally distributed T and B lymphocytes and is characterized by specificity and memory. Innate immunity was formerly thought to be a nonspecific immune response characterized by engulfment and digestion of microorganisms and foreign substances by macrophages and leukocytes. However, innate immunity has considerable specificity and is capable of discriminating between pathogens and self1,2,3. In addition, the activation of the innate immune response can be a prerequisite for the triggering of acquired immunity.
Adaptive immunity is influenced by the generation of helper T (TH) cell subsets and the consequent production of “effector” cytokines by these cells4 (Fig. 1). Naïve TH cells, when stimulated with cognate antigens by antigen-presenting cells (APCs), differentiate into two cell subsets: TH1 and TH2. TH1 cells secrete interferon-γ (IFN-γ) and promote mainly cellular immunity, whereas TH2 cells produce interleukin 4 (IL-4), IL-5, IL-10 and IL-13 and primarily promote humoral immunity. The cytokine milieu is critically involved in this step. IL-12 drives TH1 differentiation, whereas IL-4 induces TH2 differentiation. These “conditional” (or instructive) cytokines are produced in the early phase of infection. Infection by intracellular pathogens induces primarily a TH1-dominant response that protects against the majority of microorganisms, whereas helminth infection induces a TH2 response that is associated with resistance to helminths. The TH1 to TH2 balance also determines the onset and outcome of a wide variety of immune disorders that include autoimmune and allergic diseases. Autoimmune diseases, such as multiple sclerosis and Crohn's disease, are associated with vigorous TH1 responses, whereas allergic diseases seem to involve predominantly TH2 responses. Manipulation of the TH1 to TH2 balance in the immune response is considered to be one strategy with which to alter a number of disease processes.

Through the recognition of pathogens or their products, TLRs can induce the production of cytokines such as IL-12 and IL-18 in APCs. These cytokines function as “instructive” cytokines and drive naïve T cells to differentiate into TH1 cells. Pathogens are also captured in multiple ways, including phagocytosis, endocytosis or via TLRs themselves. Captured pathogens are then processed and presented to T cells as major histocompatibility complex–antigen. For expansion of antigen-specific T cell clones, antigen presentation requires concomitant up-regulated expression of costimulatory molecules on the cell surface of APCs. This up-regulation is also triggered by TLR signaling. TLR-stimulated APCs mainly induce TH1 development. It remains unclear at present whether TLRs in APCs are involved in TH2 development.
In addition to instructive cytokines, APCs use several costimulatory molecules, including CD80 and CD86, to signal T cells and to induce clonal expansion of antigen-specific T cells. Antigen presentation in the absence of costimulation leads to T cell anergy, whereas engagement by costimulatory molecules alone does not activate antigen-specific T cells. To induce effective immunogenicity, these stimuli must be provided simultaneously by APCs to T cells. Adjuvants boost APC signaling to promote immunity by enhancing antigen presenting activity and by inducing cytokine production and costimulatory molecule expression in APCs. Adjuvants are primarily derived from microbial products and include killed mycobacteria, such as complete Freund's adjuvant (killed Mycobacterium tuberculosis), and microbial components, such as Bordetella pertussis toxin, extracts of Toxoplasma gondii, Mycobacterium-derived muramyl peptides, lipopolysaccharide (LPS) or its toxic components, lipid A and CpG–rich DNA motifs. The initial recognition of these microbial pathogens is mediated by Toll-like receptors (TLRs) expressed on APCs, where they may also play critical roles as adjuvant receptors.
Mammalian Toll-like receptors
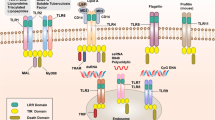
Toll receptors are type I transmembrane proteins that are evolutionarily conserved between insects and humans5. Toll was first identified as an essential molecule for embryonic patterning in Drosophila and was subsequently shown to be key in antifungal immunity6. A homologous family of Toll receptors, the so-called TLRs, exists in mammals7. Based on the similarity in the cytoplasmic portions (designated the Toll–IL-1R, or TIR, domain), TLRs are related to IL-1 receptors (IL-1Rs) (Fig. 2). However, the extracellular portions of TLRs and IL-1Rs are quite different: the extracellular portion of TLRs contains leucine-rich repeats, whereas IL-1Rs contain three immunoglobulin-like domains. More than ten members of the TLR family can be found in a search of human and mouse public databases (S. Akira et al., unpublished data) and ten members (TLRs 1–10) have been reported8,9,10,11,12. The TLR genes are dispersed throughout the genome: those encoding TLR1 and TLR6 map to human chromosome 4p14, TLR2 and TLR3 to 4q31.3-q35, TLR4 to 9q32-q33, TLR5 to 1q33.3-q42, TLR7 and TLR8 to Xp22 and TLR9 to 3p21.3.

Molecular components involved in IL-1R and TLR4 signaling are shown. Activated IL-1R1 or TLR4 associates with a cytoplasmic adaptor molecule, MyD88, through the homophilic interaction between their TIR domains. MyD88 also possess the death domain, which mediates the association with a serine-threonine kinase, IRAK. Subsequently, another adaptor molecule, TRAF6, is activated and in turn activates MAPK kinases (MKKs) and the IKK complex. MKK can lead to AP-1 activation through Jnk. The IKK complex induces phosphorylation of IκB, which renders IκB competent for being ubiquitinated and degraded. IκB degradation liberates NF-κB and allows it to translocate into the nucleus where it can induce target gene expression. P, phosphate; Ub, ubiquitin.
