
Abstract
Innate immune responses have the ability to both combat infectious microbes and drive pathological inflammation. Inflammasome complexes are a central component of these processes through their regulation of interleukin 1β (IL-1β), IL-18 and pyroptosis. Inflammasomes recognize microbial products or endogenous molecules released from damaged or dying cells both through direct binding of ligands and indirect mechanisms. The potential of the IL-1 family of cytokines to cause tissue damage and chronic inflammation emphasizes the importance of regulating inflammasomes. Many regulatory mechanisms have been identified that act as checkpoints for attenuating inflammasome signaling at multiple steps. Here we discuss the various regulatory mechanisms that have evolved to keep inflammasome signaling in check to maintain immunological balance.
Similar content being viewed by others
Basic mechanisms of inflammasome activation
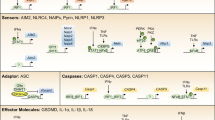
The innate immune system is composed of germline-encoded receptors that collectively serve as a sensor for monitoring the extracellular and intracellular compartments for signs of infection or tissue injury. A key component of cytosolic surveillance is the inflammasome, a large multimolecular complex that controls activation of the proteolytic enzyme caspase-1 (refs. 1,2). Caspase-1 in turn regulates maturation of the proinflammatory cytokines interleukin-1β (IL-1β) and IL-18 or the rapid inflammatory form of cell death called 'pyroptosis'. Inflammasomes come in at least two distinct 'flavors', and those identified thus far contain a member of the NLR family (nucleotide-binding-and-oligomerization domain (Nod) and leucine-rich-repeat–containing) or PYHIN family (pyrin domain (PYD) and HIN domain–containing).
The human and mouse genomes encode 23 and 34 NLRs, respectively, several of which assemble into inflammasome complexes. These complexes form in cells exposed to a range of physically and chemically diverse ligands of microbial, environmental or endogenous origin3. NLR inflammasomes contain the adaptor ASC, which is common to most inflammasome complexes, although ASC-independent complexes also form in certain cases. Although the actual ligand that engages the NLR is for the most part unknown, the understanding of how the PYHIN inflammasomes become activated is much clearer. AIM2 and IFI16 (two members of the PYHIN family) aretrue receptors, as they engage their double-stranded DNA (dsDNA) ligands directly via one or two DNA-binding HIN domains, respectively4,5,6. As for the NLRs, NLRP1, NLRP3, NLRC4 (IPAF), NLRP6 and NLRP12 have all been shown to assemble into inflammasomes. These molecules are characterized by a tripartite structure containing a central Nod (or NACHT domain), a carboxy-terminal leucine-rich-repeat domain, and an amino-terminal effector domain, which can be a baculovirus inhibitor-of-apoptosis-protein repeat, a caspase-recruitment-and-activation domain (CARD) or a PYD3. The leucine-rich-repeat domains are thought to act as the sensing component of these molecules; however, the molecular basis by which individual inflammasomes respond to specific stimuli is very poorly understood and there is no solid evidence indicating the ability of this domain to directly bind ligand.
In general, after activation, the NLRs oligomerize through their NACHT domains, and this oligomerization facilitates recruitment of pro-caspase-1 through direct interactions between the CARDs of the NLR and that of pro-caspase-1. However, most NLRs lack CARDs, and those that contain PYDs require ASC. The PYD of ASC interacts with the PYD of the NLR, whereas the CARD of ASC recruits pro-caspase-1. The binding of a single molecule of ASC to both partners is facilitated by a tandem orientation of PYDs and CARDs restrained by a flexible linker region7. ASC operates as a molecular nucleating platform for protein-protein interactions during inflammasome activation by oligomerizing into large disc-like structures7. Once pro-caspase-1 is recruited into a multiprotein inflammasome complex, the proximity-induced multimerization is suggested to induce autoproteolytic cleavage of pro-caspase-1. Initially pro-caspase-1 is cleaved into a p35 fragment (containing the CARD) and a p10 fragment. Subsequently, the p35 fragment is processed into its CARD and a p20 subunit; two molecules of p20 heterodimerize with two molecules of p10 to form an active caspase-1 enzyme. ASC is mainly sequestered in the nucleus, and its redistribution from the nucleus to the cytosol is required for inflammasome assembly8. Published reports have suggested that ASC is also present in resting cells in the cytosol, which indicates that compartmental localization of ASC may confer distinct functional abilities to this important molecule9.
Caspase-1 is an aspartate-specific cysteine protease that cleaves its substrates, IL-1β and IL-18, at recognition sites adjacent to aspartic acid residues. Activation of caspase-1 is also linked to pyroptosis10. Cytokine processing and pyroptosis seem to be mediated by distinct complexes formed in response to bacterial pathogens such as Salmonella typhimurium and Legionella pneumophila. A large ASC-containing inflammasome induces autoproteolysis of caspase-1 and processing of inflammatory cytokines, whereas an ASC-independent caspase-1 complex promotes pyroptotic cell death. The latter complex is smaller and does not facilitate caspase-1 autoproteolysis11. Activation of caspase-1 has also been linked to the release of endogenous danger signals (also called danger-associated molecular patterns) such as HMGB1, unconventional secretion of leaderless peptides, cleavage of enzymes in the glycolytic pathway, restriction of bacterial replication in infected macrophages, and augmentation of cell-repair pathways via the regulation of lipid metabolism10,12.
The caspase-1-related protease caspase-11 (caspase-4 and caspase-5 in humans) has also been linked to inflammasome signaling1. Caspase-11 is essential for the processing of caspase-1 and maturation of IL-1β and IL-18 by enteric bacteria such as Escherichia coli, Citrobacter rodentium and Vibrio cholerae. This noncanonical inflammasome pathway involving caspase-11 has both caspase-1-dependent and caspase-1-independent outcomes. Whereas caspase-11-mediated processing of IL-1β and IL-18 requires caspase-1, macrophage cell death does not involve caspase-1. Notably, NLRP3 and ASC are both required for the activation of caspase-1 by noncanonical activators such as E. coli; however, these molecules are dispensable for cell death1. The exact mechanism by which caspase-11 is activated and contributes to inflammasome function is still unclear. Noncanonical inflammasome activators such as E. coli induce cleavage of pro-caspase-11 to the caspase-11 subunit p26. It is possible that caspase-11 directly interacts with caspase-1 and promotes formation of the inflammasome complex, as both pro-caspase-11 and the p26 subunit immunoprecipitate together with caspase-1 after activation with E. coli or cholera toxin. The role of caspase-11 in other aspects of inflammasome function remains to be clearly defined.
