
Abstract
Autoinflammatory diseases were first recognized nearly 20 years ago as distinct clinical and immunological entities caused by dysregulation in the innate immune system. Since then, advances in genomic techniques have led to the identification of new monogenic disorders and their corresponding signaling pathways. Here we review these monogenic autoinflammatory diseases, ranging from periodic fever syndromes caused by dysregulated inflammasome-mediated production of the cytokine IL-1β to disorders arising from perturbations in signaling by the transcription factor NF-κB, ubiquitination, cytokine signaling, protein folding, type I interferon production and complement activation, and we further examine their molecular mechanisms. We also explore the overlap among autoinflammation, autoimmunity and immunodeficiency, and pose a series of unanswered questions that are expected to be central in autoinflammatory disease research in the coming decade.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
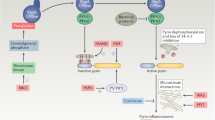
Change history
17 August 2017
In the version of this article initially published, the final sentence of the third paragraph of the first subsection ('Monogenic periodic fever syndromes') incorrectly states that "Colchicine causes microtubule destabilization, thereby facilitating pyrin-inflammasome activation independently of RhoA." That sentence should read: "Colchicine also causes microtubule destabilization, thereby inhibiting pyrin-inflammasome activation independently of RhoA." The error has been corrected in the HTML and PDF versions of the article.
18 October 2017
Nat. Immunol. 18, 832–842 (2017); published online 19 July 2017; corrected after print 17 August 2017 In the version of this article initially published, the final sentence of the third paragraph of the first subsection ('Monogenic periodic fever syndromes') incorrectly states that “Colchicine causes microtubule destabilization, thereby facilitating pyrin-inflammasome activation independently of RhoA.
References
McDermott, M.F. et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 97, 133–144 (1999).
Medzhitov, R. & Janeway, C. Jr. Innate immunity. N. Engl. J. Med. 343, 338–344 (2000).
Medzhitov, R. & Janeway, C.A. Jr. Innate immunity: the virtues of a nonclonal system of recognition. Cell 91, 295–298 (1997).
Lamkanfi, M. & Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 157, 1013–1022 (2014).
Liu, X. et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158 (2016).
Ding, J. et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116 (2016).
Kayagaki, N. et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015).
Shi, J. et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015).
Shi, J., Gao, W. & Shao, F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem. Sci. 42, 245–254 (2017).
Wang, Y. et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a Gasdermin. Nature http://dx.doi.org/10.1038/nature22393 (2017).
The International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 90, 797–807 (1997).
French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat. Genet. 17, 25–31 (1997).
Padeh, S. et al. Clinical and diagnostic value of genetic testing in 216 Israeli children with Familial Mediterranean fever. J. Rheumatol. 30, 185–190 (2003).
Booty, M.G. et al. Familial Mediterranean fever with a single MEFV mutation: where is the second hit? Arthritis Rheum. 60, 1851–1861 (2009).
Marek-Yagel, D. et al. Clinical disease among patients heterozygous for familial Mediterranean fever. Arthritis Rheum. 60, 1862–1866 (2009).
Lachmann, H.J. et al. Clinical and subclinical inflammation in patients with familial Mediterranean fever and in heterozygous carriers of MEFV mutations. Rheumatology 45, 746–750 (2006).
Chae, J.J. et al. Gain-of-function Pyrin mutations induce NLRP3 protein-independent interleukin-1β activation and severe autoinflammation in mice. Immunity 34, 755–768 (2011).
Seshadri, S., Duncan, M.D., Hart, J.M., Gavrilin, M.A. & Wewers, M.D. Pyrin levels in human monocytes and monocyte-derived macrophages regulate IL-1beta processing and release. J. Immunol. 179, 1274–1281 (2007).
Yu, J.W. et al. Cryopyrin and pyrin activate caspase-1, but not NF-kappaB, via ASC oligomerization. Cell Death Differ. 13, 236–249 (2006).
Xu, H. et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513, 237–241 (2014).
Park, Y.H., Wood, G., Kastner, D.L. & Chae, J.J. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat. Immunol. 17, 914–921 (2016). References 20 and 21 describe the signaling pathway mediated by RhoA GTPase, PKN1/2 and 14–3-3 proteins in the regulation of the pyrin inflammasome in FMF and HIDS.
Masters, S.L. et al. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Sci. Transl. Med. 8, 332ra45 (2016).