TLR family members are expressed differentially among immune cells13 and appear to respond to different stimuli. Surface expression of TLRs, as measured by monoclonal antibody (mAb) binding, seems to be very low. It corresponds to a few thousand molecules per cell in monocytes and a few hundred or less in immature dendritic cells (DCs)14. However, TLR expression is observed in a variety of other cells, including vascular endothelial cells, adipocytes, cardiac myocytes and intestinal epithelial cells. The expression of the various TLRs is also modulated in response to a variety of stimuli. Further experiments, including staining with TLR antibodies, seem necessary to clarify TLR expression patterns in various tissues.
TLR4 recognizes LPS
LPS is an integral component of the outer membranes of Gram-negative bacteria and can provoke a life-threatening condition called endotoxic shock15. LPS is a complex glycolipid composed of a hydrophilic polysaccharide and a hydrophobic domain, known as lipid A, which is responsible for the biological activity of LPS. A complex of LPS and the serum protein LPS-binding protein (LBP) initiates signals through membrane-bound CD14 in monocytes and myeloid cells. However, because of the presence, in the serum, of soluble CD14 that can substitute for membrane-bound CD14, CD14− cells such as endothelial and epithelial cells also respond to LPS. Although LBP and CD14 were identified as factors that bind LPS, other evidence suggested the presence of a coreceptor that transmits the LPS signal across the cell membrane.
This putative coreceptor was identified through analysis of a strain of mice, C3H/HeJ, that is hyporesponsive to LPS. The C3H/HeJ mouse strain carries a mis-sense point mutation within the Tlr4 gene region encoding the cytoplasmic tail and this mutation changes a highly conserved proline to histidine16. TLR4−/− mice were generated; they are as hyporesponsive to LPS as C3H/HeJ mice, which confirms that TLR4 is required for LPS signaling17 (Fig. 3). TLR4 mutations are also associated with endotoxin hyporesponsiveness in humans18.

Certain pathogen-associated molecular patterns and host-derived products utilize TLR family members as critical signal transducers. TLR2 recognizes a variety of microbial products. TLR4 is essential for signaling via LPS from Gram-negative bacteria; the exceptions are Leptospira and P. gingivalis, the LPS of which is recognized by TLR2. TLR4 recognizes not only viral and plant products but also endogenous host-derived products, such as hsp60 and fragments from fibronectin. Compared with TLR2 and TLR4, recognition by TLR5 and TLR9 is more restricted and essential for flagellin- and CpG DNA–mediated signaling, respectively.
Overexpression of TLR4 did not confer LPS responsiveness on human embryonic kidney 293 cells, which suggested that an additional molecule is required for TLR4-mediated LPS signaling; this was subsequently identified as the secreted molecule MD-219. Transfection with either TLR4 or MD-2 alone did not confer responsiveness to LPS, but cotransfection with both TLR4 and MD-2 did; MD-2 is physically associated with the extracellular domain of TLR4 on the cell surface (Fig. 3).
Several observations indicate that TLR2 is also involved in LPS signaling20,21. However, neither human nor murine TLR2 play a role in LPS signaling, so it was suggested that overexpression of TLR2 may cause cell lines to become extremely sensitive to minor non-LPS contaminations in LPS preparations22,23,24,25. Highly purified LPS does not activate cells through TLR226. Other data indicate that TLR2 may be involved in the response to LPS from Leptospira and Prophyromonas, which are structurally different from Gram-negative LPS27,28.
Taxol, a diterpene purified from the bark of the Western yew (Taxus brevifolia), is an anti-tumor agent that blocks mitosis by binding to and stabilizing microtubules. Taxol bears no apparent structural homology to LPS, but possesses many LPS-like activities. Interestingly, the LPS-mimetic action of Taxol is species-specific; it mimics the actions of LPS on murine, but not human, macrophages. The mouse TLR4–MD-2 complex mediates the signal induced by Taxol29.
TLR2 recognizes lipoproteins and glycolipids
Lipoproteins are proteins containing lipid that is covalently linked to the NH2-terminal cysteines; they are present in a variety of bacteria, including Gram-negative and Gram-positive bacteria and mycoplasmas. Lipoproteins possess immunostimulating activities that are attributed to the presence of their lipoylated NH2 termini. TLR2 mediates the responses to lipoproteins derived from M. tuberculosis, Borrelia burgdorfei, Treponema pallidium and Mycoplasma fermentans30,31,32,33 (Fig. 3). In addition, TLR2 mediates cellular responses to a wide variety of infectious pathogens and their products. These include yeast cell walls, whole mycobacteria, mycobacterial lipoarabinomannan, whole Gram-positive bacteria, peptidoglycan (PGN), Treponema glycolipid and Trypanosoma cruzi glycophosphatidylinositol anchor34,35,36,37,38,39,40,41. However for TLR2 activity, ligand specificity as well as signal transducing ability is determined by heterodimeric interactions with other TLRs, such as TLR6 and TLR142. Dimerization of the cytoplasmic domain of TLR2 does not induce cytokine production in a macrophage cell line, whereas the cytoplasmic portion of TLR2 can functionally pair with that of TLR6 or TLR1, which results in cytokine production. Expression of dominant-negative TLR2 or dominant-negative TLR6 in the macrophage cell line showed that TLR6 and TLR2 function together to detect Gram-positive bacteria, PGN and zymosan, whereas TLR2 functions either alone or with TLRs other than TLR6 to detect bacterial lipopeptides. However, responses to PGN are not abolished in TLR6−/− mice43. This discrepancy may be due to artifacts associated with overexpression of the proteins and/or to impurities in the microbial components utilized.