The NLRP3 inflammasome
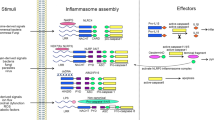
Depending on the NLR in the complex, inflammasomes are equipped with the ability to respond to a wide array of signals. The NLRP3 inflammasome is by far the best-studied member of this family and is activated by a wide range of signals of pathogenic, endogenous and environmental origin. Several pathogens, such as Staphylococcus aureus, Listeria monocytogenes, Klebsiella pneumoniae, Neisseria gonorrhoeae, Candida albicans and influenza A virus, activate NLRP3 (refs. 13,14,15,16,17,18,19). In addition, NLRP3 is alerted to the presence of endogenous danger signals—molecules released during or indicative of tissue injury, such as extracellular ATP, hyaluronan, amyloid-β fibrils and uric acid crystals20,21,22,23. Despite the growing number of activators for NLRP3 and the clear genetic evidence linking this pathway to the immunostimulatory activity of many molecules, the exact mechanism(s) by which NLRP3 is activated is (are) still unclear. There is no evidence that NLRP3 binds directly to any of its known activators. Such diversity of structure in potential ligands suggests that some shared mechanism enables NLRP3 to oligomerize and form a functionally competent inflammasome. Three distinct mechanisms have been proposed to account for NLRP3 activation: potassium efflux; the generation of mitochondria-derived reactive oxygen species (ROS); and phagolysosomal destabilization after the digestion of large particulate agonists such as monosodium urate24 (Fig. 1). Potassium efflux is induced by bacterial cytotoxins such as hemolysins that form pores on the cell and phagolysosomal membranes and by extracellular ATP, which engages the ATP-gated cation channel P2X7. Similarly, NLRP3 activation by malaria-digested hemoglobin (hemozoin), silica, asbestos and C. albicans has been linked to ROS production25. There is evidence that as a result of phagolysosomal membrane destabilization caused by crystalline and particulate ligands such as alum and silica, cathepsin proteases are released into the cytosol. This model would indicate that a cathepsin-cleavable substrate is a critical component for NLRP3 activation; however, so far no such cathepsin substrate has been identified. How NLRP3 is activated in any of these situations is still very much unknown. During infection with influenza virus, a virus-expressed intracellular M2 ion channel is important for the facilitation of NLRP3 activation. This M2 ion channel is located in the Golgi apparatus, where it enables the export of hydrogen ions from acidified Golgi, which in turn signals NLRP3 activation26. Overall, these findings indicate that changes in the intracellular ionic environment influence the activation potential of the NLRP3 inflammasome. However, a unifying mechanism that could explain how this complex is activated awaits clarification.

NLRP3 is activated by three common cellular events elicited by different stimuli: potassium efflux; the generation of ROS; and phagolysosomal destabilization and the release of endogenous mediators into the cytosol. NLRP1b is activated by anthrax lethal toxin. In the mouse, NLRC4 is activated by NAIP proteins bound to specific ligands. NAIP5 and NAIP6 bind to bacterial flagellin, whereas NAIP2 binds to the bacterial T3SS component PrgJ. These NAIP-ligand complexes subsequently bind and activate NLRC4. LF, lethal factor; PA, protective antigen.
The NLRC4 inflammasome
Inflammasomes have been best studied in the mouse system. The NLRC4 inflammasome responds to bacterial flagellin and PrgJ, a component of the type III secretion system (T3SS), and activates inflammasome assembly via direct CARD-CARD homotypic interactions with caspase-1 (ref. 27). Although ASC is not an absolute requirement, it substantially augments NLRC4-mediated inflammasome activation28,29. Studies have provided critical insights into the molecular basis for activation of the NLRC4 inflammasome by flagellin and PrgJ30,31. Those studies have demonstrated that the NAIP family of proteins, previously believed to function merely as accessory molecules for the NLRC4 inflammasome, act as the receptors (Fig. 1). There are seven genes encoding NAIP proteins in the mouse, four of which (Naip1, Naip2, Naip5 and Naip6) are expressed in C57BL/6 mice. Distinct NAIP molecules endow the NLRC4 inflammasome with the ability to differentiate among its bacterial ligands. Specifically, NAIP2 binds PrgJ, and this interaction facilitates NAIP2-NLRC4 binding, which results in activation of this inflammasome. In contrast, NAIP5 physically binds to flagellin, which results in the activation of NLRC4. Interestingly, there is only one NAIP in humans, and its role is still unclear. Human NAIP does not respond to flagellin or PrgJ, which suggests the existence of another more-unifying detection mechanism that feeds into NLRC4. Human NAIP does, however, interact with Cprl, a needle protein from the T3SS of Chromobacterium violaceum30. These findings suggest that the needle component of the T3SS is the true activator of the human NLRC4 inflammasome, which raises important questions about the effect of the sensing of bacterial flagellin by NLRC4 in human infectious diseases. The findings linking NAIP proteins to NLR activation have provided evidence that supports the notion that perhaps NLRs are merely downstream mediators of these pathways rather than being sensors themselves.
The NLRP1 inflammasome
NLRP1, the first inflammasome to be described, is activated mainly by the lethal toxin from Bacillus anthracis (Fig. 1). In contrast to human NLRP1, the mouse gene encoding NLRP1 is polymorphic, with three homologs: NLRP1a, NLRP1b and NLRP1c. Variations in NLRP1b provide sensitivity or resistance to the toxin32. Bacterial muramyl dipeptide also activates NLRP1; however, the physiological relevance of this activation is not clear. Biochemical reconstitution studies have shown that the essential components of the NLRP1 inflammasome include a sensitive NLRP1, pro-caspase-1, dNTPs and muramyl dipeptide33,34. Even though the presence of CARDs allows NLRP1 to bypass the requirement for ASC in inflammasome activation, ASC enhances the activity of NLRP1 inflammasome complexes. NLRP1 also contains a FIIND (function-to-find domain), which is also present in CARD8 (Cardinal)35. Rodents notably lack a gene encoding CARD8. Although it was identified many years ago, the role of the FIIND is still unknown. Evidence suggests that NLRP1 undergoes autoproteolytic cleavage at a conserved Ser-Phe/Ser motif (where 'Phe/Ser' indicates either phenylalanine or serine) in FIIND36. That suggests that the FIIND is a previously unrecognized type of ZU5-UPA domain, which undergoes post-translational autocleavage36. Those findings extend intramolecular autoproteolysis mediated by the ZU5-UPA motif to the NLR family of innate immunity proteins. Whether other NLRs undergo cleavage has not been addressed so far. Crosstalk between the Nod2 and NLRP1 pathways has also been proposed. Nod2 has been linked to NLRP1-dependent sensing of B. anthracis and muramyl dipeptide, where it forms a complex with NLRP1 in activated cells37. Inhibition of the mitogen-activated protein kinase p38 and the kinase Akt by lethal toxin leads to opening of the connexin channel, ATP release and P2X7-dependent signaling, which leads to NLRP1 activation38. Phagolysosomal membrane destabilization and cathepsin B release in lethal toxin-mediated NLRP1 activation have also been proposed39. These observations highlight some unifying themes in the mechanism of activation of both NLRP1 and NLRP3 inflammasomes. It will be important to understand the effect of NLRP1 cleavage in NLRP1-dependent inflammasome function and whether other NLRs undergo similar cleavage mechanisms.
NLRP6 and NLRP12 inflammasomes
In addition to the NLR inflammasomes described above, early transfection studies linked both NLRP6 (PYPAF5; ref. 40) and NLRP12 (PYPAF7 (refs. 41,42,43) or Monarch-1) to inflammasome function. NLRP6 and NLRP12 associate with ASC and induce caspase-1 dependent processing of IL-1β40,41. Exciting subsequent work has linked NLRP6 to maintenance of the microbiome44 (Fig. 1). NLRP6 is expressed mainly in nonhematopoietic compartments, where it acts to maintain intestinal homeostasis by regulating the composition of intestinal flora. Deficiency in NLRP6 in mouse colonic epithelial cells results in altered intestinal microbiota characterized by expanded representation of the bacterial phyla Bacteroidetes (specifically the family Prevotellaceae) and TM7. This aspect of NLRP6 function is dependent on its ability to nucleate an inflammasome and regulate IL-18 rather than IL-1β. Deficiency in NLRP6 or IL-18 skews the composition of the microbiome toward a colitogenic type. This imbalance induces spontaneous inflammation through production of the chemokine CCL5 and recruitment of inflammatory cells, which culminates in colitis. Together these studies indicate that perturbations in components of this inflammasome pathway, including NLRP6, ASC, caspase-1 and IL-18, may constitute a predisposing or initiating event in some cases of human inflammatory bowel disease.