Gao, W., Yang, J., Liu, W., Wang, Y. & Shao, F. Site-specific phosphorylation and microtubule dynamics control Pyrin inflammasome activation. Proc. Natl. Acad. Sci. USA 113, E4857–E4866 (2016).
Van Gorp, H. et al. Familial Mediterranean fever mutations lift the obligatory requirement for microtubules in Pyrin inflammasome activation. Proc. Natl. Acad. Sci. USA 113, 14384–14389 (2016).
Holt, B.F. III., Mackey, D. & Dangl, J.L. Recognition of pathogens by plants. Curr. Biol. 10, R5–R7 (2000).
Vance, R.E. Immunology taught by bacteria. J. Clin. Immunol. 30, 507–511 (2010).
Ratner, D. et al. The Yersinia pestis effector YopM inhibits pyrin inflammasome activation. PLoS Pathog. 12, e1006035 (2016).
Chung, L.K. et al. The Yersinia virulence factor YopM hijacks host kinases to inhibit type III effector-triggered activation of the pyrin inflammasome. Cell Host Microbe 20, 296–306 (2016). The YopE and YopT proteins from Yersinia inactivate RhoA and lead to pyrin-inflammasome activation, whereas YopM inhibits the pyrin inflammasome. YopM is critical for maintaining the virulence of Yersinia . The authors speculate that FMF-associated mutations may confer protection from Yersinia infection.
Aubert, D.F. et al. A Burkholderia type VI effector deamidates Rho GTPases to activate the pyrin inflammasome and trigger inflammation. Cell Host Microbe 19, 664–674 (2016). TecA, a type VI secretion system effector from Burkholderia cenocepacia , inactivates RhoA and leads to pyrin-inflammasome activation and lung inflammation. TecA promotes survival of mice from B. cenocepcia infection.
Liston, A. & Masters, S.L. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol. 17, 208–214 (2017).
Drenth, J.P. et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. Nat. Genet. 22, 178–181 (1999).
Houten, S.M. et al. Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nat. Genet. 22, 175–177 (1999).
Akula, M.K. et al. Control of the innate immune response by the mevalonate pathway. Nat. Immunol. 17, 922–929 (2016).
Zhang, S.-Q. et al. Exome sequencing identifies MVK mutations in disseminated superficial actinic porokeratosis. Nat. Genet. 44, 1156–1160 (2012).
Liu, Y. et al. Identification of three mutations in the MVK gene in six patients associated with disseminated superficial actinic porokeratosis. Clin. Chim. Acta 454, 124–129 (2016).
Hoffman, H.M., Mueller, J.L., Broide, D.H., Wanderer, A.A. & Kolodner, R.D. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle–Wells syndrome. Nat. Genet. 29, 301–305 (2001).
Agostini, L. et al. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle–Wells autoinflammatory disorder. Immunity 20, 319–325 (2004).
Levy, R. et al. Phenotypic and genotypic characteristics of cryopyrin-associated periodic syndrome: a series of 136 patients from the Eurofever Registry. Ann. Rheum. Dis. 74, 2043–2049 (2015).
Saito, M. et al. Somatic mosaicism of CIAS1 in a patient with chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. 52, 3579–3585 (2005).
Saito, M. et al. Disease-associated CIAS1 mutations induce monocyte death, revealing low-level mosaicism in mutation-negative cryopyrin-associated periodic syndrome patients. Blood 111, 2132–2141 (2008).
Tanaka, N. et al. High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an international multicenter collaborative study. Arthritis Rheum. 63, 3625–3632 (2011).
Zhou, Q. et al. Brief report: cryopyrin-associated periodic syndrome caused by a myeloid-restricted somatic NLRP3 mutation. Arthrtis Rheumatol. 67, 2482–2486 (2015).
Mensa-Vilaro, A. et al. Brief report: late-onset cryopyrin-associated periodic syndrome due to myeloid-restricted somatic NLRP3 mosaicism. Arthritis Rheumatol. 68, 3035–3041 (2016).
de Koning, H.D. et al. Myeloid lineage-restricted somatic mosaicism of NLRP3 mutations in patients with variant Schnitzler syndrome. J. Allergy Clin. Immunol. 135, 561–564 (2015).
Loock, J. et al. Genetic predisposition (NLRP3 V198M mutation) for IL-1-mediated inflammation in a patient with Schnitzler syndrome. J. Allergy Clin. Immunol. 125, 500–502 (2010).
Lee, G.S. et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492, 123–127 (2012).