Most lipoproteins are triacylated at the NH2-terminal cysteine residue, but mycoplasmal macrophage-activating lipopeptide 2 (MALP-2) is only diacylated. TLR2−/− cells are unresponsive to all lipoproteins, whereas TLR6−/− cells are unresponsive to MALP-2 but responsive to other lipopeptides of bacterial origin43. Coexpression of TLR2 and TLR6 is absolutely required for MALP-2 responsiveness, as shown by reconstitution experiments in TLR2−/−TLR6−/− embryonic fibroblasts. These results indicated that TLR2 and TLR6 cooperate to recognize MALP-2 and TLR6 appears to confer the ability to discriminate between the NH2-terminal lipoylated structure of MALP-2 and lipopeptides derived from other bacteria.
TLR5 recognizes flagellin
Flagellin is a 55-kD protein monomer obtained from bacterial flagella, polymeric rod-like appendages which extend from the outer membrane of Gram-negative bacteria, that propel the organism through its aqueous environment. Flagellin is also a potent proinflammatory factor. Flagellated bacteria, purified flagellin and medium conditioned by flagellated bacteria all readily induce IκBα degradation, NF-κB activation and inducible nitric oxide synthetase expression in transformed human epithelial cells and murine macrophages44,45. TLR5 recognizes bacterial flagellin from both Gram-positive and Gram-negative bactria46 (Fig. 3). TLR5-stimulating activity was purified from Listeria monocytogenes culture supernatants using Chinese hamster ovary cells expressing human TLR5 and bearing a luciferase-linked reporter to examine stimulation. This activity was identified as flagellin by tandem mass spectrometry. Expression of L. monocytogenes flagellin in nonflagellated Escherichia coli conferred the ability to activate TLR5, whereas deletion of the flagellin genes from Salmonella typhimurium abrogated TLR5-stimulating activity.
TLR9 recognizes bacterial (CpG) DNA
Bacterial DNA and certain oligonucleotides containing unmethylated CpG dinucleotides can stimulate murine and human lymphocytes, whereas eukaryotic DNA and methylated oligonucleotides cannot47,48. CpG motifs are more common in bacterial DNA than in vertebrate DNA and, when present, are more likely to be methylated in vertebrates. CpG DNA directly stimulates B cells, macrophages and DCs to secrete cytokines, especially TH1-like cytokines such as IL-12 and IL-18; the cells express costimulatory molecules and show increased antigen-presentation. The strong TH1-like response induced by CpG DNA suggests that these molecules may have promise as adjuvants in vaccines against for a wide variety of targets, including infectious agents, cancer antigens and allergens. However, we have yet to understand the molecular mechanism by which CpG DNA exerts its biological effects. Both TLR2−/− and TLR4−/− cells respond normally to CpG DNA, but cells from MyD88−/− mice do not (see later), so CpG DNA is recognized by TLRs other than TLR2 or TLR449. TLR9−/− mice are completely defective in their response to CpG DNA, including cytokine production by macrophages, B cell proliferation, maturation of DCs and CpG DNA–d-galactosamine–induced shock in vivo11. Thus, TLR9 plays an essential role in the cellular response to CpG DNA (Fig. 3). The enzyme DNA-dependent protein kinase (DNA-PK) is essential for the response to CpG DNA50. Immunostimulatory CpG DNA triggers DNA-PK activation, which, in turn, phosphorylates IκB-kinase β and leads to NF-κB activation. In addition, mice without the catalytic subunit of DNA-PK have a selectively impaired response to CpG DNA. However, the relationship between TLR9 and DNA-PK in the response to CpG DNA remains unclear. Endosomal acidification and/or maturation are prerequisites for the activation of the signaling pathways mediated by CpG DNA. This data suggests that generation of the CpG DNA signal takes place inside the endosome51. Because macrophages mediate innate immunity by phagocytosing pathogens and thereby eliciting an inflammatory immune response, some, or all, TLRs—including TLR9—may be recruited to macrophage phagosomes where they function to discern the nature of the specific invading pathogen. TLR1, TLR2 and TLR6 localize to phagosomes in macrophages42.
Viruses and TLRs
In the defense response of plants, proteins bearing the Toll–IL-1R–nucleotide-binding site–leucine-rich repeat domain confer resistance to viruses52. However, TLRs may also function in viral recognition in mammals. The innate immune response to the fusion protein of respiratory syncytial virus (RSV) is mediated by TLR4 and CD1453. Compared to normal mice, RSV persists longer in the lungs of infected TLR4-deficient mice, which indicates the importance of TLR4 in the pathogenesis of RSV disease. Poxviruses employ many strategies to evade and neutralize the host immune response. Two vaccinia virus open-reading frames, termed A46R and A52R, share amino acid sequence similarity within the TIR domain and can inhibit IL-1–, IL-18– and TLR4-mediated signal transduction. Vaccinia virus may evade host immune responses by suppressing TIR domain-dependent intracellular signaling54. Thus, activation of TLR may be involved in protecting the host from viruses. Further studies will be required to establish the roles of the TLRs in resistance to viral infection in mammals.