The role of NLRP12 in innate immunity has remained unclear, with both inflammatory and inhibitory functions ascribed to this molecule. NLRP12 was the first NLR shown to associate with ASC in transfected cells and to form an active IL-1β-maturing inflammasome40. Additional studies have suggested that NLRP12 is an antagonist of proinflammatory signals induced by Toll-like receptors (TLRs), tumor-necrosis factor (TNF) and Mycobacterium tuberculosis43. NLRP12 has also been shown to control the migration of dendritic cells (DCs) and myeloid cells to affect contact hypersensitivity45,46,47,48. Mutations in the gene encoding NLRP12, as noted for NLRP3 (discussed below), have been linked to hereditary inflammatory diseases. Patients with NLRP12 mutations have been successfully treated with antibody to IL-1 (refs. 49,50,51). A critical role for NLRP12 in maintaining intestinal homeostasis and providing protection against colorectal tumorigenesis has also been proposed52. NLRP12 acts to negatively regulate signaling via the transcription factor NF-κB and is dispensable for caspase-1 activation by a variety of activators, including ATP, Salmonella typhimurium and Listeria species52. Whether NLRP12 forms inflammasomes in response to other activators of caspase-1 remains to be determined.
PYHIN inflammasomes
A second class of inflammasomes has also been described that do not contain NLRs but instead contain members of the PYHIN family. The PYHIN proteins are encoded by a family of four human genes (IFI16, AIM2, MNDA and IFIX) and thirteen mouse genes and are characterized by the presence of a PYD and one or two HIN-200 DNA-binding domains53. So far, AIM2 and IFI16 have been shown to form caspase-1-activating inflammasomes. Unlike the NLRs, AIM2 and IFI16 bind directly to their ligand, which in both cases is dsDNA. AIM2 and IFI16 lack CARDs and therefore require ASC for the recruitment of pro-caspase-1. AIM2, the first member of this family linked to inflammasome function, is located in the cytosol and senses dsDNA of viral or bacterial origin54,55,56 (Fig. 2). The recognition of dsDNA by AIM2 depends on the length of the DNA rather the sequence; dsDNA of less than 80 base pairs in length is a poor activator of the AIM2 inflammasome (V. Hornung, E. Latz and K.A.F., unpublished data). Although AIM2 can recognize self DNA and activate inflammasome complexes, the cytosolic location of AIM2 seems to limit recognition of self DNA under steady-state conditions. However, in situations in which self DNA is inefficiently cleared from the extracellular milieu or is improperly degraded in the phagolysosomal compartment, self DNA seems to access cytosolic compartments, where it turns on transcription of genes encoding type I interferons and associated genes57,58,59,60. It is likely that under these conditions, DNA would also activate AIM2 and drive inflammation. During infection, AIM2 senses DNA from murine cytomegalovirus and vaccinia virus, as well as from intracellular bacterial pathogens such as Francisella tularensis and L. monocytogenes61,62,63.

PYHIN inflammasomes (AIM2 and IFI16) bind directly to their ligand, DNA. The AIM2 inflammasome is activated by cytosolic dsDNA from DNA viruses and cytosolic bacteria; IFI16 is activated by KSHV DNA in the nucleus.
The AIM2-like receptor IFI16 has also been linked to inflammasome activation. In contrast to AIM2, IFI16 is located mainly in the nucleus of cells, although cytosolic IFI16 has also been detected in small amounts in some cells. In the cytosol, IFI16 engages viral dsDNA to drive the production of type I interferon6; however, IFI16 has also been reported to recognize the genome of Kapsosi's sarcoma–associated herpes virus (KSHV) in the nucleus of infected cells5. In response to KSHV, IFI16 and ASC translocate from the nucleus to the cytosolic perinuclear region, where they form an inflamamsome complex with caspase-1 (ref. 5; Fig. 2). The mechanism by which IFI16 discriminates between self and viral DNA in the nucleus remains unknown. One possibility is that in the context of chromatin, self DNA is inaccessible to IFI16, although how this inaccessibility is maintained during cell division is an important issue that remains to be addressed.
Regulation of inflammasome activation
Once unleashed, the innate immune response through surveillance mechanisms such as the inflammasome has the power to both eliminate infection and 'instruct' the T cells and B cells of the adaptive immune system to establish memory after repeated exposure to the eliciting pathogen. Because the ensuing IL-1β, IL-18 and pyroptotic death responses have the potential to damage the host, tight control of these pathways is critical for the prevention of sterile inflammation. Failure to curb the activity of inflammasomes is exemplified by the finding that deregulated inflammasome activation is linked to autoinflammatory and/or autoimmune conditions such as familial Mediterranean fever or cryopyrin-associated periodic syndromes64. Cryopyrin-associated periodic syndromes such as familial cold autoinflammatory syndrome, Muckle-Wells syndrome and chronic infantile neurological cutaneous and articular syndrome–neonatal onset multisystem inflammatory disease are all linked to gain-of-function mutations in NLRP3, and all of these diseases respond to therapy with the IL-1 receptor antagonist.
Unlike the production of most inflammatory cytokines, the production of biologically active IL-1β is dependent on transcription, translation, maturation and secretion mechanisms, all of which are tightly regulated. Multiple checkpoints in this process ensure that IL-18 and IL-1β are not inappropriately induced. Many of these steps can be manipulated by microbial pathogens to subvert host defense. In addition, a growing number of cellular checkpoints have been identified that act to prevent excessive activation of IL-1β- or IL-18-driven inflammation. Cell-intrinsic and cell-extrinsic mechanisms exert rigorous control on these pathways at multiple levels.
Microbial regulation of inflammasomes
Given the importance of IL-1β, IL-18 and pyroptosis in the elimination of infectious agents, it is not surprising that pathogens have developed counter strategies to block these defenses (Fig. 3). Viruses are especially successful in evading inflammasome activation through a set of proteins homologous to host-derived inflammasome inhibitors such as cPOP and serpins (discussed below). Good examples of the viral subversion of inflammasome signaling are the poxvirus proteins M013 (from myxoma virus) and gp013L (from Shope fibroma virus) that bind ASC and inhibit inflammasome activation65,66. These proteins also inhibit transcription of the gene encoding IL-1β and thus interfere with both the transcription of the gene encoding IL-1β and the processing of IL-1β (ref. 67). Additionally, poxviruses produce serpin homologs. Serpins are serine-protease inhibitors that block the catalytic activity of several proteases. Although their main targets are serine proteases, they also inhibit cysteine proteases such as caspase-1. The cowpox virus protein CrmA (cytokine-response modifier A) and its homologs in orthopoxviruses, such as B13R (vaccinia virus), Serp2 (myxoma virus) and SPI-2 (ectromelia virus), abolish the proteolytic activity of caspase-1 (refs. 68,69,70,71). Influenza virus and baculoviruses also use NS1 and p35, respectively, to inhibit inflammasome activation72,73. KSHV expresses a homolog of NLRP1, Orf63, which interacts with NLRP1 and NLRP3, resulting in repression of inflammasome activation that enables the virus to avoid immune control and establish latency74.