Franchi, L., Muñoz-Planillo, R. & Núñez, G. Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 13, 325–332 (2012).
Hornung, V. et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856 (2008).
Martinon, F., Pétrilli, V., Mayor, A., Tardivel, A. & Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 (2006).
Dostert, C. et al. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320, 674–677 (2008).
Franchi, L., Kanneganti, T.-D., Dubyak, G.R. & Núñez, G. Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J. Biol. Chem. 282, 18810–18818 (2007).
Duewell, P. et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 (2010).
Grant, R.W. & Dixit, V.D. Mechanisms of disease: inflammasome activation and the development of type 2 diabetes. Front. Immunol. 4, 50 (2013).
Lobito, A.A. et al. Abnormal disulfide-linked oligomerization results in ER retention and altered signaling by TNFR1 mutants in TNFR1-associated periodic fever syndrome (TRAPS). Blood 108, 1320–1327 (2006).
Simon, A. et al. Concerted action of wild-type and mutant TNF receptors enhances inflammation in TNF receptor 1-associated periodic fever syndrome. Proc. Natl. Acad. Sci. USA 107, 9801–9806 (2010).
Bulua, A.C. et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 208, 519–533 (2011).
Williams, K.L. et al. The CATERPILLER protein monarch-1 is an antagonist of toll-like receptor-, tumor necrosis factor alpha-, and Mycobacterium tuberculosis-induced pro-inflammatory signals. J. Biol. Chem. 280, 39914–39924 (2005).
Lich, J.D. et al. Monarch-1 suppresses non-canonical NF-kappaB activation and p52-dependent chemokine expression in monocytes. J. Immunol. 178, 1256–1260 (2007).
Allen, I.C. et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity 36, 742–754 (2012).
Jéru, I. et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc. Natl. Acad. Sci. USA 105, 1614–1619 (2008).
Shen, M., Tang, L., Shi, X., Zeng, X. & Yao, Q. NLRP12 autoinflammatory disease: a Chinese case series and literature review. Clin. Rheumatol. http://dx.doi.org/10.1007/s10067-016-3410-y (2016).
Jéru, I. et al. Identification and functional consequences of a recurrent NLRP12 missense mutation in periodic fever syndromes. Arthritis Rheum. 63, 1459–1464 (2011).
Borghini, S. et al. Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of an NLRP12 mutation. Arthritis Rheum. 63, 830–839 (2011).
Wise, C.A. et al. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum. Mol. Genet. 11, 961–969 (2002).
Yu, J.W. et al. Pyrin activates the ASC pyroptosome in response to engagement by autoinflammatory PSTPIP1 mutants. Mol. Cell 28, 214–227 (2007).
Shoham, N.G. et al. Pyrin binds the PSTPIP1/CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway. Proc. Natl. Acad. Sci. USA 100, 13501–13506 (2003).
Starnes, T.W. et al. The F-BAR protein PSTPIP1 controls extracellular matrix degradation and filopodia formation in macrophages. Blood 123, 2703–2714 (2014).
Omenetti, A. et al. Disease activity accounts for long-term efficacy of IL-1 blockers in pyogenic sterile arthritis pyoderma gangrenosum and severe acne syndrome. Rheumatology 55, 1325–1335 (2016).
Ferguson, P.J. et al. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome). J. Med. Genet. 42, 551–557 (2005).
Al-Mosawi, Z.S., Al-Saad, K.K., Ijadi-Maghsoodi, R., El-Shanti, H.I. & Ferguson, P.J. A splice site mutation confirms the role of LPIN2 in Majeed syndrome. Arthritis Rheum. 56, 960–964 (2007).
Lordén, G. et al. Lipin-2 regulates NLRP3 inflammasome by affecting P2X7 receptor activation. J. Exp. Med. 214, 511–528 (2017).
Wen, H., Ting, J.P.Y. & O'Neill, L.A.J. A role for the NLRP3 inflammasome in metabolic diseases: did Warburg miss inflammation? Nat. Immunol. 13, 352–357 (2012).
Mills, E.L., Kelly, B. & O'Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 18, 488–498 (2017).
Aksentijevich, I. et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N. Engl. J. Med. 360, 2426–2437 (2009).
Reddy, S. et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N. Engl. J. Med. 360, 2438–2444 (2009).
Marrakchi, S. et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N. Engl. J. Med. 365, 620–628 (2011).
Onoufriadis, A. et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am. J. Hum. Genet. 89, 432–437 (2011).