TLRs and endogenous ligands
In Drosophila, fungal infection triggers protease cascades leading to the generation of the Spatzle protein, the endogenous ligand for Toll, from inactive precursors. The biologically active Spatzle then induces ligand-dependent Toll receptor dimerization55. In contrast, mammalian TLRs may recognize microbial components directly. Several studies have shown species-specificity in the response of mammalian cells to different LPS structures and this specificity is determined by the TLR. When the TLR4 from one species is expressed in cells from a different species, the response to lipid A is determined by the introduced TLR, not that of the host cells56,57. These data suggest that TLR4 functions as the species-specific lipid A receptor. Cross-linking experiments further suggested that LPS is present in close proximity to CD14, TLR4 and MD-258. However, conclusive evidence showing that LPS is directly recognized by TLR4 remains to be published.
There is no evidence yet to show that, in vertebrates, endogenous TLR ligands are generated in response to infection by pathogens, in a manner that is analogous to Drosophila Spatzle processing. However, increasing evidence suggests that endogenous ligands can stimulate TLRs and trigger an immune response. Signals from damaged or stressed cells may initiate an immune response even in the absence of infection, the so-called “danger model” of the immune response59. Such signals may be generated when cells undergo pathological cell death (necrosis), which initiates inflammation and immune responses, in contrast to physiological apoptotic cell death, which does not trigger inflammation. Such “stressed” or “damaged” cells could generate signals as a result of changes in the lipid and/or carbohydrate moieties expressed on their surfaces or by the expression of proteins not normally found in the outer cell membrane. Whether TLRs mediate immune responses triggered by tissue damage or tissue stress remains to be determined.
One potential stress signal is the increased expression of heat shock proteins (hsps). hsps are highly conserved molecules that participate in protein folding and assembly, as well as in the translocation of proteins between different cellular compartments. hsp synthesis is dramatically increased under stress conditions. Microbial and mammalian hsp60 have potent immunostimulatory properties. Necrotic, but not apoptotic, cells can release hsps. The LPS-hyporesponsive C3H/HeJ strain is resistant to hsp60-induced macrophage activation, which suggests that human hsp60 activates TLR460.
During an inflammatory response, production and degradation of various extracellular matrix (ECM) components is increased. The activation of extracellular proteases or the release of intracellular proteases generates lower molecular mass ECM components that are proinflammatory. Oligosaccharides of hyaluronan and heparan sulfate can induce maturation of DCs61,62 and cellular fibronectin fragments can activate TLR463. The inflammatory responses by other ECM components may also be mediated by TLR family members and thereby play a role in the immune response.
In ischemic heart diseases and heart failure, myocardial expression of inflammatory cytokines is increased, seemingly without any evidence of infection. Reactive oxygen species are proposed to be a major pathogenic factor for hypoxic and ischemic damage in the heart. Hydrogen peroxide can trigger NF-κB and AP-1 activation through a mechanism that involves TLR264. However, it seems unlikely that hydrogen peroxide directly interacts with TLR2 to form a classical ligand-receptor complex. One possibility may be that cell death or injury could cause the release of one or more factors that interact with and activate TLR2.
MyD88-dependent and independent pathways
Because the TLRs share sequence similarity with the IL-1R family in their cytoplasmic regions, it is not unexpected that downstream events are mediated by common components (Fig. 2). MyD88 is an adaptor protein that links the IL-1 receptor to IR-1R–associated protein kinase (IRAK), a serine-threonine kinase that is related to the Pelle kinase of Drosophila65,66. Upon binding of ligand to IL-1R, IRAK is phosphorylated, subsequently dissociated from the receptor complex and associates with tumor necrosis factor (TNF) receptor–activated factor 6 (TRAF6). This process results in the activation of two different pathways that involve the c-Jun NH2-terminal kinase (Jnk) and p38 mitogen-activated protein kinase (MAPK) family and the Rel family transcription factor NF-κB. These pathways are evolutionally conserved because Drosophila Toll activates Dorsal, a homolog of NF-κB, through an adaptor called Tube and via the Pelle pathway. MyD88−/− mice do not respond to IL-1, IL-18, LPS or other microbial cell wall components, such as PGN and lipopeptides, which shows that this molecule is indispensable for responses to these stimuli33,67,68. However, studies in MyD88−/− macrophages have suggested differences between TLR2 and TLR4 signaling33,68. In MyD88−/− macrophages, production of inflammatory cytokines such as IL-1β, TNF-α and IL-6 in response to LPS or MALP-2 is completely impaired. MALP activation of NF-κB and MAPK, which is mediated by TLR2, is completely abolished in both TLR2−/− and MyD88−/− macrophages. However, LPS activates NF-κB, Jnk or p38 in MyD88−/− macrophages, although this activation is delayed compared to wild-type macrophages68. This suggests it is a MyD88-independent pathway(s) that mediates NF-κB, Jnk or p38 activation after TLR4 signaling.