Many bacterial and viral proteins inhibit inflammasome activation. The poxvirus proteins M013 (myxoma virus) and gp013L (Shope fibroma virus) inhibit transcription of the gene encoding IL-1β and bind ASC to limit inflammasome activation. The poxvirus proteins CrmA, B13R, Serp2 and SPI-2 abolish the proteolytic activity of caspase-1. Other microbial proteins that inhibit inflammasome activation include NS1 (influenza virus), p35 (baculovirus), ExoU, ExoS (P. aeruginosa), YopE and YopT (Y. enterocolitica), Zmp1 (M. tuberculosis), and MviN and RipA (F. tularensis). In contrast, endogenous POPs such as POP1 and POP2 bind to the PYD of ASC and NLRs, respectively, and sequester them from the inflammasome complex. Similarly, endogenous COPs, such as COP1 (pseudo-ICE), INCA, ICEBERG, caspase-12, and Nod2-S, bind to CARD of caspase-1 and sequester it from inflammasome activation.
Bacterial pathogens do not seem to express anti-inflammasome molecules. However, they use strategies that manipulate inflammasome activation, in most cases interfering with the production of or recognition of bacterial ligands that trigger inflammasomes. A good example of this is YopK of Yersinia pseudotuberculosis, which interacts with the T3SS translocon, thereby blocking the inflammasome from sensing the pathogen75. Similarly, during systemic infection, Salmonellae species downregulate expression of flagellin and the SPI-1 T3SS while simultaneously upregulating the SPI-2 T3SS, which is not detected by NLRC4 (ref. 34). Several bacterial proteins with enzymatic activity also modulate inflammasome activation. For example, Pseudomonas aeruginosa expresses ExoU, a phospholipase that suppresses caspase-1, and ExoS, a Rho GTPase–activating protein that negatively regulates IL-1β processing76,77. Likewise, Yersinia enterocolitica expresses YopE, another Rho GTPase–activating protein, and YopT, a cysteine protease, which regulate oligomerization of caspase-1 and block inflammasome activation78. M. tuberculosis expresses the zinc metalloprotease Zmp1, which inhibits caspase-1 activation by NLRP3 inducers79. Similarly, F. tularensis expresses mviN, a putative lipid II phospholipid translocase, and ripA, a cytosolic membrane protein, which have been shown to suppress inflammasome activation80,81. L. pneumophila interferes with transcription of the gene encoding ASC and prevents ASC expression to block the formation of complexes82. Pneumolysin, a pore-forming cytotoxin of S. pneumoniae, is reported to have both inflammasome-activating and inflammasome-inhibitory functions. Pneumolysin inhibits inflammasome activation in human DCs83, although the role of pneumolysin in activation of the NLRP3 inflamamsome by S. pneumoniae has also been demonstrated by several groups84,85,86.
Regulation of inflammasomes by type I interferon
When the host responds to infection, waves of inflammatory gene expression are turned on in cells of the immune system. Early responses include the production of inflammatory cytokines as well as type I interferons. Many cell stimuli, including TLR activators and IL-1β itself, activate transcription of the gene encoding IL-1β. TLRs, as well as cytokine signaling pathways, also control the expression of NLRP3 at the transcriptional level. Reports have indicated important crosstalk between cytokine signaling and the inflammasome. This is particularly true for type I and II interferons. A case in point relates to the AIM2 inflammasome, for which, in some cases, type I interferon signaling is important for inflammasome activation. Activation of the AIM2 inflammasome by infection with F. tularensis requires an intact type I interferon response61,87,88. Consistent with that, in macrophages deficient in the transcription factor IRF3, which have a defect in the secretion of type I interferons, or macrophages deficient in the receptor for interferon-α (IFN-α) and IFN-β, which are unable to respond to type I interferon, infection with F. tularensis results in less-efficient activation of the AIM2 inflammasome than that in wild-type macrophages61,62,88. The precise role of interferon signaling in this setting is still a little unclear. Although AIM2 is encoded by an interferon-inducible gene, the amount of AIM2 is unaffected in IRF3-deficientcells or in cells deficient in the receptor for IFN-α and IFN-β61. Interestingly, activation of the AIM2 inflammasome by infection with mouse cytomegalovirus does not require an intact type I interferon response63. It has been proposed that type I interferons act to enhance the killing and lysis of F. tularensis in the phagosome to generate the cytosolic DNA that activates the AIM2 inflammasome61.
Although they are clearly important in some situations for inflammasome activation, there is additional evidence that the type I interferons can also antagonize these pathways. Type I interferons restrain IL-1β production by two distinct mechanisms, depending on the cell type; they act by diminishing the pool of intracellular pro-IL-1β and/or inhibiting caspase-1 activation89 (Fig. 4). The decrease in the pool of intracellular pro-IL1β is dependent on the ability of type I interferons to induce production of the anti-inflammatory cytokine IL-10. The autocrine and/or paracrine action of IL-10 acting via the STAT3 signaling pathway inhibits the synthesis of pro-IL-1β and pro-IL-1α. Additionally, type I interferons suppress caspase-1 processing by a mechanism that is dependent on the transcription factor STAT1. How STAT1 activation leads to caspase-1 blockade is unclear. The ability of type I interferons to block caspase-1 activation seems to be specific to NLRP3 and NLRP1 inflammasomes. An important consequence of this crosstalk between IL-1β and type I interferons is reflected in the diminished NLRP3 inflammasome activation observed in monocytes from patients with multiple sclerosis that are being treated with IFN-β. Consistent with an inhibitory role for type I interferons on the inflammasome, treatment of mice with inducers of IFN-β such as poly(I:C) suppresses NLRP3 activation by alum and C. albicans. The exact molecular mechanism that governs the 'preferential' inhibition of NLRP3 and NLRP1 inflammasomes by type I interferon–STAT1 signaling is yet to be identified.

Type I interferon signaling attenuates NLRP3- and NLRP1-dependent IL-1β responses by two mechanisms (NLRP1 not shown here). IFN-α and IFN-β trigger the production of IL-10, which then acts on the cells in an autocrine and/or paracrine manner to suppress the intracellular concentration of pro-IL-1β. Type I interferon signaling can also inhibit NLRP3- and NLRP1-mediated caspase-1 processing via an unknown mechanism. Autophagy regulates IL-1β production via the removal of damaged mitochondria, which are potent sources of ROS that drive NLRP3 activation. Additionally, autophagosomes sequester intracellular stores of pro-IL-1β and target it for degradation. Effector and memory T cells block NLRP3 and NLRP1 inflammasomes via a cognate mechanism mediated by interactions between ligands of the TNF superfamily and their receptors. Mitophagy, mitochondrial autophagy.
Published evidence has indicated the importance of type I interferons in suppressing IL-1 in vivo during infection with M. tuberculosis90. Both IL-1α and IL-1β are critically required for host resistance to M. tuberculosis. Inflammatory monocyte-macrophage and DC populations coexpress both IL-1α and IL-1β in lungs of M. tuberculosis–infected mice. Type I interferons regulate IL-1 production by these cells in vivo. Type I interferons inhibit the production of IL-1 by both subsets of cells. These data provide a cellular basis for both the anti-inflammatory effects of interferons and the probacterial functions of type I interferons during M. tuberculosis infection and demonstrate differences in the regulation of IL-1 production by distinct cell populations as an additional layer of complexity in the activity of IL-1 in vivo.
IFN-γ and T cell regulation of inflammasome activation
There is also emerging evidence that the CD4+ T cell–derived type II interferon IFN-γ also exerts an inhibitory effect on the production of IL-1 (ref. 90). The effect of IFN-γ seems to be somewhat transient and cell type specific; IFN-γ priming of macrophages and DCs from mice, but not of those from humans, has been shown to suppress pro-IL-1β synthesis triggered by lipopolysaccharide (LPS)91. Later, this inhibitory effect can be overcome by SOCS1, a negative regulator of IFN-γ signaling that is induced in LPS-treated cells. During infection with M. tuberculosis, IFN-γ selectively suppresses monocyte-macrophage–dependent production of both IL-1α and IL-1β (ref. 90). Precisely how IFN-γ modulates production of the IL-1 family of cytokines and its effect on inflammasome-dependent processing of caspase-1 remain to be determined. As IFN-γ is the signature cytokine of T helper type 1 and CD8+ T cell responses, it may act as a feedback regulator of inflammasome responses once the adaptive immune response has been elicited.