Gresnigt, M.S. & van de Veerdonk, F.L. Biology of IL-36 cytokines and their role in disease. Semin. Immunol. 25, 458–465 (2013).
Maeda, S. et al. Nod2 mutation in Crohn's disease potentiates NF-kappaB activity and IL-1β processing. Science 307, 734–738 (2005).
Kobayashi, K.S. et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307, 731–734 (2005).
Keestra-Gounder, A.M. et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature 532, 394–397 (2016).
Torres, J., Mehandru, S., Colombel, J.-F. & Peyrin-Biroulet, L. Crohn's disease. Lancet 389, 1741–1755 (2017).
Sidiq, T., Yoshihama, S., Downs, I. & Kobayashi, K.S. Nod2: a critical regulator of ileal microbiota and Crohn's disease. Front. Immunol. 7, 367 (2016).
Hugot, J.-P. et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature 411, 599–603 (2001).
Ogura, Y. et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 411, 603–606 (2001).
Miceli-Richard, C. et al. CARD15 mutations in Blau syndrome. Nat. Genet. 29, 19–20 (2001).
Strober, W., Murray, P.J., Kitani, A. & Watanabe, T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat. Rev. Immunol. 6, 9–20 (2006).
Kanazawa, N. et al. Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: common genetic etiology with Blau syndrome. Blood 105, 1195–1197 (2005).
Maekawa, S., Ohto, U., Shibata, T., Miyake, K. & Shimizu, T. Crystal structure of NOD2 and its implications in human disease. Nat. Commun. 7, 11813 (2016).
Mensa-Vilaro, A. et al. Brief report: first identification of intrafamilial recurrence of Blau syndrome due to gonosomal NOD2 mosaicism. Arthritis Rheumatol. 68, 1039–1044 (2016).
Jordan, C.T. et al. PSORS2 is due to mutations in CARD14. Am. J. Hum. Genet. 90, 784–795 (2012).
Fuchs-Telem, D. et al. Familial pityriasis rubra pilaris is caused by mutations in CARD14. Am. J. Hum. Genet. 91, 163–170 (2012).
Jordan, C.T. et al. Rare and common variants in CARD14, encoding an epidermal regulator of NF-kappaB, in psoriasis. Am. J. Hum. Genet. 90, 796–808 (2012).
Van Nuffel, E. et al. CARD14-mediated activation of paracaspase MALT1 in keratinocytes: implications for psoriasis. J. Invest. Dermatol. 137, 569–575 (2017).
Glocker, E.-O. et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 361, 2033–2045 (2009).
Glocker, E.O. et al. Infant colitis: it's in the genes. Lancet 376, 1272 (2010).
Kotlarz, D. et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology 143, 347–355 (2012).
Shouval, D.S. et al. Interleukin 1β mediates intestinal inflammation in mice and patients with interleukin 10 receptor deficiency. Gastroenterology 151, 1100–1104 (2016).
Warwicker, P. et al. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. 53, 836–844 (1998).
Maga, T.K., Nishimura, C.J., Weaver, A.E., Frees, K.L. & Smith, R.J. Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum. Mutat. 31, E1445–E1460 (2010).
Noris, M. et al. Familial haemolytic uraemic syndrome and an MCP mutation. Lancet 362, 1542–1547 (2003).
Delvaeye, M. et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 361, 345–357 (2009).
Baines, A.C. & Brodsky, R.A. Complementopathies. Blood Rev. http://dx.doi.org/10.1016/j.blre.2017.02.003 (2017).
Edwards, A.O. et al. Complement factor H polymorphism and age-related macular degeneration. Science 308, 421–424 (2005).
Haines, J.L. et al. Complement factor H variant increases the risk of age-related macular degeneration. Science 308, 419–421 (2005).
Klein, R.J. et al. Complement factor H polymorphism in age-related macular degeneration. Science 308, 385–389 (2005).
Calippe, B. et al. Complement factor H inhibits CD47-mediated resolution of inflammation. Immunity 46, 261–272 (2017).
Takeda, J. et al. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell 73, 703–711 (1993).
Bessler, M. et al. Paroxysmal nocturnal haemoglobinuria (PNH) is caused by somatic mutations in the PIG-A gene. EMBO J. 13, 110–117 (1994).
Zhou, Q. et al. A hypermorphic missense mutation in PLCG2, encoding phospholipase Cγ2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am. J. Hum. Genet. 91, 713–720 (2012).