Several components involved in the MyD88-independent signaling pathway have been identified (Fig. 4). Subtractive hybridization cloning identified several genes that are induced in MyD88−/− macrophages upon stimulation with LPS (S. Akira et al., unpublished data). These are the so-called IFN-γ–inducible genes, which include IP-10 (IFN-inducible protein 10), a member of the CXC chemokine family, GARG16 (glucocorticoid attenuated response gene 16) and IRG1 (IFN-regulated gene 1). Expression of these genes is TLR4-dependent, but MyD88-independent, and probably occurs through the coordinate action of IFN regulatory factor 3 (IRF3) and NF-κB. Induction of these genes is not observed in MALP2 (TLR2 ligand)-stimulated wild-type macrophages. Induction of NF-κB DNA binding activity in response to LPS is defective in both NF-κB essential modulator NEMO-deficient (IKKγ−/−) and IKKβ−/− embryonic fibroblasts69,70,71. Thus, MyD88-dependent and independent pathways must converge at the formation of the IKK complex, which leads to the activation of NF-κB.

(a) Schematic representation and (b) biological outcome of TLR4 signaling pathway. TLR4 can activate the MyD88-dependent pathway (blue arrows), which can also be stimulated by IL-1 and IL-18 and ligands for other TLR family members. TLR4 also activates MyD88-independent pathways (orange arrows). For example, NF-κB activation can be induced, with delayed kinetics, in the absence of MyD88 and leads to induction of costimulatory molecules. Phosphorylation and nuclear translocation of IRF3 can occur in a MyD88-independent manner and is involved in IFN-inducible gene expression. In addition, in Kupffer cells, caspase-1 activation can be induced independently of MyD88 and results in mature IL-18 production.
How LPS activates NF-κB in a MyD88-independent manner remains to be determined. LPS activates a number of signaling molecules, including protein kinase C (PKC), Src-type tyrosine kinases, small G proteins, phosphatidylinositol 3-kinase (PI3K) and the serine-threonine protein kinase PKB (also known as Akt), all of which can activate NF-κB. Double-stranded RNA-dependent protein kinase (PKR) can also activate NF-κB. That the MyD88-dependent pathway is essential for cytokine production by all bacterial components indicates that as yet unknown factors, in addition to NF-κB and MAPK, may be required for cytokine production.
As well as inducing many transcriptional pathways, LPS activates caspase-1 in a process that does not require transcription or protein synthesis. IL-18, produced as a biologically inactive precursor (pro-IL-18), is cleaved by caspase-1 and secreted after LPS stimulation of macrophages. This caspase-1–mediated release of active IL-18 by LPS is independent of MyD8872.
Dendritic cells and TLRs
The DC is another critical sentinel in antimicrobial immune responses73,74. DCs are professional APCs that can activate naïve T cells (Fig. 1). DC maturation can be induced by inflammatory cytokines and by microbes or—if they are ligands of TLRs—their products, including LPS, lipoproteins and CpG DNA75. DC maturation is characterized by the production of proinflammatory cytokines (IL-12 and TNF-α), up-regulation of costimulatory molecules (CD40, CD80 and CD86) and altered expression of chemokine receptors (CCR2, CCR5 and CCR7). Mature DCs show increased antigen-presenting capacity and migrate from the peripheral tissues to draining lymph nodes, where they instruct the adaptive immune response by stimulating T lymphocytes. Thus, DC maturation, which is mediated through the TLR family signaling, is a critical link between innate and adaptive immunity.
T cell polarization to TH1 or TH2 can be determined by several factors, including the DC type, the ratio of DCs to T cells and the tissue from which DCs originate76,77,78. The maturation-inducing stimuli described above are critical for regulating T cell differentiation. For example, TLR-mediated signaling initiated by LPS or CpG DNA can enhance the ability of DCs to support TH1 cell differentiation by inducing the release of cytokines such as IL-12. Of note, DCs are heterogeneous and, based on the particular cell surface markers they express, can be divided into certain subsets in both humans and mice. Although there are few published reports on the expression of TLRs on each subset, TLR4 and TLR9 are expressed on monocyte-derived DCs (MDDCs) and plasmacytoid DCs (pDC2s), respectively79. Consistent with this data, LPS can induce cytokine production from MDDCs but not from pDC2s, whereas CpG DNA can induce cytokine production from pDC2s but not from MDDCs80. Thus, DCs are also heterogeneous in terms of TLR expression. Differential expression of TLRs on DCs is presumably involved in fine-tuning host immune responses.
Stimulation of either TLR4 or TLR9 on DCs induces IL-12 production and enhances surface expression of costimulatory molecules such as CD40. MyD88 is essential for cytokine production induced by TLR4 and TLR9. However, analysis of MyD88−/− DCs has revealed that the MyD88-independent pathway is responsible for some features of DC maturation, for example, up-regulation of costimulatory molecules and allogeneic T cell activation81,82. Some TLR4 signaling is MyD88-independent, but DC maturation as well as cytokine production in response to CpG DNA is absolutely dependent on MyD88, which further suggests that TLR family signaling components are rather heterogeneous. For example, CpG DNA can stimulate IFN-α production by pDC2 cells, whereas LPS cannot stimulate IFN-α production by TLR4+ MDDCs80,83. Thus, different TLRs seem to activate similar but distinct signaling pathways. Clarification of TLR signaling is crucial to elucidate how pathogens and their products affect DC maturation.
Concluding remarks
The past two years have seen rapid progress in our understanding of how TLRs function in the recognition of pathogens. Ligands for TLR2, TLR4, TLR5, TLR6 and TLR9 have been identified, but the identity of ligands recognized by the other TLRs remain unknown. Although overexpression studies often reveal important information about the function of signal transduction molecules, their results can sometimes be misleading. The identification of microbial components that activate TLRs should preferably be done with the use of knockout mice, although a potential artifact in both overexpression and knockout studies is the purity of the microbial component used. The sample components should be highly purified and synthetic compounds used whenever possible.