In addition to IFN-γ, additional mechanisms by which T cells regulate inflammasome activation have been proposed92. Effector and memory T cells can attenuate the processing of caspase-1 and IL-1β in macrophages and DCs (Fig. 4). This inhibitory effect requires cell-to-cell contact and seems to be mediated by CD40L, OX40L and RANKL, all of which are members of the TNF superfamily of ligands expressed on activated T cells. Again, this negative feedback loop exerted by T cells targets only NLRP1 and NLRP3 inflammasomes. It is not yet clear how interactions between members of the TNF family of ligands and their receptors mediate the inhibition of caspase-1-dependent production of IL-1β. Nonetheless, activated CD4+ T cells and CD8+ T cells use cognate as well as noncognate mechanisms (that probably involve IFN-γ) to limit the magnitude of the production of IL-1β and IL-18 by antigen-presenting cells.
Regulation of inflammasome activation by autophagy
Autophagy, an evolutionarily conserved catabolic process, facilitates the recycling of damaged cellular proteins and organelles93. During autophagy, cytosolic constituents are engulfed by autophagosomes and then are delivered to the lysosomes for degradation. Growing evidence indicates that autophagic dysfunction is associated with cancer, neurodegenerative disorders and impaired host defense. Furthermore, autophagy regulates various aspects of immune responses, including antigen presentation, cell death and cytokine secretion in immune cells93. Early evidence for the involvement of autophagy in proinflammatory cytokine secretion was provided by studies of autophagocytosis-deficient Atg16−/− mice, which have exaggerated production of IL-1β and IL-18 in response to LPS and other pathogen-derived signals94. This augmented production of IL-1β and IL-18 is not due to enhanced transcription of the gene encoding pro-IL-1β but instead is due to enhanced activation of caspase-1. Cells from mice with targeted deletions in key components of the autophagy machinery or cells treated with pharmacological inhibitors of autophagy have augmented production of IL-1β and IL-18 and contribute to enhanced disease severity in dextran sodium sulfate–induced colitis and LPS-induced septic shock in vivo94. The molecular basis for the augmented caspase-1 response is the failure of autophagy-deficient cells to clear damaged mitochondria (Fig. 4). In the absence of cellular autophagy (or mitochondrial autophagy), damaged mitochondria accumulate and generate excessive ROS and release mitochondrial DNA into the cytoplasm of cells, events that trigger activation of the NLRP3 inflammasome95,96. Precisely how mitochondrial ROS and/or mitochondrial DNA enhance the inflammasome-mediated release of IL-1β and IL-18 is still unclear. Additional evidence indicates that the autophagy machinery also acts to regulate IL-1β responses in an inflammasome-independent manner; autophagosomes sequester pro-IL-1β and target it for degradation, thereby limiting the substrate available for caspase-1 to process97. In contrast to its effect in mouse cells, in human peripheral blood mononuclear cells (PBMCs), the inhibition of basal autophagy augments the transcription of pro-IL 1β mRNA in response to TLR ligands98. Contrary to the roles described above, there is also evidence that autophagy can positively regulate IL-1β responses. Mammalian cells export most proteins by a pathway dependent the endoplasmic reticulum and Golgi apparatus. However, some proteins that lack signal peptides are secreted via unconventional, poorly understood mechanisms. The latter include IL-1β and IL-18. Starvation-induced autophagy contributes to inflammasome-dependent release of IL-1β via an export pathway that depends on the autophagy regulator Atg5 and at least one of the two mammalian paralogs of Golgi reassembly stacking proteins, GRASP55 (GORASP2) and Rab8a99. Thus, the autophagic process regulates IL-1β production at various levels.
Another situation in which the autophagic machinery and inflammasome complexes may also intersect is the removal of large inflammasome complexes from cells. Autophagy facilitates the turnover of damaged proteins and protein aggregates. Although inflammasome-complex formation is associated in many cases with pyroptotic cell death, cell death does not always accompany these events. When the cell dies, inflammasome complexes, like other endogenous contents, will be released into the extracellular environment, where they are probably degraded by extracellular proteases. However, in situations in which death does not occur, these large inflammasome complexes must be removed to prevent excessive activation of the IL-1 pathway. It is conceivable that the autophagic machinery has a role in directing these events.
The TRIM family of proteins
Emerging evidence also indicates that transmembrane adaptors of the TRIM family modulate inflammasome-mediated IL-1 responses. The initial evidence linking TRIM proteins and inflammasome pathways was provided by studies of familial Mediterranean fever in which missense mutations in the sequence encoding the carboxy-terminal B30.2 domain of TRIM20 (pyrin or MEFV) is linked to disease100,101. Although the exact role of TRIM20 in inflammasome activation has remained controversial, a published study has demonstrated that a gain-of-function mutant of TRIM20 constitutively forms inflammasome complexes containing ASC and caspase-1, leading to the generation of active caspase-1 in mice102. Additional members of the TRIM family have also been linked to regulation of the inflammasome. TRIM16 (EBBP), a close homolog of TRIM20, lacks a PYD and has been shown to enhance IL-1β production by modulating the unconventional secretion of leaderless peptide103. Although biochemical data have suggested that TRIM16 could interact with pro-caspase-1 and NLRP1, the importance of these interactions is not known. TRIM30, a RING domain–containing TRIM protein that has been identified as a negative regulator of TLR signaling, specifically suppresses activation of the NLRP3 inflammasome in response to a variety of ligands, including ATP, the antibiotic nigericin, silica and monosodium urate in vitro as well as in vivo, by inhibiting ROS production104.
Pyrin-only and CARD-only proteins
Pyrin-only proteins (POPs) and CARD-only proteins (COPs) are the main regulators of the inflammasome in humans. Both POPs and COPs execute their inflammasome modulatory function through their PYDs and CARDs. The POPs include POP1 and POP2, with POP1 having a higher degree of homology with the PYD of ASC. Consequently, POP1 interacts with ASC and sequesters it from other PYD-containing proteins, such as the NLRs, which ultimately results in the inhibition of inflammasome activation105. However, POP2 (although less similar to ASC) resembles the PYD of NLRP2 and NLRP7. Therefore, POP2 is believed to bind NLRs and exclude them from inflammasome activation106.
In contrast to the POPs, the COPs have considerable similarity to the CARD of caspase-1 and seem to function as decoy inhibitors of caspase-1 by binding to its CARD, thereby sequestering it from association with inflammasome adaptors and receptors. The COP family includes COP1 (pseudo-ICE), INCA, ICEBERG, caspase-12s and Nod2-S, each with varying degrees of similarity to the CARD of caspase-1. The best studied COP, caspase-12, has been explored in detail in mice107. In mice, caspase-12 deficiency confers resistance to sepsis, and the presence of caspase-12 exerts a dominant-negative suppressive effect on caspase-1 that results in enhanced vulnerability to bacterial infection and septic mortality. With the exception of Nod2-S, the genes encoding COPs are located in the locus encoding caspase-1 in humans and seem to have resulted from a series of gene duplications that occurred after the divergence of humans and mice. It is interesting to note that with the exception of caspase-12, mice lack POPs and COPs108. The presence of COPs and POPs in humans reflects the higher degree of complexity with which inflammasomes are regulated to avoid inflammatory damage.