Chae, J.J. et al. Connecting two pathways through Ca2+ signaling: NLRP3 inflammasome activation induced by a hypermorphic PLCG2 mutation. Arthritis Rheumatol. 67, 563–567 (2015).
Ombrello, M.J. et al. Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N. Engl. J. Med. 366, 330–338 (2012).
Canna, S.W. et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 46, 1140–1146 (2014).
Romberg, N. et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet. 46, 1135–1139 (2014). References 113 and 114 describe people with gain-of-function mutations in the NLRC4 inflammasome that lead to recurrent MAS and severe enterocolitis. The disease is mediated by IL-1 and IL-18.
Vance, R.E. The NAIP/NLRC4 inflammasomes. Curr. Opin. Immunol. 32, 84–89 (2015).
Kitamura, A., Sasaki, Y., Abe, T., Kano, H. & Yasutomo, K. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J. Exp. Med. 211, 2385–2396 (2014).
Kawasaki, Y. et al. Identification of a high-frequency somatic NLRC4 mutation as a cause of autoinflammation by pluripotent cell–based phenotype dissection. Arthritis Rheumatol. 69, 447–459 (2017).
Zhong, F.L. et al. Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell 167, 187–202.e17 (2016).
Grandemange, S. et al. A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis). Ann. Rheum. Dis. http://dx.doi.org/10.1136/annrheumdis-2016-210021 (2016).
Jin, Y. et al. NALP1 in vitiligo-associated multiple autoimmune disease. N. Engl. J. Med. 356, 1216–1225 (2007).
Agarwal, A.K. et al. PSMB8 encoding the β5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am. J. Hum. Genet. 87, 866–872 (2010).
Kitamura, A.K. et al. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J. Clin. Invest. 121, 4150–4160 (2011).
Arima, K. et al. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc. Natl. Acad. Sci. USA 108, 14914–14919 (2011).
Liu, Y. et al. Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 64, 895–907 (2012).
Brehm, A. et al. Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J. Clin. Invest. 125, 4196–4211 (2015).
Liu, Y. et al. Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med. 371, 507–518 (2014). De novo mutations in TMEM173 , which encodes an indirect sensor of cytosolic DNA called STING, lead to SAVI. People with SAVI have a prominent type I interferon signature.
Sun, L., Wu, J., Du, F., Chen, X. & Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013).
Wu, J. et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830 (2013).
Rice, G.I. et al. Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat. Genet. 41, 829–832 (2009).
Rice, G.I. et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat. Genet. 46, 503–509 (2014).
Rice, G.I. et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat. Genet. 44, 1243–1248 (2012).
Rice, G.I. et al. Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. 12, 1159–1169 (2013).
Crow, Y.J. et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nat. Genet. 38, 910–916 (2006).
Crow, Y.J. et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat. Genet. 38, 917–920 (2006).
Zhou, Q. et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat. Genet. 48, 67–73 (2016). Haploinsufficiency of A20 is caused by loss-of-function mutations in TNFAIP3 , which encodes the A20 deubiquitinase. Insufficient A20 leads to accumulation of complexes of Lys63-ubiquitinated proteins that activate NF-κB and IL-1β signaling.
Vande Walle, L. et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 512, 69–73 (2014).
Duong, B.H. et al. A20 restricts ubiquitination of pro-interleukin-1β protein complexes and suppresses NLRP3 inflammasome activity. Immunity 42, 55–67 (2015).
Zilberman-Rudenko, J. et al. Recruitment of A20 by the C-terminal domain of NEMO suppresses NF-κB activation and autoinflammatory disease. Proc. Natl. Acad. Sci. USA 113, 1612–1617 (2016).
Zhou, Q. et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc. Natl. Acad. Sci. USA 113, 10127–10132 (2016).
Damgaard, R.B. et al. The deubiquitinase OTULIN is an essential negative regulator of inflammation and autoimmunity. Cell 166, 1215–1230.e20 (2016).References 139 and 140 describe people with loss-of-function mutations in the linear deubiquitinase OTULIN and subsequent activation of NF-κB signaling.
Boisson, B. et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J. Exp. Med. 212, 939–951 (2015).
Boisson, B. et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat. Immunol. 13, 1178–1186 (2012).
Chakraborty, P.K. et al. Mutations in TRNT1 cause congenital sideroblastic anemia with immunodeficiency, fevers, and developmental delay (SIFD). Blood 124, 2867–2871 (2014).