How the TLRs recognize their ligands is still unknown. A unique characteristic of ligand recognition by TLRs is that it is specific yet diverse. For example, TLR4 can recognize the entirely unrelated ligands LPS, hsp60 and Taxol. Because high-affinity ligand binding to a TLR has not been shown, it is possible that unknown coreceptors may be required for specific recognition of each ligand. Crystal structure studies will be necessary to elucidate the interaction between the particular TLR and its ligand.
In addition to their common activation of the MyD88-IRAK-TRAF pathway, individual TLRs may activate different, alternative, signaling pathways. This combined action may ultimately determine the varying gene expression and biological effects mediated by different TLRs. Characterization of the TLR signaling pathways should reveal the molecular mechanisms that link the initial recognition of pathogens and elicitation of acquired immunity.
Finally, the known role of TLRs is still expanding. TLR activation is involved not only in the recognition of M. tuberculosis but also initiates the killing of these organisms84. Another open question is whether or not abnormal TLR activation by endogenous ligands is involved in immunological disorders such as autoimmune diseases and chronic inflammatory responses. The combined use of murine models of these disorders with genetically modified mice will be of use in addressing these questions.
References
Medzhitov, R. & Janeway, C. A. Jr Innate immunity: the virtues of a nonclonal system of recognition. Cell 91, 295–298 (1997).
Hoffmann, J. A., Kafatos, F. C., Janeway, C. A. & Ezekowitz, R. A. Phylogenetic perspectives in innate immunity. Science 284, 1313–1318 (1999).
Aderem, A, & Ulevitch, R. J. Toll-like receptors in the induction of the innate immune response. Nature 406, 782–787 (2000).
Abbas, A. K., Murphy, K. M. & Sher, A. Functional diversity of helper T lymphocytes. Nature 383, 787–793 (1996).
Anderson, K. V. Toll signaling pathways in the innate immune response. Curr. Opin. Immunol. 12, 13–19 (2000).
Lemaitre, B., Nicolas, E., Michaut, L., Reichhart, J. M. & Hoffmann, J. A. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86, 973–983 (1996).
Medzhitov, R., Preston-Hurlburt, P. & Janeway, C. A. Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388, 394–397 (1997).
Rock, F. L., Hardiman, G., Timans, J. C., Kastelein, R. A. & Bazan, J. F. A family of human receptors structurally related to Drosophila Toll. Proc. Natl Acad. Sci. USA 95, 588–593 (1998).
Takeuchi, O. et al. TLR6: a novel member of an expanding toll-like receptor family. Gene 231, 59–65 (1999).
Du, X., Poltorak, A., Wei, Y., & Beutler, B. Three novel mammalian toll-like receptors: gene structure, expression, and evolution. Eur. Cytokine Network 11, 362–371 (2000).
Hemmi, H. et al. A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745 (2000).
Chuang, T.-H. & Ulevitch, R. J. Identification of hTLR10:a novel human Toll-like receptor preferentially expressed in immune cells. Biochim. Biophys. Acta 1518, 157–161 (2001).
Muzio, M. et al. Differential expression and regulation of toll-like receptors (TLR) in human leukocytes: selective expression of TLR3 in dendritic cells. J. Immunol. 164, 5998–6004 (2000).
Visintin, A. et al. Regulation of Toll-like receptors in human monocytes and dendritic cells. J. Immunol. 166, 249–255 (2001).
Ulevitch, R. J. & Tobias, P. S. Receptor-dependent mechanisms of cell stimulation by bacterial endotoxin. Annu. Rev. Immunol. 13, 437–457 (1995).
Poltorak, A. et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088 (1998).
Hoshino, K. et al. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. 162, 3749–3752 (1999).
Arbour, N. C. et al. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans Nature Genet. 25, 187–191 (2000).
Shimazu, R. et al. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 189, 1777–1782 (1999).
Yang, R. B. et al. Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signalling. Nature 395, 284–288 (1998)
Kirschning, C. J., Wesche, H., Merrill Ayres, T. & Rothe, M. Human toll-like receptor 2 confers responsiveness to bacterial lipopolysaccharide. J. Exp. Med. 188, 2091–2097 (1998).
Heine, H. et al. Cutting edge: cells that carry a null allele for toll-like receptor 2 are capable of responding to endotoxin. J. Immunol. 162, 6971–6975 (1999).
Takeuchi, O. et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11, 443–451 (1999).
Underhill, D. M. et al. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature 401, 811–815 (1999).
Faure, E. et al. Bacterial lipopolysaccharide activates NF-κB through toll-like receptor 4 (TLR-4) in cultured human dermal endothelial cells. Differential expression of TLR-4 and TLR-2 in endothelial cells. J. Biol. Chem. 275, 11058–11063 (2000).
Hirschfeld, M., Ma, Y., Weis, J. H., Vogel, S. N. & Weis, J. J. Cutting edge: repurification of lipopolysaccharide eliminates signaling through both human and murine toll-like receptor 2. J. Immunol. 165, 618–622 (2000).
Werts, C. et al. Leptospiral lipopolysaccharide activates cells through a TLR2-dependent mechanism. Nature Immunol. 2, 346–352 (2001).