Additional mechanisms
As discussed above, viruses encode serpin-like molecules such as CrmA that block caspase-1 activity. Serpins have been linked to the attenuation of inflammasome pathways. The serine-protease inhibitor PI-9 is constitutively expressed in human vascular smooth muscle cells, where it acts to inhibit caspase-1-dependent processing of IL-1β and IL-18. Notably, human atherosclerotic lesions have relatively less PI-9 but more IL-1β than normal arterial walls have109. Serpin B or the plasminogen-activation inhibitor PAI-2 also inhibits the caspase-1-dependent as well as caspase-1-independent processing of IL-1β in macrophages and neutrophils, respectively. Antiapoptotic factors such as Bcl-2 and Bcl-XL bind NLRP1 and inhibit NLRP1-dependent activation of caspase-1 and processing of IL-1β (ref. 110). This mechanism parallels inhibition of the caspase CED3 by the Bcl-2 ortholog CED-9 in C. elegans, which emphasizes the conserved nature of caspase regulation by members of the Bcl-2 family. Little is known about the role of alternatively spliced isoforms of NLR or PYHIN family members. It is likely that for the NLR and PYHIN proteins themselves, isoforms generated by alternative splicing may regulate these pathways. Alternative isoforms of ASC and NLRP1 have been reported to act as dominant-negative inhibitors of their respective inflammasome pathways111.
In addition to the NLRs, the PYHIN proteins AIM2 and IFI16 can also form inflammasome. There is evidence that these molecules can counter-regulate each other (Fig. 5). Type I and type II interferons induce the expression of IFI16 and AIM2. Although IFI16 itself can form an inflammasome in response to KSHV, there is also evidence that IFI16 can suppress the activation of caspase-1 by the AIM2 and NLRP3 inflammasomes112. IFI16 thus represents a novel mediator of the anti-inflammatory actions of the type I interferons. Of the 13 putative mouse PYHIN proteins, IFI202 (p202), which is linked to susceptibility to lupus, has two DNA-binding HIN-200 domains but lacks a PYD. IFI202 (p202) binds to dsDNA in the cytosol and sequesters it and thus prevents DNA-induced caspase-1 activation56. Alternatively spliced isoforms of AIM2 that lack a PYD have also been predicted, raising the possibility that these isoforms could mimic p202 to attenuate activation of AIM2 or IFI16 (ref. 113). Activation of AIM2 has been linked to psoriasis through its sensing of host DNA. Cathelicidin (LL-37) is an antimicrobial peptide that forms a complex with self DNA released from dying cells in psoriatic skin lesions and accelerates the intake of self DNA and activation of TLR9 in plasmacytoid DCs, which contributes to disease pathology. Interestingly, LL-37 inhibits the ability of DNA to induce IL-1β production. The tight association of LL-37 with DNA sequesters the DNA, which prevents it from engaging the AIM2 inflammasome114 (Fig. 5). This is probably due to the cationic charge of LL-37 or a conformational change in DNA structure or a combination of both.

Binding of dsDNA by IFI202 (p202) and in some cases by IFI16 can block AIM2 from gaining access to dsDNA. Additionally, LL-37, a human cathelicidin antimicrobial peptide, inhibits activation of the AIM2 inflammasome in psoriasis by forming a complex with self DNA released from the dying cells in the inflammatory milieu.
Conclusions
Given the potent proinflammatory nature of IL-1, IL-18 and pyroptotic cell-death pathways, it is not surprising that multiple positive and negative regulatory mechanisms control inflammasome function. Negative regulatory mechanisms range from those elicited by pathogens to evade recognition by inflammasomes to those of microbial molecules that subvert the formation, signaling or effector function of inflammasome complexes. In the case of host control mechanisms, modulation by T cells, cytokines of either innate or adaptive origin, endogenous intracellular host molecules and cell-intrinsic mechanisms such as autophagy all act as a 'rheostat' to reset these pathways back to baseline after activation. A hallmark of NLR or PYHIN signaling is ligand selectivity and, therefore, functional specificity. Whereas some of the negative regulators selectively inhibit one or more inflammasome complexes, others target common components of all of these pathways. Many of the regulators block ASC or caspase-1 itself, which might shut down not only the entire NLR family but also the PYHIN inflammasomes. NLRP3 seems to be the most heavily regulated of all the NLRs. One possibility for this might relate to the fact that NLRP3 is activated by such a diversity of microbial, host and environmental triggers it is therefore important to ensure this pathway is not misregulated. The existence of multiple and apparently nonredundant negative regulators of the inflammasome raises an important question about the relative importance of these molecules. Such control at multiple checkpoints could suggest that individually these molecules are not sufficient to control a particular inflammasome pathway, but a concerted effort by multiple regulators is required. Elucidating the crosstalk between the NLR and PYHIN inflammasomes and their negative regulators is an exciting and important challenge. Better understanding of the regulatory control of inflammasomes is critical for the understanding of host defenses and of the growing number of diseases in which inflammasomes are involved. Inflammasome-driven disease could be a consequence of either overstimulation of the inflammasome pathways or defective negative regulation. Given the emerging role of inflammasomes in infectious, autoinflammatory and autoimmune diseases, understanding negative regulation is crucial for the identification of new targets for the treatment of infectious and inflammatory diseases.
References
Kayagaki, N. et al. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011).
Martinon, F., Burns, K. & Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 10, 417–426 (2002).
Kanneganti, T. Central roles of NLRs and inflammasomes in viral infection. Nat. Rev. Immunol. 10, 688–698 (2010).
Hornung, V. & Latz, E. Intracellular DNA recognition. Nat. Rev. Immunol. 10, 123–130 (2010).
Kerur, N. et al. IFI16 Acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi sarcoma-associated herpesvirus infection. Cell Host Microbe 9, 363–375 (2011).
Unterholzner, L. et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11, 997–1004 (2010).
de Alba, E. Structure and interdomain dynamics of apoptosis-associated speck-like protein containing a CARD (ASC). J. Biol. Chem. 284, 32932–32941 (2009).
Bryan, N.B., Dorfleutner, A., Rojanasakul, Y. & Stehlik, C. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J. Immunol. 182, 3173–3182 (2009).
Ippagunta, S.K. et al. The inflammasome adaptor ASC regulates the function of adaptive immune cells by controlling Dock2-mediated Rac activation and actin polymerization. Nat. Immunol. 12, 1010–1016 (2011).
Lamkanfi, M. Emerging inflammasome effector mechanisms. Nat. Rev. Immunol. 11, 213–220 (2011).
Broz, P., von Moltke, J., Jones, J.W., Vance, R.E. & Monack, D.M. Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8, 471–483 (2010).
Keller, M., Ruegg, A., Werner, S. & Beer, H.D. Active caspase-1 is a regulator of unconventional protein secretion. Cell 132, 818–831 (2008).
Munoz-Planillo, R., Franchi, L., Miller, L.S. & Nunez, G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the NLRP3 inflammasome. J. Immunol. 183, 3942–3948 (2009).
Meixenberger, K. et al. Listeria monocytogenes-infected human peripheral blood mononuclear cells produce IL-1β, depending on listeriolysin O and NLRP3. J. Immunol. 184, 922–930 (2010).
Kim, S. et al. Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur. J. Immunol. 40, 1545–1551 (2010).
Willingham, S.B. et al. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J. Immunol. 183, 2008–2015 (2009).
Duncan, J.A. et al. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J. Immunol. 182, 6460–6469 (2009).
Hise, A.G. et al. An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 5, 487–497 (2009).
Allen, I.C. et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30, 556–565 (2009).