Standing, A.S. et al. Autoinflammatory periodic fever, immunodeficiency, and thrombocytopenia (PFIT) caused by mutation in actin-regulatory gene WDR1. J. Exp. Med. 2041, 59–71 (2017).
Kim, M.L. et al. Aberrant actin depolymerization triggers the pyrin inflammasome and autoinflammatory disease that is dependent on IL-18, not IL-1β. J. Exp. Med. 212, 927–938 (2015).
Zhou, Q. et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N. Engl. J. Med. 370, 911–920 (2014).
Navon Elkan, P. et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N. Engl. J. Med. 370, 921–931 (2014).
Van Montfrans, J.M. et al. Phenotypic variability in patients with ADA2 deficiency due to identical homozygous R169Q mutations. Rheumatology 55, 902–910 (2016).
Boyden, S.E. et al. Vibratory urticaria associated with a missense variant in ADGRE2. N. Engl. J. Med. 374, 656–663 (2016).
Goldbach-Mansky, R. et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1β inhibition. N. Engl. J. Med. 355, 581–592 (2006).
Lachmann, H.J. et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N. Engl. J. Med. 360, 2416–2425 (2009). Canakinumab, an anti-IL-1β monoclonal antibody, is shown in this double-blind, placebo-controlled trial to significantly decrease disease activity in people with CAPS.
Sibley, C.H. et al. A 24-month open-label study of canakinumab in neonatal-onset multisystem inflammatory disease. Ann. Rheum. Dis. 74, 1714–1719 (2015).
Goldbach-Mansky, R. et al. A pilot study to evaluate the safety and efficacy of the long-acting interleukin-1 inhibitor rilonacept (interleukin-1 Trap) in patients with familial cold autoinflammatory syndrome. Arthritis Rheum. 58, 2432–2442 (2008).
Hoffman, H.M. et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum. 58, 2443–2452 (2008).
Canna, S.W. et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J. Allergy Clin. Immunol. 139, 1698–1701 (2017).
Chae, J.J. et al. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1β production. Proc. Natl. Acad. Sci. USA 103, 9982–9987 (2006).
Hashkes, P.J. et al. Rilonacept for colchicine-resistant or -intolerant familial Mediterranean fever: a randomized trial. Ann. Intern. Med. 157, 533–541 (2012).
König, N. et al. Familial chilblain lupus due to a gain-of-function mutation in STING. Ann. Rheum. Dis. 76, 468–472 (2017).
Van Eyck, L. Jr. et al. Hematopoietic stem cell transplantation rescues the immunologic phenotype and prevents vasculopathy in patients with adenosine deaminase 2 deficiency. J. Allergy Clin. Immunol. 135, 283–287.e5 (2015).
Neven, B. et al. Allogeneic bone marrow transplantation in mevalonic aciduria. N. Engl. J. Med. 356, 2700–2703 (2007).
Giardino, S. et al. Long-term outcome of a successful cord blood stem cell transplant in mevalonate kinase deficiency. Pediatrics 135, e211–e215 (2015).
Rodero, M.P. & Crow, Y.J. Type I interferon-mediated monogenic autoinflammation: the type I interferonopathies, a conceptual overview. J. Exp. Med. 213, 2527–2538 (2016).
Canna, S.W. & Goldbach-Mansky, R. New monogenic autoinflammatory diseases–a clinical overview. Semin. Immunopathol. 37, 387–394 (2015).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Manthiram, K., Zhou, Q., Aksentijevich, I. et al. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat Immunol 18, 832–842 (2017). https://doi.org/10.1038/ni.3777
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ni.3777
This article is cited by
-
Canakinumab treatment real world evidence in 3 monogenic periodic fever syndromes in 2009–2022: an interim analysis using the French JIR cohort database
Arthritis Research & Therapy (2024)
-
Human SHARPIN deficiency is linked to inborn errors of cell death
Nature Immunology (2024)
-
Pyrin Inflammasome Activation Defines Colchicine-Responsive SURF Patients from FMF and Other Recurrent Fevers
Journal of Clinical Immunology (2024)
-
Inflammatory comorbidities ın the largest pediatric Familial Mediterranean fever cohort: a multicenter retrospective study of Pediatric Rheumatology Academy (PeRA)-Research Group (RG)
Clinical Rheumatology (2024)
-
“Deficiency in ELF4, X-Linked”: a Monogenic Disease Entity Resembling Behçet’s Syndrome and Inflammatory Bowel Disease
Journal of Clinical Immunology (2024)