Hirschfeld, M. et al. Signaling by toll-like receptor 2 and 4 agonists results in differential gene expression in murine macrophages. Infect. Immun. 69, 1477–1482 (2001).
Kawasaki, K. et al. Mouse toll-like receptor 4.MD-2 complex mediates lipopolysaccharide-mimetic signal transduction by Taxol. J. Biol. Chem. 275, 2251–2254 (2000).
Aliprantis, A. O. et al. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science 285, 736–739 (1999).
Brightbill, H. D. et al. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science 285, 732–736 (1999).
Hirschfeld, M. et al. Cutting edge: inflammatory signaling by Borrelia burgdorferi lipoproteins is mediated by toll-like receptor 2. J. Immunol. 163, 2382–2386 (1999).
Takeuchi, O. et al. Cutting edge: preferentially the R-stereoisomer of the mycoplasmal lipopeptide macrophage-activating lipopeptide-2 activates immune cells through a toll-like receptor 2- and MyD88-dependent signaling pathway. J. Immunol. 164, 554–557 (2000).
Lien, E. et al. Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J. Biol. Chem. 274, 33419–33425 (1999).
Yoshimura, A. et al. Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J. Immunol. 163, 1–5 (1999).
Schwandner, R., Dziarski, R., Wesche, H., Rothe, M., Kirschning, C. J. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol. Chem. 274, 17406–17409 (1999).
Means, T. K. et al. Human toll-like receptors mediate cellular activation by Mycobacterium tuberculosis. J. Immunol. 163, 3920–3927 (1999).
Flo, T. H. et al. Human toll-like receptor 2 mediates monocyte activation by Listeria monocytogenes, but not by group B streptococci or lipopolysaccharide. J. Immunol. 164, 2064–2069 (2000).
Takeuchi, O. et al. Cellular responses to bacterial cell wall components are mediated through MyD88-dependent signaling cascades. Int. Immunol. 12, 113–117 (2000).
Opitz, B. et al. Toll-like receptor (TLR)-2 mediates Treponema Glycolipid and lipoteichoic acid (LTA)-induced NF-κB translocation. J. Biol. Chem. 276, 22041–22047 (2001).
Marco, A. S. et al. Activation of toll-like receptor-2 by glycosylphosphatidylinositol anchors from a protozoan parasite. J. Immunol. 167, 416–423 (2001).
Ozinsky, A. et al. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc. Natl Acad. Sci. USA 97, 13766–13771 (2000).
Takeuchi, O. et al. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol. 13, 933–940 (2001).
Steiner, T. S., Nataro, J. P., Poteet-Smith, C. E., Smith, J. A. & Guerrant, R. L. Enteroaggregative Escherichia coli expresses a novel flagellin that causes IL-8 release from intestinal epithelial cells. J. Clin. Invest. 105, 1769–1777 (2000).
Eaves-Pyles, T. et al. Flagellin, a novel mediator of Salmonella-induced epithelial activation and systemic inflammation: IκBα degradation, induction of nitric oxide synthase, induction of proinflammatory mediators, and cardiovascular dysfunction. J. Immunol. 166, 1248–1260 (2001).
Hayashi, F. et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor-5. Nature 410, 1099–1103 (2001).
Tokunaga, T., Yamamoto, T. & Yamamoto, S. How BCG led to the discovery of immunostimulatory DNA. Jpn J. Infect. Dis. 52, 1–11 (1999).
Krieg, A. M. & Wagner, H. Causing a commotion in the blood: immunotherapy progresses from bacteria to bacterial DNA. Immunol. Today 21, 521–526 (2000).
Hacker, H. et al. Immune Cell Activation by Bacterial CpG-DNA through Myeloid Differentiation Marker 88 and Tumor Necrosis Factor Receptor-Associated Factor (TRAF)6. J. Exp. Med. 192, 595–600 (2000).
Chu, W. et al. DNA-PKcs is required for activation of innate immunity by immunostimulatory DNA. Cell 103, 909–918 (2000).
Hacker, H. et al. Cell type-specific activation of mitogen-activated protein kinases by CpG-DNA controls interleukin-12 release from antigen-presenting cells. EMBO J. 18, 6973–6982 (1999).
Whitham, S. et al. The product of the tobacco mosaic virus resistance gene N: similarity to toll and the interleukin-1 receptor. Cell 78, 1101–1115 (1994).
Kurt-Jones, E. A. et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nature Immunol. 1, 398–401 (2000).
Bowie, A. et al. A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc. Natl Acad. Sci. USA 97, 10162–10167 (2000).
Levashina, E. A. et al. Constitutive activation of toll-mediated antifungal defense in serpin-deficient Drosophila. Science 285, 1917–1919 (1999).
Poltorak, A., Ricciardi-Castagnoli, P., Citterio, S. & Beutler, B. Physical contact between lipopolysaccharide and toll-like receptor 4 revealed by genetic complementation. Proc. Natl Acad. Sci. USA 97, 2163–2167 (2000).
Lien, E. et al. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J. Clin. Invest. 105, 497–504 (2000).
da Silva Correia, J., Soldau, K., Christen, U., Tobias, P. S. & Ulevitch, R. J. Lipopolysaccharide is in close proximity to each of the proteins in its membrane receptor complex: transfer from CD14 to TLR4 and MD-2. J. Biol. Chem. 276, 21129–21135 (2001).