Mariathasan, S. et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232 (2006).
Yamasaki, K. et al. NLRP3/cryopyrin is necessary for interleukin-1β (IL-1β) release in response to hyaluronan, an endogenous trigger of inflammation in response to injury. J. Biol. Chem. 284, 12762–12771 (2009).
Halle, A. et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 9, 857–865 (2008).
Gasse, P. et al. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am. J. Respir. Crit. Care Med. 179, 903–913 (2009).
Jin, C. & Flavell, R.A. Molecular mechanism of NLRP3 inflammasome activation. J. Clin. Immunol. 30, 628–631 (2010).
Hornung, V. et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856 (2008).
Ichinohe, T., Pang, I.K. & Iwasaki, A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 11, 404–410 (2010).
Miao, E.A. et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. USA 107, 3076–3080 (2010).
Mariathasan, S. et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218 (2004).
Miao, E.A. et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1β via Ipaf. Nat. Immunol. 7, 569–575 (2006).
Zhao, Y. et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477, 596–600 (2011).
Kofoed, E.M. & Vance, R.E. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477, 592–595 (2011).
Newman, Z.L. et al. Susceptibility to anthrax lethal toxin-induced rat death is controlled by a single chromosome 10 locus that includes rNLRP1. PLoS Pathog. 6, e1000906 (2010).
Faustin, B. et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol. Cell 25, 713–724 (2007).
Broz, P. & Monack, D.M. Molecular mechanisms of inflammasome activation during microbial infections. Immunol. Rev. 243, 174–190 (2011).
Tschopp, J., Martinon, F. & Burns, K. NALPs: a novel protein family involved in inflammation. Nat. Rev. Mol. Cell Biol. 4, 95–104 (2003).
D'Osualdo, A. et al. CARD8 and NLRP1 Undergo Autoproteolytic Processing through a ZU5-Like Domain. PLoS ONE 6, e27396 (2011).
Hsu, L.C. et al. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1β secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc. Natl. Acad. Sci. USA 105, 7803–7808 (2008).
Ali, S.R. et al. Anthrax toxin induces macrophage death by p38 MAPK inhibition but leads to inflammasome activation via ATP leakage. Immunity 35, 34–44 (2011).
Newman, Z.L., Leppla, S.H. & Moayeri, M. CA-074Me protection against anthrax lethal toxin. Infect. Immun. 77, 4327–4336 (2009).
Wang, L. et al. PYPAF7, a novel PYRIN-containing Apaf1-like protein that regulates activation of NF-κB and caspase-1-dependent cytokine processing. J. Biol. Chem. 277, 29874–29880 (2002).
Grenier, J.M. et al. Functional screening of five PYPAF family members identifies PYPAF5 as a novel regulator of NF-κB and caspase-1. FEBS Lett. 530, 73–78 (2002).
Lich, J.D. et al. Monarch-1 suppresses non-canonical NF-κB activation and p52-dependent chemokine expression in monocytes. J. Immunol. 178, 1256–1260 (2007).
Williams, K.L. et al. The CATERPILLER protein monarch-1 is an antagonist of toll-like receptor-, tumor necrosis factor α-, and Mycobacterium tuberculosis-induced pro-inflammatory signals. J. Biol. Chem. 280, 39914–39924 (2005).
Elinav, E. et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757 (2011).
Arthur, J.C. et al. Cutting edge: NLRP12 controls dendritic and myeloid cell migration to affect contact hypersensitivity. J. Immunol. 185, 4515–4519 (2010).
Lich, J.D. & Ting, J.P. Monarch-1/PYPAF7 and other CATERPILLER (CLR, NOD, NLR) proteins with negative regulatory functions. Microbes Infect. 9, 672–676 (2007).
Lich, J.D. et al. Monarch-1 suppresses non-canonical NF-κB activation and p52-dependent chemokine expression in monocytes. J. Immunol. 178, 1256–1260 (2007).
Jéru, I. et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc. Natl. Acad. Sci. USA 105, 1614–1619 (2008).
Jéru, I. et al. Role of interleukin-1β in NLRP12-associated autoinflammatory disorders and resistance to anti-interleukin-1 therapy. Arthritis Rheum. 63, 2142–2148 (2011).
Lachmann, H.J. et al. In vivo regulation of interleukin 1β in patients with cryopyrin-associated periodic syndromes. J Exp. Med. 206, 1029–1036 (2009).
Hawkins, P.N., Lachmann, H.J. & McDermott, M.F. Interleukin-1-receptor antagonist in the Muckle-Wells syndrome. N. Engl. J. Med. 348, 2583–2584 (2003).
Zaki, M.H. et al. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 20, 649–660 (2011).
Schattgen, S.A. & Fitzgerald, K.A. The PYHIN protein family as mediators of host defenses. Immunol. Rev. 243, 109–118 (2011).
Burckstummer, T. et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 10, 266–272 (2009).
Hornung, V. et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518 (2009).
Roberts, T.L. et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323, 1057–1060 (2009).
Stetson, D.B., Ko, J.S., Heidmann, T. & Medzhitov, R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598 (2008).
Yoshida, H., Okabe, Y., Kawane, K., Fukuyama, H. & Nagata, S. Lethal anemia caused by interferon-β produced in mouse embryos carrying undigested DNA. Nat. Immunol. 6, 49–56 (2004).
Okabe, Y., Kawane, K., Akira, S., Taniguchi, T. & Nagata, S. Toll-like receptor–independent gene induction program activated by mammalian DNA escaped from apoptotic DNA degradation. J. Exp. Med. 202, 1333–1339 (2005).
Kawane, K. et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 443, 998–1002 (2006).
Fernandes-Alnemri, T. et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 11, 385–393 (2010).
Jones, J.W. et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. USA 107, 9771–9776 (2010).
Rathinam, V.A. et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11, 395–402 (2010).
Kastner, D.L. & Hoffman, H.M. CATERPILLERs, pyrin and hereditary immunological disorders. Nat. Rev. Immunol. 6, 183–195 (2006).
Johnston, J.B. et al. A poxvirus-encoded pyrin domain protein interacts with ASC-1 to inhibit host inflammatory and apoptotic responses to infection. Immunity 23, 587–598 (2005).
Dorfleutner, A. et al. Cellular pyrin domain-only protein 2 is a candidate regulator of inflammasome activation. Infect. Immun. 75, 1484–1492 (2007).
Rahman, M.M., Mohamed, M.R., Kim, M., Smallwood, S. & McFadden, G. Co-regulation of NF-κB and inflammasome-mediated inflammatory responses by myxoma virus pyrin domain-containing protein M013. PLoS Pathog. 5, e1000635 (2009).
Young, J.L. et al. The serpin proteinase inhibitor 9 is an endogenous inhibitor of interleukin 1β-converting enzyme (caspase-1) activity in human vascular smooth muscle cells. J. Exp. Med. 191, 1535–1544 (2000).
Ray, C.A. et al. Viral inhibition of inflammation: cowpox virus encodes an inhibitor of the interleukin-1β converting enzyme. Cell 69, 597–604 (1992).
Kettle, S. et al. Vaccinia virus serpin B13R (SPI-2) inhibits interleukin-1β-converting enzyme and protects virus-infected cells from TNF- and Fas-mediated apoptosis, but does not prevent IL-1β-induced fever. J. Gen. Virol. 78, 677–685 (1997).
Petit, F. et al. Characterization of a myxoma virus-encoded serpin-like protein with activity against interleukin-1β-converting enzyme. J. Virol. 70, 5860–5866 (1996).