Matzinger, P. An innate sense of danger. Semin. Immunol. 10, 399–415 (1998).
Ohashi, K., Burkart, V., Flohe, S. & Kolb, H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J. Immunol. 164, 558–561 (2000).
Termeer, C. C. et al. Oligosaccharides of hyaluronan are potent activators of dendritic cells. J. Immunol. 165, 1863–1870 (2000).
Kodaira, Y., Nair, S. K., Wrenshall, L. E., Gilboa, E. & Platt, J. L. Phenotypic and functional maturation of dendritic cells mediated by heparan sulfate. J. Immunol. 165, 1599–1604 (2000).
Okamura, Y. et al. The EDA domain of fibronectin activates toll-like receptor 4. J. Biol. Chem. 276, 10229–10233 (2001).
Frantz, S., Kelly, R. A. & Bourcier, T. Role of TLR-2 in the Activation of Nuclear Factor B by Oxidative Stress in Cardiac Myocytes. J. Biol. Chem. 276, 5197–5203 (2001).
Muzio, M., Ni, J., Feng, P. & Dixit, V. M. IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science 278, 1612–1615 (1997).
Medzhitov, R. et al. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol. Cell 2, 253–258 (1998).
Adachi, O. et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL- 18-mediated function. Immunity 9, 143–150 (1998).
Kawai, T., Adachi, O., Ogawa, T., Takeda, K. & Akira, S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11, 115–122 (1999).
Rudolph, D. et al., Severe liver degeneration and lack of NF-κB activation in NEMO/IKKγ-deficient mice. Genes Dev. 14, 854–862 (2000).
Schmidt-Supprian, M. et al. NEMO/IKKγ-deficient mice model incontinentia pigmenti. Mol. Cell 6, 981–992 (2000).
Chu, W. M. et al. JNK2 and IKKβ are required for activating the innate response to viral infection. Immunity 11, 721–731 (1999).
Seki, E. et al. Lipopolysaccharide-induced IL-18 secretion from murine kupffer cells independently of myeloid differentiation factor 88 that is critically involved in induction of production of IL-12 and IL-1β. J. Immunol. 166, 2651–2657 (2001).
Steinman, R. M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 9, 271–296 (1991).
Banchereau, J. & Steinman, R. M. Dendritic cells and the control of immunity. Nature 392, 245–252 (1998).
Reis e Sousa, C., Sher, A. & Kaye, P. The role of dendritic cells in the induction and regulation of immunity to microbial infection. Curr. Opin. Immunol. 11, 392–399 (1999).
Moser, M. & Murphy, K. M. Dendritic cell regulation of TH1-TH2 development. Nature Immunol. 1, 199–205 (2000).
Pulendran, B., Banchereau, J., Maraskovsky, E. & Maliszewski, C. Modulating the immune response with dendritic cells and their growth factors. Trends Immunol. 22, 41–47 (2001).
Liu, Y. J., Kanzler, H., Soumelis, V. & Gilliet, M. Dendritic cell lineage, plasticity and cross-regulation. Nature Immunol. 2, 585–589 (2001).
Bauer, S. et al. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG-motif recognition. Proc. Natl Acad. Sci. USA (2001, in the press).
Bauer, M. et al. Bacterial CpG-DNA triggers activation and maturation of human CD11c(−), CD123(+) dendritic cells. J. Immunol. 166, 5000–5007 (2001).
Kaisho, T. & Akira, S. Dendritic cell function in Toll-like receptor- and MyD88-knockout mice. Trends Immunol. 22, 78–83 (2001).
Kaisho, T., Takeuchi, O., Kawai, T., Hoshino, K. & Akira, S. Endotoxin-induced maturation of MyD88-deficient dendritic cells. J. Immunol. 166, 5688–5694 (2001).
Kadowaki, N., Antonenko, S. & Liu, Y. J. Distinct CpG DNA and polyinosinic-polycytidylic acid double-stranded RNA, respectively, stimulate CD11c(−) type 2 dendritic cell precursors and CD11c(+) dendritic cells to produce type I IFN. J. Immunol. 166, 2291–2295 (2001).
Thoma-Uszynski, S. et al. Induction of direct antimicrobial activity through mammalian toll-like receptors. Science 291, 1544–1547 (2001).
Acknowledgements
We thank lab members for useful discussions and M. Lamphier for critical reading of the manuscript. Supported by grants from the Ministry of Education, Culture, Sports, Science and Technology in Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Akira, S., Takeda, K. & Kaisho, T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol 2, 675–680 (2001). https://doi.org/10.1038/90609
Issue Date:
DOI: https://doi.org/10.1038/90609
This article is cited by
-
Evaluation of the TLR3 involvement during Schistosoma japonicum-induced pathology
BMC Immunology (2024)
-
Inflammasomes in neurological disorders — mechanisms and therapeutic potential
Nature Reviews Neurology (2024)
-
Auraptene Mitigates Colitis Induced by Dextran Sulfate Sodium in Mice by Regulating Specific Intestinal Flora and Repairing the Intestinal Barrier
Inflammation (2024)
-
Pathogen recognition pathway gene variants and inflammasome sensors gene expression in tuberculosis patients under treatment
Molecular Biology Reports (2024)
-
Toll-like receptor 4 (TLR4): new insight immune and aging
Immunity & Ageing (2023)