Stasakova, J. et al. Influenza A mutant viruses with altered NS1 protein function provoke caspase-1 activation in primary human macrophages, resulting in fast apoptosis and release of high levels of interleukins 1β and 18. J. Gen. Virol. 86, 185–195 (2005).
Bump, N.J. et al. Inhibition of ICE family proteases by baculovirus antiapoptotic protein p35. Science 269, 1885–1888 (1995).
Gregory, S.M. et al. Discovery of a viral NLR homolog that inhibits the inflammasome. Science 331, 330–334 (2011).
Brodsky, I.E. et al. A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe 7, 376–387 (2010).
Sutterwala, F.S. et al. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med. 204, 3235–3245 (2007).
Galle, M. et al. The Pseudomonas aeruginosa Type III secretion system plays a dual role in the regulation of caspase-1 mediated IL-1β maturation. J. Cell Mol. Med. 12, 1767–1776 (2008).
Schotte, P. et al. Targeting Rac1 by the Yersinia effector protein YopE inhibits caspase-1-mediated maturation and release of interleukin-1β. J. Biol. Chem. 279, 25134–25142 (2004).
Master, S.S. et al. Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe 3, 224–232 (2008).
Huang, M.T. et al. Deletion of ripA alleviates suppression of the inflammasome and MAPK by Francisella tularensis. J. Immunol. 185, 5476–5485 (2010).
Ulland, T.K. et al. Cutting edge: mutation of Francisella tularensis mviN leads to increased macrophage absent in melanoma 2 inflammasome activation and a loss of virulence. J. Immunol. 185, 2670–2674 (2010).
Abdelaziz, D.H. et al. Apoptosis-associated speck-like protein (ASC) controls Legionella pneumophila infection in human monocytes. J. Biol. Chem. 286, 3203–3208 (2011).
Littmann, M. et al. Streptococcus pneumoniae evades human dendritic cell surveillance by pneumolysin expression. EMBO Mol Med 1, 211–222 (2009).
McNeela, E.A. et al. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog. 6, e1001191 (2010).
Witzenrath, M. et al. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J. Immunol. 187, 434–440 (2011).
Fang, R. et al. Critical Roles of ASC inflammasomes in caspase-1 activation and host innate resistance to Streptococcus pneumoniae infection. J. Immunol. 187, 4890–4899 (2011).
Jiang, F. et al. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature 479, 423–427 (2011).
Henry, T., Brotcke, A., Weiss, D.S., Thompson, L.J. & Monack, D.M. Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J. Exp. Med. 204, 987 (2007).
Guarda, G. et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34, 213–223 (2011).
Mayer-Barber, K.D. et al. Innate and adaptive interferons suppress IL-1α and IL-1β production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity 35, 1023–1034 (2011).
Masters, S.L. et al. Regulation of interleukin-1β by interferon-γ is species specific, limited by suppressor of cytokine signalling 1 and influences interleukin-17 production. EMBO Rep. 11, 640–646 (2010).
Guarda, G. et al. T cells dampen innate immune responses through inhibition of NLRP1 and NLRP3 inflammasomes. Nature 460, 269–273 (2009).
Levine, B., Mizushima, N. & Virgin, H.W. Autophagy in immunity and inflammation. Nature 469, 323–335 (2011).
Saitoh, T. et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 456, 264–268 (2008).
Zhou, R., Yazdi, A.S., Menu, P. & Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2010).
Nakahira, K. et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230 (2010).
Harris, J. et al. Autophagy controls IL-1β secretion by targeting pro-IL-1β for degradation. J. Biol. Chem. 286, 9587 (2011).
Crisan, T.O. et al. Inflammasome-independent modulation of cytokine response by autophagy in human cells. PLoS ONE 6, e18666 (2011).
Dupont, N. et al. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 30, 4701–4711 (2011).
The International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 90, 797–807 (1997).
A candidate gene for familial Mediterranean fever. Nat. Genet. 17, 25–31 (1997).
Chae, J.J. et al. Gain-of-function Pyrin mutations induce NLRP3 protein-independent interleukin-1β activation and severe autoinflammation in mice. Immunity 34, 755–768 (2011).
Munding, C. et al. The estrogen-responsive B box protein: a novel enhancer of interleukin-1β secretion. Cell Death Differ. 13, 1938–1949 (2006).
Hu, Y. et al. Tripartite-motif protein 30 negatively regulates NLRP3 inflammasome activation by modulating reactive oxygen species production. J. Immunol. 185, 7699 (2010).
Stehlik, C. et al. The PAAD/PYRIN-only protein POP1/ASC2 is a modulator of ASC-mediated nuclear-factor-κB and pro-caspase-1 regulation. Biochem. J. 373, 101 (2003).
Bedoya, F., Sandler, L.L. & Harton, J.A. Pyrin-only protein 2 modulates NF-κB and disrupts ASC:CLR interactions. J. Immunol. 178, 3837 (2007).
Saleh, M. et al. Enhanced bacterial clearance and sepsis resistance in caspase-12-deficient mice. Nature 440, 1064–1068 (2006).
da Cunha, J.P.C., Galante, P.A.F. & de Souza, S.J. Different evolutionary strategies for the origin of caspase-1 inhibitors. J. Mol. Evol. 66, 591–597 (2008).
Young, J.L. et al. The Serpin proteinase inhibitor 9 is an endogenous inhibitor of interleukin 1β–converting enzyme (caspase-1) activity in human vascular smooth muscle cells. J. Exp. Med. 191, 1535 (2000).
Bruey, J.M. et al. Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell 129, 45–56 (2007).
Andrea, D., Sara, K., Chawon, Y., Yon, R. & Christian, S. Differential splicing of the apoptosis-associated speck like protein containing a caspase recruitment domain (ASC) regulates inflammasomes. J. Inflamm. 7, 7–23 (2010).
Veeranki, S., Duan, X., Panchanathan, R., Liu, H. & Choubey, D. IFI16 protein mediates the anti-inflammatory actions of the type-I interferons through suppression of activation of caspase-1 by inflammasomes. PLoS ONE 6, e27040 (2011).
Choubey, D. et al. Interferon-inducible p200-family proteins as novel sensors of cytoplasmic DNA: role in inflammation and autoimmunity. J. Interferon Cytokine Res. 30, 371–380 (2010).
Dombrowski, Y. et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci. Transl. Med. 3, 82ra38–82ra38 (2011).
Acknowledgements
Supported by the US National Institutes of Health (AI083713, AI079293 and AI067497 to K.A.F.) and the New England Regional Center of Excellence for Biodefense and Emerging Infectious Diseases (US National Institutes of Health, National Institute of Allergy and Infectious Diseases U54 AI057159 to V.A.K.R. and S.K.V.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Rathinam, V., Vanaja, S. & Fitzgerald, K. Regulation of inflammasome signaling. Nat Immunol 13, 333–342 (2012). https://doi.org/10.1038/ni.2237
Published:
Issue Date:
DOI: https://doi.org/10.1038/ni.2237
This article is cited by
-
Gasdermin D in macrophages drives orchitis by regulating inflammation and antigen presentation processes
EMBO Molecular Medicine (2024)
-
Immunosuppressive effects of the mycotoxin patulin in macrophages
Archives of Microbiology (2024)
-
NRF2 transcriptionally regulates Caspase-11 expression to activate HMGB1 release by Autophagy-deficient hepatocytes
Cell Death Discovery (2023)
-
Epithelial IFNγ signalling and compartmentalized antigen presentation orchestrate gut immunity
Nature (2023)
-
Targeting LGSN restores sensitivity to chemotherapy in gastric cancer stem cells by triggering pyroptosis
Cell Death & Disease (2023)
